

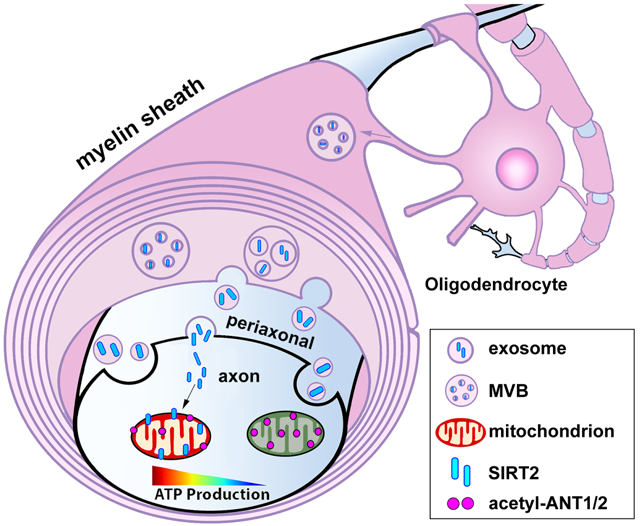
Summary
Neurons require mechanisms to maintain ATP homeostasis in axons, which are highly vulnerable to bioenergetic failure. Here, we elucidate a transcellular signaling mechanism by which oligodendrocytes support axonal energy metabolism via transcellular delivery of NAD-dependent deacetylase SIRT2. SIRT2 is undetectable in neurons but enriched in oligodendrocytes and released within exosomes. By deleting sirt2, knocking down SIRT2, or blocking exosome release, we demonstrate that transcellular delivery of SIRT2 is critical for axonal energy enhancement. Mass spectrometry and acetylation analyses indicate that neurons treated with oligodendrocyte-conditioned media from WT, but not sirt2 knockout mice, exhibit strong deacetylation of mitochondrial adenine nucleotide translocases 1 and 2 (ANT1/2). In vivo delivery of SIRT2-filled exosomes into myelinated axons rescues mitochondrial integrity in sirt2 knockout mouse spinal cords. Thus, our study reveals an oligodendrocyte-to-axon delivery of SIRT2, which enhances ATP production by deacetylating mitochondrial proteins, providing a target for boosting axonal bioenergetic metabolism in neurological disorders.
Keywords: axonal energetics, oligodendrocyte, energy metabolism, myelin, axonal mitochondria, acetylation, sirtuin 2
Graphical Abstract
eTOC Blurb
Neurons require mechanisms to maintain axonal ATP. Chamberlain and Huang et al. identify an exosome-mediated transcellular pathway through which oligodendrocyte-derived sirtuin-2 is delivered to axons, enhancing bioenergetics by deacetylation of mitochondrial proteins for increased ATP generation. Revealing this pathway advances understanding of axonal energy maintenance in health and neurological disorders.
INTRODUCTION
Neurons are highly specialized cells that generate action potentials, drive organelle transport, preserve ionic gradients, and maintain neurotransmission, all of which require significant energetic resources in the form of adenosine triphosphate (ATP) (Devine and Kittler, 2018; Harris et al., 2012; Sheng, 2017). As ATP diffusion is not efficient within long axons that can extend to several feet for some peripheral nerves (Hubley et al., 1996; Sun et al., 2013; Zhou et al., 2016), neurons require mechanisms maintaining local ATP supply in distal axons and synapses (Huang et al., 2021; Li et al., 2020), which are particularly vulnerable to bioenergetic failure clinically relevant to axonal pathology and disease progression in neurodegenerative diseases including Alzheimer’s, Parkinson’s, and Huntington’s diseases, as well as amyotrophic lateral sclerosis and multiple sclerosis (MS) (Camandola and Mattson, 2017; Pathak et al., 2013; Sheng and Cai, 2012; Smith et al., 2019). Thus, revealing mechanisms maintaining axonal energy supply is an emerging frontier for therapeutic investigation.
Approximately 86 billion neurons and 84 billion glial cells (Azevedo et al., 2009) all wire together into intricate networks in the human brain. Thus, a comprehensive model of axonal energy maintenance and regulation must include the contribution of glial cells, including microglia, astrocytes, and oligodendrocytes (OLs) (MacVicar and Choi, 2017; Thorn et al., 2015). OLs serve as myelinating cells surrounding axons of the central nervous system (CNS). Myelination isolates axonal internodes from the extracellular milieu, in contrast to nodes of Ranvier, which are exposed to the extracellular environment (Black and Waxman, 1988; Raine, 1984). This unique structure ideally positions OLs to support axonal energy metabolism beyond their role as passive electrical insulators (Edgar et al., 2010; Edgar et al., 2009; Lappe-Siefke et al., 2003; Luders et al., 2017; Nave and Trapp, 2008; Rasband et al., 2005; Saab et al., 2013; Snaidero et al., 2017). This notion was supported by a study that isolated functions of OLs from myelin sheath insulation via targeted removal of OL cell bodies, revealing axonopathy despite no overt demyelination (Oluich et al., 2012). Demyelinated axons in MS contain significantly more mitochondria (Campbell et al., 2011; Kiryu-Seo et al., 2010; Mahad et al., 2009; Ohno et al., 2014; Witte et al., 2009; Zambonin et al., 2011). Similarly, mice deficient in OL proteolipid protein 1 (PLP1), which causes hereditary spastic paraplegia 2 when mutated, exhibit accumulations of dysfunctional axonal mitochondria despite preserved myelin (Edgar et al., 2004; Griffiths et al., 1998; Klugmann et al., 1997; Yin et al., 2016). These studies highlight a potential link between axon degeneration, energy crisis, and OL dysfunction (Chamberlain and Sheng, 2019; Ettle et al., 2016).
OLs deliver lactate to axons (Funfschilling et al., 2012; Lee et al., 2012; Saab et al., 2016; Trevisiol et al., 2017) and support neuronal activity by delivering energy substrates to axons when neurons are deprived of energy substrates during aglycemia (Meyer et al., 2018). However, whether OLs actively modulate axonal bioenergetics under homeostatic conditions, in which glucose is freely available, remains largely unknown. Signaling pathways that maintain axonal mitochondrial bioenergetics have emerged as crucial regulators of axonal integrity and function. Addressing this gap will fundamentally advance our understanding of how mitochondrial energy metabolism in axons is maintained and boosted in response to changes in local energy demands under physiological and pathological conditions.
In this study, we reveal a transcellular signaling mechanism through which OLs promote axonal mitochondrial ATP production. Using a state-of-the-art ATP sensor in live-imaging assays in microfluidic devices, we demonstrate that OLs increase energy availability within the axonal compartment by altering acetylation of key mitochondrial proteins. NAD-dependent deacetylase sirtuin 2 (SIRT2) is absent in neurons but highly expressed in mature OLs and released within exosomes. Exogenous expression of SIRT2 in neurons enhances axonal ATP level. By deleting sirt2 in mouse, knocking down SIRT2 in OLs, or blocking exosome release from OLs, we consistently demonstrate that transcellular OL-to-axon delivery of SIRT2 enhances axonal ATP availability. Although cell-to-cell mitochondrial transfer has surfaced as a possible avenue for therapeutic development (Torralba et al., 2016), our findings suggest that exosome-mediated transcellular signaling is an efficient and robust mechanism for boosting mitochondrial energetic capacity within axons.
RESULTS
OLs Enhance Axon Mitochondrial Energetics with Neuron Maturation
To address whether OLs play a critical role in enhancing axon energetics independent of myelination, we modeled a unique myelination-independent physiological interaction between neuronal axons and OLs. Primary cortical neurons from embryonic day 18 (E18) mice were cultured in the somatodendritic chamber of microfluidic devices. Due to the physical parameters of the central microgrooves, axonal processes are exclusively capable of growing into the axonal chamber, thereby allowing for physical and fluidic isolation of MAP2-labeled neuronal cell bodies and dendrites from their axons labeled by βIII-tubulin (Figure 1A). Primary OLs isolated from postnatal days 3-5 (P3-5) mice via magnetic activated cell sorting (MACS) were added to the axonal chamber 3-4 days in vitro (DIV3-4) after neuronal plating to establish an OL-axon co-culture system. Mature OLs are labeled by myelin basic protein (MBP) and make contacts with distal axons (Figure 1B). However, these short-term co-cultures do not yield bona fide myelination, offering a platform for studying myelination-independent axonal energetic support.
Figure 1. OLs Enhance Axonal Energetics with Neuron Maturation.

(A) Microfluidic devices separate the somatodendritic chamber (1) from the axonal chamber (2) by the microgroove region (450 μm long) (3). Immunostaining of DIV7 cortical neurons reveals that MAP2-labeled somas/dendrites (green) are restricted to the somatodendritic chamber, while axons labeled with βIII-tubulin (red) extend into axonal chamber.
(B) Immunostaining of axonal chambers culturing YFP-labeled axons alone and co-culturing axons and MBP-labeled OLs (magenta). Cells were fixed for immunostaining at DIV7.
(C, D) Schematic diagram of the GO-ATeam2 ATP sensor (C) and heatmap index (D) depicting ratiometric GO-ATeam2 signal intensity.
(E, F) Images (E) and quantification (F) showing that axons co-cultured with OLs exhibit increased ATP levels compared to axons cultured alone (control).
(G) Quantification of axonal ATP levels (upper) and images (lower) showing that DIV8-11 axons treated with OL-CM for 24 hours exhibit higher ATP levels compared to axon control (Ctrl).
(H) OCR traces during a single Seahorse Extracellular Flux experiment. DIV8 neurons were treated with control media or OL-CM for 24 hours, followed by sequential injections of oligomycin (1μM), FCCP (1.5μM), and Rotenone/AA (0.5μM).
(I-K) Quantification of mitochondrial stress tests revealing that neurons treated with OL-CM exhibit increased basal respiration (I), ATP production (J), and extracellular acidification rate (ECAR, K).
Data were quantified from the total number of axons (F, G) or neuronal wells (I-K) indicated within bars (F, G) or under bars (I-K) from more than three biological replicates and expressed as mean ± SD. Statistical analyses were performed using a two-way ANOVA test with Sidak’s multiple comparisons test (F), an unpaired Student’s t-test (G), or a one-way ANOVA test with Tukey’s multiple comparisons test (I-K). Scale bars, 50 μm (A), 200 μm (B), 5 μm (E, G).
See also Figures S1 and S2.
To characterize the impact of local OLs on axonal energy level, we infected neurons with Forster Resonance Energy Transfer (FRET)-based ATP sensor GO-ATeam2 that emits at 560nm (OFP) when bound to ATP and 510nm (GFP) when free from ATP (Nakano et al., 2011) (Figure 1C). Higher 560nm/510nm ratios indicate higher cytosolic ATP levels and are visually depicted by hotter colored heatmaps throughout this study (Figure 1D). GO-ATeam2 permits real-time measurement of ATP without the necessity of pH monitoring and calibrating. Axons co-cultured with OLs exhibited higher ATP levels at DIV6-7 (P = 0.0001), DIV8-9 (P < 0.0001), DIV10-11 (P = 0.0048), and DIV14-15 (P < 0.0001) compared to isolated axons maintained in identical media (Figures 1E and F). These results lead to our first conclusion: proximal OLs similarly enhanced axonal ATP levels from DIV6-11, while they had a stronger beneficial impact on more mature axons at DIV14-15. It is possible that “aging” axons in vitro undergo energy stress and thus may rely more on bioenergetic support from OLs. To avoid the confounding variable of this “aging effect,” we chose to investigate OL-axon transcellular signaling at younger ages from DIV6-11.
A previous ATP titration demonstrated a non-linear correlation between the FRET/GFP ratio of GO-ATeam2 and ATP levels in vitro (Nakano et al., 2011). We characterized the significance of the FRET/GFP ratio change in neurons by plotting the FRET/GFP ratio vs ATP content measured by a luciferase-based luminescence assay (Yang et al., 2015). Cellular [ATP] was mildly suppressed in neurons with antimycin-A (AA, 1 nM), or elevated by phosphocreatine (PCr, 500 μM) for 24 hours, respectively. A small reduction (12%) or increase (10%) of the FRET/GFP ratio is correlated with a larger (~30%) decline or increase (20%) in ATP content (μM/105 neurons) (Figure S1A and S1B), confirming a non-linear correlation between the FRET/GFP ratio and cellular [ATP] in neurons. We further examined the sensitivity of GO-ATeam2 and showed that the sensor signal effectively conveys relative [ATP] in both axons and somatodendritic regions treated with 5 nM AA or 500 μM PCr for 24 hours (Figures S1C-S1F). As a control, GO-ATeam3, a R122K/R126K mutant of GO-ATeam2, did not respond to changes in [ATP] induced by PCr treatment (Figures S1G and S1H).
To test whether OLs enhance axonal energetics in a contact-independent manner, conditioned media harvested from purified OL cultures was added directly to the axonal chamber for 24 hours. OL-conditioned media (OL-CM) increased axonal ATP levels (P < 0.0001) to the same extent as co-cultured OLs (Figure 1G). We further measured neuronal energetics using Seahorse Extracellular Flux Analysis. By monitoring oxygen consumption rate (OCR) during sequential injections of chemicals targeting the mitochondrial electron transport chain, the Seahorse XF Cell Mito Stress Test is a precise measurement of energy metabolism. Several control experiments were performed to optimize cell density, FCCP concentration, and glucose concentration (Figures S2A-S2F) for robust metabolic readouts in our neuron model. DIV7-10 neurons treated with OL-CM for 24 hours exhibited significantly enhanced metabolism, including basal respiration (P = 0.0002) (Figures 1H and 1I) and mitochondrial ATP production (P < 0.0001) (Figures 1H and 1J). Importantly, neurons pre-treated with 100 nM AA exhibited reduced basal respiration and ATP production. As expected, neurons pre-treated with AA compensated for reduced mitochondrial energy with a robust increase in glycolysis (P < 0.0001), as measured by extracellular acidification rate (ECAR) (Figure 1K). ECAR reflects the sum of glycolytic acidification in the form of lactate and respiratory acidification in the form of carbon dioxide generated in the citric acid cycle (Mookerjee et al., 2015). Thus, a small ECAR increase in neurons treated with OL-CM likely reflects increased oxidative phosphorylation (OXPHOS) rather than a robust increase in lactate production. Together, these results support our second conclusion that OL-secreted factors enhance axonal mitochondrial energetic capacity.
Lactate Does not Mediate the Main Effect of OLs on Enhanced Axonal ATP
We next aimed to identify which components of OL-CM mediate the beneficial effect on axonal energy metabolism. Previous studies suggested that OL-derived lactate is transported into axons, where it is converted into pyruvate as a substrate for mitochondrial OXPHOS (Funfschilling et al., 2012; Lee et al., 2012). We thought to examine if L-lactate mediates the effect of OLs on enhanced axonal ATP level in our co-culture system. If OL-secreted lactate mediates the increase in axonal ATP, then lactate addition to media would be expected to recapitulate the increase in axonal energy level. To our surprise, addition of 10 mM lactate for 24 hours to the axonal chamber of cortical neurons at DIV7 did not alter axonal ATP levels (P = 0.1758) (Figures S3A and S3B). Considering the high level of glucose (25 mM) in the media, we measured axonal ATP levels in DIV8 neurons maintained in glucose-free (0 mM) medium for 3 hr. Indeed, while axons cultured in 0 mM glucose exhibited significantly reduced ATP level compared to controls (P < 0.0001), ATP levels in axons treated with 10 mM lactate was not different from axons cultured in 25 mM glucose (Figures S3C and S3D), demonstrating that neurons utilize lactate as an energy substrate when glucose is depleted. We also tracked axonal ATP levels over 60 min, during which lactate concentration was progressively increased from 2 to 20 mM. No differences in axonal ATP levels were observed between control axons and those supplemented with increased concentration of lactate (Figure S3E). Furthermore, adding 3.2 μM AR-C155858 (AR), a potent inhibitor of lactate transporter, to the axonal chamber for 24 hours had no impact on OL-mediated ATP enhancement (Figure S3F). These experiments suggest that OLs enhance axonal energetics through a different mechanism.
OL-Derived Exosomes Stimulate Axon Mitochondrial ATP Production
Extracellular vesicles (EVs) are membrane-delimited secreted cargos that mediate cell communication critical for neural development, maintenance, and regeneration (Budnik et al., 2016; Kramer-Albers and Hill, 2016). Exosomes are a major group of EVs and are released from the lumen of endosomal multivesicular bodies (MVBs). OL-derived exosomes (OL-EXOs) have been demonstrated to increase neuronal survival, maintain axonal transport in nutrient-deprived neurons, and promote long-term axonal health (Fruhbeis et al., 2013; Fruhbeis et al., 2020). Proteomic analysis of OL-EXOs revealed an enrichment of metabolic enzymes (Frohlich et al., 2014; Kramer-Albers et al., 2007). We chose to investigate if exosomes mediate the effect of OLs on axonal energetic enhancement. OL-EXOs were isolated from OL-CM by ultracentrifugation, followed by gradient fractionation (Chiou and Ansel, 2016). As a negative control, OL-CM was collected from cultured OLs treated with DMSO or GW4869 (EI), a potent inhibitor of neutral sphingomyelinases that blocks exosome biogenesis and release with few off-target effects (Catalano and O'Driscoll, 2020; Essandoh et al., 2015). Exosomes were analyzed for size and concentration via Nanoparticle Tracking Analysis (NTA). Plotting the size distribution revealed a clustering of nanoparticles around 100 nanometers (nm) in OLs treated with DMSO (Figure S4A), which was largely reduced in EI-treated OLs (Figure S4B). Plotting the number of nanoparticles (particles per ml) within binned size ranges along the x-axis (Figure S4C) revealed that the majority of nanoparticles were within the 50-150nm size range, consistent with exosomes (Frohlich et al., 2014). As expected, particle count was robustly reduced when exosomes were isolated from EI-treated OLs. To trace axonal uptake of OL-EXOs, we labeled OL-EXOs with the exosome membrane labeling dye ExoGlow (Sundar et al., 2019) (Figure 2A). Labeled OL-EXOs were incubated with cortical neurons at DIV8 for 2 hours, followed by immunostaining of βIII-tubulin. OL-EXOs were readily detected in somatodendritic regions and axons (Figure 2B), indicating effective uptake of OL-EXOs into neurons.
Figure 2. OL-EXOs Are Internalized into Neurons and Enhance Axonal Mitochondrial Energetics.

(A, B) Imaging of ExoGlow-labeled OL-EXOs (arrows, A) and their uptake by cultured neurons (B). ExoGlow-OL-EXOs (green) were incubated with cortical neurons at DIV8 for 2 hours, followed by immunostaining of βIII-tubulin (red).
(C, D) Neurons treated with OL-EXOs exhibit increased basal respiration (C) and ATP production (D). As a negative control, mitochondrial respiration was blocked by AA (100 nM, 24 hours).
(E, F) Analyses of ATP content (E, μM/105 cells) using luciferase-based assay and FRET-based ATP sensor (F) in DIV8 neurons cultured with OL-EXOs for 24 hours.
(G, H) Heatmap images (G) and quantification (H) of normalized axonal ATP levels in DIV8 axons cultured alone (Ctrl), co-cultured with OLs, co-cultured with OLs in the presence of exosome inhibitor GW4869 (EI, 1 μM) (OL+EI), or cultured in the presence of EI alone.
(I, J) Heatmap images (I) and quantification (J) of normalized axonal ATP levels in DIV8 axons treated with control media (Ctrl) or purified OL-EXOs (EXO) for the time indicated. Data were collected from five biological replicates.
(K, L) Images (K) and quantification (L) of mitochondrial ATP levels within the inner mitochondrial matrix using mitochondria-targeted ATP sensor GO-ATeam2-Mito. DIV8 neuronal axons were incubated with OL-EXOs for 24 hours.
Data were quantified from the total number of neuronal wells (C-E) or the total number of neuron (F) or axon images (H, J, L) indicated within or above bars from more than three biological replicates and expressed as mean ± SD. Statistical analyses were performed using a one-way ANOVA test with Tukey’s multiple comparisons test (C, D, H) or Dunnett’s multiple comparisons test (E, F), a two-way ANOVA test with Sidak’s multiple comparisons test (J), or an unpaired Student’s t-test (L). Scale bars, 10 μm (A, B, K), 5 μm (G, I).
See also Figures S3 and S4.
To determine whether OL-EXOs are sufficient to enhance neuronal energy metabolism, we conducted a Seahorse XF Cell Mito Stress Test. When compared with controls, DIV7-9 neurons treated with OL-EXOs for 24 hours exhibit enhanced bioenergetic metabolism, including basal respiration (P < 0.0001) and ATP production (P = 0.0002) (Figures 2C and 2D). As expected, pre-treating neurons with mitochondrial complex III inhibitor AA (100 nM) abolished basal respiration and mitochondrial ATP production, and increased ECAR (Figures S4D and S4E). To confirm enhanced ATP production following incubation of OL-EXOs, we monitored neuronal ATP levels using both luciferase-based luminescence assay and FRET-based assay. A 24-hour incubation with OL-EXOs increased both ATP content (μM/105 neurons) (P = 0.0134) and the FRET/GFP ratio (P < 0.001) (Figures 2E and 2F). As a negative control, treatment with AA (5 nM) depleted ATP content (P = 0.0019) and reduces the FRET/GFP ratio (P < 0.001). Thus, both assays display a correlated increase or decline in neuronal ATP levels, thus supporting the notion that OL-EXOs are sufficient to enhance neuronal energetics.
As both Seahorse analysis and luminescence assay do not allow for measurement of local axonal energy metabolism, we instead assessed axonal ATP levels by conducting live imaging of GO-ATeam2 using the microfluidic chamber paradigm. EI (1 μM) was added to the axon/OL co-culture chamber for 24 hours to block exosome biogenesis and release. Consistently, OLs significantly enhanced axonal ATP level (P < 0.0001) compared to controls (Figures 2G and 2H). However, this effect was abolished by EI treatment (P = 0.9741, OL+EI vs Ctrl), suggesting that biogenesis or release of exosomes from OLs is integral to the enhancement of axonal energy metabolism. Importantly, EI treatment had no effect on the energy level of axons cultured alone (P = 0.9376, EI vs Ctrl). We next examined if direct application of purified exosomes was sufficient to increase axonal ATP level. Isolated OL-EXOs were added to the axonal chamber of microfluidic cultures for 30 min, 2 or 3 hours on DIV7-8. While no difference was observed at 30 min (P = 0.8388), axons treated with OL-EXOs for 2 (P = 0.0078) or 3 hours (P < 0.0001) exhibited enhanced ATP levels compared to controls (Figures 2I and 2J).
To determine whether the observed energetic effect was mediated by enhanced axonal mitochondrial function, we performed two lines of experiments. First, we used the mitochondria-targeted GO-ATeam2-Mito, which harbors a sequence of cytochrome c oxidase subunit 8 (COX8), to monitor ATP synthesis within mitochondrial matrix following incubation with OL-EXOs. The ratiometric intensity was significantly increased after OL-EXO incubation for 24 hours (P < 0.001) (Figures 2K and 2L), suggesting enhanced mitochondrial ATP synthesis. Second, we measured mitochondrial membrane potential (ψm), a driving force of ATP synthesis. CMTMRos, a fluorescent dye that accumulates inside mitochondria as a function of ψm, was utilized as a readout. OL-EXOs were added to the axonal chamber of DIV7-9 cultures for 24 hours, followed by loading CMTMRos (20nM) for 30 min, similar to our previous studies (Lin et al., 2017; Zhou et al., 2016). Live imaging revealed increased ψm in axons treated with OL-EXOs (P < 0.0001) compared to control (Figures S4F and S4G). These results support our third conclusion: OL-EXOs are both sufficient and necessary to enhance neuronal mitochondrial ATP production.
Sirtuin-2 Is Enriched in OLs and Released within Exosomes
To identify candidate components of OL-EXOs in regulating neuronal mitochondrial energetics, we revisited the proteomic array of OL-EXOs (Kramer-Albers et al., 2007). Among several enzymes involved in mitochondrial energy metabolism, we chose to investigate NAD-dependent deacetylase sirtuin 2 (SIRT2) based on the following five rationales. First, SIRT2 is enriched in OL-EXOs (Kramer-Albers et al., 2007). Second, an RNA-sequencing transcriptome database (https://www.brainrnaseq.org/) of mouse cerebral cortex demonstrated SIRT2 expression at ∼40X higher in OLs compared to neurons (Zhang et al., 2014). Relatively low neuronal SIRT2 suggests a physiological impact on neuronal energy metabolism upon delivery of OL-derived SIRT2 to axons. Third, in contrast to cytosolic distribution of SIRT2 (Michishita et al., 2005; North et al., 2003), recent immunogold transmission electron microscopic (iTEM) images of mouse brains showed that SIRT2 also localizes to mitochondria, where it deacetylates mitochondrial proteins (Liu et al., 2017). Fourth, major enzymes involved in cellular energy metabolism are regulated by deacetylation of lysine residues (Wang et al., 2010; Zhao et al., 2010). It was estimated that ∼63% of mitochondrial proteins contain lysine acetylation sites (Baeza et al., 2016) and 35% of mitochondrial proteins are acetylated (Anderson and Hirschey, 2012). Finally, mitochondrial acetylation is a known regulator of ATP production; mice lacking sirt2 display reduced cellular ATP levels and increased oxidative stress (Liu et al., 2017). These findings prompted us to propose a working model: transcellular delivery of SIRT2 from OLs to axons via exosomes enhances energetic metabolism by deacetylation of mitochondrial proteins.
To begin testing our working model, we first assessed relative SIRT2 expression in cortical neurons and OLs by immunostaining of cortical cultures at DIV7. While hardly detectable in βIII-tubulin-labeled neurons, SIRT2 is abundantly expressed in MBP-labeled OLs (Figure 3A). Next, we examined developmental expression of SIRT2 in mouse cortical homogenates. SIRT2 expression starts at postnatal day 14 (D14), coinciding with MBP expression during postnatal myelination (Figures 3B and 3C). In white matter-enriched cervical spinal cord, SIRT2 and MBP expression are substantially increased 7 days (D7) after birth (Figure 3D and 3E), consistent with earlier myelination of spinal cord compared to cortex (Foran and Peterson, 1992) and high expression of SIRT2 in OLs (Li et al., 2007).
Figure 3. SIRT2 Is Undetectable in Neurons but Enriched in OLs and Released within Exosomes.

(A) Selective expression of SIRT2 in OLs but not in neurons. Mouse cortical cells at DIV7-8 were co-immunostained for SIRT2 (green), myelin basic protein (MBP, magenta), and neuron-specific βIII-tubulin (red).
(B, C) Immunoblots (B) and bar graph (C) showing development-associated expression of SIRT2 and MBP in mouse brain cortex. Brain cortical tissues were isolated from mice at indicated postnatal days (D) or months (M) of age. 10-μg homogenates were loaded and immunoblotted with the indicated antibodies (n = 2).
(D, E) Immunoblots (D) and bar graph (E) showing development-associated expression of SIRT2 and MBP in mouse cervical spinal cords. 10-μg homogenates were loaded and immunoblotted with the indicated antibodies (n = 2).
(F, G) Immunogold electron micrographs showing SIRT2-labeled MVBs (arrows) in both in vitro and in vivo OLs. Cultured primary OLs at DIV5 (F) or mouse T4 spinal cord dorsal white matter
(G) were labeled by anti-SIRT2 immunogold particles. Note a SIRT2-containing MVB in the adjacent myelin sheath (G).
(H) SIRT2-filled exosomes are released from WT but not sirt2 KO OLs. Purified OL-EXOs were co-immunostained with antibodies against SIRT2 (red) and exosome marker HSP70 (green). Right panels show an enlarged exosome.
Scale bars, 25 μm (A), 200 nm (F, G), 5 μm (H) and 500 nm (H, enlarged boxes).
See also Figure S5.
Next, we aimed to build upon the proteomic findings of Kramer-Albers et al (2007) by analyzing SIRT2 distribution in OL-EXOs at the ultrastructural level. Using anti-SIRT2 iTEM, we observed SIRT2 expression within primary OLs at DIV5. Importantly, SIRT2 puncta were visible within MVBs (Figure 3F), where exosome biogenesis occurs via ESCRT-mediated invagination of endosomal membranes to form intraluminal vesicles (Hessvik and Llorente, 2018). MVBs have been observed in non-compacted myelin sheath directly adjacent to axons, serving as the pathway for exosome release into the periaxonal space (Fruhbeis et al., 2013). We confirmed SIRT2-labeled MVBs located in the adjacent myelin sheath in OLs from mouse spinal cord dorsal white matter (Figure 3G). To validate the specificity of the anti-SIRT2 antibody, we obtained a sirt2 knockout (KO) mouse strain (Sirt2tm1.1Fwa; The Jackson Laboratory) in which exons 5-6 and part of exon 7 were replaced. SIRT2 immunostaining was robust in DIV10 MBP-labeled OLs isolated from WT, but not sirt2 KO mice (Figure S5A). Western blot analysis also confirmed lack of SIRT2 in mature sirt2 KO OLs (Figure S5B).
To determine if SIRT2 is present within exosomes following their release into the extracellular environment, exosomes were purified from conditioned media harvested from DIV1-5 primary WT and sirt2 KO OLs and co-labeled with antibodies against SIRT2 and heat shock protein 70 (HSP70), a protein frequently packaged within exosomes and identified in a proteomic array of OL-EXOs (Kramer-Albers et al., 2007). Confocal images demonstrated that the majority (62.56%) of HSP70-labeled exosomes (n = 195) released from WT OLs contain SIRT2 (Figure 3H). As a negative control, SIRT2 was not detected in a total of 207 HSP70-positive exosomes released from sirt2 KO OLs. Exosome size in fluorescent imaging (diameter ∼300 nm) was consistent with a previous report (Mondal et al., 2019). Altogether, these data indicate that SIRT2 is highly expressed in OLs and released within exosomes, thus supporting our model of transcellular delivery of SIRT2 from OLs to axons.
Increased SIRT2 Expression in Neurons Is Associated with Enhanced ATP Supply
Given that SIRT2 is undetectable in neurons, we asked whether elevating SIRT2 expression in neurons contributes to enhanced ATP production by co-transfecting DIV4 neurons with GFP and Flag-tag (vector) or SIRT2-Flag. Immunostaining revealed robust exogenous SIRT2 expression in GFP-labeled transfected neurons, while SIRT2 was undetectable in control neurons expressing GFP alone (Figure 4A). Live imaging of the ATP probe at DIV7-8 revealed enhanced somatic (P < 0.0001) and axonal (P = 0.0011) ATP levels in neurons overexpressing SIRT2 (Figures 4B-4E). These findings support our fourth conclusion that elevated neuronal SIRT2 is sufficient to enhance ATP supply.
Figure 4. Elevated SIRT2 Expression in Neurons Increases ATP Production.

(A) Images showing exogenous expression of SIRT2 (red) in cortical neurons at DIV7. Neurons were co-transfected with GFP and Flag-tag (vector) or SIRT2-Flag at DIV4. Note that SIRT2 is undetectable in control neurons but overexpressed in SIRT2-transfected neurons (SIRT2 OE).
(B-E) Representative images and quantification showing relative ATP levels in neuronal somas (B, D) and axons (C, E) in DIV7-8 neurons co-transfected with GO-ATeam2 and Flag-tag (vector) or SIRT2-Flag at DIV4.
Data were quantified from the total number of somas (D) or axons (E) indicated in the bars from three biological replicates and expressed as mean ± SD. Statistical analyses were performed using an unpaired Student’s t-test. Scale bars, 20 μm (A, B), 5 μm (C).
SIRT2-deficient OLs Fail to Enhance Axonal ATP
To test whether OL-derived SIRT2 plays a key role in enhancing axonal energetics, we depleted SIRT2 in OLs with small interfering ribonucleic acid (siRNA; Invitrogen™). Target specificity of SIRT2-siRNA was confirmed utilizing an siRNA-resistant SIRT2. SIRT2-siRNA effectively depleted SIRT2 (P = 0.0001), but not siRNA-resistant SIRT2 mutant (Figures S5C-S5E). To test whether SIRT2-deficient OLs fail to boost axonal ATP levels, SIRT2 was knocked down in OLs at DIV1; immunostaining after 72 hours transfection revealed a significant reduction in SIRT2 expression in OLs transfected with SIRT2-siRNA compared to Ctrl-siRNA (P < 0.0001) (Figures 5A and 5B). Live imaging revealed that axons co-cultured with control OLs (Ctrl-siRNA) display increased axonal ATP levels (P = 0.0033) (Figures 5C and 5D). In contrast, axons co-cultured with SIRT2-deficient OLs (SIRT2-siRNA) exhibit ATP levels comparable to axons maintained alone (P = 0.2254). Thus, proper SIRT2 expression in OLs is required for the transcellular signaling that mediates the enhancement of axonal ATP supply.
Figure 5. SIRT2-Deficient OLs Abrogates Axonal ATP Increase.

(A, B) Images (A) and quantification (B) showing relative SIRT2 expression in DIV4 OLs transfected with Ctrl-siRNA or SIRT2-siRNA for 72 hours. Intensity of SIRT2 immunolabeling (red) was normalized to MBP (green).
(C, D) Heatmap images (C) and quantification (D) of normalized ATP levels from DIV7-8 axons cultured alone (Ctrl), co-cultured with OLs transfected with Ctrl-siRNA or SIRT2-siRNA.
(E) Representative DIC images demonstrating that sirt2 WT and KO OLs are morphologically similar, grow extensively in the axonal compartment, and make contacts with axons (green).
(F) Particles per milliliter (ml) detected during Nanosight tracking analysis (NTA) of exosomes purified from WT and sirt2 KO OLs cultured at the same density. Note that OLs release similar exosomes per ml regardless of sirt2 deletion.
(G, H) Heatmap images (G) and quantification (H) of normalized ATP levels from DIV7-10 axons cultured alone (Ctrl), co-cultured with WT or sirt2 KO OLs.
Data were quantified from the total number of cells (B), axons (D, H), or Nanosight recordings (F) indicated in or above bars from two (B) or three (D, F, H) biological replicates and expressed as mean ± SD. Statistical analyses were performed using an unpaired Student’s t-test (B) or a one-way ANOVA test with Tukey’s multiple comparisons test (D, F, H). Scale bars, 20 μm (A), 10 μm (C), 25 μm (E), 5 μm (G).
See also Figure S5.
Acute siRNA knockdown may potentially impact OL health status. We alternatively co-cultured cortical neuronal axons with primary OLs isolated from WT or sirt2 KO mice. Deleting sirt2 did not affect overall morphology of primary OLs or their growth with axons in microfluidic devices (Figure 5E), or exosome biogenesis and release (Figure 5F). While co-culture with WT OLs enhanced axonal ATP (P < 0.0001) (Figures 5H and 5G), co-culture with sirt2 KO OLs abolished enhancement of axonal ATP level (P = 0.9729). Altogether, studies from SIRT2 knockdown and sirt2 deletion support our fifth conclusion that OL-derived SIRT2 boosts axonal energy metabolism.
OL-Derived SIRT2 Induces Deacetylation of Neuronal Mitochondrial Proteins
As SIRT2 exhibits robust deacetylase activity (Feldman et al., 2013), and deacetylation is associated with enhanced mitochondrial ATP production capacity (Liu et al., 2017; Nie et al., 2011), we hypothesized that mitochondrial ATP levels would be higher in axons co-cultured with OLs compared to axons cultured alone. To test this, neurons were plated in microfluidic devices and infected with lentiviruses expressing mitochondria-targeted GO-ATeam2-Mito. Enhanced mitochondrial ATP levels were observed in axons co-cultured with WT OLs (P = 0.0013), but not with sirt2 KO OLs (P = 0.9442), compared to axons cultured alone (Figures 6A and 6B). As expected, axonal mitochondrial ATP levels were significantly reduced after adding AA (5 nM) for 24 hours (Figure S6A and S6B). These results suggest that OL-derived SIRT2 stimulates mitochondrial ATP production capacity in axons, likely via deacetylation of mitochondrial proteins.
Figure 6. SIRT2 Regulates Axonal Energetics by Deacetylation of Mitochondrial Proteins.

(A, B) Images (A) and quantification (B) of the mitochondrial-targeted ATP sensor GO-ATeam2-Mito, reflecting ATP levels in the inner mitochondrial matrix, in DIV8 neuronal axons cultured alone (Ctrl), co-cultured with WT or sirt2 KO OLs.
(C) Experimental workflow depicting mitochondrial fractionation from DIV7 cortical neurons for detection of acetylated peptides via mass spectrometry (MS).
(D) Graphical depiction of MS revealing the categorization of a total of 446 mitochondrial proteins detected.
(E, F) Immunoblot (E) and normalized ratios of acetylation signal (F) showing a reduced acetylation of mitochondrial proteins in neurons treated with OL-CM for 24 hours. Mitochondria were prepared by fractionation and acetylated proteins were detected with an anti-Pan-acetylation antibody. Acetylated signal was calibrated with TOM40 levels and normalized to neurons treated with control media (n = 9).
(G, H) Immunoblots (G) and quantification (H) of acetylation assay of 5 mitochondrial proteins as indicated. Neurons at DIV8 were treated with control media (Ctrl), or OL-CM derived from WT or sirt2 KO mice for 24 hours, then lysed and incubated with Acetyl-Lysine affinity beads and analyzed by immunoblots with the indicated antibodies. The protein of interest was normalized to IgG level and subsequently normalized to control group (n = 3). Note that acetylation levels of ANT1 and ANT2 are largely reduced by OL-derived SIRT2.
Data were quantified from the total number of axons (B) indicated in bars and statistical analysis was performed on data collected from three or nine biological replicates using paired Student’s t-test (F) or one-way ANOVA test with Tukey’s multiple comparisons test (B, H). Scale bars: 5 μm.
We next characterized mitochondrial protein acetylation in cortical neurons according to the workflow (Figure 6C). Neurons at DIV7 were lysed and differential centrifugation was utilized to isolate the mitochondrial fraction, which was subjected to liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis. Of a total of 2229 proteins identified in the mitochondrial fraction, 446 mitochondrial proteins exhibit the strongest enrichment score (113.75) (Figure 6D, S6C and S6D). Further mass spectrometry analysis revealed 9 mitochondrial proteins associated with a minimum of one peptide acetylated on lysine residues (Figure S6E, Table S1). Thus, these proteins were considered candidates for deacetylation through transcellular delivery of SIRT2.
To test if OLs affect post-translational acetylation, mitochondria were isolated from DIV8-9 neurons pretreated with control media or OL-CM for 24 hr. Relative purity of mitochondrial fractionation was confirmed by western blot analysis (Figure S6F). Total lysine acetylation was analyzed with an anti-Pan-acetylation antibody. Neurons pretreated with OL-CM exhibited reduced acetylation of mitochondrial proteins (P = 0.0006) compared to control (Figure 6E and 6F). Next, we performed in vitro affinity enriched acetylation assay. Neurons at DIV8 were treated with control media, OL-CM from WT or sirt2 KO OLs for 24 hours before immunoprecipitation with Acetyl-Lysine affinity beads coated with a mixture of two anti-acetyl lysine antibodies (Clones 7B5A1 and 10C4B2.1). Based on our MS results, we chose three acetylated mitochondrial proteins, adenine nucleotide translocase 1 and 2 (ANT1 and ANT2) and Prohibitin-2 (PHB2), as candidates for deacetylation through transcellular delivery of OL-derived SIRT2. ANT1 and ANT2 (also known as Slc25a4 and Slc25a5) are mitochondrial carrier proteins that exchange ADP/ATP across the inner mitochondrial membrane for oxidative phosphorylation. Strong deacetylation of both ANT1 and ANT2, but not PHB2, was observed in neurons pretreated with OL-CM from WT mice (P = 0.005 and P = 0.005, respectively) (Figures 6G and 6H). Such deacetylation was abolished when neurons were pretreated with OL-CM from sirt2 KO mice (P = 0.9842 and P = 0.2931, respectively). However, two acetylated proteins, ATP synthase subunit alpha (ATP5A) and NADH dehydrogenase 1 alpha subcomplex subunit 5 (NDUFA5) (Liu et al., 2017), failed to be deacetylated by OL-derived SIRT2; it is likely that mitochondrial proteins are deacetylated by different members of the sirtuin family (Cheng et al., 2016). These acetylation data support the notion that OL-derived SIRT2 modulates neuronal mitochondrial energetic capacity by selective deacetylation of ANT1 and ANT2. Our iTEM observations in OLs revealed a strong association of SIRT2 with mitochondria, in addition to its cytosolic distribution (Figure S6G). ANT1 and ANT2 are acetylated at Lys10 and ANT1 is also acetylated at Lys23 (Table S1). Interestingly, deacetylation of Lys23 robustly enhances ANT1 for ADP binding affinity, thus facilitating conversion of ADP to ATP (Mielke et al., 2014). These findings support that OL-derived SIRT2 regulates axonal mitochondrial energetics by deacetylating key mitochondrial proteins.
In vivo Delivery of OL-EXOs Rescues Axonal Mitochondrial Integrity in sirt2 KO Mice
We validated this transcellular signaling in myelinated axons of mouse spinal cord dorsal white matter by examining mitochondrial membrane potential (ψm) as a readout of bioenergetic capacity following delivery of OL-EXOs. We first examined mitochondrial density in WT and sirt2 KO mouse spinal cord by immunostaining of axonal mitochondrial marker syntaphilin (SNPH) (Lin et al., 2017). SNPH intensity was not different in WT and sirt2 KO dorsal white matter (P = 0.7479, Figures 7A, 7B), We next compared axonal mitochondrial ψm in WT and sirt2 KO spinal cord. The mitochondrial ψm dye CMTMRos and myelin sheath dye FluoroMyelin™ Green were co-injected (Figures 7C, 7E, S7A, S7B). CMTMRos signal intensity within axon bundles was significantly higher in WT mice compared to sirt2 KO mice (P < 0.001, Figures 7D, 7F, 7K), suggesting reduced bioenergetic capacity in axonal mitochondria of sirt2 KO mouse spinal cords, consistent with a study showing that loss of sirt2 decreases mitochondrial ATP production (Liu et al., 2017).
Figure 7. In vivo Delivery of WT but not sirt2 KO OL-EXOs Increases Axonal Mitochondrial Integrity in sirt2 KO Mouse Spinal Cord by Deacetylating ANT1/2.

(A, B) Representative images (A) and quantitative analysis (B) showing similar density of axonal mitochondria labeled by SNPH in spinal cord dorsal white matter of WT and sirt2 KO mice (P = 0.7479, n = 81 or 82 images from 5 pairs of mice at P60-70).
(C-F, K) Schematic injection of the dye cocktail (mitochondrial potential dye CMTMRos and Myelin Green, C, E), representative images of myelinated axons (D, F), and quantification (K) showing relative mitochondrial membrane potential in spinal cord dorsal white matter of WT and sirt2 KO mice at P60-70. Note that axonal mitochondrial integrity, reflected by CMTMRos intensity, significantly declines in the spinal cord dorsal white matter of sirt2 KO mice compared to WT mice (P < 0.001, n > 60 images from 6 pairs of mice) (K).
(G-J, K) Schematic co-injection of the dye cocktail and purified OL-EXOs (G, I), representative images of myelinated axons (H, J), and quantification (K) showing rescued mitochondrial membrane potential in sirt2 KO spinal cord dorsal white matter by in vivo delivery of purified exosomes released from WT, but not from sirt2 KO, mouse OLs.
(L, M) Schematic (L) and representative immunoelectron micrographs (M) showing SIRT2-labeled vesicles readily detected at periaxonal sites in myelinated axons of spinal cord dorsal white matter from WT mouse. Insets depict enlarged views of the boxed areas. M1: a SIRT2-labeled MVB targets the inner OL adaxonal cytoplasmic loop in close proximity to the axon; M2, M3: SIRT2-labeled exosomes contact the axonal surface within the periaxonal space; M4, M5: SIRT2 vesicles undergo internalization into axons.
(N) Representative immunoelectron micrographs showing delivery of exosomal SIRT2 into sirt2 KO myelinated axons of spinal cord dorsal white matter following injection of WT OL-EXOs. Arrows point to mitochondrial targeted SIRT2.
(O, P) Representative immunoblots (O) and quantification (P) of in vivo acetylation assay of mitochondrial proteins in sirt2 KO mouse spinal cord dorsal white matter after injection of OL-EXOs released from WT or sirt2 KO mice. The white matter-enriched area around the injection site was collected and lysed, mixed with Acetyl-Lysine affinity beads, and then analyzed by immunoblot with the indicated antibodies. Protein levels were normalized to those in non-injected sirt2 KO group. Note that acetylation levels of ANT1 and ANT2 were decreased by injection of WT OL-EXOs, but not sirt2 KO OL-EXOs (n = 4 mouse replicates).
Data were quantified from the total number of images indicated under the bar graphs, expressed as mean ± SEM, and analyzed by an unpaired Student’s t-test (B) or one-way ANOVA test with Tukey’s multiple comparisons test (K, P). Scale bars, 10 μm (A, D, F, H, J); 200 nm (M, N).
See also Figure S7.
To confirm OL-EXO uptake in vivo, we injected ExoGlow-labeled OL-EXOs into mouse spinal cord dorsal horn (Figures S7A and S7B). Due to the broad emission spectrum of ExoGlow dye, co-labeling with myelin dyes is not feasible. Instead, we immunostained SNPH as an axonal marker. After injection for 30 min, labeled OL-EXOs (green) were readily found in the myelinated axon bundles and cell bodies within white matter-enriched dorsal funiculus area (Figure S7C). We next examined whether in vivo delivery of OL-EXOs rescues axonal mitochondrial deficiency in sirt2 KO mice. OL-EXOs were isolated from conditioned media of WT or sirt2 KO mouse OL cultures on DIV7 and purified by gradient ultracentrifugation. OL-EXOs (5 μl) were mixed with CMTMRos and MyelinGreen and co-injected into T2 spinal cord (Figures 7G and 7I). Injection of WT OL-EXOs, but not sirt2 KO OL-EXOs, efficiently enhanced CMTMRos signal intensity within axon bundles of spinal cord dorsal white matter in sirt2 KO mice (P = 0.002, Figures 7H, 7J, 7K).
To provide in vivo ultrastructural evidence showing SIRT2 delivery into myelinated axons, we performed iTEM analyses. The periaxonal space has been reported as the predominant site for myelinated axons to internalize exosomes (Figure 7L) (Fruhbeis et al., 2013; Fruhbeis et al., 2020). SIRT2-labeled vesicles were readily found at periaxonal sites in myelinated axons and likely delivered into axons via the following steps: (1) SIRT2-MVBs target the inner cytoplasmic loop close to the axon (periaxonal or adaxonal space) to release exosomes; (2) exosomes contact the axonal surface within the periaxonal space; and (3) exosomes undergo internalization into axons (Figure 7M). We further showed in vivo delivery of exosomal SIRT2 into sirt2 KO myelinated axons following injection of WT OL-EXOs (Figure 7N). Such SIRT2 immunogold particles could not be observed before injection of OL-EXOs (Figures S7D and S7E).
To confirm SIRT2-mediated deacetylation in vivo, we performed an acetylation assay by injecting WT or sirt2 KO OL-EXOs into spinal cord dorsal horn of sirt2 KO mice for at least 30 min before tissue collection for immunoprecipitation with Acetyl-Lysine affinity beads. Both ANT1 and ANT2 showed declined acetylation levels in white matter-enriched tissues after injecting WT OL-EXOs (P = 0.0024 and P = 0.0072, respectively), but not sirt2 KO OL-EXOs (P = 0.9166 and P = 0.5947, respectively) (Figures 7O and 7P). In summary, these in vivo results support our conclusion that OL-derived SIRT2 is delivered to the myelinated axonal compartment, where SIRT2 rescues axonal mitochondrial deficiency in sirt2 KO mice.
DISCUSSION
Axon degeneration, mitochondrial stress, energy crisis, and OL dysfunction are pathological features of major neurological disorders. Elucidating mechanisms underlying OL-mediated support of axonal energetics is an important step towards therapeutic development. In this study, we reveal an energetic transcellular signaling pathway through which OL-derived SIRT2 boosts axonal energy metabolism by deacetylation of neuronal mitochondrial proteins. We provide seven lines of evidence to support our model. First, proximal OLs enhance local energy availability within the distal axonal compartment. Second, addition of OL-CM or OL-EXOs is sufficient and necessary to enhance axonal energy level and mitochondrial energetic function. Third, SIRT2 is absent in neurons but highly enriched in mature OLs and released within exosomes. Fourth, exogenous expression of SIRT2 in neurons enhances axonal ATP level, recapitulating the phenotype observed when axons are co-cultured with OLs. Fifth, SIRT2 knockdown or deletion of sirt2 gene abolishes the role of OLs in mediating axonal energetic enhancement. Sixth, OL-derived SIRT2 boosts axonal bioenergetic capacity by deacetylation of mitochondrial proteins ANT1/ANT2. Finally, injection of OL-EXOs isolated from WT, but not from sirt2 KO mice, rescues axonal mitochondrial deficiency in the spinal cord of sirt2 KO mice. Our study advances understanding of neurological diseases characterized by OL dysfunction and axonal energy failure.
OLs Stimulate Axonal Mitochondrial Energetics
Axons are particularly vulnerable to early neurodegeneration (Salvadores et al., 2017). A detailed energy budget based on theoretical calculations of the fully myelinated optic nerve determined that small diameter axonal fibers have a 38% shortage in ATP supply compared to ATP demand (Harris and Attwell, 2012). As neurons have a polarized morphology and ATP diffusion is not efficient within long axons (Hubley et al., 1996; Sun et al., 2013), these findings highlight the importance of incorporating the contribution of local glial cells in an all-inclusive model of axonal energetic maintenance. With increasing evidence of mitochondrial dysfunction and energy deficits in the progression of neurodegenerative diseases (see reveiws by Chamberlain and Sheng, 2019; Cunnane et al., 2020; Pathak et al., 2013), it is an important emerging frontier to reveal glia-mediated transcellular pathways that regulate local energy metabolism in axons in response to changes in both energy supply and demand.
Considering that the myelin sheath physically isolates axons from the extracellular milieu, OLs may energetically support the axon by supplying energy substrates. OLs can ‘sense’ nearby neuronal activity levels via N-methyl-D-aspartate receptors (NMDARs) that respond to extracellular glutamate via calcium waves (Karadottir et al., 2005; Micu et al., 2006; Salter and Fern, 2005). NMDAR activation in OLs induces glucose transporter 1 (GLUT1) expression in the myelin sheath (Saab et al., 2016) and stimulates OL production of glycolytic lactate (Funfschilling et al., 2012), thus supporting activity-dependent axonal energy demand. While lactate shuttling is clearly important for maintaining brain function (Lee et al., 2012), these studies rely on models in which neurons or OLs are energetically challenged via oxygen-glucose deprivation (Meyer et al., 2018). We found that lactate does not primarily underlie the beneficial effect in our system where glucose is available. Rather, enhanced axonal ATP in the presence of OLs is reliant on release of exosomes, which contain several proteins related to energy metabolism (Frohlich et al., 2014). While our findings demonstrate that OLs actively modulate or boost axonal energy metabolism through transcellular OL-to-axon signaling, precisely how exosome release from OLs is regulated remains unknown. OLs release exosomes when cultured in isolation, suggesting a constant, homeostatic nature. Our study showed that the majority (62.56%) of HSP70-labeled exosomes contain SIRT2, supporting the notion that SIRT2-filled exosomes are constantly secreted by OLs. It was reported that OL-EXO release is upregulated by a Ca2+-dependent mechanism (Fruhbeis et al., 2013). Given that exosomes are heterogenous populations of extracellular vesicles, future work is necessary to determine whether release of SIRT2-filled EXOs from myelin-forming OLs is a response to changes in neuronal activity or local energy demands.
We demonstrate that OLs enhance neuronal ATP levels, with a particularly strong effect on maturing axons, which are highly energy demanding to maintain axonal and synaptic functions, including transport of various organelles, generation of action potentials, and synaptic vesicle recycling. Neuronal development influences OL maturation (Chamberlain et al., 2016; Piaton et al., 2010); OLs also alter axonal structure and affect the propagation of electrical signals via myelination (Doretto et al., 2011; Piaton et al., 2010). Thus, the large energetic effect of OLs on mature neurons is likely due to the increasing symbiotic reliance of neurons and OLs on one another.
Transcellular SIRT2 Delivery Mediates OL-Axon Energetic Signaling
We chose to investigate SIRT2 as a possible mediator of OL-axon energetic signaling for several reasons. First, we and others have demonstrated that SIRT2 is highly expressed in mature OLs and undetectable in mature neurons, thus building a foundation for transcellular regulation of neuronal mitochondrial function by OL-derived SIRT2. Second, we demonstrate that SIRT2 is released within exosomes, thus revealing OL-to-axon delivery of SIRT2. Third, although SIRT3-5 are the mitochondrial-localized sirtuins (Verdin et al., 2010), SIRT2 deacetylates mitochondrial proteins in the brain and genetic deletion of sirt2 is associated with reduced cellular ATP and axon degeneration (Fourcade et al., 2017; Liu et al., 2017). SIRT2 inhibition is also associated with reduced ATP levels in PC12 cells (Nie et al., 2011), consistently supporting a key role of SIRT2 in regulating mitochondrial energetic function. Fourth, sirtuin-mediated deacetylation of mitochondrial proteins is associated with robust increases in cellular respiration (Kim et al., 2006; Lombard et al., 2007; Vassilopoulos et al., 2014; Yang et al., 2016); SIRT2 is one of three sirtuin proteins (SIRT1-3) with strong deacetylase activity (Feldman et al., 2013). Fifth, only SIRT1 and SIRT2 are abundantly expressed in brain tissues, with SIRT1 being highly expressed in the nucleus of neuronal cells (Jayasena et al., 2016) and SIRT2 primarily enriched in mature OLs (Liu et al., 2019; Zhang et al., 2020). Interestingly, SIRT2 expression changes over time, accumulating in the aging CNS (Maxwell et al., 2011). Our study demonstrates that SIRT2-deficent OLs fail to mediate their beneficial effect on axonal energy enhancement. However, overexpressing SIRT2 in cortical neurons is sufficient to enhance axonal ATP levels, consistent with a previous study in SH-SY5Y neuroblastoma cells where SIRT2 overexpression is protective against oxidative stress (Singh et al., 2017). Interestingly, transport of SIRT2 into myelin membranes requires the OL-specific protein PLP (Werner et al., 2007; Zhu et al., 2012). PLP-deficient mice exhibit impaired energy status and disrupted mitochondrial morphology in neuronal axons (Yin et al., 2016), thus supporting the assumption that loss of either PLP or SIRT2 in the myelin sheath contributes to these phenotypes.
We further showed that injected OL-EXOs can be readily internalized into myelinated axons of mouse spinal cord dorsal horn in vivo. More importantly, mitochondrial membrane potential, a proxy for ATP production, is significantly declined within myelinated axons of sirt2 KO mice. Injection of OL-EXOs isolated from WT, but not from sirt2 KO mice, rescues axonal mitochondrial deficiency. Furthermore, acetylation levels of both mitochondrial ANT1 and ANT2 are significantly decreased by injecting WT OL-EXOs. These findings add to our body of in vitro and in vivo evidence showing that exosome-mediated transcellular delivery of SIRT2 from OLs to myelinated axons enhances mitochondrial energy metabolism by deacetylation of neuronal mitochondrial proteins.
SIRT2-Mediated Deacetylation of ANT1/2
Reversible acetylation and deacetylation have emerged as key post-translational modifications in cell metabolism; deacetylation is associated with enhanced mitochondrial ATP production capacity (Liu et al., 2017; Nie et al., 2011). Our mass spectrometry analysis of neuronal mitochondria reveals two primary deacetylation targets of SIRT2, ANT1 and ANT2, that display strong deacetylation after neurons are pretreated with OL-CM from WT but not sirt2 KO mice. ANT1 and ANT2 play an essential role for mitochondrial bioenergetics by exchanging ADP and ATP across the mitochondrial inner membrane, therefore converting ADP to ATP and catalyzing the export of mitochondrial ATP. How does deacetylation affect mitochondrial function? Acetylation increases hydrophobicity and neutralizes positive charges that alter protein interaction and function (Kim et al., 2006). Both ANT1 and ANT2 are acetylated at Lys10 and ANT1 is also acetylated at Lys23, a region critical for ADP binding. Deacetylation of Lys23 robustly enhances ANT1 for ADP binding affinity, thus facilitating conversion of ADP to ATP (Mielke et al., 2014). SIRT2 was reported to target to inner mitochondrial membranes in brain cells (Liu et al., 2017). Our iTEM images consistently show robust association of SIRT2 with mitochondria in OLs and internalized SIRT2 in myelinated axons also associates with mitochondria. Thus, SIRT2 targets mitochondria to exert its post-translational modification.
It was reported that approximately 20% of mitochondrial proteins in murine liver are acetylated (Kim et al., 2006). The relatively lower acetylation in neuronal mitochondria may reflect cell type specific patterns of lysine acetylation and bioenergetic status under our experimental conditions (Lundby et al., 2012). A mass spectrometry study identified 11 acetylated proteins in human muscle mitochondria, of which ANT1 was found to be abundantly acetylated (Mielke et al., 2014). Although we cannot exclude other mitochondrial proteins as targets for SIRT2-mediated deacetylation, ANT1/2 are two important targets of SIRT2 in boosting neuronal mitochondrial energy metabolism.
STAR * METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zu-Hang Sheng (shengz@ninds.nih.gov).
MATERIALS AVAILABILITY
This study did not generate any new reagents.
DATA AND CODE AVAILABILITY
List of acetylated peptides of neuronal mitochondrial proteins identified by mass spectrometry is shown in Table S1. The datasets supporting the current study are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Lines and Care
Sirt2tm1.1Fwa mice were purchased from The Jackson Laboratory. Timed-pregnant C57BL/6 female mice were purchased from Charles River Laboratories. For preparing cortical neuron cultures, male and female E18 embryonic mice were used. For preparing primary OL cultures, sirt2+/+ and sirt2−/− male and female P3-5 postnatal mice (sex: random) were used. For in vivo experiments, sirt2+/+ and sirt2−/− P60-70 postnatal mice (sex: random) were used. No sex-dependent influence was observed in both in vitro and in vivo experiments. All mice were maintained in the National Institute of Neurological Disorders and Stroke (NINDS) animal facility and housed in a 12-hour light/dark cycle. All animal procedures were performed according to National Institutes of Health (NIH) guidelines and were approved by the Animal Care and Use Committee of NINDS/National Institute on Deafness and Other Communication Disorders (NIDCD).
Cell lines
Human embryonic kidney cells (HEK293T, ATCC) cells were plated in 10 cm cell culture dishes, maintained in complete growth medium comprised of 1 × DMEM with 10% fetal bovine serum (FBS), and used for experiments at 80%-90% confluency. For freezing, 5% dimethyl sulfide (DMSO) was added to complete growth media and cryovials were maintained at −80°C.
METHOD DETAILS
DNA Constructs
GFP-Mito was a gift from Dr. R. Youle (NINDS, NIH, MD). GO-ATeam2 and 3 were a gift from Dr. Hiromi Imamura (Kyoto University, Kyoto, Japan). The viral envelope and packaging constructs pMD2.G and psPAX2 were a gift from Dr. Didier Trono (EPFL, Switzerland). The mApple-Lamp1 construct was obtained from Addgene (Cat# 54627). The SIRT2 overexpression construct, pCDH-SIRT2-Flag (Cat# 102624) and the empty vector p3xFLAG-CMV-7.1 (Cat# E4026) were obtained from Addgene. The pHAGE-YFP viral construct was previously generated in the Sheng Lab (Zhou et al., 2016).
RNAi Oligonucleotides
SIRT2 stealth siRNA (Cat# 1320001) and the medium G-C content stealth RNAi siRNA negative control (Cat# 12935300) were both obtained from Thermo Fisher Scientific.
Primary Neuron Culture
Cortices were dissected from male and female E18-19 mouse embryos and kept on ice-cold dissection buffer containing HBSS (without CaCl2, without MgCl2), 1M HEPES, and 1% Pen/Strep (v/v). A single cell suspension was prepared via papain dissociation as previously described (Kang et al., 2008). Cells were resuspended in neuronal plating media comprised of neurobasal media supplemented with 2% B27, 0.5mM L-GlutaMAX, 55 μM 2-mercaptoethanol (all Thermo Fisher Scientific), 30 μg/mL insulin (Sigma Aldrich), and 10% fetal bovine serum (FBS) (Hyclone). For microfluidic chambers, 200,000 cells were centrifuged at 700 rpm for 6 min and resuspended in 10 μL of plating media, briefly incubated with virus (if applicable), and added to the somatic compartment previously coated with poly-ornithine (Sigma-Aldrich; 1:4 in PBS) and laminin (Roche; 1:400 in PBS). Neurons were allowed to adhere for 30 min, after which plating media was added to the top of chamber reservoirs (∼400 μL total). After 24 hours, plating media was replaced with co-culture media comprised of DMEM-F12 (Thermo Fisher Scientific Cat# 12500-652 supplemented with 2.1 g/L NaHCO3), 1% N2, 1% B27, 1% Pen/Strep, 50 ng/mL insulin, and 40 ng/mL T3 according to a previously published protocol (Dincman et al., 2012). Neuronal culture media was replaced every three days. For mass spectrometry and western blot experiments, 10 × 106 neurons were added to a T75 flask double coated with poly-ornithine and laminin. Neurons were transfected with various constructs using either Lipofectamine 2000 or Lipofectamine RNAiMAX (Thermo Fisher Scientific) or infected with lenti-virus at plating and imaged via confocal microscopy at the indicated time point.
Primary Oligodendrocyte Culture
Primary OLs were obtained from male and female P3-P5 mouse cortices via magnetic activated cell sorting (MACS) using the Neural Tissue Dissociation Kit for Postnatal Neurons (Miltenyi, Catalog #130-094-802), Mouse FcR Blocking Reagent (Miltenyi, Catalog #130-092-575), and anti-O4 Microbeads (Miltenyi, Catalog #130-094-543) according to the manufacturers protocol. To co-culture OLs with DIV3 neuronal axons, media in the axonal compartment was gently removed and replaced with 10 μL of OLs suspension containing 100,000 cells. OLs were allowed to adhere for 30 min, after which co-culture media was added to the top of the axonal chamber reservoir. To culture primary OLs alone, 50,000 cells were resuspended in 300 μL co-culture media and seeded onto a 12-mm coverslip (Deckgläser) double coated with poly-ornithine (Sigma-Aldrich; 1:4 in PBS) and laminin (Roche; 1:400 in PBS).
Immunofluorescence and Fixed-Cell Imaging
Cells in microfluidic devices or coverslips were fixed in 4% paraformaldehyde (PFA) and 120 mM sucrose for 20 min and then washed three times with PBS. Cells were permeabilized in 0.25% Triton X-100 in PBS for 10 min and then washed once with PBS. For blocking, cells were incubated in PBS containing 0.1% (v/v) Triton X-100 and 10% (w/v) bovine serum albumin (BSA) for 1 hour at room temperature (RT). Primary antibodies were diluted in PBS containing 0.1% (v/v) Triton X-100 and 1.0% (w/v) BSA. Cells were incubated in primary antibody in a dark, humidified chamber overnight at 4°C. Primary antibodies were used as follows: MAP2 (BD Pharmingen, 1:1000), βIII-Tubulin (Covance, 1:500), MBP (Biorad, 1:500), SIRT2 (ProteinTech, 1:100).
After three-time washes in PBS, cells were incubated in AlexaFluor® 488, 546, 594, or 633-conjugated secondary antibodies (Thermo Fisher Scientific) diluted at 1:700 in PBS containing 0.1% (v/v) Triton X-100 and 1.0% (w/v) BSA in a humidified chamber for 1 hour at RT. After three-time washes in PBS, nuclei were stained with Hoechst solution for 3 min at RT. After two-time washes in PBS, cells on microfluidic devices were imaged directly, while coverslips were mounted on glass slides (Globe Scientific) and allowed to dry at RT overnight. Confocal images were obtained on a Zeiss 880 confocal microscope with a 1.45 NA 63 × objective.
Labeling of exosomes
To immuno-label exosomes, a previously published protocol was followed (Mondal et al., 2019). First, the isolated exosomes released from WT or sirt2 KO OLs were permeabilized using 0.001% Triton X-100 for 5 min. Second, PEG10000 was added to the exosomes at a final concentration of 10%, followed by centrifugation at 3,000 g at RT for 5 min. The pellet was dissolved in PBS and incubated with primary antibodies against SIRT2 (ProteinTech, 1:100) and HSP70 (GeneTex, 1:100) overnight at 4°C. Third, 20% PEG10000 was added to the exosome suspension and centrifuged at 3,000 g at RT for 5 min for three times to remove excess antibodies, and then the exosome suspension was incubated with AlexaFluor® 488 and 594-conjugated secondary antibodies (Thermo Fisher Scientific) diluted at 1:100 in PBS for 1 hour at RT. Fourth, the exosome suspension was washed with PEG10000 and centrifuged at 3,000 g three times at RT for 5 min, and passed through Sephadex G-25 column (G2580, Sigma Aldrich) to remove unbound antibodies. Fifth, the exosome pellets were dissolved in 20 μL PBS, placed on microscope slides, covered by a coverslip and kept drying for 5-10 min. Finally, the slides were imaged by a Zeiss 880 confocal microscope with a 1.45 NA 63 × objective.
Live imaging analysis
To image living neurons, microfluidic chambers were equilibrated with Hibernate A low fluorescence media (BrainBits) supplemented with 2% B27, 0.5 mM L-Glutamax, and 1% Pen/Strep. Chambers were imaged on a Zeiss 880 confocal microscope with Airyscan using a 63 × oil objective (Zeiss). The temperature was maintained at 37°C during the image duration. For ATP analysis, the GO-ATeam2/3 sensors were excited using the 488 filter and two emissions were collected, one at 505-550 nm (GFP) and one long pass above 545 nm (OFP).
Lentivirus production and infection
YFP and GFP-Mito vectors (Addgene), as well as the GO-ATeam2 and GO-ATeam3 vectors (gift from H. Imamura, Kyoto University, Kyoto, Japan), were cloned into the pFUGW vector (Zhou et al., 2016). To produce virus, HEK293T cells were transfected with vector, psPAX2 plasmid, and pMD2G plasmid. After 24 hours, transfection medium was replaced with UltraCULTURE™ medium containing 1% (w/v) L-Glutamine, 1% (w/v) Sodium Pyruvate, and 1% (w/v) Sodium Bicarbonate (all from Lonza). Virus-containing media was collected after 48 hours and centrifuged for 5 min at 1,000 rpm to remove cell debris. The supernatant was passed through a 0.45 μm filter and ultra-centrifuged at 25,000 rpm at 4°C for 2 hours. After removing the supernatant, the viral pellets were resuspended in PBS and stored at −80°C. For infection, 200,000 freshly dissociated cortical neurons suspended in 10 μL were incubated with 0.5 μL purified virus for 5 min and then plated in a microfluidic chamber device.
Mitochondrial fractionation
Primary cortical neurons were seeded at 10 × 106 in a T75 flask and treated with control or OL-CM for 24 hours on DIV6-7. Mitochondria were isolated as previously described with minor modifications (Rogers et al., 2019). Neurons were collected on DIV7-8, washed in ice-cold PBS, and briefly centrifuged. Pellets were resuspended in 100 μL of 5 × cytosolic extraction buffer (250 mM sucrose, 70 mM KCl, 137 mM NaCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, 300 μg/mL digitonin, protease inhibitor cocktail) and incubated on ice for 5 min. Samples were centrifuged at 3,000 g at 4°C for 10 min, after which the supernatant was collected as the cytosolic fraction. The pellet was resuspended in 50 μL of 5 × mitochondrial lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.2% Triton X-100, 0.3% NP-40, 2.5 mM Vorinostat (LC Laboratories) and protease inhibitor cocktail (Roche)), pipetted vigorously, and incubated on ice for 30 min. Samples were then centrifuged at 14,000 g at 4°C for 10 min. The supernatant was collected as the mitochondrial fraction for further analysis by mass spectrometry or immunoblot.
Mass spectrometry
Mitochondrial fraction samples for protein identification were reduced with TCEP, alkylated with NEM, and digested with trypsin. Peptides were desalted using Oasis HLB μElution plate (Waters) before being injected into HPLC. A system with an UltiMate 3000 RSLC-nano HPLC (Thermo Fisher Scientific) coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) was used for data acquisition. LC-MS/MS experiments were performed in data-dependent acquisition (DDA) mode. Peptides were separated on a nano-ES802 column. The mobile phase B (98% acetonitrile, 0.1% formic acid) amount was increased from 5% to 36 % over 152 mins at a flow rate of 300 nL/min. The MS resolution is 120K at m/z 400, MS scan range is 350-1500 m/z, the automated gain control (AGC) target is 2 × 10e5. The quadrupole isolation window is 1.6 m/z. Precursors with charge states 2-6 and intensity higher than 1 × 10e4 within a 3 sec cycle between MS1 scans were selected for MS/MS acquisition in the linear ion trap.
For TMT experiment, samples were digested using the same method described above. Digests were labeled with TMT11plex isobaric label reagent set (Thermo Fisher). After labeling, samples were combined and fractionated with high pH reverse phase HPLC separation kit (Thermo Fisher). Eight fractions were collected and used for LC-MS/MS data acquisition. Peptides were separated on an ES802 nano-column over 136 min. TMT MS2 method was used. Both MS1 and MS2 scans were performed in orbitrap. The resolution for MS1 and MS2 scans were 120K and 50K, respectively. Peptides were fragmented using HCD method with collision energy fixed at 32%. The precursor isolation window is 1.2 Da.
The protein identification, quantitation and acetylation analysis were performed with Proteome Discoverer 2.4 software. The search parameters included: trypsin digestion with full specificity; 4 missed cleavages allowed; N-ethylmaleimide on cysteines as fixed modification; acetylation (K, protein N-term), oxidation (M) as variable modifications, the mass tolerance is 10 ppm for precursor ions; for fragment ions, it is 0.6 Da and 20 ppm for ID and TMT data, respectively. The false discovery rate for peptide-spectrum matches (PSMs) was set to 0.01 using Percolator.
Immunoblots
Mouse cortical neurons, OLs, or HEK293T cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% DOC, 2 mM EDTA) supplemented with Protease Inhibitor Cocktail (Roche) and when applicable, 1 μM of the histone deacetylase inhibitor Vorinostat (LC Laboratories). Protein concentration was determined via BCA Assay (Thermo Fisher Scientific) and equal amounts of proteins were loaded and analyzed by 4%-12% Bris-Tris NuPAGE (Thermo Fisher Scientific) and western blot. Primary antibodies were used as follows: SIRT2 (ProteinTech, 1:500), MBP (BioRad, 1:500), GAPDH (Millipore, 1:2000), Panacetyl (Cytoskeleton, 1:500), PLP (Abcam 1:500), TOM20 (Millipore, 1:500), and TOM40 (Santa Cruz Biotechnology, 1:500).
Microfluidic chamber preparation
Polydimethylsiloxane (PDMS) microfluidic chambers were made in-house using a silicon wafer patterned out of SU-8 by photolithography. SYLGARD™ 184 silicone elastomer base was mixed with the curing agent at a ratio of 10:1. Next, the PDMS was mixed using a THINKY mixer ARF-310 at 2,000 rpm for 4 min, followed by a de-foaming at 2200 rpm for 4 min. After mixing, ∼35 grams of PDMS were poured onto the silicon wafer and placed into a Bel-Art vacuum desiccator for 2.5 hours. After all bubbles were removed, the wafers were moved to a −80°C oven for 2 hours to cure. Once cooled, chambers were punched out and washed via sonification. To prepare for culture, microfluidic chambers and glass coverslips were placed in a PDC-32G plasma cleaner for 1 min and then bonded together. Chamber reservoirs were coated with poly-ornithine (Sigma; 1:4 in PBS) for 72 hours at 37°C, washed three times with water, coated with laminin (Roche; 1:500 in PBS) for 1 hour at 37°C, and washed once with media prior to cell plating.
Primary cell transfection
For overexpression of SIRT2, primary neurons were transfected on DIV7 using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturers protocol. To transfect 0.2 × 106 neurons located within the somatodendritic compartment of microfluidic devices, 100 ng of each DNA construct and 0.6 μL of Lipofectamine 2000 was diluted in 100 μL of co-culture media and added to neurons for 3 hours. For knockdown of SIRT2, primary OLs in the axonal compartment were transfected on DIV1 using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturers protocol. Briefly, to transfect 0.1 × 106 OLs located within the axonal compartment, 3 pmoles of control or SIRT2 Stealth RNAi (Thermo Fisher Scientific) and 0.6 μL of Lipofectamine RNAiMAX were diluted in 100 μL of co-culture media and added to OLs for 3 hours.
GO-ATeam2 ATP signal analysis
To quantify ATP levels, ImageJ (NIH) was used to create a selection over the axon region. Within this selection, OFP signal was divided by GFP signal to generate ratiometric intensities for quantification. To create a visual heat map of axonal ratiometric signal, non-axonal background signal was removed by first using the ImageJ “clear outside” function to set the non-axonal signal to 0. The image calculator function was then used to divide OFP signal by GFP signal, producing a representative image which was converted into a heat map using the ImageJ “fire” lookup table. Within experiment, all side-by-side heat maps are presented with the same brightness and contrast settings.
ATP measurement by luminescence assay
To measure neuronal ATP content, ATPlite Luminescence Assay System (PerkinElmer) was used according to the manufacturer’s protocol. Briefly, 100,000 cortical neurons were seeded and cultured in one well for 7 days. After 24 hours treatment with AA or PCr, or OL-EXOs incubation, neurons were lysed and subjected to ATP measurement by Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek). Five to eight wells of cultures were included for each condition.
Immuno-electron microscopy
For detection of SIRT2 and HSP70 in vivo, mice were anesthetized and transcardially perfused with freshly made EM fixative (0.05% glutaraldehyde and 4% paraformaldehyde). Spinal cord samples are collected and kept in fixative for 30 min before sectioning into 100 μm sections in 1 × PBS using a vibratome (Leica VT1200 S). For detection of SIRT2 in primary OL cultures, OLs were plated on glass coverslips at 30,000 cells/12 mm. Cells were fixed at DIV3 in fresh 4% paraformaldehyde (PFA) in 1 × PBS for 30 min. After four 5 min washes in 1 × PBS, spinal cord section or cell coverslip samples were blocked and permeabilized in 1 × PBS with 5% normal goat serum (NGS) and 0.1% saponin for 30 min. Samples were then incubated in anti-SIRT2 (ProteinTech, 1:100) or anti-HSP70 (Thermo Fisher, 1:400) primary antibody diluted in 1 × PBS containing 5% NGS and 0.05% saponin for 1 hour at room temperature (RT). After four 5-min wash in 1 × PBS, secondary antibody nanogold Fab’ conjugate (1:200, Nanoprobe) diluted in 1 × PBS containing 5% NGS and 0.05% saponin was added for 1 hour at RT. Samples were washed four times for 5 min each in 1 × PBS and post-fixed in 2% glutaraldehyde overnight, then sent to the NINDS EM facility for processing. Samples were subjected for staining and silver enhancement. After dehydration, embedding and sectioning, and images were taken using an electron microscope (JEM-1400, JEOL).
Nanoparticle tracking analysis (NTA)
NTA was performed using a NanoSight NS300 418 running NTA software version 3.1 (Malvern Panalytical). The concentration and size distribution of exosomal vesicles was measured by continuously pushing samples through a fluidics flow chamber using a syringe pump (Harvard Apparatus). Vesicles were illuminated with a 488 nm laser and the scattered light was recorded using a sCMOS camera (30 frames per second, 3-5 recordings, 60 seconds per recording). Camera sensitivity settings were held constant within each experiment and the threshold for particle analysis was set to 3.
Seahorse extracellular flux analysis
Oxygen consumption rate (OCR) of primary neurons was measured using a Seahorse Bioscience Extracellular Flux Analyzer (XFe96) and the Seahorse XF Cell Mito Stress Test (Agilent 103015-100) according to the manufacturers protocol. Briefly, primary neurons were seeded between 80,000-100,000 cells per well, a plating density selected through cell titration experiments. Neurons were maintained in co-culture media, which was carefully exchanged every 3 days. DIV6-11 neurons were treated with OL-CM or OL-EXOs for 24 hours. Pre-treatment with the potent inhibitor of mitochondrial respiration, antimycin-A (AA, 100 nM), for 24 hours served as a negative control. Prior to the assay, neurons were washed with pre-warmed Seahorse assay media (120 mM NaCl, 3.5 mM KCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1 mM MgCl2, 5 mM HEPES, 10 mM Sodium Pyruvate, and 10 mM Glucose in dH2O, pH 7.4) and maintained in a CO2-free 37°C incubator for 1 hour. To perform the stress test, OCR was measured at basal level and following sequential injections of oligomycin, FCCP, and rotenone/antimycin-A at a final concentration of 1.0 μM, 1.5 μM, and 0.5 μM, respectively. The Seahorse XF Cell Mito Stress Test Report Generator (Agilent) was utilized to analyze the resulting data, generating values for basal respiration, ATP production, and extracellular acidification rate. Similar to previously published methodology (Nicholas et al., 2017), replicates having OCR or ECAR measurements below zero were assumed to be in error and thus, these wells were entirely excluded from downstream analysis. Note that data were normalized to cell number at the time of plating, as attempts to normalize data based on cell number in an equivalently plated 96-well plate were not useful due to variation in plating materials. In addition, 10-12 wells were used for each condition to account for well-to-well variability in cell number, allowing for adequate statistical power after removal of wells exhibiting a significantly skewed metabolic profile within group.
Measurement of mitochondrial membrane potential (ψm)
To measure mitochondrial membrane potential, the axonal chamber of microfluidic chamber devices was loaded with 20 nM MitoTracker™ Orange CMTMRos (Thermo Fisher), a fixable ψm-dependent dye, for 20 min at 37°C. Chambers were then rinsed three times with pre-warmed live-cell imaging buffer (Hibernate A low fluorescence media (BrainBits) supplemented with 2% B27, 0.5mM L-Glutamax, and 1% Pen/Strep) and imaged on a Zeiss 880 confocal microscope. For analysis, the integrated density of mitochondrial CMTMRos signal was measured within a 100 μm region of the distal axon and normalized to control.
Exosome purification
Conditioned media (OL-CM) was collected from coverslips containing 50,000 primary O4+ OLs isolated via MACS from DIV2-6. OL-CM (5 mL) was further processed for exosome isolation according to a previously published protocol (Chiou and Ansel, 2016) or a commercial exosome purification kit (ExoQuich-TC, System Biosciences). First, OL-CM was centrifuged for 10 min at 1,000 g (4°C) to remove intact cells. The supernatant was then removed and centrifuged for 20 min at 2,000 g to remove cell debris (4°C). The supernatant from this second spin was removed and centrifuged for 30 min at 10,000 g (4°C) to remove microvesicles. Finally, the supernatant was transferred to a 17 mL Beckman Ultra-Clear™ tube, topped off with sterile PBS, and ultra-centrifuged for 2 hours at 100,000 g (4°C). The crude exosome pellet was resuspended in 100 μL PBS, which was further processed by density gradient purification. Briefly, OptiPrep™ (60% w/v) solution was used to prepare a discontinuous iodixanol gradient composed of 40%, 20%, 10%, and 5% OptiPrep diluted in 0.25 M sucrose and 10 mM Tris solution, pH 7.5. The gradient was generated by sequentially layering 3 mL of 40% (bottom), 20%, and 10% iodixanol solutions, followed by 2.5 mL of 5% iodixanol solution, to a 17 mL Beckman Ultra-Clear™ centrifuge tube, being careful not to displace the layers. Lastly, the 100 μL crude exosome sample was overlaid on the gradient, which was centrifuged for 16 hours at 100,000 g (4°C). Fractions of 1 mL were collected from the top and exosome-enriched fractions #6 and #7 were collected, diluted to 17 mL in PBS, and ultra-centrifuged again for 2 hours at 100,000 g (4°C). The final pellet was resuspended in 100 μL PBS and stored at −20°C until further use. To test the effect of OL-EXOs in culture, 15 μL of the purified exosome sample was diluted in 1 mL of culture media. To test the effect of OL-EXOs in vivo, exosomes were prepared from 40 mL of OL-CM by using Exosome Precipitation Solution (ExoQuich-TC, System Biosciences), and resuspended in 50 μL of PBS.
Cell treatment
To inhibit exosome biogenesis and release within OLs, the neutral sphingomyelinase inhibitor GW-4869 (Sigma-Aldrich) was diluted to 5 mM in DMSO and then further diluted to 1 μM in co-culture media, which was added to the axonal compartment for 24 hours. To inhibit cellular respiration, primary neurons were treated with Antimycin A (AA) (Sigma-Aldrich) for 24 hours. AA was diluted to 2 mM in DMSO and then further diluted to 100 nM for Seahorse Extracellular Flux Analysis, 20 nM for live-cell imaging of GO-ATeam2mito, or 5 nM for live-cell imaging of GO-ATeam2. To enhance intracellular ATP buffering, phosphocreatine (PCr) (Sigma-Aldrich) was diluted to 10 mM in water and then further diluted to 500 μM in co-culture media, which was added to primary neurons for 24 hours. To inhibit lactate transport through monocarboxylate transporters 1 and 2, AR-C155858 (AR) (Tocris) was diluted to 1 mM in DMSO and then further diluted to 3.2 μM in co-culture media and added to primary neurons for either 24 hours or 10 min, as indicated in figure legends. Primary neurons were treated with 2-10 mM L-lactate (Sigma-Aldrich) diluted in water for either 10 min or 24 hours, as indicated in figure legends.
Protein acetylation assay
To detect acetylation of mitochondrial proteins, immunoprecipitation assay was performed using anti-Acetyl-Lysine (mAb mix) affinity beads. Cortical neurons were seeded at 20 × 106 in a T75 flask and treated with control, SIRT2 WT or KO OL-CM for 24 hours on DIV6-7. Neurons were collected on DIV7-8, washed with PBS, and lysed by lysis buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% NP-40, 5% glycerol) and protease inhibitor cocktail (Roche) on ice for 30 min. Samples were centrifuged at 14, 000 g at 4° C for 15 min. Cell lysate (1 mg) was incubated at 4° C for 3 hours with Anti-Acetyl-Lysine (mAb mix) affinity beads (Cytoskeleton, Inc) that were coated with a mixture of two anti-acetyl lysine antibodies (Clones 7B5A1 and 10C4B2.1), then spun down and washed three times by cold lysis buffer. 50 μL 2 × sample buffer with 20 mM DTT was added into each sample and boiled for 5 min prior to loading for immunoblots. Primary antibodies were used as follows: ANT1 (1:500, Sigma), ANT2 (1:500 Cell Signaling), ATP5A (1:1000, Santa Cruz), NDUFA5 (1:1000, Thermo), PHB2 (1:1000, Proteintech). VeriBlot for IP detection reagent (1:2000, Abcam) was used as a secondary antibody to avoid IgG signals in the IP samples.
Spinal cord injections and in vivo delivery of exosomes
The procedure for spinal cord injection was described in our previous study (Han et al., 2020) with minor modifications. All surgical procedures were performed under sterile conditions. Briefly, animals were deeply anesthetized using a cocktail of ketamine (100 mg/kg) and xylazine (10 mg/kg) via intraperitoneal injection. Each animal also received a subcutaneous injection of meloxicam (10 mg/kg) to minimize post-operative pain. Anesthesia was judged by the absence of hind paw pinch reflexes. Anesthetized animals were placed on a heating pad to maintain body temperature at 37°C ± 0.5°C. For spinal cord injections, a small incision was made in the skin and the underlying musculature or adipose tissue was teased apart to reveal the vertebral column. Tissue joining the dorsal processes of consecutive vertebrae was removed and the vertebral surfaces were cleaned with fine forceps and gently separated to reveal the dorsal surface of the spinal cord. The dura was punctured by pinching with sharp forceps to facilitate smooth entry of the needle. The dye cocktail of CMTMRos (10 μM) and FluoroMyelin™ Green (Myelin Green, 1:300 dilution) alone or mixed with purified sirt2 WT or KO OL-EXOs (5 μL) was pressure injected using a 10-μL Hamilton syringe with a 30-gauge needle (point style 4, 30-degree bevel). Three injections were performed on both the left and right sides at a depth of 700–800 μm from the dorsal surface and a total 3 μL of the cocktail was released at a rate of 500 nL/min. Following injections, the overlying muscle was sutured, and the skin incision was closed using a wound clip. After 30 min, mice were anesthetized again followed by 4% PFA perfusion. Spinal cord samples from both the control and experimental group were parallelly embedded in OCT medium, followed by cryostat sectioning. After DAPI or immunohistochemistry staining of SNPH, sections were observed using a Zeiss LSM-880 confocal microscope equipped with a 63 × oil lens. The observer was blind to the treatment group when taking the images and measuring the fluorescent intensity. Data were collected from 5-6 pairs of animals.
QUANTIFICATION AND STATISTICAL ANALYSIS
All quantifications were performed not blinded. Statistical parameters including the definitions and exact value of n (e.g., number of experiments, replications, axons, microgrooves, neurons, mitochondria, images, animals, etc), deviations, P values, and the types of the statistical tests are shown in Figures and Figure Legends. Statistical analyses were performed using Prism 8 (GraphPad Software). Two groups were compared using Student’s t test or Mann-Whitney test and comparisons between three or more groups were performed using either a one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test for post hoc testing or a two-way ANOVA with Sidak’s multiple comparisons test for post hoc testing. Data were expressed as mean ± SD or mean ± SEM indicated in figure legends. Differences were considered significant with P < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-MAP2 | BD Pharmingen | Cat# 556320, RRID:AB_396359 |
| Rabbit Anti-Tubulin, beta | Covance | Cat# PRB-435P-100, RRID:AB_291637 |
| Rat Anti-MBP | Bio-Rad | Cat# MCA409S, RRID:AB_325004 |
| Rabbit Anti-SIRT2 | ProteinTech | Cat# 19655-1-AP, RRID:N/A |
| Rabbit Anti-GAPDH | Millipore | Cat# CB1001, RRID: AB_2107426 |
| Rabbit Anti-HSP70 | Thermo Fisher Scientific | Cat# PA5-28003, RRID:AB_2545479 |
| Mouse Anti-HSP70 | GeneTex | Cat# GTX22787, RRID:AB_384850 |
| Rabbit Anti-PLP | Abcam | Cat# ab28486, RRID:AB_776593 |
| Rabbit Anti-SNPH | Abcam | Cat# 192605 |
| Rabbit Anti-TOM40 | Santa Cruz Biotechnology | Cat# sc-11414, RRID:AB_793274 |
| Mouse Anti-TOM20 | Millipore | Cat# MABT166, RRID: N/A |
| Mouse Anti-Acetyl Lysine | Cytoskeleton | Cat# 19C4B2.1, RRID: N/A |
| Mouse IgG, HRP-linked | GE Healthcare | Cat# NA931, RRID: AB_772210 |
| Rabbit IgG, HRP-linked | GE Healthcare | Cat# na934, RRID: AB_772206 |
| Goat Anti-Mouse, Alexa 488 Conjugate | Thermo Fisher Scientific | Cat# 11017, RRID: AB_2534084 |
| Donkey Anti-Rabbit, Alexa 488 Conjugate | Thermo Fisher Scientific | Cat# 21206, RRID: AB_2535792 |
| Goat Anti-Mouse, Alexa 546 Conjugate | Thermo Fisher Scientific | Cat# 11018, RRID: AB_2534085 |
| Goat Anti-Rabbit, Alexa 546 Conjugate | Thermo Fisher Scientific | Cat# 11035, RRID: AB_2534093 |
| Goat Anti-Rabbit, Alexa 594 Conjugate | Thermo Fisher Scientific | Cat# 11037, RRID: AB_2534095 |
| Goat Anti-Rabbit, Alexa 633 Conjugate | Thermo Fisher Scientific | Cat# 21052, RRID: AB_2535720 |
| Goat Anti-Rabbit IgG (H+L) (AH) | Ted Pella | Cat# 15727 |
| Nanogold Fab' Goat Anti-Rabbit IgG (H+L) | Nanoprobes | Cat# 2004 |
| Vectors and Virus Strains | ||
| pFUW685-CMVGrp78-mitoGoATeam | Zhou et al., 2016 | N/A |
| pFUW685-CMVGrp78-GoATeam2 | Zhou et al., 2016 | N/A |
| pHAGE-YFP | Zhou et al., 2016 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Antimycin A | Sigma-Aldrich | Cat# A8674 |
| Phosphocreatine disodium salt hydrate | Sigma-Aldrich | Cat# P7936 |
| AR-C155858 | Tocris | Cat# 4960 |
| Sodium L-lactate | Sigma-Aldrich | Cat# L7022 |
| ExoQuick-TC Exosome Precipitation Solution | System Biosciences | Cat# EXOTC10A-1 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 111668019 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | Cat# 13778075 |
| FluoroMyelin™ Green Flourescent Myelin Stain | Thermo Fisher Scientific | Cat# F34651 |
| MitoTracker Orange CMTMRos | Thermo Fisher Scientific | Cat# M7510 |
| GW4869 | Sigma-Aldrich | Cat# D1692 |
| DMEM/F-12, powder | Thermo Fisher Scientific | Cat# 12500062 |
| Sodium bicarbonate | Sigma-Aldrich | Cat# 792519 |
| N2 Supplement | Thermo Fisher Scientific | Cat# 17502048 |
| B27 Supplement | Thermo Fisher Scientific | Cat# 17504044 |
| T3 | Sigma-Aldrich | Cat# T6397 |
| Insulin | Sigma-Aldrich | Cat# I5500 |
| Papain | Worthington | Cat# LS003122 |
| GlutaMAX | Thermo Fisher Scientific | Cat# 35050-61 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985-023 |
| Fetal Bovine Serum | HyClone | Cat# SH30071.03 |
| Poly-ornithine | Sigma-Aldrich | Cat# P4957 |
| Laminin | Roche | Cat# 11243217001 |
| Neural Tissue Dissociation Kit-Postnatal Neurons | Miltenyi | Cat# 130-094-802 |
| Mouse FcR Blocking Reagent | Miltenyi | Cat# 130-092-575 |
| anti-o4 Microbeads | Miltenyi | Cat# 130-094-543 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat# A2153 |
| Goat anti-mouse, Alexa 488 Conjugate | Thermo Fisher Scientific | Cat# 11017, RRID: AB_2534084 |
| Donkey anti-rabbit, Alexa 488 Conjugate | Thermo Fisher Scientific | Cat# 21206, RRID: AB_2535792 |
| Goat anti-mouse, Alexa 546 Conjugate | Thermo Fisher Scientific | Cat# 11018, RRID: AB_2534085 |
| Goat anti-rabbit, Alexa 546 Conjugate | Thermo Fisher Scientific | Cat# 11035, RRID: AB_2534093 |
| Goat anti-rabbit, Alexa 594 Conjugate | Thermo Fisher Scientific | Cat# 11037, RRID: AB_2534095 |
| Goat anti-rabbit, Alexa 633 Conjugate | Thermo Fisher Scientific | Cat# 21052, RRID: AB_2535720 |
| Mouse IgG, HRP-linked | GE Healthcare | Cat# NA931, RRID: AB_772210 |
| Rabbit IgG, HRP-linked | GE Healthcare | Cat# na934, RRID: AB_772206 |
| Low Fluorescence Hibernate A Media | BrainBits | Cat# HALF |
| UltraCULTURE | Lonza | Cat# 12-725F |
| Sodium Bicarbonate | Lonza | Cat# 17-613E |
| Sodium Pyruvate | Lonza | Cat# 13-115E |
| L-Glutamine | Lonza | Cat# 17-605E |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Vorinostat | LC Laboratories | V-8477 |
| DMEM | Thermo Fisher Scientific | Cat# 11995-065 |
| Goat Serum | Sigma-Aldrich | Cat# G9023 |
| D-Glucose | Sigma-Aldrich | Cat# G8769 |
| Critical Commercial Assays | ||
| Seahorse XF Cell Mito Stress Test | Agilent | Cat# 103708-100 |
| ATPlite Luminescence Assay System | PerkinElmer | Cat# 6016943 |
| ExoGlow-Membrane EV Labeling Kit | System Biosciences | Cat#EXOGM600A-1 |
| Pierce BCA Protein Assay | Thermo Fisher Scientific | Cat# 23225 |
| Deposited Data | ||
| Brain RNA-Seq Database | Zhang et al., 2014 | https://www.brainrnaseq.org/ |
| Experimental Models: Cell Lines | ||
| HEK 293T | ATCC | Cat# CRL-3216 |
| Experimental Models: Mouse Strains | ||
| SIRT2 KO/B6.129-Sirt2 | The Jackson Laboratory | Cat# 012772 |
| C57BL/6J | Charles River Laboratories | Cat# 000664 |
| Oligonucleotides | ||
| SIRT2 Stealth RNAi | Thermo Fisher Scientific | 1320001 |
| Stealth RNAi siRNA Negative Control, Med GC | Thermo Fisher Scientific | 12935300 |
| Recombinant DNA | ||
| GO-ATeam2 | Nakano et al., 2011 | N/A |
| GO-ATeam2-mito | Nakano et al., 2011 | N/A |
| GO-ATeam3 | Nakano et al., 2011 | N/A |
| pMD2.G | Gift from Dr. Didier Trono (unpublished) | Addgene Plasmid #12259 |
| psPAX2 | Gift from Dr. Didier Trono (unpublished) | Addgene Plasmid #12260 |
| pCDH-SIRT2-Flag | Jing et al., 2016 | Addgene Plasmid #102624 |
| p3xFLAG-CMV-7.1 | Sigma-Aldrich | Cat #E7533 |
| pEGFP-C2 | Clontech | Cat #6083-1 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij, RRID: SCR_003070 |
| Prism 8 | GraphPad Software | https://www.graphpad.com/, RRID: SCR_002798 |
| Adobe Photoshop CC | Adobe | http://www.adobe.com/, RRID:SCR_014199 |
Highlights:
Oligodendrocyte-to-neuron signaling enhances axonal mitochondria ATP production
Elevating neuronal deacetylase SIRT2 facilitates mitochondria ATP production
SIRT2 is transcellularly delivered from oligodendrocytes to axons via exosomes
In vivo delivery of OL-EXOs rescues axonal mitochondrial integrity in spinal cords
Acknowledgments
We thank Sheng lab members for technical assistance and discussion; X.-T. Cheng for model illustration; S. Cheng, S. Lara (NINDS Electron Microscopy Facility) for EM assistance; N. Morgan (NIBIB, NIH) for microfluidic device templates; C. Smith (NINDS Light Imaging Facility) and V. Schram (NICHD Microscopy and Imaging Core) for imaging support; T. Costain, M. Barzik, and L. Cunningham (NIDCD, NIH) for assistance with NTA, and H. Imamura (Kyoto University) for GO-ATeam2/3. This work was supported by the Intramural Research Program of NINDS, NIH ZIA NS003029 and ZIA NS002946 (Z-H.S.), and NINDS Competitive Fellowship Award (K.A.C.).
Footnotes
COMPETING INTERESTS STATMENT
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson KA, and Hirschey MD (2012). Mitochondrial protein acetylation regulates metabolism. Essays Biochem 52, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, Jacob Filho W, Lent R, and Herculano-Houzel S (2009). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 513, 532–541. [DOI] [PubMed] [Google Scholar]
- Baeza J, Smallegan MJ, and Denu JM (2016). Mechanisms and Dynamics of Protein Acetylation in Mitochondria. Trends Biochem Sci 41, 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, and Waxman SG (1988). The perinodal astrocyte. Glia 1, 169–183. [DOI] [PubMed] [Google Scholar]
- Budnik V, Ruiz-Canada C, and Wendler F (2016). Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 17, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camandola S, and Mattson MP (2017). Brain metabolism in health, aging, and neurodegeneration. EMBO J 36, 1474–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM, and Mahad DJ (2011). Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 69, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano M, and O'Driscoll L (2020). Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J Extracell Vesicles 9, 1703244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain KA, Nanescu SE, Psachoulia K, and Huang JK (2016). Oligodendrocyte regeneration: Its significance in myelin replacement and neuroprotection in multiple sclerosis. Neuropharmacology 110, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain KA, and Sheng ZH (2019). Mechanisms for the maintenance and regulation of axonal energy supply. J Neurosci Res 97, 897–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, et al. (2016). Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab 23, 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou N-T, and Ansel KM (2016). Improved exosome isolation by sucrose gradient fractionation of ultracentrifuged crude exosome pellets. Protocol Exchange 1–4. [Google Scholar]
- Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, Beal MF, Bergersen LH, Brinton RD, de la Monte S, et al. (2020). Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov 19, 609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, and Kittler JT (2018). Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci 19, 63–80. [DOI] [PubMed] [Google Scholar]
- Dincman TA, Beare JE, Ohri SS, and Whittemore SR (2012). Isolation of cortical mouse oligodendrocyte precursor cells. J Neurosci Methods 209, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doretto S, Malerba M, Ramos M, Ikrar T, Kinoshita C, De Mei C, Tirotta E, Xu X, and Borrelli E (2011). Oligodendrocytes as regulators of neuronal networks during early postnatal development. PLoS One 6, e19849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McCulloch MC, Montague P, Brown AM, Thilemann S, Pratola L, Gruenenfelder FI, Griffiths IR, and Nave KA (2010). Demyelination and axonal preservation in a transgenic mouse model of Pelizaeus-Merzbacher disease. EMBO Mol Med 2, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Werner HB, McCulloch MC, Barrie JA, Brown A, Faichney AB, Snaidero N, Nave KA, and Griffiths IR (2009). Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia 57, 1815–1824. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, et al. (2004). Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol 166, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essandoh K, Yang L, Wang X, Huang W, Qin D, Hao J, Wang Y, Zingarelli B, Peng T, and Fan GC (2015). Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim Biophys Acta 1852, 2362–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettle B, Schlachetzki JCM, and Winkler J (2016). Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Mol Neurobiol 53, 3046–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Baeza J, and Denu JM (2013). Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem 288, 31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran DR, and Peterson AC (1992). Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. J Neurosci 12, 4890–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcade S, Morato L, Parameswaran J, Ruiz M, Ruiz-Cortes T, Jove M, Naudi A, Martinez-Redondo P, Dierssen M, Ferrer I, et al. (2017). Loss of SIRT2 leads to axonal degeneration and locomotor disability associated with redox and energy imbalance. Aging Cell 16, 1404–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich D, Kuo WP, Fruhbeis C, Sun JJ, Zehendner CM, Luhmann HJ, Pinto S, Toedling J, Trotter J, and Kramer-Albers EM (2014). Multifaceted effects of oligodendroglial exosomes on neurons: impact on neuronal firing rate, signal transduction and gene regulation. Philos Trans R Soc Lond B Biol Sci 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruhbeis C, Frohlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, Kirchhoff F, Mobius W, Goebbels S, Nave KA, et al. (2013). Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol 11, e1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruhbeis C, Kuo-Elsner WP, Muller C, Barth K, Peris L, Tenzer S, Mobius W, Werner HB, Nave KA, Frohlich D, et al. (2020). Oligodendrocytes support axonal transport and maintenance via exosome secretion. PLoS Biol 18, e3000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Mobius W, et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, et al. (1998). Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280, 1610–1613. [DOI] [PubMed] [Google Scholar]
- Han Q, Xie Y, Ordaz JD, Huh AJ, Huang N, Wu W, Liu N, Chamberlain KA, Sheng ZH, and Xu XM (2020). Restoring Cellular Energetics Promotes Axonal Regeneration and Functional Recovery after Spinal Cord Injury. Cell Metab 31, 623–641 e628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, and Attwell D (2012). The energetics of CNS white matter. J Neurosci 32, 356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, and Attwell D (2012). Synaptic energy use and supply. Neuron 75, 762–777. [DOI] [PubMed] [Google Scholar]
- Hessvik NP, and Llorente A (2018). Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 75, 193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Li S, Xie Y, Han Q, Xu XM, and Sheng ZH (2021). Reprogramming an energetic AKT-PAK5 axis boosts axon energy supply and facilitates neuron survival and regeneration after injury and ischemia. Curr Biol 31, 3098–3114 e3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubley MJ, Locke BR, and Moerland TS (1996). The effects of temperature, pH, and magnesium on the diffusion coefficient of ATP in solutions of physiological ionic strength. Biochim Biophys Acta 1291, 115–121. [DOI] [PubMed] [Google Scholar]
- Jayasena T, Poljak A, Braidy N, Zhong L, Rowlands B, Muenchhoff J, Grant R, Smythe G, Teo C, Raftery M, et al. (2016). Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci Rep 6, 35391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H, Hu J, He B, Negron Abril YL, Stupinski J, Weiser K, Carbonaro M, Chiang YL, Southard T, Giannakakou P, et al. (2016). A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 29, 767–768. [DOI] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, and Sheng ZH (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadottir R, Cavelier P, Bergersen LH, and Attwell D (2005). NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438, 1162–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23, 607–618. [DOI] [PubMed] [Google Scholar]
- Kiryu-Seo S, Ohno N, Kidd GJ, Komuro H, and Trapp BD (2010). Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci 30, 6658–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugmann M, Schwab MH, Puhlhofer A, Schneider A, Zimmermann F, Griffiths IR, and Nave KA (1997). Assembly of CNS myelin in the absence of proteolipid protein. Neuron 18, 59–70. [DOI] [PubMed] [Google Scholar]
- Kramer-Albers EM, Bretz N, Tenzer S, Winterstein C, Mobius W, Berger H, Nave KA, Schild H, and Trotter J (2007). Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: Trophic support for axons? Proteomics Clin Appl 1, 1446–1461. [DOI] [PubMed] [Google Scholar]
- Kramer-Albers EM, and Hill AF (2016). Extracellular vesicles: interneural shuttles of complex messages. Curr Opin Neurobiol 39, 101–107. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, and Nave KA (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33, 366–374. [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Xiong GJ, Huang N, and Sheng ZH (2020). The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat Metab 2, 1077–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang B, Tang J, Cao Q, Wu Y, Wu C, Guo J, Ling EA, and Liang F (2007). Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J Neurosci 27, 2606–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, and Sheng ZH (2017). Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 94, 595–610 e596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Park SH, Imbesi M, Nathan WJ, Zou X, Zhu Y, Jiang H, Parisiadou L, and Gius D (2017). Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid Redox Signal 26, 849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Zhou Z, Zhang L, Meng S, Li S, and Wang X (2019). Inhibition of SIRT2 by Targeting GSK3beta-Mediated Phosphorylation Alleviates SIRT2 Toxicity in SH-SY5Y Cells. Front Cell Neurosci 13, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luders KA, Patzig J, Simons M, Nave KA, and Werner HB (2017). Genetic dissection of oligodendroglial and neuronal Plp1 function in a novel mouse model of spastic paraplegia type 2. Glia 65, 1762–1776. [DOI] [PubMed] [Google Scholar]
- Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, et al. (2012). Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep 2, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA, and Choi HB (2017). Astrocytes Provide Metabolic Support for Neuronal Synaptic Function in Response to Extracellular K(). Neurochem Res 42, 2588–2594. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, Lassmann H, and Turnbull DM (2009). Mitochondrial changes within axons in multiple sclerosis. Brain 132, 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell MM, Tomkinson EM, Nobles J, Wizeman JW, Amore AM, Quinti L, Chopra V, Hersch SM, and Kazantsev AG (2011). The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum Mol Genet 20, 3986–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, Richter N, Fan Z, Siemonsmeier G, Pivneva T, Jordan P, Steinhauser C, Semtner M, Nolte C, and Kettenmann H (2018). Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep 22, 2383–2394. [DOI] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, and Horikawa I (2005). Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16, 4623–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, et al. (2006). NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439, 988–992. [DOI] [PubMed] [Google Scholar]
- Mielke C, Lefort N, McLean CG, Cordova JM, Langlais PR, Bordner AJ, Te JA, Ozkan SB, Willis WT, and Mandarino LJ (2014). Adenine nucleotide translocase is acetylated in vivo in human muscle: Modeling predicts a decreased ADP affinity and altered control of oxidative phosphorylation. Biochemistry 53, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal A, Ashiq KA, Phulpagar P, Singh DK, and Shiras A (2019). Effective Visualization and Easy Tracking of Extracellular Vesicles in Glioma Cells. Biol Proced Online 21, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, and Brand MD (2015). The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta 1847, 171–181. [DOI] [PubMed] [Google Scholar]
- Nakano M, Imamura H, Nagai T, and Noji H (2011). Ca(2)(+) regulation of mitochondrial ATP synthesis visualized at the single cell level. ACS Chem Biol 6, 709–715. [DOI] [PubMed] [Google Scholar]
- Nave KA, and Trapp BD (2008). Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci 31, 535–561. [DOI] [PubMed] [Google Scholar]
- Nicholas D, Proctor EA, Raval FM, Ip BC, Habib C, Ritou E, Grammatopoulos TN, Steenkamp D, Dooms H, Apovian CM, et al. (2017). Advances in the quantification of mitochondrial function in primary human immune cells through extracellular flux analysis. PLoS One 12, e0170975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H, Chen H, Han J, Hong Y, Ma Y, Xia W, and Ying W (2011). Silencing of SIRT2 induces cell death and a decrease in the intracellular ATP level of PC12 cells. Int J Physiol Pathophysiol Pharmacol 3, 65–70. [PMC free article] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, and Verdin E (2003). The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11, 437–444. [DOI] [PubMed] [Google Scholar]
- Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, Ransohoff RM, Sheng ZH, Komuro H, and Trapp BD (2014). Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci U S A 111, 9953–9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oluich LJ, Stratton JA, Xing YL, Ng SW, Cate HS, Sah P, Windels F, Kilpatrick TJ, and Merson TD (2012). Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J Neurosci 32, 8317–8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak D, Berthet A, and Nakamura K (2013). Energy failure: does it contribute to neurodegeneration? Ann Neurol 74, 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piaton G, Gould RM, and Lubetzki C (2010). Axon-oligodendrocyte interactions during developmental myelination, demyelination and repair. J Neurochem 114, 1243–1260. [DOI] [PubMed] [Google Scholar]
- Raine CS (1984). On the association between perinodal astrocytic processes and the node of Ranvier in the C.N.S. J Neurocytol 13, 21–27. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Tayler J, Kaga Y, Yang Y, Lappe-Siefke C, Nave KA, and Bansal R (2005). CNP is required for maintenance of axon-glia interactions at nodes of Ranvier in the CNS. Glia 50, 86–90. [DOI] [PubMed] [Google Scholar]
- Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, and Alnemri ES (2019). Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10, 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab AS, Tzvetanova ID, and Nave KA (2013). The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol 23, 1065–1072. [DOI] [PubMed] [Google Scholar]
- Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Mobius W, Goetze B, Jahn HM, Huang W, et al. (2016). Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 91, 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MG, and Fern R (2005). NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 438, 1167–1171. [DOI] [PubMed] [Google Scholar]
- Salvadores N, Sanhueza M, Manque P, and Court FA (2017). Axonal Degeneration during Aging and Its Functional Role in Neurodegenerative Disorders. Front Neurosci 11, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH (2017). The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol 27, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, and Cai Q (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Hanson PS, and Morris CM (2017). Sirtuin-2 Protects Neural Cells from Oxidative Stress and Is Elevated in Neurodegeneration. Parkinsons Dis 2017, 2643587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EF, Shaw PJ, and De Vos KJ (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett 710, 132933. [DOI] [PubMed] [Google Scholar]
- Snaidero N, Velte C, Myllykoski M, Raasakka A, Ignatev A, Werner HB, Erwig MS, Mobius W, Kursula P, Nave KA, et al. (2017). Antagonistic Functions of MBP and CNP Establish Cytosolic Channels in CNS Myelin. Cell Rep 18, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan PY, Chen Y, and Sheng ZH (2013). Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep 4, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar IK, Li D, and Rahman I (2019). Small RNA-sequence analysis of plasma-derived extracellular vesicle miRNAs in smokers and patients with chronic obstructive pulmonary disease as circulating biomarkers. J Extracell Vesicles 8, 1684816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn TL, He Y, Jackman NA, Lobner D, Hewett JA, and Hewett SJ (2015). A Cytotoxic, Co-operative Interaction Between Energy Deprivation and Glutamate Release From System xc- Mediates Aglycemic Neuronal Cell Death. ASN Neuro 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torralba D, Baixauli F, and Sanchez-Madrid F (2016). Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front Cell Dev Biol 4, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisiol A, Saab AS, Winkler U, Marx G, Imamura H, Mobius W, Kusch K, Nave KA, and Hirrlinger J (2017). Monitoring ATP dynamics in electrically active white matter tracts. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilopoulos A, Pennington JD, Andresson T, Rees DM, Bosley AD, Fearnley IM, Ham A, Flynn CR, Hill S, Rose KL, et al. (2014). SIRT3 deacetylates ATP synthase F1 complex proteins in response to nutrient- and exercise-induced stress. Antioxid Redox Signal 21, 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Hirschey MD, Finley LW, and Haigis MC (2010). Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35, 669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. (2010). Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner HB, Kuhlmann K, Shen S, Uecker M, Schardt A, Dimova K, Orfaniotou F, Dhaunchak A, Brinkmann BG, Mobius W, et al. (2007). Proteolipid protein is required for transport of sirtuin 2 into CNS myelin. J Neurosci 27, 7717–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte ME, Bo L, Rodenburg RJ, Belien JA, Musters R, Hazes T, Wintjes LT, Smeitink JA, Geurts JJ, De Vries HE, et al. (2009). Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol 219, 193–204. [DOI] [PubMed] [Google Scholar]
- Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, and Tessier-Lavigne M (2015). Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160, 161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S, Hayes SD, Jedrychowski MP, Vyas FS, Zaganjor E, et al. (2016). Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 167, 985–1000 e1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Kidd GJ, Ohno N, Perkins GA, Ellisman MH, Bastian C, Brunet S, Baltan S, and Trapp BD (2016). Proteolipid protein-deficient myelin promotes axonal mitochondrial dysfunction via altered metabolic coupling. J Cell Biol 215, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambonin JL, Zhao C, Ohno N, Campbell GR, Engeham S, Ziabreva I, Schwarz N, Lee SE, Frischer JM, Turnbull DM, et al. (2011). Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain 134, 1901–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Du W, Acklin S, Jin S, and Xia F (2020). SIRT2 protects peripheral neurons from cisplatin-induced injury by enhancing nucleotide excision repair. J Clin Invest 130, 2953–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. (2010). Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin MY, Sun T, Chen Y, and Sheng ZH (2016). Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol 214, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Zhao L, Wang E, Dimova N, Liu G, Feng Y, and Cambi F (2012). The QKI-PLP pathway controls SIRT2 abundance in CNS myelin. Glia 60, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
List of acetylated peptides of neuronal mitochondrial proteins identified by mass spectrometry is shown in Table S1. The datasets supporting the current study are available from the Lead Contact upon request.