

Abstract
M6620, a selective ATP-competitive inhibitor of the ATM and RAD3-related (ATR) kinase, is currently under investigation with radiation in patients with non-small cell lung cancer (NSCLC) brain metastases. We evaluated the DNA damage response (DDR) pathway profile of NSCLC and assessed the radiosensitizing effects of M6620 in a preclinical NSCLC brain metastasis model. Mutation analysis and transcriptome profiling of DDR genes and pathways was performed on NSCLC patient samples. NSCLC cell lines were assessed with proliferation, clonogenic survival, apoptosis, cell cycle, and DNA damage signaling and repair assays. NSCLC brain metastasis patient-derived xenograft models were used to assess intracranial response and overall survival. In vivo immunohistochemistry was performed to confirm in vitro results. A significant portion of NSCLC patient tumors demonstrated enrichment of DDR pathways. DDR pathways correlated with lung squamous cell histology; and mutations in ATR, ATM, BRCA1, BRCA2, CHEK1, and CHEK2 correlated with enrichment of DDR pathways in lung adenocarcinomas. M6620 reduced colony formation after radiotherapy and resulted in inhibition of DNA DSB repair, abrogation of the radiation-induced G2 cell checkpoint, and formation of dysfunctional micronuclei, leading to enhanced radiation-induced mitotic death. The combination of M6620 and radiation resulted in improved overall survival in mice compared to radiation alone. In vivo immunohistochemistry revealed inhibition of pChk1 in the radiation plus M6620 group. M6620 enhances the effect of radiation in our preclinical NSCLC brain metastasis models, supporting the ongoing clinical trial (NCT02589522) evaluating M6620 in combination with whole brain irradiation in patients with NSCLC brain metastases.
Keywords: ATR, non-small cell lung cancer, radiation, M6620, VX-970
Introduction
Non-small cell lung cancer (NSCLC) remains the leading cause of cancer-related mortality worldwide (1). Approximately 30% of patients with NSCLC will develop brain metastases during the course of their disease leading to significant morbidity and portend poor prognosis (2,3). Based on a large database series, the median overall survival (OS) of patients with NSCLC with brain metastases is approximately 7 months (4). Radiation therapy plays a major role in the treatment of brain metastases, since cytotoxic chemotherapy has poor penetrance across the blood-brain barrier and only a subset of patients are candidates for oncogene-directed therapy that has effective intracranial activity. Stereotactic radiotherapy and/or whole brain radiotherapy (WBRT) is standard of care as first-line or salvage treatment for many patients with brain metastases (5,6). Despite the use of radiation, many patients have disease progression in the brain and die from their uncontrolled intracranial disease.
Combining drugs to enhance the effect of radiation is one approach to improve the effectiveness of radiotherapy. Currently, there are no approved drugs delivered concurrently with WBRT. Multiple clinical studies investigating radiosensitizers in patients with brain metastases have been performed and have failed to show a benefit (7–11). Some studies have even shown a survival detriment with the addition of drugs to WBRT(10,11). There are multiple reasons for the failure of these studies including lack of preclinical data, the use of inappropriate preclinical models, selecting agents that have poor penetration in the brain, and designing studies with unselective patient populations (12).
One clinical study underway is investigating the safety and effectiveness of M6620 (formerly VX-970 and also known as berzosertib and VE-822) in combination with WBRT in patients with NSCLC brain metastases in the phase I setting (NCT02589522). M6620 is a first-in-class small molecule ATP-competitive inhibitor of ATR (13–15) that has been shown to have penetrance across the blood-brain barrier in pre-clinical studies in GBM (16). M6620 enhances antitumor effects of multiple DNA-damaging agents including radiation, temozolomide, cisplatin, gemcitabine, and irinotecan (14,15,17–20). NSCLC tumors reportedly have significant deficiencies in DNA damage response (DDR) genes (21), therefore they may benefit from ATR inhibition. In this study, we assess the DDR pathway landscape in NSCLC patient tumors and demonstrate a radiosensitizing effect of M6620, using preclinical patient-derived NSCLC brain metastasis models, suggesting the beneficial effect of combining ATR inhibition with radiation in NSCLC brain metastases. These data provide direct support for the ongoing clinical trial (NCT02589522).
Materials and Methods
Mutation Analysis
Mutation profiles from 1,144 NSCLC tumors from The Cancer Genome Atlas (TCGA) (22,23) were queried using the cBioPortal (24,25). A total of 53 genes classified as being related to DDR (21) were assessed. Both known driver mutations determined by the OncoKB annotation database (26) and mutations of unknown significance were included.
Pathway Profiling using Gene Set Variation Analysis
TCGA NSCLC gene expression data was obtained from The University of California Santa Cruz Xena Functional Genomics Explorer (xena.ucsc.edu). The dataset is combined from TCGA lung squamous cell carcinoma and lung adenocarcinoma datasets. Expression data was analyzed using Gene Set Variation Analysis (GSVA)(27) in R 4.0.3 against 12 GO terms for DNA damage response, DNA maintenance, and cell cycle regulation (Supplemental Table 1). Heat maps were then created with the significant pathways with associated metadata in R 4.0.3 using heatmap.3 (https://github.com/obigriffith/biostar-tutorials/blob/master/Heatmaps/heatmap.3.R). Heatmap.3 requires packages gplots (28) and devtools (29). Row and column dendrograms were generated using Euclidean measure to obtain distance matrix and complete agglomeration method for clustering. Expression of the significant pathways between lung squamous cell carcinoma and lung adenocarcinoma can be compared in each of the pathways along with relevant gene mutations and metadata. Statistical analysis of these 12 pathways between squamous cell carcinoma and lung adenocarcinoma and relevant gene mutations was performed using a Welch two sample t-test and the P values were adjusted using the Benjamini-Hochberg method.
Cell lines and drug
Three NSCLC cell lines—A549 derived from a lung adenocarcinoma, and NCI-H226 and NCI-H520 from lung squamous cell carcinomas, were obtained from American Type Culture Collection, tested for mycoplasma, cultured in standard conditions, kept at low passage and their identity confirmed via short tandem repeat testing (Supplemental Table 2). UW-lung-16 cell line was derived from a human lung adenocarcinoma brain metastasis and transfected with luciferase for use in the in vivo intracranial experiments (30). M6620 was purchased from Selleck Chemicals (Houston, TX, USA).
Cell proliferation
Cells were plated in 96-well plates at densities ranging from 2,000 to 5,000 cells per well according to the cell-type growth rate. Twenty-four hours post-plating, cells were treated with indicated doses of M6620 or DMSO control and incubated for 72 h. Once control wells neared full confluence, Cell Counting Kit-8 reagent was added (Dojindo Molecular Technologies) and absorbance measured at 450 nm on a SpectraMax i3 plate reader (Molecular Devices). The absorbance of treated wells was normalized to control wells and the half-maximal inhibitory concentration (IC50) values were calculated.
Western blot analysis
Cells were plated in 10 cm dishes, and treated with either DMSO or 40 nM M6620 after overnight incubation. Mock or 10 Gy irradiation was applied 1 h after DMSO or M6620 treatment, and cells were harvested 4 h or 24 h after radiation or mock treatment. The procedure was performed as described previously (31,32). Specific antibodies and sources are listed in Supplementary Table 3.
Irradiation
Cells were irradiated with a Xstrahl X-ray System, Model RS225 (Xstrahl, UK) at a dose rate of 3.27 Gy/min at 30 cm FSD, tube voltage of 160 kVp, current of 4 mA, and filtration with 3 mm Al ranging from 2–10 Gy. For the subcutaneous tumor experiments, animals were irradiated with a Precision Xray XRAD 320 with 1.14 Gy/min delivered at 320 kV and 12.5 mA at 50 cm FSD with a beam-hardening filter with a half-value layer of 4 mm Cu. The delivered dose rate was confirmed monthly by the ionization chamber. Mice were shielded with custom-built lead jigs to isolate exposure to the rear quarter of the body. For the intracranial tumor experiments, animals were irradiated with an Xstrahl Small Animal Radiation Research Platform (SARRP) (Xstrahl, UK). Mice were anesthetized in a chamber with 3–5% isoflurane at 1–2 L/min O2 and moved to a flatbed with a nosecone within the SARRP. Animals were placed in the prone position with the forelimbs tucked underneath to prevent any unintentionally absorbed dose in the paws. Using the MuriPlan software, a Cone-Beam CT image was acquired with the X-ray tube operating at 60 kV and 0.8 mA with aluminum filtration and a protocol was established for administering a total of 2.5Gy split between two beams at 90 and −90 degrees to the animals’ head to equally affect both hemispheres. A 10×10mm fixed collimator was used to target as much of the brain as possible, while sparing other tissues in the region. Delivery of 2.5 Gy dose for the whole brain irradiation was applied by operating at 220 kV and 13 mA with copper filtration. The dose rate was 2.68 Gy/min.
Clonogenic survival assay
Cells were seeded into 6-well plates at specific densities, incubated overnight, and then irradiated as indicated after 1 h of M6620 or DSMO treatment and media refreshed the next day. Once colonies averaged 50 or more cells (12–20 days) in the control wells, plates were fixed and stained with 1% (w/v) crystal violet in methanol, imaged, and colonies of 50 or more cells were counted. Survival curves were generated after normalizing for the amount of M6620-induced cell death. The clonogenic survival curve for each condition was fitted to a linear quadratic model (Y = e−[A * X + B * X2]) according to a least-squares fit, weighted to minimize the relative distances squared, and compared using the extra sum-of-squares F test. Each point represents the mean surviving fraction calculated from 6 replicates and the assays have repeated at least 3 times; error bars represent the standard error (SE). Radiation dose enhancement factor (DEF) was calculated at 10% survival levels by dividing the mean radiation dose for control conditions by the mean radiation dose after drug exposure. A value > 1.0 indicates enhancement of radiosensitivity.
Cell cycle analysis
Cells were plated in 6-well plates at densities ranging from 200,000–300,000 cells/well depending on the cell type and growth rate. Twenty-four hours post-plating, cells were treated with the indicated dose of M6620 or DMSO control, irradiated with 2 Gy (Xstrahl RS225), trypsinized and collected at 24 h. Cells were spun down at 500 g for 10 min, washed with PBS, spun down again, and a single cell suspension was created in 0.5 mL PBS. 4.5 mL chilled 70% methanol was forcefully added to each tube and cells were stored at 4°C. Twenty-four hours later, cells were centrifuged at 500 g for 10 minutes, and a single cells suspension was created in 1 mL solution of propidium iodide (Molecular probes #p-3566 – 1mg/mL), Triton X-100(0.1%) (Sigma #T9284), and RNase A, DNase and protease-free solution (Thermoscientific #EN0531) in 1X Phosphate-buffered Saline(PBS): for 20 mL total = 100ul TritonX (0.1%) + 1 mL 20X propidium iodide + 100uL RNaseA + 18.8 mL 1X PBS. The samples were analyzed using the Attune NxT flow cytometer, and the cell cycle distribution was calculated using ModFIT software (Verity Software House, Top-sham, ME, USA).
Apoptosis
Apoptosis was measured by using Caspase-Glo 3/7 assay (Promega, Madison WI). Cells were seeded into 96-well plates at defined densities, incubated overnight, treated with DMSO or indicated doses of M6620, and irradiated with 2 Gy or 10 Gy one h later. Thirty h post-treatment, caspase activity was measured by manufacturer’s instruction and luminescence was detected by SpectraMax i3 plate reader (Molecular Devices). Results were reported as relative light units (RLU). As a positive control, 1 uM of staurosporine was added for the last 6 h of treatment.
γH2AX Assay
In vivo and in vitro γH2AX immunofluorescence assays were performed as described previously (31,32). In vitro slides were imaged at 60x magnification using a NiKon A1RS inverted point scanning confocal microscope. γH2AX foci were counted and data presented as number of foci per cell. For each treatment condition, γH2AX foci per cell were counted by a blinded observer in at least 150 cells from three independent experiments.
Mitotic Catastrophe
The presence of fragmented micronuclei was used as the criteria for defining cells undergoing mitotic cell death. Cells were plated in 8-well chamber slides at densities of 7,000 cells/well for each cell line. Twenty-four hours post-plating cells were treated with 40 nM M6620 or DMSO control, incubated for 1 h, irradiated with 2 Gy (Xstrahl RS225), fixed with 100% methanol at indicated time points, and stored in PBS. Cells were then permeabilized with 0.1% Triton X-100 in TBST for 10 min, blocked with SuperBlock Blocking Buffer for 1 h at room temperature, and then cover-slipped with VECTASHIELD Mounting medium with DAPI. The next day, slides were imaged at 40x magnification using an Olympus BX-41 microscope with a Thorlabs Kiralux CMOS scientific camera. In three fields of ≥50 cells, the number of cells were first counted using the DAPI image. Then the number of fragmented micronuclei were counted using the DAPI image. The number of micronuclei per nucleus was then calculated and plotted.
Xenograft growth delay studies
Six- to eight-week old female Hsd:athymic Nude-Foxn1nu mice (Envigo) were used for growth delay studies. Mice were kept in the Association for Assessment and Accreditation of Laboratory Animal Care-approved Wisconsin Institute for Medical Research Animal Care Facility. All animal studies were conducted in accordance with, and with the approval of the University of Wisconsin Institutional Animal Care and Use Committee (IACUC).Animals were housed in specific pathogen-free rooms, and their clinical health was evaluated weekly. Studies involving the mice were carried out in accordance with an animal protocol approved by the University of Wisconsin. Tumors were minced finely in culture media (Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 2.5 μg/mL amphotericin), mixed 1:1 with Matrigel (BD Biosciences), and injected subcutaneously into bilateral flanks of nude mice with an 18-gauge needle. Tumor volume was measured twice weekly with Vernier calipers and calculated according to the relationship . Once the average tumor size reached 200 mm3, mice were randomized into treatment groups. Each experimental group contained 8–10 mice.
In vivo and in vitro immunohistochemistry
Tumors were processed for immunohistochemistry (IHC) as described previously (31). For the quantification of p-Chk1 (Ser345) IHC, images were obtained at 10× magnification and quantified in a blinded manner with ImageJ software (http://rsb.info.nih.gov/ij). The IHC Image Analysis Toolbox Plug tool was first used to identify areas of positive staining (33). Images were converted to the RGB scale, and the threshold tool was used to calculate the percentage area positively stained. For quantification of Cleaved Caspase-3 (CC3), images were obtained at 10× magnification and the number of positive cells were counted per high power field in a blinded manner. Three high power fields were counted and averaged.
Intracranial Implants
Intracranial implants were performed as previously reported (30). UW-Lung-16 cells transfected with luciferase were trypsinized to single cells, and 2×105 cells were suspended in 5 μL of phosphate-buffered saline. Using a Hamilton syringe, the cells were stereotactically injected under sterile conditions into the right striatum of anesthetized athymic nude mice at 1 μL/min at the following coordinates referenced from bregma: 0 mm antero-posterior, +2.5 mm medio-lateral, and −3.5 mm dorso-ventral. The scalp was closed with 6–0 polypropylene suture. Mice were placed on a heating pad in sterile cages and allowed to awaken from anesthesia. Three days after implantation, animals were randomized to either: M6620 alone or M6620 plus irradiation for five consecutive days. M6620 was administered at a dose of 60 mg/kg by daily oral gavage. Radiation was administered as 2.5 Gy daily fractions for 5 days. M6620 was given 1 h before radiation. Radiation was directed as whole brain irradiation. Each experimental group contained ten mice. The day of tumor implantation was assigned as day zero.
In vivo bioluminescence Imaging
Tumor growth in the brain was quantified using IVIS Spectrum bioluminescence imaging system (Perkin Elmer) starting on day 3. Prior to the in vivo imaging, the mice were anesthetized with isoflurane. D-Luciferen firefly/potassium salt solution (150 mg/kg) was injected 18 min before each scan. The IVIS system detects the emitted light with a CCD camera, yielding a quantitative readout expressed as radiance (photon/sec/cm2/sr). Tumor volume was calculated as total flux (photon/s), after the same region of interest (ROI) was automatically segmented on the bioluminescent image of each animal with Living Image software 4.7 (Perkin Elmer).
Statistical analyses
Association of mutated genes and gene transcription pathway analysis was determined with Welch’s two sample t-test. In vitro experiments were repeated three times and statistical analyses were carried out using a Student’s t-test or ANOVA. Extra sum-of-squares F test was used to determine if clonogenic survival curves were statistically different. Data are presented as the mean ± SE. A P-value of < 0.05 was considered significant. All graphs and analyses were performed and graphed using GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA, USA). To analyze the longitudinal xenograft tumor growth volumes, generalized linear mixed models (with a log-link function) were fit to the data using the ‘lme4’ package in R (V 4.0.3) (34). The fixed-effect portion of the models was parameterized such that all main and interactive effects between treatment groups (vehicle, radiation, M6620 and M6620+radiation) and time were estimated, while the random-effect structure accommodated for intra-tumor correlation via random intercepts. Model assumptions (Gaussian random effects and model error) were assessed via a graphical analysis of the model residuals. The synergistic effect between the two treatments was assessed via inference about the three-way interaction between time, M6620, and radiation. For the bioluminescence analysis, a student t-test was used to determine the statistical significance of the total flux of each treatment group at each time point. Kaplan-Meier curves were used for calculating survival for the intracranial experiments and statistical significance was determined by the Gehan-Breslow-Wilcoxon test. Mice were randomized at the beginning of the experiment and the endpoint of the experiment was overall survival. Any mice alive at the end of the experiment without neurologic symptoms were euthanized. These mice were censored in the Kaplan Meier analysis.
Results
Mutational and transcriptomic analysis of DDR genes and pathways was performed to determine the proportion of NSCLC patients who could potentially benefit from ATR inhibition and ATR-induced radiosensitization. Utilizing patient data from the TCGA, we analyzed the mutation status of 53 DDR genes on 1,144 patient NSCLC samples. Overall, 11.8% of patients with adenocarcinoma and 10.8% of patients with squamous cell carcinoma harbored an oncogenic driver mutation in a DDR gene (Fig 1A). The most common mutations were in ATM (3.0%), ATR (1.2%), BRCA1 (0.9%) and BRCA2 (0.8%), indicating that the DDR pathways are negligibly disrupted by mutation (Fig 1B).
Figure 1.

DNA damage and repair mutation and transcriptional analysis in non-small cell lung cancer patients. A, Percent of DDR mutations in lung adenocarcinoma and squamous cell carcinomas. B, Breakdown of mutations in 53 genes DDR genes. C, Heatmap of pathways and molecular processes (GO terms) involved in DNA maintenance and cell cycle regulation activated in DNA damage response. Age, gender, histological subtype is annotated as well as mutation status of important DDR and clinically relevant oncogenic genes.
DDR transcriptome profiling was applied across 980 NSCLC patient tumors from the TCGA dataset. Canonical DDR pathway gene sets curated from Reactome were used as a fair representation of these pathways. Unsupervised hierarchical clustering of patient-level DDR pathway enrichment profiles revealed distinct clusters of patterns of DDR pathway enrichment (Fig 1C). There was a significant correlation between all DDR pathways and squamous cell carcinoma histology (all P values < 0.001; Supplemental Table 4). In addition, mutations in ATM, ATR, BRCA1, BRCA2, CHEK1, and CHEK2 in lung adenocarcinomas correlated with enrichment of eight DDR pathways (all P values < 0.5, Supplemental Table 5). Mutations in ATM, ATR, BRCA1, BRCA2, CHEK1, and CHEK2 did not correlate with enrichment of any DDR pathways in the squamous cell histology (Supplemental Table 6). In the adenocarcinomas, mutations in KRAS also correlated with enrichment of 10 out of the 12 DDR pathways (all P values < 0.5, Supplemental Table 7). Taken together, these data indicate that there is a substantial population of patients with NSCLC that have dysregulation of DDR pathways.
To determine sensitivity to ATR inhibition, a cell viability assay was performed on the three NSCLC cell lines after 48–72 h exposure to varying doses of M6620. Fig. 2 A shows the full dose-response curves with corresponding IC50s for the three cell lines. IC50 proliferation values were in the 1–4 μM range. The ability of M6620 to cause in vitro radiation sensitization was then assessed in these three NSCLC cell lines with clonogenic survival assays. Doses of 40 nM and 80 nM were selected given previous studies demonstrating radiation sensitization (13, 17, 18). Administration of M6620 1 h before radiation significantly decreased the surviving fraction of cells in all three NSCLC cell lines (P = 0.001; Fig. 2 B). The DEF at a surviving fraction of 0.1 for A549, NCI-H226, and NCI-H520 cells exposed to 40 nM and 80 nM of M6620 were 1.59 and 1.65; 1.75 and 1.74; and 2.05 and 2.26, respectively. The impact of M6620 on radiation-induced ATR and ATM was assessed by western blot (Fig. 2C). Phosporylation of ATM (Ser1981) is activated as an early response for radiation and so was activated at 4 h and reduced at 24 h. Addition of M6620 did not show any effect for ATM response. However, M6620 diminished radiation-induced activation of p-ATR (Thr1989) in the cell lines even at 24 h.
Figure 2.

Response to ATR inhibitor M6620 and radiosensitivity effects in three NSCLC cell lines. A, Sensitivity to M6620 determined by proliferation assay in three NSCLC cell lines A549, NCI-H226 and NCI-H520 following varying doses of M6620. B, In vitro radiosensitizing effects of M6620 on A549, NCI-H226, and NCI-H520. Cell cultures were treated with 40 or 80 nM M6620 for 1 h before irradiation and maintained in the medium after irradiation. Colony-forming efficiency was determined 14–21 days later, and survival curves were generated. C, Western blot was used to assess ATR, p-ATR (Thr1989), ATM and p-ATM (Ser1981) in M6620-treated and irradiated cell lines. Cells were pretreated with vehicle (DMSO) or M6620 (40 nM) for 1 h and either collected after (no radiation) or exposed to radiation (10 Gy) at the indicated time points. Points, mean; Bars, SE (n=3).
To address the mechanisms of M6620-induced radiosensitization in vitro, we focused on A549 cells treated with 40 nM of M6620 given 1 h prior to irradiation. Because radiation-induced ATR signaling activates Chk1, leading cells to arrest in S-phase and G2/M phase, flow cytometry was used to determine the cell cycle phase distribution of cells exposed to 40 nM of M6620, radiation (2 Gy), or both M6620 and radiation at 16 h after treatment. The addition of M6620 1 h prior to radiation significantly reduced the numbers of cells in G2/M and shifted cells into G1 (P< 0.01, Fig. 3A) compared to control, M6620 alone and radiation alone. To determine whether apoptosis is involved in the M6620-induced radiosensitization, caspase-3/7 activity was assessed in A549 30 h following M6620 (40 nM or 80 nM), irradiation (2 Gy or 10 Gy), or both M6620 and radiation. There was no increase in apoptosis when M6620 was combined with 2 Gy, indicating that apoptosis is not the central mechanism of radiation-induced cell death at 2 Gy. However, an increase in apoptosis was seen when M6620 (40 and 80 nM) was combined with 10 Gy radiation, compared to radiation alone (Fig. 3B, P <0.001). Apoptosis was confirmed on western blot with increased cleaved-PARP after M6620 and radiation combination therapy (Supplemental Figure).
Figure 3.
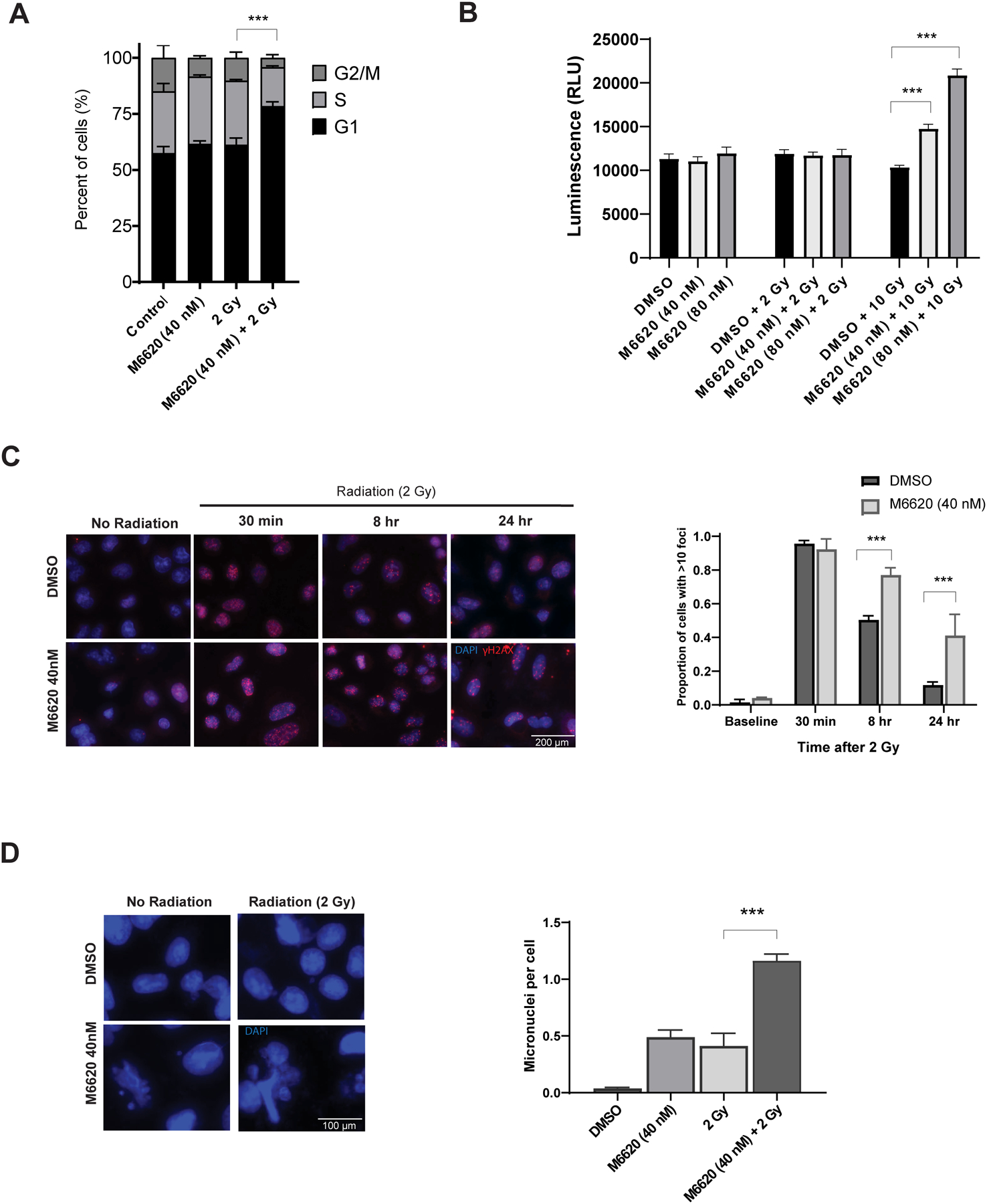
Mechanism of cell death after combined radiation and M6620 treatment in A549 NSCLC cells. A, Cell cycle, B, Apoptosis, C, γH2AX foci and D, mitotic cell death analyses. Cells were treated with vehicle (DMSO), M6620 (40 nM or 80 nM), 2 Gy or 10 Gy, or M6620 1 h prior to radiation and analyzed at designated time points. For γH2AX, cells with >10 γH2AX foci were counted and plotted at the time points following 2 Gy radiation. Representative DAPI stained immunofluorescence images and micronuclei quantification are shown. Columns, mean; bars, SE, n=3. ***, P < 0.001, according to t-test (radiation versus M6620 plus radiation).
To determine whether M6620 inhibits DNA damage repair, γH2AX expression was evaluated in the in vitro setting. The combination of M6620 plus irradiation in A549 cells resulted in a statistically significant increase in the number of γH2AX foci at 8 h (P <0.001) and at 24 h (P = 0.016) compared with cells treated with irradiation alone (Fig. 3C). Since M6620 inhibits DSB repair and did not cause a significant increase in radiation-induced apoptosis in A549 cells at 2 Gy, we hypothesized that M6620-induced radiosensitization involves an enhancement of cells undergoing mitotic catastrophe. Mitotic cell death was determined according to the number of cells with abnormal fragmented micronuclei. A statistically significant increase in the number of cells undergoing mitotic catastrophe after the treatment with irradiation and M6620 was observed at 72 h post-treatment compared to M6620 alone or radiation alone (P ≤ 0.001) (Fig. 3D). These results suggest that the M6620 induced radiosensitization at 2 Gy is mediated by an inhibition of DSB repair, resulting in increased mitotic cell death. Additional analysis did not support necrosis or ferroptosis as mechanisms of radiation induced cell death (Supplemental Figure).
Given our in vitro findings, we examined the ability of M6620 to enhance tumor cell radiosensitivity in an in vivo tumor growth delay setting using two NSCLC brain metastasis PDXs: UW-lung-16 and UW-lung-18. UW-lung-16 harbors KRAS (G12C), TP53, and STK11 mutations, and UW-lung-18 has KRAS (G13C) and TP53 mutations. Mice bearing subcutaneous NSCLC brain metastasis tumors were randomized into four groups: vehicle, M6620, radiation, or M6620 plus radiation. M6620 (60 mg/kg) was given by p.o. gavage 1 h before local tumor irradiation (2 Gy × 10 daily fractions). The combination of M6620 and radiation delayed the growth of UW-lung-16 and UW-lung-18 tumors compared to vehicle control, drug alone, or radiation alone (Fig. 4A and B). In both experiments, the growth delay following the combination treatment was more than the sum of the growth delays caused by individual treatments. In the UW-lung-16 tumors, the slope of the estimated tumor growth curve for the combination treatment group was significantly lower than the slope of the curves for both the radiation treatment group (P <0.0001) and the M6620 only group (P <0.0001). In addition, there was evidence of a synergistic effect between radiation and M6620 (P = 0.002). Similar results were seen for UW-lung-18 tumors as well. The combination treatment group’s estimated growth curve was significantly lower than the radiation only treatment group (P <= 0.0001) and the M6620 only group (p<0.0001). Once again, a synergistic effect between radiation and M6620 was observed (p<0.0001). The DEF, obtained by dividing the normalized tumor growth delay in mice treated with both M6620 and radiation by the absolute growth delay in mice treated with radiation alone, was 1.8 in the UW-lung-16 tumors and 1.4 in the UW-lung-18 tumors.
Figure 4.

The effects of M6620 on radiation-induced patient-derived xenograft tumor growth delay. Tumor growth delay curves of A, UW-Lung-16 and UW-Lung-18 subcutaneous flank xenografts treated with vehicle (5%DMSO+45%PEG300+water), M6620 (60 mg/kg), radiation (2 Gy × 10) or M6620 (60 mg/kg) plus radiation (2 Gy × 10). M6620 was administered daily 1 h before radiation fraction. Points, mean tumor volume in mice after treatment; bars, SE. P < 0.001. IHC analysis of B, UW-Lung-16 and UW-Lung-18 tumors showing H&E, phospho-Chk1 (Ser345), and Cleaved Caspase-3 (CC3) expression and quantification. Tumors were harvested at 8 h following a single dose of vehicle, M6620 (60 mg/kg), radiation (2 Gy) and M6620 plus radiation. The scale bar represents 50 mm and all images are to the same scale. C, In-vivo γH2AX foci analysis after combined radiation and M6620 treatment in UW-Lung-16 and UW-Lung-18. UW-lung-16 and UW-lung-18 tumors were harvested at 8 h following a single dose of vehicle, M6620 (60 mg/kg), radiation (2 Gy) and M6620 plus radiation. γH2AX foci were counted in 50 cells in each section and plotted (n=3). Columns, mean (n=3); bars, SE; ***, P < 0.05 by ANOVA for all graphs (radiation versus M6620 plus radiation).
IHC performed 8 h post-treatment on both UW-lung-16 and UW-lung-18 tumors demonstrated inhibition of phospho-Chk1 (Ser345) with M6620 alone or M6620 plus radiation compared to control and radiation alone (P<0.01, Fig. 4B). In addition, CC-3 as a marker of apoptosis was increased in the combination group compared to the other groups (P<0.05, Fig. 4B). γH2AX foci were also assessed in vivo in both UW-lung-16 and UW-lung-18 tumors 8 h post-treatment (Fig. 4C). There were significantly more foci in the M6620 plus radiation group compared to the other treatment groups in both PDXs (P<0.01).
To further evaluate the clinical potential of the M6620 plus radiation combination, we extended the in vivo observations to a translationally-relevant intracranial metastasis implant xenograft survival model (25). Three days after undergoing intracranial implantation of UW-lung-16 luciferase transfected cells, groups of 10 mice were randomized based on bioluminescence intensity to irradiation alone (2.5 Gy × 5 fractions) or M6620 (60 mg/kg) plus irradiation (2.5 Gy × 5 fractions) groups. Tumors were imaged and total flux was calculated. Fig. 5A shows representative images of tumors pre-treatment at Day 3 and post-treatment. On day 25 post-implant the total flux started to diverge between the radiation alone group and the radiation plus M6620 group. By days 32 and 40 post implant, the difference in bioluminescence was statistically significant (P <0.01) (Fig. 5B). Mice treated with M6620 and radiation had significantly improved survival compared to radiation alone (Fig. 5C). The median survival in the irradiation alone group was 67 days compared to 95 days in the group receiving M6620 and radiation (P = 0.022, by Gehan-Breslow-Wilcoxon test). There was no significant difference in weights between the two groups during and 40 days post-treatment (Fig. 5D).
Figure 5.

The addition of M6620 to radiation improves overall survival in mice bearing NSCLC brain metastases. Mice bearing UW-lung-16 NSCLC brain metastasis tumors were randomized 3 days post implant based on bioluminescence signal to either irradiation (2.5 Gy × 5), or M6620 (60 mg/kg) plus irradiation (2.5 Gy × 5). A single dose of M6620 was delivered as p.o. gavage 1 h before receiving each daily radiation treatment. Each group contained 10 mice. A, Representative bioluminescence imaging of mice post implant, showing pre-treatment day 3 and post-treatment day 11 and day 40. B, Total flux of implanted brain metastases over time. Average total flux between the two groups was statistically significant at day 32 and 40 post implantation (P = 0.01, Students t-test). C, Kaplan-Meier curve showing survival of mice with implanted intracranial UW-lung-16 NSCLC brain metastasis tumors (P = 0.022, Gehan-Breslow-Wilcoxon test). D, Weights between the two treatment groups during and 40 days post-treatment.
Discussion
DDR is regulated by a complex network of genes with both ATM (ataxia-telangiectasia mutated) and ATR (ATM and Rad3 related) kinases playing important roles. ATM is primarily recruited to double-strand breaks (DSBs), whereas ATR is activated predominately by single-strand breaks from replicative stress (35,36). ATR may also be activated by other types of DNA damage including DSBs, base adducts and crosslinks (35). When DNA damage occurs, ATR phosphorylates multiple downstream targets, including CHK1 and the tumor suppressor p53. ATR–CHK1 signaling ultimately leads to G2/M and intra-S checkpoint arrest, replication fork stabilization, and origin firing allowing for DNA repair (35,36).
Many cancer cells have deficiencies in ATM signaling, either by direct loss or silencing of ATM or through the loss of P53, and rely on the ATR repair pathway for survival after DNA damage (37). In NSCLC, reduction or loss of ATM protein expression has been reported to be present in approximately 18–40% (38–40) of tumors, and TP53 is frequently mutated in greater than 50% (22,23). In addition, DDR genes are often mutated in NSCLC and have been associated with improved clinical outcomes in patients with NSCLC treated with programmed death-ligand 1 (PD-L1) blockade (21). In this study, we have found 10% of NSCLC patient tumors harbor an oncogenic mutation in a DDR gene. Focusing on DDR pathways (DNA maintenance, DNA replication, and cell cycle) directly involved in ATR signaling; we found clusters of distinct DDR pathway patterns across a large cohort of NSCLC tumors. These DDR pathways correlated with histology and mutations in ATM, ATR, BRCA1, BRCA2, CHEK1, and CHEK2 in adenocarcinomas.
Given the proportion of patients who develop brain metastases from NSCLC and the challenges in treating these tumors, findings ways to improve the therapeutic ratio of whole brain radiotherapy with molecular-directed therapies remains an ongoing endeavor. The high level of DNA replicative stress found in NSCLC cells and the reliance on ATR to maintain genome stability makes ATR inhibition an attractive target for radiosensitization.
In the current study, we provide preclinical evidence of radiosensitization with the ATR inhibitor M6620 in NSCLC brain metastases. The combination of M6620 with radiation resulted in reduced DNA DSB repair, abrogation of the radiation-induced G2 cell checkpoint, and formation of dysfunctional micronuclei, indicative of mitotic cell death. Using clinically meaningful doses of whole brain irradiation, M6620 combined with radiation delayed tumor growth and improved overall survival in our intracranial NSCLC brain metastasis PDX model compared to radiation alone. These results directly support the ongoing phase I clinical study evaluating M6620 in combination with whole brain irradiation (37.5 Gy in 10 fractions) (NCT02589522). M6620 has already undergone evaluation as monotherapy and in combination with carboplatin or topotecan in two phase I studies and has demonstrated safety and preliminary anti-tumor activity (41,42).
While our data support further clinical evaluation, there remain open questions regarding combining M6620 with radiation in NSCLC brain metastases, including whether all NSCLC tumors will respond and will the combination increase neurotoxicity. It is unclear if M6620 will radiosensitize ATM proficient tumors to the same degree as ATM-deficient tumors. Our data demonstrates preclinical radiosensitization in ATM proficient tumors. We evaluated cell lines and PDXs that were P53-mutated and/or KRAS-mutated. A549 cell line harbors a KRAS mutation and is P53 wild type, while NCI-H520 and NCI-H226 cell lines are both KRAS wild type but harbor TP53 mutations. All three cell lines, which demonstrated in vitro radiosensitization, have also been reported to express ATM and have been defined as ATM-proficient cell lines (39,43–45).
Defining DDR dysregulation in order to exploit ATR inhibition and radiosensitization needs to be further explored. Much of the focus is on ATM however; DDR and ATR pathways are complex and controlled by multiple DNA repair proteins.(46). For example while ATM signaling is mediated by P53, P53 status alone does not predict for the ability of ATR inhibitors to radiosensitize tumors (47,48). The role of KRAS mutations also needs to be further investigated. Studies have shown that KRAS-mutated tumors have an enhanced dependence on ATR functionality for the maintenance of genomic stability (49). We did see correlation between KRAS mutations and enrichment of DDR pathways in our patient analysis. It is also unknown if ATR inhibition will radiosensitize NSCLC brain metastases with EGFR-, ALK-, MET-, or RET-driven tumors. Another limitation of our study is that neurotoxicity was not assessed. It has been proposed that normal cells are able to tolerate ATR inhibition because of low replicative stress levels and the presence of compensatory DNA repair pathways, including the ATM pathway. However, this will need to be studied further as it will be important not to increase the known neurotoxic effects of WBRT in patients.
In summary, we have demonstrated that DDR genes and pathways are altered in NSCLC patient tumors and have shown in vitro and in vivo radiation enhancement by combining the ATR inhibitor M6620 with clinically relevant doses of radiotherapy in a NSCLC brain metastasis preclinical model. These data provide support for the ongoing phase I clinical trial (NCT02589522) evaluating M6620 in combination with whole brain irradiation in patients with NSCLC brain metastases.
Supplementary Material
Funding:
This project was supported in part by grants from the American Cancer Society (RSG-16-091-01-TBG), UW Paul P. Carbone Young Investigator Award and the University of Wisconsin Carbone Cancer Center Support Grant (P30 CA014520), and pilot award from the University of Wisconsin Lung Disease Oriented Team.
Disclosures:
TL: Advisory board, Jazz Pharmaceuticals Inc, AstraZeneca, EMD Serono, Merck, Boehringer-Ingelheim, Blueprint, Debio, Bayer; Consulting: Boehringer-Ingelheim, Genentech, AstraZeneca, Lilly; non-financial disclosure: Steering Committee Mirati SAPPHIRE study
RJK: commercial research grant from Ignyta and is consultant/advisory board relationship member for Galera Therapeutics.
Footnotes
No potential conflicts of interest were disclosed by the other authors.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: A Cancer Journal for Clinicians. 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Waqar SN, Samson PP, Robinson CG, Bradley J, Devarakonda S, Du L, et al. Non-small-cell Lung Cancer With Brain Metastasis at Presentation. Clin Lung Cancer. 2018/03/13 ed. 2018;19:e373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun A, Hu C, Wong SJ, Gore E, Videtic G, Dutta S, et al. Prophylactic Cranial Irradiation vs Observation in Patients With Locally Advanced Non-Small Cell Lung Cancer: A Long-term Update of the NRG Oncology/RTOG 0214 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2019/03/15 ed. 2019;5:847–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sperduto PW, Yang TJ, Beal K, Pan H, Brown PD, Bangdiwala A, et al. Estimating Survival in Patients With Lung Cancer and Brain Metastases: An Update of the Graded Prognostic Assessment for Lung Cancer Using Molecular Markers (Lung-molGPA). JAMA Oncol. 2016/11/29 ed. 2017;3:827–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoyama H, Shirato H, Tago M, Nakagawa K, Toyoda T, Hatano K, et al. Stereotactic Radiosurgery Plus Whole-Brain Radiation Therapy vs Stereotactic Radiosurgery Alone for Treatment of Brain Metastases: A Randomized Controlled Trial. JAMA. American Medical Association; 2006;295:2483–91. [DOI] [PubMed] [Google Scholar]
- 6.Baschnagel A, Wolters PL, Camphausen K. Neuropsychological testing and biomarkers in the management of brain metastases. Radiat Oncol. 2008;3:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips TL, Scott CB, Leibel SA, Rotman M, Weigensberg IJ. Results of a randomized comparison of radiotherapy and bromodeoxyuridine with radiotherapy alone for brain metastases: Report of RTOG trial 89–05. International Journal of Radiation Oncology*Biology*Physics. 1995;33:339–48. [DOI] [PubMed] [Google Scholar]
- 8.Mehta MP, Rodrigus P, Terhaard CHJ, Rao A, Suh J, Roa W, et al. Survival and neurologic outcomes in a randomized trial of motexafin gadolinium and whole-brain radiation therapy in brain metastases. J Clin Oncol. 2003;21:2529–36. [DOI] [PubMed] [Google Scholar]
- 9.Suh JH, Stea B, Nabid A, Kresl JJ, Fortin A, Mercier J-P, et al. Phase III Study of Efaproxiral As an Adjunct to Whole-Brain Radiation Therapy for Brain Metastases. JCO. American Society of Clinical Oncology; 2006;24:106–14. [DOI] [PubMed] [Google Scholar]
- 10.Knisely JPS, Berkey B, Chakravarti A, Yung AWK, Curran WJ, Robins HI, et al. A Phase III Study of Conventional Radiation Therapy Plus Thalidomide Versus Conventional Radiation Therapy for Multiple Brain Metastases (RTOG 0118). International Journal of Radiation Oncology, Biology, Physics. Elsevier; 2008;71:79–86. [DOI] [PubMed] [Google Scholar]
- 11.Sperduto PW, Wang M, Robins HI, Schell MC, Werner-Wasik M, Komaki R, et al. A Phase 3 Trial of Whole Brain Radiation Therapy and Stereotactic Radiosurgery Alone Versus WBRT and SRS With Temozolomide or Erlotinib for Non-Small Cell Lung Cancer and 1 to 3 Brain Metastases: Radiation Therapy Oncology Group 0320. International Journal of Radiation Oncology, Biology, Physics. Elsevier; 2013;85:1312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stone HB, Bernhard EJ, Coleman CN, Deye J, Capala J, Mitchell JB, et al. Preclinical Data on Efficacy of 10 Drug-Radiation Combinations: Evaluations, Concerns, and Recommendations. Translational Oncology. 2016;9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charrier J-D, Durrant SJ, Golec JMC, Kay DP, Knegtel RMA, MacCormick S, et al. Discovery of Potent and Selective Inhibitors of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Protein Kinase as Potential Anticancer Agents. J Med Chem. American Chemical Society; 2011;54:2320–30. [DOI] [PubMed] [Google Scholar]
- 14.Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death & Disease. Nature Publishing Group; 2012;3:e441–e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. Impact Journals; 2014;5:5674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talele S, Mohammad A, Kim M, Burgenske D, Tuma ACM, Sarkaria JN, et al. Abstract 4858: CNS delivery of VX-970: A selective ATR inhibitor for radiosensitization in GBM. Cancer Res. American Association for Cancer Research; 2019;79:4858–4858. [Google Scholar]
- 17.Jossé R, Martin SE, Guha R, Ormanoglu P, Pfister TD, Reaper PM, et al. ATR Inhibitors VE-821 and VX-970 Sensitize Cancer Cells to Topoisomerase I Inhibitors by Disabling DNA Replication Initiation and Fork Elongation Responses. Cancer Res. American Association for Cancer Research; 2014;74:6968–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leszczynska KB, Dobrynin G, Leslie RE, Ient J, Boumelha AJ, Senra JM, et al. Preclinical testing of an Atr inhibitor demonstrates improved response to standard therapies for esophageal cancer. Radiother Oncol. 2016;121:232–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tu X, Kahila MM, Zhou Q, Yu J, Kalari KR, Wang L, et al. ATR Inhibition Is a Promising Radiosensitizing Strategy for Triple-Negative Breast Cancer. Mol Cancer Ther. American Association for Cancer Research; 2018;17:2462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson CB, Noorbakhsh SI, Sundaram RK, Kalathil AN, Ganesa S, Jia L, et al. Temozolomide Sensitizes MGMT-Deficient Tumor Cells to ATR Inhibitors. Cancer Res. American Association for Cancer Research; 2019;79:4331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricciuti B, Recondo G, Spurr LF, Li YY, Lamberti G, Venkatraman D, et al. Impact of DNA Damage Response and Repair (DDR) Gene Mutations on Efficacy of PD-(L)1 Immune Checkpoint Inhibition in Non–Small Cell Lung Cancer. Clin Cancer Res. American Association for Cancer Research; 2020;26:4135–42. [DOI] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014/08/01 ed. 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. Nature Publishing Group; 2012;489:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. American Association for Cancer Research; 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precision Oncology. Wolters Kluwer; 2017;1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, et al. gplots: Various R Programming Tools for Plotting Data [Internet]. 2020. [cited 2021 Mar 9]. Available from: https://CRAN.R-project.org/package=gplots
- 29.Wickham H, Hester J, Chang W, RStudio, R) RC team (Some namespace and vignette code extracted from base. devtools: Tools to Make Developing R Packages Easier [Internet]. 2020. [cited 2021 Mar 9]. Available from: https://CRAN.R-project.org/package=devtools
- 30.Baschnagel AM, Kaushik S, Durmaz A, Goldstein S, Ong IM, Abel L, et al. Development and characterization of patient-derived xenografts from non-small cell lung cancer brain metastases. Scientific Reports. Nature Publishing Group; 2021;11:2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.SenthilKumar G, Fisher MM, Skiba JH, Miller MC, Brennan SR, Kaushik S, et al. FGFR inhibition enhances sensitivity to radiation in non-small cell lung cancer. Mol Cancer Ther. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fisher MM, SenthilKumar G, Hu R, Goldstein S, Ong I, Miller M, et al. Fibroblast Growth Factor Receptors as Targets for Radiosensitization in Head and Neck Squamous Cell Carcinomas. Int J Radiat Oncol Biol Phys. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shu J, Dolman GE, Duan J, Qiu G, Ilyas M. Statistical colour models: an automated digital image analysis method for quantification of histological biomarkers. Biomed Eng Online [Internet]. 2016. [cited 2021 Mar 4];15. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4848853/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bates D, Mächler M, Bolker B, Walker S. Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software. 2015;67:1–48. [Google Scholar]
- 35.Shiotani B, Zou L. ATR signaling at a glance. Journal of Cell Science. The Company of Biologists Ltd; 2009;122:301–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cimprich KA, Cortez D. ATR: An Essential Regulator of Genome Integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cremona CA, Behrens A. ATM signalling and cancer. Oncogene. Nature Publishing Group; 2014;33:3351–60. [DOI] [PubMed] [Google Scholar]
- 38.Villaruz LC, Jones H, Dacic S, Abberbock S, Kurland BF, Stabile LP, et al. ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget. Impact Journals; 2016;7:57714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber AM, Drobnitzky N, Devery AM, Bokobza SM, Adams RA, Maughan TS, et al. Phenotypic consequences of somatic mutations in the ataxia-telangiectasia mutated gene in non-small cell lung cancer. Oncotarget. 2016;7:60807–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petersen LF, Klimowicz AC, Otsuka S, Elegbede AA, Petrillo SK, Williamson T, et al. Loss of tumour-specific ATM protein expression is an independent prognostic factor in early resected NSCLC. Oncotarget. Impact Journals; 2017;8:38326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yap TA, O’Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. JCO. American Society of Clinical Oncology; 2020;38:3195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas A, Redon CE, Sciuto L, Padiernos E, Ji J, Lee M-J, et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J Clin Oncol. 2018;36:1594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lloyd RL, Wijnhoven PWG, Ramos-Montoya A, Wilson Z, Illuzzi G, Falenta K, et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene. Nature Publishing Group; 2020;39:4869–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai M-Y, Dunn CE, Chen W, Kochupurakkal BS, Nguyen H, Moreau LA, et al. Cooperation of the ATM and Fanconi Anemia/BRCA Pathways in Double-Strand Break End Resection. Cell Reports. 2020;30:2402–2415.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng X, Li H, Kornaga EN, Dean M, Lees-Miller SP, Riabowol K, et al. Low Ki67/high ATM protein expression in malignant tumors predicts favorable prognosis in a retrospective study of early stage hormone receptor positive breast cancer. Oncotarget. 2016;7:85798–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang R-X, Zhou P-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduction and Targeted Therapy. Nature Publishing Group; 2020;5:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, et al. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther. 2017;16:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pires IM, Olcina MM, Anbalagan S, Pollard JR, Reaper PM, Charlton PA, et al. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br J Cancer. 2012;107:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grabocka E, Pylayeva-Gupta Y, Jones MJK, Lubkov V, Yemanaberhan E, Taylor L, et al. Wild-Type H- and N-Ras Promote Mutant K-Ras-Driven Tumorigenesis by Modulating the DNA Damage Response. Cancer Cell. 2014;25:243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.