

Abstract
Lymph nodes (LNs) are tissues of the immune system that house leukocytes, making them targets of interest for a variety of therapeutic immunomodulation applications. However, achieving accumulation of a therapeutic in the LN does not guarantee equal access to all leukocyte subsets. LNs are structured to enable sampling of lymph draining from peripheral tissues in a highly spatiotemporally regulated fashion in order to facilitate optimal adaptive immune responses. This structure results in restricted nanoscale drug delivery carrier access to specific leukocyte targets within the LN parenchyma. Herein, a framework is presented to assess the manner in which lymph-derived macromolecules and particles are sampled in the LN to reveal new insights into how therapeutic strategies or drug delivery systems may be designed to improve access to dLN-resident leukocytes. This summary analysis of previous reports from our group assesses model nanoscale fluorescent tracer association with various leukocyte populations across relevant time periods post administration, studies the effects of bioactive molecule NO on access of lymph-borne solutes to dLN leukocytes, and illustrates the benefits to leukocyte access afforded by lymphatic-targeted multistage drug delivery systems. Results reveal trends consistent with the consensus view of how lymph is sampled by LN leukocytes resulting from tissue structural barriers that regulate inter-LN transport and demonstrate how novel, engineered delivery systems may be designed to overcome these barriers to unlock the therapeutic potential of LN-resident cells as drug delivery targets.
Keywords: Lymph node, lymphatic system, transport barrier, transport mechanism, drug delivery
Graphical Abstract
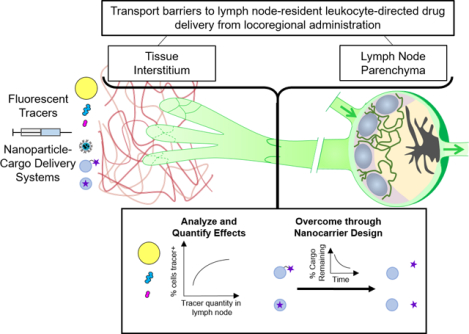
Introduction
Lymph nodes (LNs) are tissues that mediate and organize the congregation of immune cells, and are important in the priming of the adaptive immune response [1,2]. As such, LNs are tissues of significant interest for delivery of a variety of therapeutics that act on cells of the immune system, including but not limited to immunotherapies, which can benefit from direct access to the immune cell subsets they are intended to act upon [3,4]. As with any therapeutic, immunotherapy efficacy depends upon achieving sufficient accumulation at a target site [5]. Approaches that enhance delivery to cells that reside within LNs therefore have the potential to improve drug effects in the therapeutic management of a wide range of pathologies [3,6].
The LN structure is highly regulated to control antigen access to specific immune cell subsets, and just as this structure regulates the distribution of cells, immune complexes, and antigens within LNs, it also has implications on the access of therapeutic agents [7,8], including small molecules, biologics, and nano- or micro-scale drug delivery technologies. Locoregional routes of administration, which involve injection into the interstitium of peripheral tissues via intradermal (i.d.), subcutaneous, or intramuscular injection, amongst others, are commonly used for vaccination and therapeutic approaches whose effects are elicited at least in part within lymphatic tissues draining the injection site [9]. The understanding of how drug formulation may influence uptake into the lymphatic vasculature has developed substantially over the past two decades [10–12]. Their influence on distribution within the LNs, however, has only more recently been investigated. In particular, it is now well appreciated within the field of LN immune physiology that particulate and molecular access is highly regulated in a manner to coordinate optimal immune responses [13]. The influences of transport barriers on delivery have yet to be systematically assessed in the context of drug delivery systems seeking access to the cells involved in these adaptive immune responses, however. In the immunotherapy era for the treatment of a variety of diseases, the need to delineate how various immune cell subpopulations within LNs, including antigen presenting cells as well as lymphocytes, can be accessed using drug carriers continues to increase in urgency.
Here, we assess relationships between quantity of material present in the LN draining the site of injection (draining LN, dLN) and association of that material with dLN-resident leukocyte subsets through analysis of model fluorescent nanoscale material (tracer) and nanoparticle (NP) drug delivery system data sets generated and previously reported by our group [14–16]. In the first of these analyses, the effects of size-restriction at the tissue interstitium injection site and its dLN were assessed with the use of a fluorescent tracer panel [14], and results show that the influence of total tracer quantity in the dLN on cell association varies by cell subtype. Furthermore, this influence is weaker for immune cell subsets that reside farther from the subcapsular sinus (SCS) barrier. After quantifying the effects of these transport barriers, further analyses illustrate two approaches to modulate or overcome them by leveraging a NP drug delivery system with attributes optimal for prodigious uptake into and drainage by the lymphatic vasculature after locoregional administration [15–18]. The first approach explores the capacity to potentiate the effects of bioactive small molecule nitric oxide (NO), which has multiple lymphatic modulatory functions, through a lymph-draining NO delivery system in order to enhance access of lymph-borne molecular solutes to SCS-proximal lymphocytes [15]. The second demonstrates the effects of co-formulation for subsequent time-dependent in vivo release of small molecular cargo in NP-drug carrier systems that temporally optimize transport during transit through lymphatic tissues [16]. Our results establish how lymph-draining delivery systems designed to either harness specific pathways of lymphatic transport and/or disturb or overcome barriers to lymph sampling can make dLN-resident cells more “druggable”, either indirectly or directly. These results underscore how drug delivery innovations have the capacity to unlock the underleveraged therapeutic potential of LNs.
Results
Lymphatic transport mechanism regulates extent and efficiency of association with dLN leukocytes
Multiple barriers to lymphatic transport restrict access of molecules and particulates from peripheral tissues to cells resident within dLNs (Fig. 1a). Uptake into the lymphatic vasculature is affected by the competing influences of extracellular matrix pore size, permeability of the local capillary bed, and retention time in the tissue interstitium (Fig. 1a). LN structure is also organized to facilitate locally resident cells sampling lymph that is formed from interstitial fluid that drains from peripheral tissues [19–21]. This transpires in a highly regulated manner, with barrier macrophages and lymphatic endothelial cells shielding the LN parenchyma from the afferent lymph-filled subcapsular sinus, and dendritic cell populations in close proximity to the sinus to facilitate antigen sampling [22–24]. Conduits, which form channels comprised of collagen fibers ensheathed in fibroblastic reticular cells and span the inferior boundary of the SCS into the LN parenchyma, restrict access of larger lymph-borne solutes, but allow passage of small molecular solutes less than 70 kDa [25,26] (Fig. 1a).
Fig. 1. Lymphatic tissue transport barriers and analysis of their effects on access by dLN resident cells to lymph-derived molecules and particles.
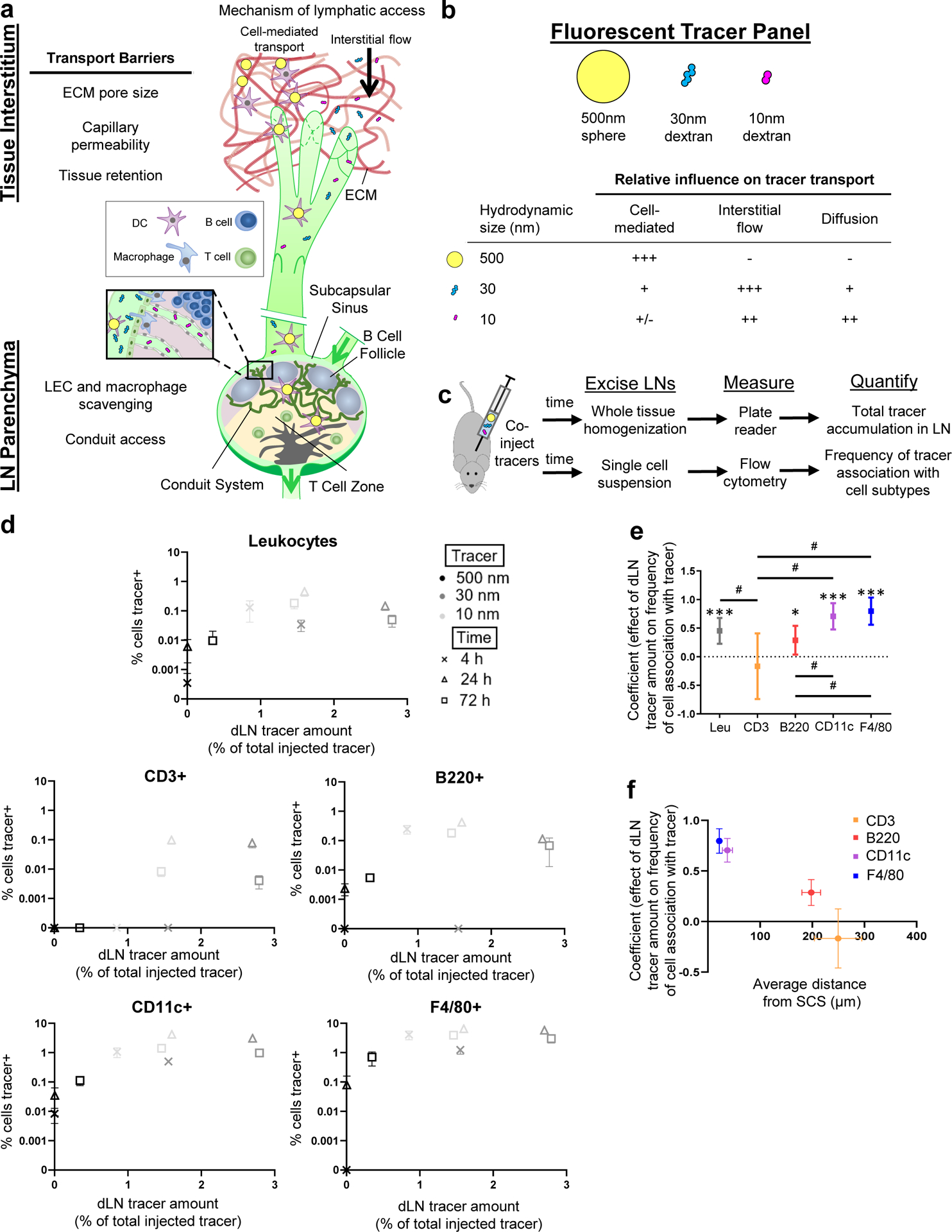
a) Schematic of transport barriers regulating access by dLN resident leukocytes to lymph-transported molecules and particles. b) Tracer panel used to quantify extents of lymphatic transport and access by dLN cells to species transited through lymphatic system by different mechanisms. c) Experimental framework used to assess total tracer accumulation in the LN and association of each tracer with leukocyte subtypes expressing various differentiation markers. d) Frequency of tracer association with dLN leukocytes expressing the indicated marker and total extent of tracer accumulation within the dLN 4, 24, and 72 h post-injection. Data are presented as mean ± standard error. e) Coefficients from beta regression analyses of d. * indicates significant difference from zero, # indicates significant difference between coefficients as determined by unpaired two-sample t test with Bonferroni correction. Coefficients are presented as mean ± standard error in confidence interval format. f) Relationship between regression coefficient relating dLN tracer accumulation with extent of tracer association with individual marker-expressing cell subtypes versus average distance of cell type from the LN subcapsular sinus (SCS), quantified by pixel distance in (n = 3) microscopy images of LN sections immunohistochemically stained with the indicated markers. Data are presented as mean ± standard error. Data in (d) to (f) are from n = 6 animals at each time point.
Our group previously reported the extent of total dLN accumulation and association with dLN-resident leukocytes of tracers fluorescently labeled to have minimal spectral overlap (Fig. 1b, Suppl. Table 1, Suppl. Table 3) simultaneously injected in the naïve skin of mice [14,27]. Tracers comprised of polymers resistant to degradation and varying by two orders of magnitude in hydrodynamic size were used as model molecules, and were restricted to uptake by the lymphatic vasculature by different transport mechanisms (Fig. 1b). Polystyrene spheres 500 nm in diameter that were too large to move freely through pores in the extracellular matrix were used to evaluate the extent of trafficking by cells from peripheral tissues to dLNs (Fig. 1b). In contrast, 500 and 40 kDa dextrans that have hydrodynamic diameters of 30 and 10 nm, respectively, were sufficiently large to be impermeable to healthy blood capillaries but small enough to move through spaces in the extracellular matrix at the injection site (Fig. 1b). Their differing access to cells resident within the dLN parenchyma resulted from the size restriction of the LN SCS and its conduits [14].
With this system, we assessed the relationships between the frequency of cell subtypes, which reside within LNs in a spatially regulated fashion [14,28], that were associated with tracer at various times post-injection and the amount of total tracer within the LN (Fig. 1c). The objective was to test the hypothesis that dLN-resident leukocyte association with lymph-derived macromolecules and/or particulates correlates with the extent of accumulation within LNs and in a manner that varies by cell subtype. Flow cytometry results for tracer association were plotted against the total amount of tracer present in the LN at each analysis time point (4, 24, and 72 h post-injection), quantified through tissue homogenate fluorescence (Fig. 1c–d).
Tracer association with dLN leukocytes (CD45+) increased with the level of total tracer that accumulated within the dLN (Fig. 1d), a result confirmed by regression (Fig. 1e, Suppl. Fig. 1a). dLN leukocytes were defined into subsets by surface markers, including macrophage marker F4/80, dendritic cell marker CD11c, and B and T lymphocyte markers B220 and CD3, respectively, revealing further correlations between accumulation and cell association (Fig. 1e, Suppl. Fig. 1a). As the data in these analyses were non-normal (Suppl. Fig. 1b), beta regression was used given its strength at regressing over continuous proportion data [29] and the superior residuals in beta compared to linear regression (Suppl. Fig. 2c–d). In so doing, dLN cells expressing F4/80, CD11c, and B220 were found to exhibit increased tracer association as tracer accumulation within dLNs increased, whereas CD3+ cells did not (Fig. 1e). We have previously hypothesized that localization within the LN tissue and proximity to the lymph-filled SCS would influence access by dLN-resident leukocytes to lymph-derived macromolecules and particles [14–16,30]. Consistent with this, the strength of the dependency of tracer association on dLN tracer amount was inversely related to distance from the SCS (R2 = 0.94, Fig. 1f). Analysis of tracer association with various leukocyte populations after 4 h in vitro cultures with cells isolated from murine LNs demonstrated that despite differences in association extent between cell types (Suppl. Fig. 2a, Suppl. Table 4), tracer association levels increased for all cell subtypes, save 10 nm tracer by highly phagocytic DCs that exhibit high tracer association levels at even low tracer concentrations (Suppl. Fig. 2b), and in particular occurring in CD3 expressing cells, in contrast to the response observed in vivo. Differences in extents of association measured in vivo thus do not appear necessarily driven by the intrinsic capacity of tracers to associate with various cell subtypes, but rather are regulated by their access to these cells within the dLN.
These relationships were further assessed by normalizing the frequency of cell-tracer association by the total amount of each tracer within the LNs, producing a metric of “efficiency” of tracer association with dLN cells. This ratio, when calculated for cell association at each measured time point at which corresponding tracer accumulation levels are measured, thus allows time, tracer, and cell subtype effects to be more easily distinguishable. Efficiency of tracer association changed for all dLN leukocytes with time post-injection to only modest, non-statistically significant extents, (Fig. 2a). Association efficiency also varied less markedly with time post-injection overall amongst individual leukocyte subtypes, and instead varied most substantially by leukocyte subtype (Fig. 2a). As would be expected due to their overall lower extents of tracer association seen in vitro (Suppl. Fig. 2), lymphocytes (identified by B220 or CD3 positivity within CD45+ parent gate) displayed a lower overall efficiency of tracer association compared to F4/80+ cells (Fig. 2a). While this difference was generally true across all time points of analysis, the effect reached statistical significance only at 24 h (Fig. 2a).
Fig. 2. Extent and efficiency of lymph-derived molecule and particle association with dLN leukocyte subtypes is time and size dependent.
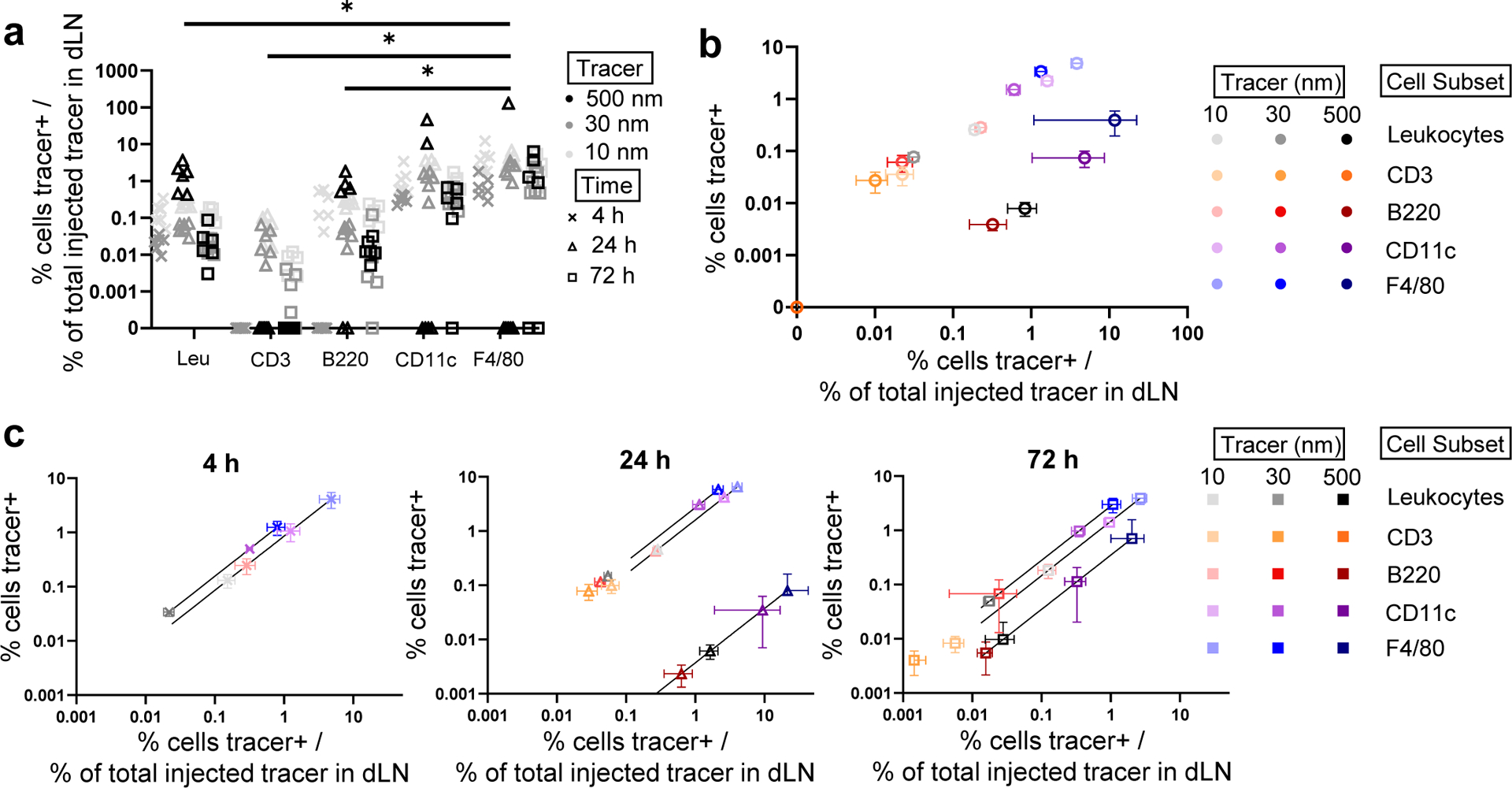
a) Efficiency of tracer association with each marker-expressing cell subset, measured as frequency of marker+ cells associated with tracer divided by total quantity of tracer present in the LN. * indicates significant difference (p < 0.05) between CD45+F4/80+ cells and all leukocytes (CD45+), as well as between CD45+F4/80+ cells and lymphocytes (CD45+CD3+ and CD45+B220+) 24 h post-injection determined by two-way ANOVA with Tukey’s comparison. b) Frequency of marker-expressing cells associated with various tracers versus the efficiency of tracer association with each marker-expressing cell subset, averaged across all analyzed times (4, 24, 72 h) post-administration. Data are presented as mean ± standard error. c) Frequency and efficiency of tracer association across all analyzed marker-expressing cell subsets at each analysis time point after injection. Data are presented as mean ± standard error. Data in (d) to (i) are from n = 6 animals at each time point.
When averaged over all measured times, the percent of tracer positive cells plotted versus the calculated efficiency of association was found to exhibit trends distinguishing the tracers and leukocyte subtypes from one another (Fig. 2b). Most notably, the 10 and 30 nm tracers associated with total dLN leukocytes to similar extents that exceeded that of the 500 nm tracer, but also demonstrated lower efficiencies than the 500 nm tracer (grayscale group, Fig. 2b). When analyzed by leukocyte subtype, these general tracer-dependent trends held, but magnitudes varied greatly (Fig. 2b). In particular, leukocytes expressing F4/80 and CD11c (blue and purple, respectively) exhibited the highest extents and efficiencies of tracer association, in sharp contrast to B220 and CD3 (red and orange, respectively) expressing dLN cells (Fig. 2b). Across all cell subsets, tracer types clustered together in their relationship between total association and efficiency of association based on their mechanism of LN access: the tracers transported to dLN via lymph drainage associated with dLN-resident cells to greater extents, whereas the tracer transported by cell-mediated trafficking, the 500 nm tracer, associated with cells to low extents but at high efficiency (Fig. 2b). These trends from data averaged across all time points (Fig. 2b) were generally consistent at each individual time point of analysis (Fig. 2c), though we note that levels of 500 nm tracer measured within dLNs at 4 h post injection were not statistically above background so no efficiencies could be calculated. Additionally, whereas the efficiency of cellular association of the 500 nm tracer at 24 h post-injection exceeded that of the 10 and 30 nm tracers, it became nearly identical at 72 h (Fig. 2c). These analyses highlight key differences in leukocyte association with model materials injected into the peripheral skin that result from different transport barriers and mechanisms in both the interstitial space and LN, and together show that the total tracer association and efficiency of tracer association with LN-resident cells is dependent on both the mechanism of tracer transport and the location of the cell subset within the LN.
Influence of lymphatic targeting of NO on lymph-borne macromolecule/particulate access to dLN leukocytes
NO, which is highly reactive and has a short half-life, is known to play a role in many in vivo processes including regulation of lymphatic vessel pumping [31–33] and vascular permeability [34,35]. Its therapeutic potential to modulate the functions of lymphatic tissues has been limited to date given that existing NO donors have low propensity to accumulate in lymph due to the poor efficiency of lymphatic uptake of small molecules. For exogenously delivered NO to take effect in the LN, a delivery system is thus required.
To this end, our group recently reported a delivery system based on S-nitrosated NP (SNO-NP) that increases NO delivery into lymphatic tissues compared to a conventional small molecule NO donor SNAP commonly used in preclinical NO research [15,18]. Effects of lymph-directed NO on association with LN-resident leukocytes of co-injected fluorescent tracers similar to the ones described above (Suppl. Table 2) were thus assessed [15]. The potential mechanisms through which lymph-directed NO may influence barriers to lymphatic uptake and dLN parenchyma access are multifaceted and span the length of lymphatic transit (Fig. 3a). At the injection site within the tissue interstitium, lymph-borne solute transport may be altered due to inflammation and modulation of capillary permeability induced by NO [34,35]; in addition, throughout lymphatic transit, pumping function of lymphatic vessels may be impacted by NO [31–33], and access within the LN may be modified due to changes in barrier cell function and immune cell activation [36,37] (Fig 3a).
Fig. 3. Lymph node-directed nitric oxide (NO) modulation of dLN leukocyte access to lymph transported tracers.
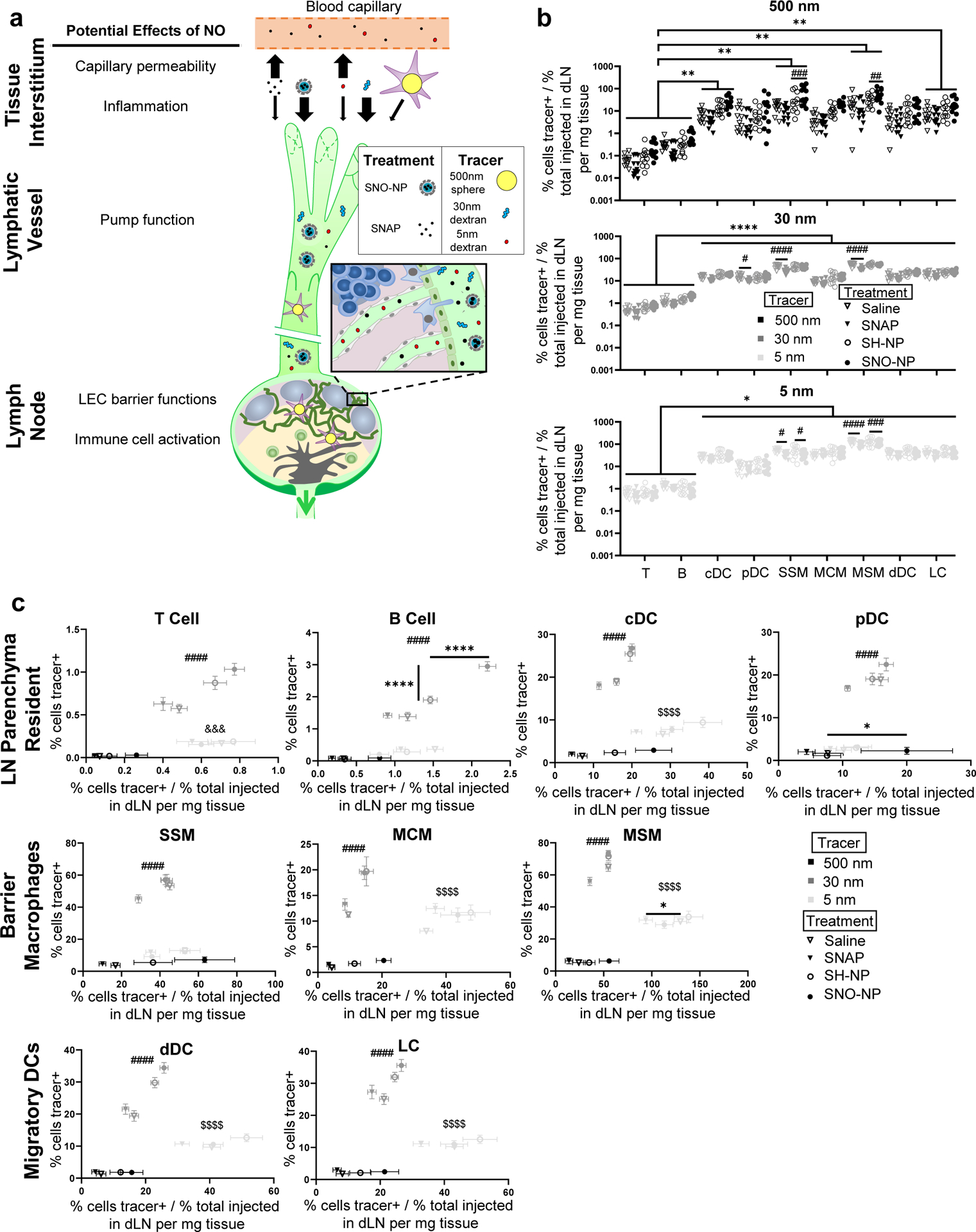
a) Schematic of potential NO effects on interstitial and lymphatic tissues that may influence the lymphatic-mediated transport of tracers to dLN from the peripheral tissue interstitum. b) Efficiency of tracer association with each dLN leukocyte subtype, measured as frequency of tracer+ cells divided by total quantity of tracer present in the LN, for each treatment. Each data point indicates an individually measured data point of tracer association normalized by the average amount of tracer accumulated within the dLN. *, **, and **** (p < 0.05, p < 0.01 and p < 0.0001, respectively) indicate trends of significantly lower efficiency of tracer association with lymphocytes than other leukocytes investigated, and #, ###, and #### (p < 0.05, p < 0.001 and p < 0.0001, respectively) indicate differences between treatment groups determined by two-way ANOVA with Tukey’s comparison. c) Frequency of tracer+ cells as a function of efficiency of tracer association with each cell type after treatment. Efficiency of tracer association is measured as average frequency of tracer association normalized by total quantity of tracer accumulated within the dLN, data are presented as mean ± standard error from n = 12 animals. * and **** indicate difference in tracer association between treatment groups (p < 0.05 and p < 0.0001, respectively). #### (p < 0.0001) indicates significant difference for 30nm vs. 5 and 500nm tracer in extent of association. &&& (p < 0.001) indicates significant difference for 5nm vs. 500nm tracer in extent and efficiency of association. $$$$ (p < 0.0001) indicates significant difference for 5nm vs. 30 and 500nm tracer in extent and efficiency of association. Significance determined by one-way ANOVA with Tukey’s comparison.
Effects of SNO-NP or small molecule NO donor (SNAP) on total levels of tracer accumulation within the dLN per LN tissue weight as well as association with dLN-resident leukocyte subtypes were compared to relevant controls (Suppl. Fig. 3), with leukocyte subsets being differentiated more granularly in this data set with respect to antigen presenting cell subtypes. These cell subtypes included LN parenchyma-resident DCs [conventional and plasmacytoid DCs (cDCs and pDCs)], barrier macrophages [SCS, medullary cord, and medullary sinus macrophages (SSM, MCM, MSM)], and migratory cells [dermal DCs and Langerhans cells (dDCs and LCs)] (Suppl. Table 5) [15]. As expected [15], the 30 nm tracer associated the most extensively with all analyzed cell types, followed by the 5 and 500 nm tracers, respectively (Suppl. Fig. 3). However, compared to these differences by tracer type, the effects of SNO-NP and SNAP treatments were far less substantial, as neither NO treatment significantly influenced extents of tracer association when compared across all cell types (Suppl. Fig. 3). Similarly, although there was a trend of SNAP treatment increasing dLN accumulation per tissue weight of all tracer sizes relative to controls, this increase was not statistically significant, and in fact neither NO treatment significantly influenced tracer accumulation within the dLN (Suppl. Fig. 3). When quantified as efficiency of total association, lymphocytes exhibited levels of association efficiency that were lower than other dLN leukocytes for the 5 nm and 30 nm tracer (Fig. 3b), consistent with our other study (Fig. 2a). This trend was generally observed for the 500 nm tracer as well, however, differences from lymphocytes were statistically significant only for cDCs, SSMs, MSMs, and LCs (Fig. 3b). When grouped by tracer type across all cell types, NO treatment (resulting from either SNO-NP or SNAP co-injection) was found to have negligible effects on the efficiency of access to tracers for the majority of dLN leukocytes; however, effects were observed for SSMs and MSMs for all tracer sizes (Fig. 3b). SNAP treatment was found to reduce efficiency of 5 nm and 30 nm tracer association with these cell types, and similarly reduced efficiency of 30 nm tracer with pDCs (Fig. 3b middle, bottom). For the 5 nm tracer, reduced efficiency of delivery was also observed for SNO-NP treatment. In contrast to the smaller tracers, SNO-NP treatment actually increased efficiency of 500 nm tracer association with SSMs and MSMs (Fig. 3b, top). Save for these differences in efficiency of delivery to barrier macrophages, considered broadly across all cell types, changes in extents and efficiencies of tracer association with leukocytes and changes in total dLN accumulation due to NO treatment thus were minimal, warranting more focused analysis controlling for leukocyte subset to probe more specific changes.
When controlling for leukocyte subtype, additional differences in the propensity for each tracer type to distribute to individual cell types as well as the subtle effects of lymph targeted or small molecule NO donor co-injection on these association profiles became more apparent (Fig. 3c). The 30 nm tracer associated the most extensively with all analyzed cell types, and while both the 5 and 500 nm tracer displayed lower extents of association, extents of 5 nm tracer association significantly exceeded that of the 500 nm tracer for all analyzed cells save B lymphocytes, pDCs, and SSMs (Fig. 3c). With respect to efficiency, the 5 nm tracer associated the most efficiently with cDCs, MCMs, MSMs, and migratory DCs, but displayed similar efficiencies of association to the 30 nm tracer in dLN lymphocytes, pDCs and SSMs (Fig. 3c). Association of the 5 nm tracer with T lymphocytes, MSMs, MCMs, and even dDCs and LCs thus exceeded that of the 500 nm tracer in not only extent but also efficiency (Fig. 3c). With respect to NO treatment effects, the extent of tracer access remained unchanged for both the 500 and 5 nm tracers as a result of either SNAP or SNO-NP treatment compared to vehicle controls for all analyzed cell types (Fig. 3c). The efficiency of 500 nm tracer association with all analyzed cell types was subtly increased overall as a result of SNO-NP relative to SH-NP control treatment, but to a statistically significant extent for pDCs only (Fig. 3c). SNAP treatment resulted in no effect on the efficiency of 500 nm tracer association with any analyzed cell type (Fig. 3c). With respect to the 30nm tracer, both the extent and efficiency of association with B lymphocytes increased for SNO-NP treatment relative to controls, while this benefit was not observed for SNAP treatment (Fig. 3c, top row). T lymphocyte, pDC, dDC, and LC association with the 30 nm tracer also subtly increased with SNO-NP treatment, but not to statistically significant extents (Fig. 3c). Extents of leukocyte association with lymph-draining tracers were not observed to change for any analyzed cell type as a result of SNAP co-injection (Fig. 3c). However, this treatment did reduce the efficiency of association of 5 nm tracer with MSMs relative to saline control (Fig. 3c, middle row). The mechanism of clearance out of the tissue site of injection thus influences how lymph-directed NO effects tracer association extents and efficiencies with dLN-resident leukocytes.
Multistage delivery increases efficiency of delivery to LN parenchyma-resident leukocytes
Our group recently described the benefits afforded by lymph draining multistage delivery systems to modulate the extent, timing, and access to subpopulations of dLN-resident leukocytes [16]. In these systems, cargo is incorporated into a lymphatic draining NP nearly identical to that implemented in analysis of the effects of lymph-directed NO evaluated in Fig. 3. Rather than assessing the effects of released NO cargo on association of other co-administered agents with cells in the dLN, however, access to leukocytes of the molecular cargo itself released from NPs in a time-dependent manner (Fig. 4a) was measured. The first of these release mechanisms included a covalent linker comprised of oxanorbornadiene (OND) that decayed by retro-Diels Alder fragmentation in a manner determined by the OND chemical composition (Fig. 4a, top) [16,38,39]. The second utilized a conventional encapsulation and release mechanism driven by diffusion from the cross-linked polymer core of the NP (Fig. 4a, bottom) [16,17]. These systems were used in order to leverage the benefit of the NP system to access lymph and the benefit of the released cargo’s higher diffusivity and capacity to access the LN conduit system relative to the conduit-excluded NP upon arrival in the LN (Fig. 4b). In our previous report, we demonstrated the benefit of this multistage delivery principle on extent of small molecule delivery to the dLN and its association with lymphocytes compared to NPs that are primarily restricted to barrier macrophages and other phagocytes (Suppl. Table 6) [16]. These benefits are further delineated here by directly considering the extents of delivery to LN leukocyte subsets as a function of their approximate distance from the SCS, which shows the substantial benefit of cargo access to cells at greater distances within the LN parenchyma as compared to access of the NP carrier (Fig. 4c). This effect is more striking in the OND fragmentation-mediated multistage delivery system, but benefits to extent of lymphocyte association are evident in both systems (Fig. 4c). When considering the spatial distribution of various dLN leukocytes within LNs, this suggests that the small molecular nature of released cargo, which enables both conduit access and more rapid diffusion, confers access to cells resident within the LN parenchyma to extents far greater than the NP. However, by virtue of the unfavorable effects small molecular size has on tissue retention and therefore lymphatic uptake, overall levels of cargo fluorophore accumulating within dLNs as a fraction of total injected amount when formulated as a multistage system is lower for the low molecular weight cargo (here either rhodamine or Cy5.5) compared to the NP (Fig. 4d, abscissa). Despite this, greater access to LN parenchyma-resident leukocytes was observed for the small molecule cargo even as less of the total percent of injected cargo quantity reached the lymph node as compared to the NP carrier (Fig. 4d). B and T lymphocytes as well as pDCs displayed this effect consistently (Fig. 4d).
Fig. 4. Effects of multistage delivery into lymph on access by dLN leukocytes to particulate and small molecular cargo.
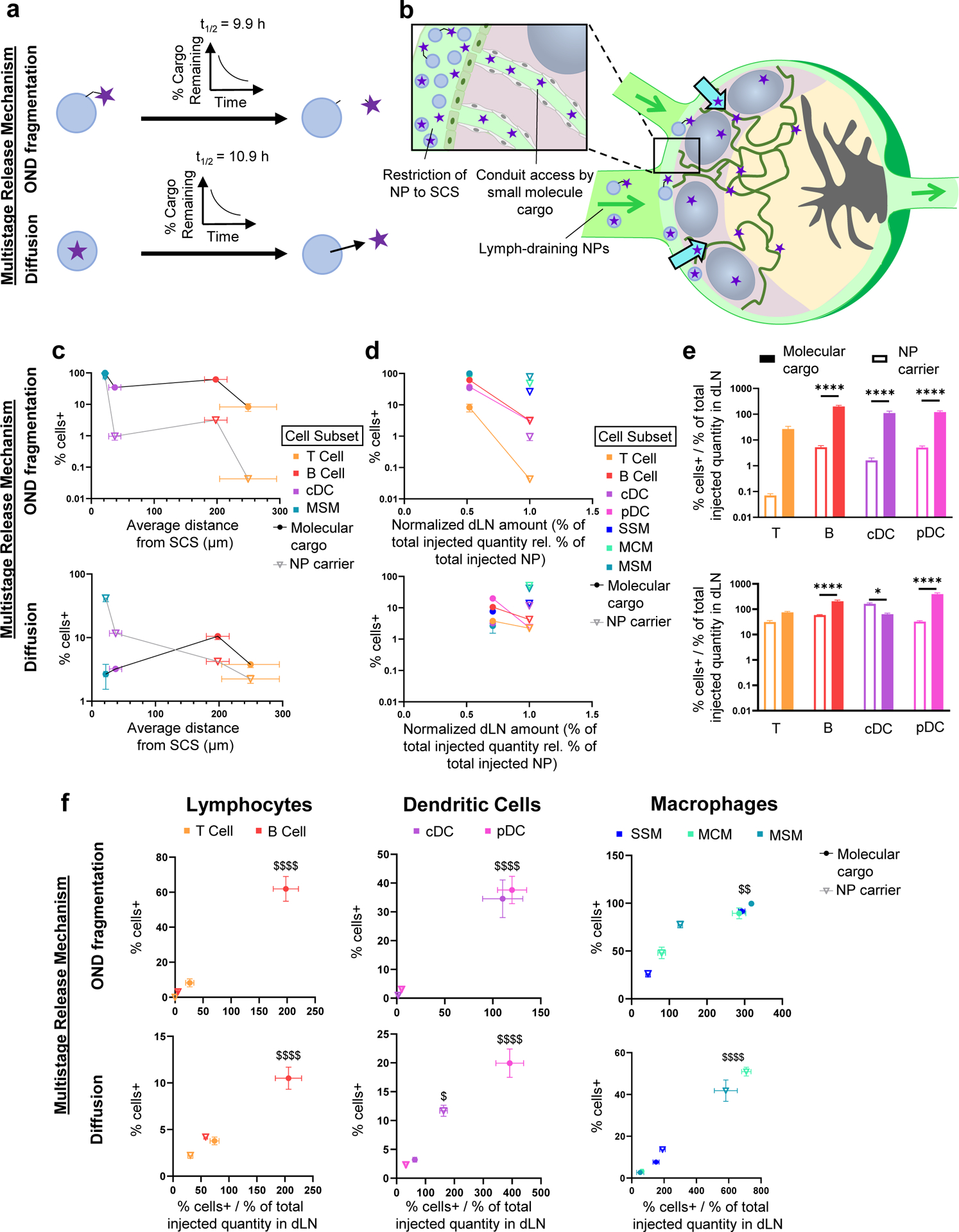
a) Schematic of multistage release systems employed with different methods of time-dependent release of small molecular cargo co-formulated using a reversible linker (OND-NP) or encapsulation (Cy5.5-NP), OND fragmentation or Diffusion, respectively. b) Schematic of multistage release systems leveraging lymph-draining NP overcoming transport barriers to delivery to dLN leukocytes resident within the LN parenchyma through cargo release within lymph. c) Frequency of fluorescently labelled NP and cargo association as a function of distance of each cell type from the SCS for each tested multistage delivery system. Data are presented as mean ± standard error. n = 6 for cell association data and 3 for distance data from SCS. d) Frequency of fluorescently labelled NP+ and cargo+ dLN leukocyte subsets versus the quantity of total NP and cargo within the dLN 24 h post-injection of the multistage release system. Lines connect the frequencies of NP and cargo uptake for LN parenchyma-resident B and T lymphocytes and pDCs. Data are presented as mean ± standard error from n = 6 animals. e) Efficiency of association of NP or small molecular cargo delivered in multistage formulation. Data are presented as mean ± standard error from n = 6 animals. Significance between groups indicated by * (p < 0.05) or **** (p < 0.0001) as determined by one-way ANOVA with Tukey’s comparison. f) Frequency vs. efficiency of association plots for groups of cell types taking up NP and multistage-released OND cargo. $, $$, and $$$$ indicate significance for molecular cargo vs. NP in extent and efficiency of association (p < 0.05, p < 0.01 and p < 0.0001, respectively) as determined by one-way ANOVA with Tukey’s comparison.
With the framework explored herein, the effects of multistage delivery on the efficiency of lymph-draining NP and co-formulated fluorophore cargo association with LN parenchyma-resident leukocyte subsets were assessed (Fig. 4e). Not only was the extent of delivery to these leukocytes higher (Fig. 4d), it was also significantly more efficient, save for efficiency of cDC association in the case of Cy5.5-NP and T lymphocyte association for both multistage systems (Fig. 4e). Specifically, multistage delivery of small molecular cargo into lymph via the lymphatic-draining NP released either via OND fragmentation or diffusion improved both the extent and efficiency of cargo as compared to NP association with dLN leukocytes in the context of delivery to B lymphocytes and pDCs, and to cDCs and barrier macrophages in the case of OND-mediated NP release (Fig. 4f). These results demonstrate the effectiveness of multistage delivery platforms in improving access of small molecule cargo to lymphocytes residing in the LN parenchyma farther from the SCS.
Discussion
Improving drug delivery to LNs has long been assumed to augment delivery to cells localized within this tissue. Evidence mounting over several years and underscored recently with the demonstrated benefit of engineered delivery systems on LN drug delivery have highlighted the limits to this assumption [16]. Leveraging two standard methods of in vivo biodistribution analysis used in tandem, relationships between delivery extent and efficiency of association of tracers or drug delivery systems and their cargo were evaluated. Multiple studies with varying objectives but structured with a similar underlying hypothesis – that various mechanisms of interstitial and lymphatic transport interdependently contribute to molecular and/or particulate distribution patterns within dLNs – were elaborated. Our analyses expand the perspective on how drug delivery innovations may be implemented to improve delivery to dLN-resident leukocytes.
Two new analyses of biodistribution data were implemented in this work to quantitate competing but related design considerations surrounding how materials are sampled by cells within LNs. Each offers differing insights and should be interpreted as a qualitative assessment of material access to various dLN leukocytes that can be used to inform design criteria governing distribution patterns in the LN on a comparative basis. Put another way, calculated values relative to one another can be more meaningful than the values themselves. First, the regression method interrogates the relationship of cell association extent with an administered material to its total level of accumulation within the LN. While analyzed data are fractions and thus bounded, regressed data would differ substantially if studies had been performed using different total administered tracer amounts. Calculated regression coefficients thus provide meaning only to assess whether a relationship exists and to assess the strength of said relationship relative to coefficients for other cell subsets. Second, the extent of tracer association with individual dLN leukocytes are normalized to quantity of tracer reaching the LN to account for how accumulation extents by materials within LNs may differ between various cargos or analysis time points. This efficiency metric thus approaches zero under conditions when either levels of tracer association with cells or total accumulation extents within the LN are low. It is also hypothetically unbounded, as the upper limit is determined by the limit of detection within tissue homogenates, which varies by fluorophore/tracer pair, but can reach down to picomolar values. Conditions under which both cell association and total accumulation extent are low would thus yield values that would indicate highly efficient delivery, despite total delivery being lowly effective. These efficiency calculations are presented in tandem with the total delivery extent to individual cell types to contextualize the competing design considerations of efficiency with effectiveness. We also note that the denominator for each calculated efficiency is the same for each cell type under each time point and tracer condition. Calculated efficiencies thus quantitate how the relative efficiency of access amongst various cell types change over time or material type. These analyses provide insight into the relative extent of delivery to leukocyte subsets within the LN for lymph-derived materials and identify influences and limitations on delivery to leukocytes for consideration in drug delivery design.
Leveraging three different tracer types designed to be transited out of the site of injection and variably distribute within the dLN after injection, we demonstrate that accumulation within the dLN as a result of lymphatic transport increases tracer association with leukocytes resident within the dLN. The dependency of this association on proximity of resident cells to the lymph-filled SCS, however, was revealed by subsetting dLN cells by flow cytometrically analyzed expression of various leukocyte markers. Specifically, this dependency was found to exist for F4/80, CD11c, and B220 but not CD3 expressing cells. These trends could be interpreted to be consistent with the former trio’s general phagocytic and antigen presenting functions [40–42]. However, in vitro, association across all tested cell subtypes increased with tracer amount, suggestive of these differences not being solely dependent on their biology. Instead, the strength of the relationship between tracer association and concentration within dLNs in vivo scaled with the mean subpopulation distance from the SCS, suggesting that this deficit is related to transport barriers that exist within the dLN that restrict free access of lymph contents to T lymphocytes. While total tracer association with lymphocytes is lower than that of DCs and macrophages in vitro, lymphocytes are still able to show appreciable increases in tracer association as incubated tracer quantity increases because their access to tracer is not inhibited by the presence of LN transport barriers. These results support the concept that T lymphocytes are poorly accessible by lymph-borne species and, given their high importance in numerous immunomodulatory therapeutic approaches, motivate the need for drug delivery innovations to effectively target this cell type.
Our results illustrate that the mechanism of transport dramatically influences not only the extent of delivery but also efficiency of dLN leukocyte access by lymphatic transported tracers, findings relevant to the design of immunotherapeutic drug carriers as well as immune physiology. Specifically, tracers that accumulate within the lymphatic vasculature via passive drainage accumulate to the greatest extent compared to tracers that are trafficked via antigen presenting cells. As a result, the efficiency of delivery with respect to the frequency of cells that are positive for tracers transited in the latter manner is far greater. This suggests that drug carrier systems that rely on uptake in peripheral tissues by antigen presenting cells for delivery to LNs may be far more specific in the types of dLN-resident cells they can mediate direct drug signaling to due to the lower overall quantity of these carriers reaching the LN, the nature of cell-guided transport, and the exchange of trafficked cargo that occurs once delivered into the LN. The total extent of delivery will be far lower, however, compared to that which can be achieved by carriers that drain into lymph directly. In various applications, the benefits of selectivity versus extent of delivery may be differently balanced. It also suggests that antigen presenting cells that traffic to dLN that we have recently shown to be highly efficacious in eliciting robust CD8+ T cell immunity [14] elicit their immunomodulatory effects in a manner that may be interpreted as being far more efficient compared to antigen cues delivered into lymph directly. Put another way, antigen presenting cells that migrate to the dLN do so at low levels but nevertheless elicit robust effects. This may be beneficially leveraged in immunotherapy contexts to further support or even counter immunomodulatory signals co-draining to the same LN but directly sampled from lymph by resident cells.
We note that in the studies whose further analyses are presented in Figures 1–2 and Figure 3, O’Melia et al [14] and Sestito et al [15], respectively, the smallest tracers used were implemented to achieve different ends. In the former, the objective was to assess influence of conduit access on distribution patterns of lymph-draining tracers on association. A tracer 10 nm in hydrodynamic size was hence utilized as it is larger than the permeability limit of the blood capillaries of healthy tissues (~5 nm) but small enough (<70 kDa) to access the LN conduit system [9,26]. In the latter, a 5 nm tracer that is permeable to the blood capillaries was used in order to evaluate the effects of NO on the capillary function at the tissue site of injection. As a result of their differing abilities to cross the capillary barrier and therefore capacity to be retained at the tissue site post-injection [43], the overall extents of 5 versus 10 nm tracer accumulation within dLNs differed substantially, yet their access to dLN leukocyte populations were similar. As such, the 10 nm tracer tended to exhibit high effectiveness of dLN leukocyte association that exceeded that of 30 nm tracer that is perhaps best studied for its lymph draining properties [14]. The 5 nm tracer was likewise relatively efficient in these patterns of association, resulting not from superior access necessarily but instead by its low levels of dLN accumulation since, overall, its extent of delivery was poorer than the 30 nm tracer [15]. Future studies should further elaborate how leukocyte access within LNs is influenced by differences in cargo or carrier diffusivity independently from total levels of accumulation with dLNs.
Our analyses reveal that mechanisms of clearance out of the tissue site of injection result in distinct profiles of tracer association extents and efficiencies with LN leukocytes in a manner that can be modulated by NO co-injection, but only when NO is targeted into lymph. Specifically, 30 nm tracers that transit the interstitium in a manner governed by interstitial flow associate with leukocytes resident within dLN to the greatest extents but not the greatest efficiencies compared to tracers that are highly diffuse and freely permeable to the endothelium or are trafficked to the LN via migratory antigen presenting cells (5 and 500 nm tracers, respectively). Between the highly diffuse 5 nm tracer and cell-mediated 500 nm tracer, efficiencies of association also differed in some cell types, with the efficiency of 5 nm tracer association significantly higher for T lymphocytes, cDCs, MCMs, MSMs, and migratory DCs, likely influenced most heavily by their very low total access to these cell subsets. Lymph-directed NO was also found to increase the efficiency of cell-trafficked payloads to pDCs, SSMs, and MSMs, and the efficiency of lymph-draining payloads to B cells within dLNs. Targeting NO into lymphatic tissues therefore modulates barriers intrinsic to both antigen presenting cell trafficking and passive lymphatic transport. These findings therefore support the concept that modulation of the barrier and transport functions of lymphatic tissues is an approach that can be leveraged to influence signaling of dLN cells in response to lymph-borne molecules or drug carriers [44].
We have recently proposed a general drug delivery principle to overcome barriers to delivery to the LN parenchyma based on formulation of molecular cargo into lymph-draining NP via multistage formulations [16]. In so doing, these methods dramatically improve access of small molecules to these LN parenchyma-resident cells in a temporally regulated manner dependent on release half-life [16]. This translates into not only an improvement in the extent of small molecule delivery, but also an improvement in efficiency compared to that achieved by a particulate formulation. NPs similar in hydrodynamic diameter to 30 nm tracers exhibit similar patterns of decreasing access to LN regions containing lymphocytes which are further from the lymph-laden SCS, a phenomenon discussed in Figure 1 as more likely related to dLN transport barriers than intrinsic cell uptake/association ability. In contrast, substantial association of small molecule cargo with T and B lymphocytes farthest from the SCS underscores the ability of multistage drug delivery systems to overcome transport barriers restricting both significant uptake of small molecules into lymphatic vessels at the tissue interstitium, and of larger particulates in the SCS from freely accessing conduits. These results demonstrate the principle that optimizing cargo transport mechanism during its lymphatic transit can control access to varying cell types and thus enhance potential efficacy, or eliminate undesired interactions, of delivered therapeutics.
Our findings expand upon existing work in the field of LN drug delivery by quantifying limitations on distribution of lymph-derived materials to lymphocytes and delineating the influences of transport mechanisms on this delivery. Nanomaterial formulations have been applied in LN delivery applications by many groups in recent years, and these approaches have been shown to enhance specific intended treatment effects [45–47]. In the majority of cases, the intended target of treatment is antigen presenting cells such as DCs [8,48], and given that subsets of these cell types are generally accessible in the more peripheral LN regions, enhancing total LN accumulation can be sufficient to improve treatment outcomes for these approaches. Our findings contextualize these outcomes and are in agreement with DC and macrophage delivery being favorable for lymph-derived particulates. However, for treatments intended to directly access lymphocytes, the restrictive nature of LN structure [23,25,26] can be a substantial obstacle to delivery treatment effects [16]. Specifically, T lymphocyte targeted treatment approaches are far less prevalent and may require active targeting approaches to be successful [49,50]. By leveraging an understanding of the transport limitations to T lymphocytes quantified in this work and engineering drug delivery systems around this, our results provide an effective lymphatic delivery approach for enhanced lymphocyte access that serves as an additional option for lymphocyte targeted therapeutics.
Limitations of this study include the restriction of our data to timeframes extending only to 72 h post administration, and so does not account for transport patterns within the LN that may occur as a result of longer term retention [51]. These results are instructive for delivery of small molecule drugs intended to act over the course of days, but do not capture effects of cell association over longer periods of time, which may be of additional interest in vaccination applications [52]. In addition, differences in lymph-borne solute access to LN leukocytes may be expected for delivery methods that have properties intended to activate or target specific cell types [53]. Put more simply, effects of retention have not been elaborated. Therefore, while the patterns we observe in model tracer material association with leukocyte subsets generally hold true, drug delivery systems with specific cell targets are expected to deviate from this to some degree, but nevertheless be governed at a first order by the delivery principles explored herein. We also note that no exhaustive analyses on the effects of cargo hydrophobicity has been conducted, which may dramatically influence lymphatic uptake and distribution in lymph [54]. Thus, effects of release from NP as mediated by OND fragmentation versus diffusion should not be compared, as the doses, molecular weights, and structures of cargo differed substantially between the analyzed studies.
In summary, this study includes new, unpublished cross-comparisons of data from multiple studies, having different input materials but a conserved animal experimental model enabling comparable results. In so doing, new detailed insight into the relationship between material delivery extent to LNs versus the cells that reside within them is generated. As with the complex nature of drug delivery now widely appreciated in the field of tumor-directed delivery, LNs represent intricate, highly regulated structures that house cell types of high interest in the current immunotherapy era. We quantitatively illustrate the significance of transport barriers to important cell targets for immunomodulatory treatments within the LN, and show that these transport barriers may be overcome. These findings further fortify general principles regulating the access to dLN-resident cells and highlight opportunities for drug delivery innovations to enable new advances in therapeutic efficacy and greater spatiotemporal control over the immune response and immunomodulatory treatments.
Methods
Fluorescent tracers
For tracer experiments analyzed in Fig. 1 and 2 and described in a prior report [14], red and yellow-green carboxylate modified 0.5 μm Fluospheres were purchased from Thermo Fisher. Tetramethylrhodamine isothiocyanate (TRITC) 40 kDa dextran was purchased from Sigma-Aldrich, and 500 kDa amino-dextran was purchased from Thermo Fisher and labeled with either Alexa Fluor 700 or Alexa Fluor 647 N-hydroxysuccinimide (NHS) ester dye. 500 kDa dextran-dye conjugates made in-house were cleaned of unreacted dye by size exclusion chromatography on a Sepharose CL-6B column [14,27]. Injected tracer quantities and fluorophore labels used in analyses presented in Fig. 1 and 2 are summarized in Suppl. Table 1.
For tracer experiments involving treatment with SNO-NP, SH-NP, and SNAP analyzed in Fig. 3 and described in a prior report [15], 500 kDa dextran was either purchased pre-labeled with TRITC from Thermo Fisher or 500 kDa amino-dextran was purchased from Thermo Fisher and conjugated with Alexa Fluor 647 NHS ester dye. 10 kDa dextran was either purchased pre-labeled with Alexa Fluor 647 NHS ester dye or 10 kDa amino-dextran was purchased from Thermo Fisher and conjugated with Alexa Fluor 610 NHS ester dye. Tracers made in-house were cleaned of unreacted dye by size exclusion chromatography on a Sepharose CL-6B column [15,27]. Details of injected quantities and fluorophore labels for each tracer, as well as quantities of each treatment used in data shown in Fig. 3 are summarized in Suppl. Table 2.
NP synthesis
Poly(propylene sulfide) NPs (PPS-NPs) used in the data presented in Fig. 3 and 4 were synthesized as described previously, yielding a crosslinked hydrophobic polymer core containing reactive thiol groups which is stabilized by a Pluronic corona [30]. PPS-NPs were characterized by dynamic light scattering (DLS) for size analysis (Malvern Panalytical), NP concentration was determined by lyophilizing a known volume of NPs, and thiol concentration was determined using Ellman’s assay (Sigma). PPS-NPs were further manipulated to render SNO-NP, OND-NP, or Cy5.5-NP, described in the following sections.
SNO-NP Synthesis
In NO experiments presented in Fig. 3, SNO-NP were synthesized as previously described [15] to enable lymph-directed NO delivery. Briefly, PPS-NP were nitrosated by addition of sodium nitrite in 0.5M HCl. Excess nitrite was quenched by addition of ammonium sulfamate, and SNO-NP were cleaned on a 7 kDa Zeba column. SNO-NP were nitrosated immediately before use. SNO-NP did not contain a fluorescent label, and were injected alongside fluorescent tracers as described below.
OND-NP Conjugation
Prior to reaction with OND-cargo, NPs were core-labeled to subsaturating extents with 10mM Alexa Fluor 488 or 647 purchased from Thermo Fisher for 4 h in pH 7.4 PBS, then cleaned of free dye by CL-6B size exclusion chromatography and concentrated back to the original concentration (40 mg/mL) using 30kDa molecular weight cut-off spin filters (Millipore). Fluorophore labeled PPS-NPs (typically 1mL NPs containing ~0.02mmol remaining reactive thiols) were reacted with oxanorbornadiene (OND) linker (typically 5×10−4 mmol), which cleaves in a time-dependent manner to release fluorescent Rhodamine or Dansyl multistage cargo, for 1h at RT, pH 7.4 with mixing [16]. For OND-NPs bearing Dansyl cargo, no further purification was required given that fluorescence was only present after conjugation. For OND-NPs bearing rhodamine cargo, unreacted small molecule was cleaned from OND-NPs via PD-10 size exclusion chromatography, and the resulting purified OND-NPs were concentrated using 10kDa molecular weight cut-off spin filters (Millipore). This yields NPs that are both reversibly linked to fluorescent small molecule cargo via OND, and stably linked to fluorophore via a maleimide linkage in order to detect NP cell uptake or tissue accumulation. NP conjugations were dose-matched based on relative fluorescence prior to injection.
Encapsulation of Cy5.5 into PPS-NP Core
Prior to encapsulation of Cy5.5, NPs were core-labeled with 10mM Alexa Fluor 488 maleimide (cytometry analysis) or Alexa Fluor 555 maleimide (bulk tissue homogenate analysis) purchased from Thermo Fisher for 4 h in pH 7.4 PBS, then cleaned of free dye by CL-6B size exclusion chromatography and concentrated back to the original concentration (40 mg/mL) using 30kDa spin filters (Millipore). Cy5.5-NPs were synthesized through encapsulation of Cy5.5 dye within the hydrophobic NP polymer core by mixing 5mg/mL Cy5.5 (DMSO) at 10%v/v with 40mg/mL PPS-NP (aqueous), then cleaned of free Cy5.5 dye using three successive 7kDa molecular weight cut-off Zeba spin desalting columns [16]. The resulting Cy5.5-NPs were injected i.d. into the mouse forelimb.
In vivo biodistribution studies
Institutional guidelines and approval for the care and use of laboratory animals were followed. For tracer experiments presented in Figures 1–2, a cocktail of 500 nm sphere (19 pM), 500 kDa dextran (4.8 μM), and 40 kDa dextran (4.8 μM) was co-infused into the dorsal skin of C57Bl/6 mice (purchased at 6 weeks of age and used within two weeks of receipt) by syringe pump at ~300 nL/s in a total injection volume of 10 μL of saline (Suppl. Table 1). Mice were maintained under anesthesia via isoflurane throughout the injection process. Mice were sacrificed either 4, 24, or 72 h after injection and axial and brachial LNs were resected for tissue homogenate or flow cytometry analysis.
For tracer experiments involving treatment with SNO-NP, SH-NP, or SNAP presented in Figure 3, a fluorescent tracer cocktail of 500 nm sphere (1.2 × 108 spheres), 500 kDa dextran (23.8 μg), and 10 kDa dextran (3 μg) was co-injected into the forelimb skin with SNO-NP delivering 800 nml NO, dose-matched SH-NP, or SNAP delivering 800 nmol NO. All three fluorescent tracers and NO treatment were administered within a single bolus 40 μL injection in the forelimb (Suppl. Table 2). Mice were maintained under anesthesia via isoflurane throughout the injection process, and sacrificed 72 h after injection, at which point axillary and brachial LNs were resected for tissue homogenate or flow cytometry analysis of the fluorescent tracers.
For multistage delivery experiments presented in Fig. 4, OND-NP or Cy5.5-NP were separately injected into the forelimb skin of C57Bl/6 mice purchased at 6 weeks of age in 30 μL total volume of saline. The NP carrier core of OND-NPs were covalently labeled with Alexa Fluor 647 for cell association (flow cytometry) experiments and Alexa Fluor 488 for bulk LN fluorescence (tissue homogenate) experiments, whereas OND cargo was fluorophores Rhodamine or Dansyl in each experimental cohort, respectively. For Cy5.5-NP, the NP core was labeled with Alexa Fluor 488 in flow cytometry experiments and Alexa Fluor 555 in tissue homogenate experiments, while the encapsulated cargo in both of these experiments was Cy5.5 dye. In all experiments, mice were maintained under anesthesia via isoflurane throughout the injection process and sacrificed 24 h after injection, at which point axillary and brachial LNs were resected for tissue homogenate or flow cytometry analysis.
Quantification of total fluorescence within tissue homogenates
LN homogenate analyses were performed as previously described [14,15]. Briefly, at the designated time of sacrifice after administration of fluorescent material, animals were euthanized, and axillary and brachial LNs were harvested and pooled into 1.4 mm acid-washed zirconium bead homogenization tubes containing PBS. LNs were homogenized with a FastPrep-24 automated homogenizer, then transferred to a 96-well plate to measure the fluorescence of each material in the LN homogenate. Fluorescence readings were used to calculate total percent of injected quantity of material within the LN using fluorescence standard curves in tissue homogenate. Fluorescent labels of co-injected tracers, as well as fluorescent labels covalently linked to PPS-NP core thiols and fluorescent multistage OND or Cy5.5 cargo, were selected with minimal spectral overlap, and spectral overlap was adjusted for through fluorescence compensation, such that LN accumulation of each of the injected components (tracers of different size in Fig. 1–3, or NP and released multistage cargo in Fig. 4) could be quantified independent of other co-injected components.
Flow cytometry analysis
LN processing and staining for flow cytometry analysis has been described previously for each data set [14–16]. Briefly, excised axillary and brachial LNs were pooled and incubated in collagenase D (1 mg/mL) for 1 h before being passed through a 70 μm cell strainer, transferred to a 96 well plate, Fc receptors blocked using 2.4G2 (Tonbo Biosciences, San Diego, CA), cells stained with Zombie Live/Dead fixable stain (BioLegend, San Diego, CA), stained with fluorescent antibodies, and fixed for flow cytometry. All antibodies were purchased from BioLegend. Antibody staining panels are described in each of the respective previously published reports [14–16].
Microscope image analysis
Naïve LN were frozen in OCT, sliced into 10 μm sections, immunohistochemically stained for either CD3, F4/80, B220, and CD11c, and imaged as previously described [14,16]. The average distance of each marker population from the LN SCS in immunohistochemically stained and imaged sections was measured using ImageJ and a custom MATLAB script [15].
Statistical analysis
For regression analysis applied in this manuscript, data from independent mouse studies assessing fluorescent tracer association with LN cell subsets were analyzed using beta regression implemented in Rstudio (version 1.2.5033) with the “betareg” package as well as using linear regression in Rstudio with the “lm” package. The response variable was frequency data obtained through flow cytometry analysis of signal positivity for specific cell types presented as a fraction ranging from 0 to 1. The predictor variable was tracer quantity in the dLN (measured as percent of total injected quantity). For our data sets, linear regression is inappropriate and presented only for demonstration, as beta regression is a better statistical model when the response variables are in the range of [0,1]. Data sets were split by cell type and coefficients for dLN concentration were extracted for each subset of data. Finally, statistical tests for significance of regression coefficients for each subset were performed by z-test, and significance between regression coefficients of different subsets was determined by unpaired two-sample t-test with Bonferroni correction, all in Rstudio. Unpaired two-sample t-tests with Bonferroni corrections were selected for comparisons between regression coefficients because each of the coefficients comes from a regression performed on a separate data set, making individual pairwise comparisons appropriate to adjust the unequal variance across different groups. All other statistical tests for unpaired t tests, one or two way ANOVA, and the linear regression analysis in Fig. 1f and Fig. 2c were performed in GraphPad Prism 8 (GraphPad Software Inc., La Jolla, CA). P values are denoted as follows: * = p < 0.05, ** = p < .01, *** = p < 0.001, **** = p < 0.0001, and significance was considered p < 0.05. All data are expressed as mean ± SEM unless otherwise noted.
Supplementary Material
Suppl. Fig. 3 Nitric oxide treatment influences on extent of tracer association with cells and tracer accumulation within the lymph node per tissue weight. Extent of tracer association with various lymph node leukocyte subtypes versus their measured quantity within the dLN 72 h post i.d. co-injection with vehicle controls or NO donors. Data are presented as mean ± standard error from n = 12 animals.
Suppl. Fig. 1 Regression coefficients, data distributions and regression fits of tracer frequency data sets. a) Linear regression coefficients representing the influence of increasing quantity of tracer within the dLN on tracer association with each marker-expressing cell subset. Coefficients are presented as mean ± standard error in confidence interval format. * indicates significant difference from zero (* and *** indicate p < 0.05 and p < 0.001, respectively). # indicates significant difference between coefficients as determined by unpaired two-sample t test with Bonferroni correction, p < 0.05. b) Histograms displaying distribution of the frequency of tracer+ signal in fractional form in each marker expressing cell subset. c) Q-Q plots from linear regression on each cell subset displaying deviation of standardized linear regression residuals from normality. d) Half normal plots from beta regression displaying adherence of standardized weighted beta regression residuals to confidence bands indicative of quality fit.
Suppl. Fig. 2 Immune cell association with tracer after in vitro co-incubation. a) Influence of co-incubated tracer amount on frequency of tracer association with various marker-expressing cells isolated from murine LNs incubated at 37C for 4 h. Data are presented as mean ± standard error from n = 3 replicates. b) Beta regression coefficients quantifying the relationship between increasing incubated tracer quantity and tracer association with each cell type in vitro. *** indicates coefficient significantly different from zero, p < 0.001. Coefficients are presented as mean ± standard error in confidence interval format.
Acknowledgements
This work was supported by National Institutes of Health (NIH) Grants R01CA207619 (SNT), R01CA247484 (SNT), U01CA214354 (SNT), T32GM008433 (MJO, NAR), and T32EB006343 (LFS) and the Curci Foundation (SNT). M.P.M. was supported by a National Science Foundation Graduate Research Fellowship. LFS and AS were American Heart Association Pre-doctoral Fellows. We thank Jared P. Beyersdorf for technical assistance.
Footnotes
Conflict of interest/Competing interests
Authors Archer, Sestito, Manspeaker, O’Melia, Rohner, Schudel, Mei, and Thomas declare that they have no conflict of interest in this publication.
Availability of data and code
Data and code can be made available upon request
Ethics approval
All institutional national guidelines for the care and use of laboratory animals were followed.
References
- [1].Gasteiger G, Ataide M, Kastenmüller W. Lymph node - An organ for T-cell activation and pathogen defense. Immunol Rev 2016;271:200–20. 10.1111/imr.12399. [DOI] [PubMed] [Google Scholar]
- [2].Moussion C, Girard JP. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature 2011;479:542–6. 10.1038/nature10540. [DOI] [PubMed] [Google Scholar]
- [3].Schudel A, Francis DM, Thomas SN. Material design for lymph node drug delivery. Nat Rev Mater 2019;4. 10.1038/s41578-019-0110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ryan GM, Kaminskas LM, Porter CJH. Nano-chemotherapeutics: Maximising lymphatic drug exposure to improve the treatment of lymph-metastatic cancers. J Control Release 2014;193:241–56. 10.1016/j.jconrel.2014.04.051. [DOI] [PubMed] [Google Scholar]
- [5].Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol 2020;20:321–34. 10.1038/s41577-019-0269-6.Enhancing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Thomas SN, Rohner NA, Edwards EE. Implications of Lymphatic Transport to Lymph Nodes in Immunity and Immunotherapy. Annu Rev Biomed Eng 2016;18:207–33. 10.1146/annurev-bioeng-101515-014413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang YN, Poon W, Sefton E, Chan WCW. Suppressing Subcapsular Sinus Macrophages Enhances Transport of Nanovaccines to Lymph Node Follicles for Robust Humoral Immunity. ACS Nano 2020;14:9478–90. 10.1021/acsnano.0c02240. [DOI] [PubMed] [Google Scholar]
- [8].Gerner MY, Torabi-Parizi P, Germain RN. Strategically Localized Dendritic Cells Promote Rapid T Cell Responses to Lymph-Borne Particulate Antigens. Immunity 2015;42:172–85. 10.1016/j.immuni.2014.12.024. [DOI] [PubMed] [Google Scholar]
- [9].Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev 2001;50:3–20. 10.1016/S0169-409X(01)00150-8. [DOI] [PubMed] [Google Scholar]
- [10].Gause KT, Wheatley AK, Cui J, Yan Y, Kent SJ, Caruso F. Immunological Principles Guiding the Rational Design of Particles for Vaccine Delivery. ACS Nano 2017;11:54–68. 10.1021/acsnano.6b07343. [DOI] [PubMed] [Google Scholar]
- [11].Moynihan KD, Holden RL, Mehta NK, Wang C, Karver MR, Dinter J, et al. Enhancement of peptide vaccine immunogenicity by increasing lymphatic drainage and boosting serum stability. Cancer Immunol Res 2018;6:1025–38. 10.1158/2326-6066.CIR-17-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Reddy ST, Van Der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol 2007;25:1159–64. 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- [13].Grant SM, Lou M, Yao L, Germain RN, Radtke AJ. The lymph node at a glance - how spatial organization optimizes the immune response. J Cell Sci 2020;133:1–7. 10.1242/jcs.241828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].O’Melia MJ, Rohner NA, Manspeaker MP, Francis DM, Kissick HT, Thomas SN. Quality of CD8+ T cell immunity evoked in lymph nodes is compartmentalized by route of antigen transport and functional in tumor context. Sci Adv 2020;6:1–17. 10.1126/sciadv.abd7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sestito LF, Thomas SN. Lymph-directed nitric oxide increases immune cell access to lymph-borne nanoscale solutes. Biomaterials 2020;265:120411. 10.1016/j.biomaterials.2020.120411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schudel A, Chapman AP, Yau MK, Higginson CJ, Francis DM, Manspeaker MP, et al. Programmable multistage drug delivery to lymph nodes. Nat Nanotechnol 2020;15. 10.1038/s41565-020-0679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 2014;35:814–24. 10.1016/j.biomaterials.2013.10.003. [DOI] [PubMed] [Google Scholar]
- [18].Schudel A, Sestito LF, Thomas SN. Winner of the society for biomaterials young investigator award for the annual meeting of the society for biomaterials, April 11–14, 2018, Atlanta, GA: S-nitrosated poly(propylene sulfide) nanoparticles for enhanced nitric oxide delivery to lymphatic tiss. J Biomed Mater Res - Part A 2018;106:1463–75. 10.1002/jbm.a.36348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jalkanen S, Salmi M. Lymphatic endothelial cells of the lymph node. Nat Rev Immunol 2020;20:566–78. 10.1038/s41577-020-0281-x. [DOI] [PubMed] [Google Scholar]
- [20].Moran I, Grootveld AK, Nguyen A, Phan TG. Subcapsular Sinus Macrophages: The Seat of Innate and Adaptive Memory in Murine Lymph Nodes. Trends Immunol 2019;40:35–48. 10.1016/j.it.2018.11.004. [DOI] [PubMed] [Google Scholar]
- [21].Cyster JG. B cell follicles and antigen encounters of the third kind. Nat Immunol 2010;11:989–96. 10.1038/ni.1946. [DOI] [PubMed] [Google Scholar]
- [22].Qi H, Kastenmüller W, Germain RN. Spatiotemporal Basis of Innate and Adaptive Immunity in Secondary Lymphoid Tissue. Annu Rev Cell Dev Biol 2014;30:141–67. 10.1146/annurev-cellbio-100913-013254. [DOI] [PubMed] [Google Scholar]
- [23].Gretz JE, Norbury CC, Anderson AO, Proudfoot AEI, Shaw S. Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med 2000;192:1425–39. 10.1084/jem.192.10.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gerner M, Kastenmuller W, Ifrim I, Kabat J, Germain R. Histo-Cytometry: in situ multiplex cell phenotyping, quantification, and spatial analysis applied to dendritic cell subset micro-anatomy in lymph nodes 2012;37:364–76. 10.1016/j.immuni.2012.07.011.Histo-Cytometry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, et al. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity 2009;30:264–76. 10.1016/j.immuni.2008.12.014.Conduits. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, et al. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 2005;22:19–29. 10.1016/j.immuni.2004.11.013. [DOI] [PubMed] [Google Scholar]
- [27].Rohner NA, Thomas SN. Melanoma growth effects on molecular clearance from tumors and biodistribution into systemic tissues versus draining lymph nodes. J Control Release 2016;223:99–108. 10.1016/j.jconrel.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schudel A, Chapman AP, Yau MK, Higginson CJ, Francis DM, Manspeaker MP, et al. Programmable multistage drug delivery to lymph nodes. Nat Nanotechnol 2020;15:491–9. 10.1038/s41565-020-0679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ferrari SLP, Cribari-Neto F. Beta regression for modelling rates and proportions. J Appl Stat 2004;31:799–815. 10.1080/0266476042000214501. [DOI] [Google Scholar]
- [30].Schudel A, Sestito LF, Thomas SN. nitric oxide delivery to lymphatic tissues 2019;106:1463–75. 10.1002/jbm.a.36348.S-nitrosated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Weiler M, Kassis T, Dixon JB. Sensitivity analysis of near-infrared functional lymphatic imaging. J Biomed Opt 2012;17:066019. 10.1117/1.jbo.17.6.066019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bohlen HG, Gasheva OY, Zawieja DC. Nitric oxide formation by lymphatic bulb and valves is a major regulatory component of lymphatic pumping. Am J Physiol - Hear Circ Physiol 2011;301:1897–906. 10.1152/ajpheart.00260.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, et al. Impaired lymphatic contraction associated with immunosuppression. Proc Natl Acad Sci U S A 2016;113:E5992. 10.1073/pnas.1614689113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Durán WN, Beuve AV., Sánchez FA. Nitric oxide, S-Nitrosation, and endothelial permeability. IUBMB Life 2013;65:819–26. 10.1002/iub.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, et al. S-nitrosylation of β-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell 2010;39:468–76. 10.1016/j.molcel.2010.07.013. [DOI] [PubMed] [Google Scholar]
- [36].Scallan JP, Hill MA, Davis MJ. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc Res 2015;107:89–97. 10.1093/cvr/cvv117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, et al. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol 2011;12:1096–104. 10.1038/ni.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kislukhin AA, Higginson CJ, Hong VP, Finn MG. Degradable conjugates from oxanorbornadiene reagents. J Am Chem Soc 2012;134:6491–7. 10.1021/ja301491h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kislukhin AA, Higginson CJ, Finn MG. Aqueous-phase deactivation and intramolecular [2 + 2 + 2] cycloaddition of oxanorbornadiene esters. Org Lett 2011;13:1832–5. 10.1021/ol103153f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bellomo A, Gentek R, Bajénoff M, Baratin M. Lymph node macrophages: Scavengers, immune sentinels and trophic effectors. Cell Immunol 2018;330:168–74. 10.1016/j.cellimm.2018.01.010. [DOI] [PubMed] [Google Scholar]
- [41].Savina A, Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev 2007;219:143–56. 10.1111/j.1600-065X.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- [42].Martínez‐Riaño A, Bovolenta ER, Mendoza P, Oeste CL, Martín‐Bermejo MJ, Bovolenta P, et al. Antigen phagocytosis by B cells is required for a potent humoral response. EMBO Rep 2018;19:1–15. 10.15252/embr.201846016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rohner NA, Thomas SN. Flexible Macromolecule versus Rigid Particle Retention in the Injected Skin and Accumulation in Draining Lymph Nodes Are Differentially Influenced by Hydrodynamic Size. ACS Biomater Sci Eng 2017;3:153–9. 10.1021/acsbiomaterials.6b00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sestito LF, Thomas SN. Biomaterials for Modulating Lymphatic Function in Immunoengineering. ACS Pharmacol Transl Sci 2019;2:293–310. 10.1021/acsptsci.9b00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater 2017;16:489–98. 10.1038/NMAT4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim SY, Noh YW, Kang TH, Kim JE, Kim S, Um SH, et al. Synthetic vaccine nanoparticles target to lymph node triggering enhanced innate and adaptive antitumor immunity. Biomaterials 2017;130:56–66. 10.1016/j.biomaterials.2017.03.034. [DOI] [PubMed] [Google Scholar]
- [47].Jeanbart L, Ballester M, De Titta A, Corthésy P, Romero P, Hubbell JA, et al. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol Res 2014;2:436–47. 10.1158/2326-6066.CIR-14-0019-T. [DOI] [PubMed] [Google Scholar]
- [48].Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Control Release 2006;112:26–34. 10.1016/j.jconrel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- [49].Azzi J, Yin Q, Uehara M, Ohori S, Abdi R. Targeted Delivery of Immunomodulators to Lymph Nodes 2016;46:1247–62. 10.1002/jmri.25711.PET/MRI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mchugh MD, Park J, Uhrich R, Gao W, Horwitz DA, Fahmy TM. Biomaterials Paracrine co-delivery of TGF- b and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells. Biomaterials 2015;59:172–81. 10.1016/j.biomaterials.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhang YN, Lazarovits J, Poon W, Ouyang B, Nguyen LNM, Kingston BR, et al. Nanoparticle Size Influences Antigen Retention and Presentation in Lymph Node Follicles for Humoral Immunity. Nano Lett 2019;19:7226–35. 10.1021/acs.nanolett.9b02834. [DOI] [PubMed] [Google Scholar]
- [52].Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019;177:524–40. 10.1016/j.cell.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tokatlian T, Read BJ, Jones CA, Kulp DW, Menis S, Chang JYH, et al. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science (80-) 2019;363:649–54. 10.1126/science.aat9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ke X, Howard GP, Tang H, Cheng B, Saung MT, Santos JL, et al. Physical and chemical profiles of nanoparticles for lymphatic targeting. Adv Drug Deliv Rev 2019. 10.1016/j.addr.2019.09.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig. 3 Nitric oxide treatment influences on extent of tracer association with cells and tracer accumulation within the lymph node per tissue weight. Extent of tracer association with various lymph node leukocyte subtypes versus their measured quantity within the dLN 72 h post i.d. co-injection with vehicle controls or NO donors. Data are presented as mean ± standard error from n = 12 animals.
Suppl. Fig. 1 Regression coefficients, data distributions and regression fits of tracer frequency data sets. a) Linear regression coefficients representing the influence of increasing quantity of tracer within the dLN on tracer association with each marker-expressing cell subset. Coefficients are presented as mean ± standard error in confidence interval format. * indicates significant difference from zero (* and *** indicate p < 0.05 and p < 0.001, respectively). # indicates significant difference between coefficients as determined by unpaired two-sample t test with Bonferroni correction, p < 0.05. b) Histograms displaying distribution of the frequency of tracer+ signal in fractional form in each marker expressing cell subset. c) Q-Q plots from linear regression on each cell subset displaying deviation of standardized linear regression residuals from normality. d) Half normal plots from beta regression displaying adherence of standardized weighted beta regression residuals to confidence bands indicative of quality fit.
Suppl. Fig. 2 Immune cell association with tracer after in vitro co-incubation. a) Influence of co-incubated tracer amount on frequency of tracer association with various marker-expressing cells isolated from murine LNs incubated at 37C for 4 h. Data are presented as mean ± standard error from n = 3 replicates. b) Beta regression coefficients quantifying the relationship between increasing incubated tracer quantity and tracer association with each cell type in vitro. *** indicates coefficient significantly different from zero, p < 0.001. Coefficients are presented as mean ± standard error in confidence interval format.