

Abstract
Ovarian cancers include several distinct malignancies which differ with respect to clinicopathologic features and prognosis. High-grade serous cancer is the most common histologic subtype and accounts for most ovarian cancer–related deaths. High-grade serous ovarian cancer (HGSOC) is treated with surgery and platinum-based chemotherapy, but most patients relapse and succumb to chemoresistant disease. The genetic concept of synthetic lethality, in which the synergy of mutations in multiple genes results in cell death, provides a framework to design novel therapeutic approaches to overcome chemoresistance in ovarian cancer. Recent progress in understanding the genomic architecture and hereditary drivers of ovarian cancer has shown potential for synthetic lethality strategies designed around homologous DNA repair. Clinical trials have validated high response rates for PARP inhibitors in patients with BRCA1 or BRCA2 mutations. Here we discuss the biological rationale behind targeting BRCA–PARP synthetic lethality based on genetic context in ovarian cancer and how this approach is being assessed in the clinic. Applying the concept of synthetic lethality to target non–BRCA-mutant cancers is an ongoing challenge, and we discuss novel approaches to target ovarian cancer using synthetic lethality in combination with and beyond PARP inhibitors. This review will also describe obstacles for synthetic lethality in ovarian cancer and new opportunities to develop potent targeted drugs for patients with ovarian cancer.
Introduction
In 2021, approximately 21,410 American women will receive a new diagnosis of ovarian cancer and about 13,770 women will die from the disease, according to estimates from the American Cancer Society (1). Although, ovarian cancers account for only 2.5% of all malignancies among females, most women present at initial diagnosis with advanced stage (corresponding to stages III and IV) where 5-year survival rates range 26%–42%
Epithelial ovarian carcinoma (EOC) is a heterogeneous disease classified into several major morphologic subtypes: serous carcinomas, endometrioid carcinomas, mucinous carcinomas, and clear cell carcinomas (CCC), with distinct epidemiology, etiology, morphology, and prognosis (2). Diverse genetic and epigenetic alterations distinguish tumorigenesis and disease progression among the various subtypes of EOC. High-grade serous ovarian cancers (HGSOC) are the most common and aggressive major EOC subtype, accounting for 70% to 80% of all cases (3). According to recent studies, the majority of HGSOC arise from abnormal fallopian tube epithelial (FTE) cells that undergo early genetic and epigenetic changes, which induce migration to and colonization of the ovary, usually eventually manifesting as an ovarian tumor (4). Despite tumor heterogeneity across subtypes and within single tumors, insights into the molecular biology of EOC has produced new drug targeting strategies in recent years, especially for the most common serous subtype.
Standard treatment for newly diagnosed advanced ovarian cancer incorporates cytoreductive surgery and platinum-based chemotherapy, usually combined with a taxane, and with or without concurrent and maintenance bevacizumab, a clinically approved humanized mAb against VEGF (5). The assessment and integration of predictive biomarkers to achieve optimal debulking with no residual disease (R0 resection) continues to need extensive research, also indicating that it is essential to consider tumor biology when determining choice and sequence of surgery/systemic therapy. Patients with HGSOC respond very well to initial chemotherapy, with several randomized clinical trials evaluating dose, dose density, choice of platinum and/or a taxane, schedule, route of administration, and sequence of chemotherapy agents (6). Bevacizumab has been used as an adjunct to standard chemotherapy for first-line treatment of advanced ovarian cancer and has also been utilized in second-line regimens for platinum-sensitive and platinum-resistant disease (7). While most patients respond to this treatment paradigm, advanced disease cases typically relapse after a chemotherapy-free interval ranging from 10 to 26 months, whereupon development of chemoresistant tumors is a concern. While clinical trials of molecular targeted agents have mostly focused on treatment of recurrent disease, recent trials have increasingly looked at a role for incorporating precision oncology into upfront therapy as well. (8–11). This approach uses small-molecule inhibitors to target “oncogene addictions” or oncogenic pathways integral to the survival of tumor cells to prevent the outgrowth of resistant clones to cytotoxic chemotherapy.
Over the past decade, PARP inhibitors (PARPi) have led the way in new targeted approaches in the treatment of ovarian cancer. PARP 1 and 2 enzymes play critical roles in the repair of single-strand DNA breaks (SSB) and maintenance of genomic integrity predominantly through the base excision repair (BER) pathway (12). PARPi block this DNA repair pathway by taking advantage of concomitant defects in homologous recombination repair (HRR), a pathway responsible for the high-fidelity repair of double-strand breaks (DSB) in DNA. By binding to and trapping PARP1 and PARP2 on DNA at the sites of SSBs, PARPi produce persistent SSB and collapsed replication forks, which then turn into unresolvable DSB and cause cell death. Initially, PARPi were presumed to function exclusively by inhibiting the catalytic activity of PARP1/PARP2. Later, the activity of PARP inhibitors in locking on to damaged DNA and “PARP-trapping” thereby providing an additional mechanism to catalytic inhibition that resulted in cytotoxicity was described. Select inhibitors were subsequently demonstrated to trap PARP1 and PARP2 on sites of DNA damage by means of a toxic allosteric effect (13, 14). Trapped and stabilized PARP–DNA complexes prevent DNA repair and allow degeneration of stalled replication forks into double-stranded DNA breaks, thereby killing cancer cells more effectively than catalytic inhibition. However, not all clinical-stage PARPi have equivalent PARP-trapping activity despite displaying identical capacities to inhibit PARP catalytic activity. The ability to trap PARP closely correlates with each drug's ability to kill cancer cells. Clinical PARPi can be ranked by their ability to trap PARP that parallels their cytotoxic potency (from the most to the least potent): talazoparib >> niraparib > olaparib = rucaparib >> veliparib (13, 14).
Breast related cancer antigen (BRCA) proteins BRCA1 and BRCA2 are essential to the homologous recombination (HR) pathway and inheritance of one defective copy of either of these genes predisposes individuals to breast and ovarian cancers (15). In HR-deficient cells, PARP inhibition leads to termination of two functional DNA repair pathways (HRR and BER) resulting in a reliance on the error-prone non-homologous end joining (NHEJ) for DNA repair, accumulation of genetic damage, and ultimately cell death. Approximately 15% to 30% of women with ovarian cancer carry a germline or somatic BRCA mutation, and PARPi have shown benefit among these populations in both the first- and second-line maintenance settings and treatment of recurrence (16). With our improving understanding of the HR pathway and its role in gynecologic malignancies, the potential applications of PARPi continue to expand. Currently, three FDA-approved PARPi are used in women with ovarian cancer: olaparib, rucaparib, and niraparib. Multiple completed and ongoing studies employing other PARPi such as veliparib and talazoparib have also shown encouraging results in clinical trials (NCT01472783, NCT02470585, NCT01540565, NCT01286987). The clinical success of PARPi show the potential of “synthetic lethality,” that is, when concurrent perturbation of two genes produces a lethal outcome while perturbation of either single-gene results in viable outcomes (17). Identification and characterization of robust synthetic lethal gene pairs is fundamental to exploiting synthetic lethality in cancer treatment. This review summarizes the relevance and ramifications of synthetic lethality as an engine for ovarian cancer drug target discovery and discusses potential applications for exploiting this concept for personalized medicine.
Origins of Synthetic Lethality
In 1922, Calvin Bridges made an interesting observation that simultaneous mutation of a pair of genes in a fruit fly would result in death, but mutation of either gene alone did not significantly affect survival (18). Two decades later, a similar phenomenon was confirmed by Theodore Dobzhansky (19) and John Lucchesi, also in Drosophila (20). Together, these findings contributed to the evolution of the term synthetic lethality, prompting genetic studies in yeast to understand the mechanism. In the context of cancer, a genetic alteration, such as a defect in a tumor suppressor gene, causes a second gene to be indispensable for tumor cell viability; therefore, inhibition of the second gene product should be lethal to tumor cells without affecting healthy and nonmalignant cells (Fig. 1). A synthetic lethal combination that includes a cancer-specific mutation is occasionally referred to as “nononcogene addiction” because the mutant tumor cell requires functionality of the synthetic lethal partner gene for survival (21).
Figure 1.
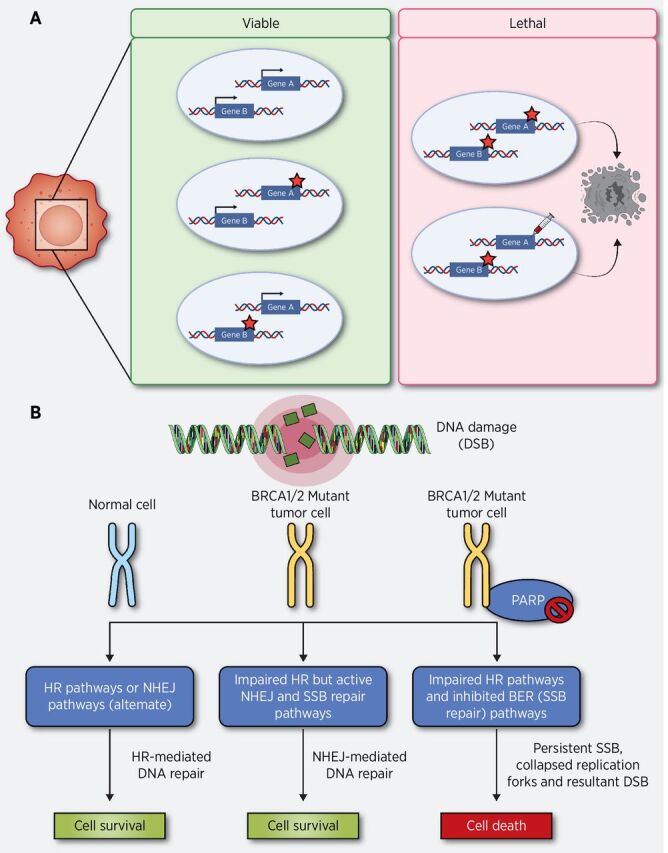
The concept of synthetic lethality and exploiting the synthetic lethal relationship between PARP1 and BRCA1/2 for the targeted treatment of HR-deficient human tumors. A, The loss, inhibition or the overexpression (either of which is denoted by a red star) of either of the protein products of gene A or B alone does not affect cell viability. Mutation, pharmacologic inhibition, or overexpression of the protein product of gene B in cells with previous mutations in gene A results in synthetic lethality-based cell death. B, HR can serve as a backup DNA repair pathway to resolve DSBs resulting from replication fork collapse. In normal cells, base modifications are repaired using base excision repair prior to S-phase entry. The newly synthesized sister chromatid can serve as a template for HR-mediated repair in BRCA1/2-proficient cells. In BRCA1/2-deficient cancer cells HR-mediated repair of PARPi-induced DSBs in not available. In this condition, tumors could still rely on the alternative error prone NHEJ pathway to repair DSBs and survive. Use of PARPi will cause unrepaired SSBs, progressive genomic instability, and synthetic lethality–based cell death in BRCA-mutated cancers.
Genetic Context in Ovarian Cancer Therapy: Advent of PARP Inhibitors
Ovarian cancer presents several challenges to precision oncology approaches to therapy. As noted, patients usually present with late-stage disease, and there is great heterogeneity both within and among tumor types. The disease is characterized by recurrent copy-number alterations, low frequency of somatic oncogenic mutations, and highly complex genomic profiles (4, 22). In addition, rather than an orderly progression from localized to systemic disease, advanced ovarian cancer also propagates via a multitude of independent “shedding” events within the peritoneal cavity (23). This heterogeneity leads to a sporadic distribution of genetically unique subpopulations across and within the tumor (spatial heterogeneity) as well as temporal alterations in the molecular architecture of cancer cells (temporal heterogeneity). For these reasons, establishing common features of the ovarian cancer genomic landscape has been essential for designing widely applicable treatment strategies.
Genomic instability is one of the common themes among HGSOC. Genomic instability in HGSOC drives the development of further variants and increases development of genetically diverse subclones within the tumor. Genomic instability is associated with treatment resistance and poor prognosis if subclones develop prosurvival characteristics (24). A mutator phenotype in cancer cells refers to mutations in genes that function to maintain genomic stability and result in accumulating unrepaired DNA damage (25). Higher levels of genomic instability are actually linked to increased response rates to platinum-based chemotherapy and PARPi and improved survival outcomes. Cells attempt to repair damaged DNA and single-strand breaks (SSB) through several mechanisms including BER, nucleotide excision repair, mismatch repair, NHEJ, and HR pathways (26), with NHEJ and HR mechanisms targeting and repairing DSBs. NHEJ causes binding proteins to attach to open ends of the DNA resulting in stabilization and induces rejoining, albeit without regard for the reading frame (27), making it error-prone. SSB repair requires the engagement of several proteins, including PARP1 and PARP2 that catalyze the addition of PAR from NAD+ molecules to proteins and to PARP1/2 themselves. PARP1 is responsible for the majority of PARylation in malignant and nonmalignant cells, which is critical to several cellular functions including DNA repair and chromatin regulation (28). In 2011, comprehensive analysis performed by The Cancer Genome Atlas (TCGA) consortium revealed that the BRCA1/2 genes play a role in up to 30% of HGSOCs, irrespective of germline status (29). Further analysis of DNA damage repair mechanisms revealed at least one mutation in genes related to HR pathways, exhibiting a “mutator phenotype” in nearly half of the tested tumors suggesting these tumors might also benefit from PARPi as a potential therapy.
Current genomic stratification of ovarian tumors reveals that about 20% of HGSOC possess a germline or somatic mutation in BRCA1/2 with smaller contributions from mutations or epigenetic silencing of other genes related to HR pathways (22). BRCA1 is known to play an important role in DSB repair via the HR pathway; BRCA1-deficient cells display impaired HR and possess an inability to repair defective chromosomes (30). In a similar manner, BRCA2 has also been implicated in HR through formation of complexes with RAD51 to provide genomic stability. Defective HR repair mechanisms, arising from deficiencies in key repair proteins such as BRCA1/2, RAD51, and CHK1, cause cells to be highly reliant on PARP activity and therefore highly sensitive to PARP inhibition (31). PARP inhibition compromises SSB repair and BER, and, in cells lacking intact HR mechanisms (e.g., BRCA1 and BRCA2 mutants), these amplify into DSBs and persistent DNA lesions, resulting in cell death. BRCA1- or BRCA2-deficient cells are, respectively, 57- and 133-fold more sensitive to PARP inhibition than wild-type cells (32). Thus, HR-deficient cells can be targeted with PARPi to further impair DNA repair mechanisms, resulting in synthetic lethality (Fig. 1). PARPi were originally proposed to function as chemosensitizers in combination with other agents; however, the level of BRCA selectivity attained with PARPi alone in preclinical studies (BRCA-mutant tumor cells were as much as 1,000 times more sensitive to PARPi than BRCA wild-type cells) provided the impetus for PARPi testing as single agents in clinical trials (32).
PARP Inhibitors as Maintenance Therapy in Recurrent Ovarian Cancer
Homologous recombination deficiency (HRD) is a pivotal determinant of platinum sensitivity in HGSOC and sensitivity to platinum correlates with sensitivity to olaparib (33). In 2014, the European Medical Agency approved olaparib as maintenance therapy for patients with platinum-sensitive recurrent serous ovarian cancer based on a series of trials that started with study NCT00753545, also called Study 19 (34). Of the 265 patients who underwent randomization, 136 were assigned to the olaparib group. Progression-free survival was significantly improved with olaparib treatment versus the placebo (8.4 months vs. 4.8 months from randomization upon completion of chemotherapy). It was also observed that regardless of subgroup, patients in the olaparib group had a lower risk of progression. The olaparib treatment cohort also reported the following adverse effects at grade 1 or 2: nausea, fatigue, vomiting, and anemia. This was followed by three large randomized phase III trials that confirmed the progression-free survival (PFS) benefit shown in Study 19 with the use of maintenance PARPi following response to platinum therapy in the recurrent setting. These studies resulted in FDA and European Medicines Agency approvals for olaparib, niraparib, and rucaparib.
Olaparib
FDA approval of olaparib was primarily based on a nonrandomized clinical trial in 298 patients with deleterious or suspected deleterious germline BRCA mutation–associated cancer and recurrent cancers, that included 193 patients with ovarian cancer (35). Patients were treated with olaparib capsules at a dose of 400 mg orally twice daily until disease progression or intolerance to therapy. Median PFS was 7 months and the proportion of patients who were progression-free at 6 months was 54.6%. After a median follow-up of 6.5 years, there was also a clinically significant improvement in overall survival (OS) in the study population (HR, 0.73; 95% CI, 0.55–0.95; nominal P = 0.021) and in patients with a BRCA mutations (HR, 0.62; 95% CI, 0.42–0.93; P = 0.021). Furthermore, responses to olaparib were seen in 30% of patients who had platinum-resistant disease, suggesting that there is not always cross resistance between platinum-resistant tumors and PARP inhibition.
Subsequent studies have expanded on the utility of olaparib in the recurrent setting. An adaptive study, Study 24 (NCT00777582), enrolling patients with advanced solid BRCA-mutated ovarian and breast tumors was carried out to determine the optimal dose of the olaparib tablet formulation for use in phase III trials and determined that the olaparib 300 mg twice daily matched or surpassed the dose of 400 mg twice daily capsules (36). The use of olaparib as maintenance therapy was demonstrated by the SOLO2 phase III trial (NCT01874353) study wherein a significant PFS improvement [19.1 months with maintenance olaparib vs. 5.5 months with placebo, HR = 0.30 (95% CI, 0.22–0.41)] with no detrimental effect on quality of life and low-grade, manageable toxicities was observed in patients with platinum-sensitive, relapsed ovarian cancer and a germline BRCA1/2 mutation who had previously received at least two rounds of chemotherapy (37). Patients were randomly assigned olaparib (300 mg in two 150-mg tablets, twice daily) or placebo tablets using an interactive voice and web response system and stratification was based on response to prior platinum-based chemotherapy (complete vs. partial) and length of platinum-free interval (>6–12 months vs. >12 months). In SOLO3, a phase III trial of olaparib tablets versus non-platinum chemotherapy in patients with germline BRCA-mutated platinum-sensitive relapsed ovarian cancer who had received at least 2 prior lines of platinum-based chemotherapy, olaparib was associated with statistically significant and clinically relevant improvements in ORR and PFS (38). Olaparib has also been tested in combination with other agents to determine efficacy as therapy for recurrent ovarian cancers. These include platinum agents such as carboplatin (39–41) and antiangiogenic agents such as cediranib (42). There are a multitude of ongoing clinical studies that aim to test usage of several FDA-approved drugs or agents in combination with olaparib for platinum-sensitive and resistant recurrent ovarian cancers.
Niraparib
Approval for niraparib maintenance therapy was based on the findings from the NOVA trial (NCT01847274; refs. 43–45). Of 203 patients with a germline BRCA mutation (gBRCAm), 99 had a partial response and 104 had a complete response to their last platinum-based therapy; of 350 patients without a confirmed gBRCAm (non-gBRCAm), 173 had a partial response and 177 had a complete response. In this randomized, double-blind, phase III trial, patients received niraparib 300 mg or placebo once daily until progression of disease or death. While there was a more substantial benefit in the germline BRCA-mutated cohort (21.0 vs. 5.5 months, HR = 0.26; 95% CI, 0.17–0.41), patients treated with niraparib in the non-gBRCAm cohort had a PFS advantage of almost 6 months as well (9.3 vs. 3.9 months, HR = 0.45; 95% CI, 0.34–0.61). Patients achieved clinical benefit from maintenance treatment with niraparib regardless of response to the last platinum-based therapy and the incidence of any-grade and grade 3 or greater adverse events was manageable. Following the NOVA trial, the principal findings were replicated among a Chinese population in the NORA trial (46). In this study, 265 patients were randomized 2:1 to receive niraparib or placebo. Notably, most patients received a dose of 200 mg daily, which was found to be a better tolerated dose than 300 mg in the NOVA trial. Again, PFS was significantly longer [18.3 vs. 5.4 months, HR = 0.32 (95% CI, 0.23–0.45)].
Rucaparib
Finally, ARIEL3 (NCT01968213) demonstrated a PFS advantage among all patients who received rucaparib maintenance therapy versus placebo [10.8 vs. 5.4 months, HR = 0·36; (95% CI, 0.30–0.45)], with a significantly pronounced effect in women with HRD [13.6 vs. 5.4 months, HR = 0.32; (95% CI, 0.24–0.32); ref. 47]. In this randomized, double-blind, placebo-controlled, phase III trial, 375 of 564 patients were allocated to oral rucaparib 600 mg twice daily in 28-day cycles using a computer-generated sequence, stratified by HR repair gene mutation status, progression-free interval after the penultimate platinum-based regimen, and best response to the most recent platinum-based regimen. The most common treatment-based adverse effects were anemia and decreased hemoglobin concentration.
The potency of PARPi as maintenance therapy for recurrent ovarian cancer was consistently demonstrated in these studies. Moreover, the overall tolerability of these drugs was determined, with familiar rare but severe adverse events such as secondary malignancies occurring at a rate of <1.5% which was equivalent across all trials. Currently, the National Comprehensive Cancer Network guidelines recommend the consideration of PARPi for all patients with platinum-sensitive recurrent ovarian cancer irrespective of BRCA status (48) and there are several studies underway testing combinations of synthetic lethality–based PARPi in varying stages of ovarian cancer (Table 1). Benefits of PARPi appear strongest in women with gBRCAm with platinum-sensitive disease. In addition, HRD causes characteristic genomic scar signatures, and the “HRD score” is the sum of these scar signature scores which correlates with sensitivity to PARPi (49–51). The HRD score is defined by determining mutations of BRCA1 and BRCA2, the methylation of their promoters, the level of expression of BRCA1 at the mRNA level, and loss of heterozygosity affecting the loci coding for BRCA1 and BRCA2. Studies are needed to validate HRD assays as predictive biomarkers for benefit to PARPi to determine and personalize the type of drug that will be the most beneficial to each patient and predict when during the treatment regimen to use them.
Table 1.
Ongoing clinical trials related to synthetic lethal interactions involving PARPi, combination of PARPi with immunotherapies, and targeting other synthetic lethal interactions in OC beyond PARPi.
| Ongoing clinical trials related to synthetic lethal interactions involving PARPi for OC | |||
|---|---|---|---|
| Trial | Phase | Design | |
| Olaparib | |||
| NCT02282020 SOLO3 | III | Randomized olaparib tablets po bid vs. physician choice single agent non–platinum-based chemo for gBRCAm platinum-sensitive relapsed HGSOC/EOC following ≥2 platinum-based chemo with progression ≥6 months after last platinum | |
| NCT02446600 | III | Randomized platinum-based chemo (carboplatin + paclitaxel; carboplatin + gemcitabine; carboplatin + PLD) vs. olaparib vs. olaparib + cediranib for platinum-sensitive relapsed HGSOC/EOC or gBRCAm HGSOC with any number of platinum-based chemo and ≤1 non-platinum therapy with CR to last platinum | |
| NCT03106987 OReO | III | Randomized olaparib vs. placebo maintenance re-treatment for relapsed nonmucinous EOC, who have had disease progression following maintenance therapy with a PARPi and a CR/PR to subsequent platinum-based chemotherapy | |
| NCT03278717 ICON 9 | III | Randomized maintenance olaparib + cediranib vs. olaparib alone for relapsed OC with disease progressed more than 6 months after first line chemotherapy or CR/PR to ≥4 cycles of platinum-based chemotherapy | |
| NCT03402841 OPINION | IIIb | Nonrandomized, noncomparative olaparib maintenance for platinum-sensitive relapsed non gBRCAm HGSOC/EOC | |
| NCT02502266 | II/III | Randomized physician choice chemo (paclitaxel; PLD; topotecan) vs. olaparib + cediranib vs. olaparib vs. cediranib for platinum-resistant or refractory relapsed, HGSOC/EOC non-gBRCAm or HGSOC gBRCAm with ≤3 prior regimens and ≤1 non-platinum | |
| NCT02340611 | II | Nonrandomized, noncomparative cediranib + olaparib after disease progression on olaparib alone in OC | |
| NCT03117933 OCTOVA | II | Randomized olaparib vs. olaparib + cediranib vs. weekly paclitaxel for BRCAm platinum-resistant OC | |
| NCT03470805 | II | Nonrandomized, noncomparative olaparib maintenance after response to trabectedin-PLD in recurrent gBRCAm or sBRCAm HGSOC/EOC | |
| NCT03161132 ROLANDO | II | Nonrandomized, noncomparative olaparib + PLD for platinum resistant advanced OC | |
| NCT03314740 BAROCCO | II | Randomized weekly paclitaxel vs. cediranib-olaparib with continuous schedule vs. cediranib-olaparib with intermittent schedule for platinum refractory or resistant recurrent HGSOC | |
| NCT03462342 CAPRI | II | Nonrandomized, noncomparative ATR inhibitor AZD6738 + olaparib for recurrent HGSOC (platinum-sensitive or -resistant) | |
| NCT03579316 | II | Randomized noncomparative adavosertib AZD1775 alone or with olaparib for recurrent OC during olaparib progression | |
| NCT04065269 ATARI | II | ATR inhibitor in combination with olaparib in gynecologic cancers with ARID1A loss | |
| NCT02345265 | II | Nonrandomized, noncomparative olaparib + cediranib for recurrent OC | |
| NCT04261465 NUVOLA | II | Neoadjuvant chemotherapy in unresectable OC with olaparib and weekly carboplatin plus paclitaxel | |
| NCT02489006 NEO | II | Randomized, neoadjuvant olaparib for platinum sensitive recurrent HGSOC prior to surgery and chemotherapy | |
| NCT02983799 | II | Nonrandomized, noncomparative olaparib for platinum-sensitive or partially platinum-sensitive, relapsed, HGSOC/EOC with at least 1 prior line of platinum-based chemotherapy, in gBRCAm, sBRCAm, or HRD subgroups | |
| NCT02889900 CONCERTO | IIb | Nonrandomized, noncomparative cediranib + olaparib for recurrent platinum resistant OC without gBRCAm | |
| NCT01116648 | I/II | To determine the safety and best dose of cediranib + olaparib for recurrent OC | |
| NCT02855697 MOLTO | I | Nonrandomized, noncomparative multi-maintenance olaparib for platinum-sensitive relapsed gBRCAm HGSOC/EOC with 2 or more courses of maintenance olaparib | |
| NCT01445418 | I | Nonrandomized, noncomparative olaparib + carboplatin for gBRCAm and sporadic OC | |
| NCT03162627 | I | Evaluation of the combination of selumetinib and olaparib in OC with Ras pathway alterations and tumors with PARP resistance | |
| NCT01623349 | I | To determine the safety of oral PI3K inhibitor BKM120 or BYL719 + olaparib for recurrent HGSOC | |
| NCT02898207 | I | To determine the safety and best dose of olaparib + HSP90 inhibitor onalespib for recurrent OC | |
| NCT01650376 | Ib | To determine the MTD of olaparib + weekly carboplatin and paclitaxel in relapsed OC | |
| NCT02208375 | Ib | To determine the MTD of olaparib + oral mTORC1/2 inhibitor AZD2014 or AKT inhibitor AZD5363 for recurrent OC | |
| NCT04586335 | Ib | Study to evaluate the safety, tolerability, pharmacokinetics and clinical activity of CYH33, a PI3K inhibitor in combination with olaparib in advanced solid tumors | |
| Niraparib | |||
| NCT02655016 PRIMA | III | Randomized niraparib po qd vs. placebo for first-line maintenance HGSOC/EOC stage III–IV with CR/PR to front-line platinum-based chemo | |
| NCT03602859 | III | Randomized comparison of platinum-based therapy with TSR-042 and niraparib vs. standard-of-care platinum-based therapy as first-line of treatment in stage III or IV EOC | |
| NCT04217798 | II | Study to evaluate the efficacy and safety of niraparib combined with oral etoposide in platinum resistant/refractory recurrent OC | |
| NCT04284852 NEOPRIMA | II | Niraparib maintenance in patients with advanced OC at neoadjuvant setting | |
| NCT04507841 | II | Niraparib for the neoadjuvant treatment of unresectable OC | |
| NCT02354586 QUADRA | II | Nonrandomized, noncomparative niraparib po qd for platinum-resistant or heavily pretreated HGSOC following 3 or 4 prior regimens, a response ≥6ms to first-line platinum-based chemo | |
| NCT04376073 ANNIE | II | Niraparib in combination with anlotinib in platinum-resistant recurrent OC | |
| NCT04826198 REVOCAN | I/II | Study to assess the safety and efficacy of AsiDNATM, a DNA repair inhibitor and niraparib in relapsed platinum-sensitive OC | |
| NCT04502602 iNNOVATE | I/Ib | Niraparib and neratinib in advanced solid tumors with an expansion cohort in platinum-resistant OC | |
| NCT03154281 | I | Evaluation of the safety and tolerability of niraparib in combination with everolimus in advanced OC | |
| NCT03586661 | I | Niraparib with the PI3K inhibitor copanlisib for recurrent OC | |
| NCT04267939 | Ib | To determine the maximum tolerated and/or recommended phase 2 dose of the ATR inhibitor BAY 1895344 in combination with niraparib in patients with recurrent OC | |
| Rucaparib | |||
| NCT02855944 ARIEL 4 | III | Randomized rucaparib po bid vs. chemotherapy (carboplatin/paclitaxel, carboplatin/gemcitabine, cisplatin/gemcitabine, paclitaxel, carboplatin, cisplatin) for relapsed or progressing BRCAm HGSOC ≥2 prior regimens | |
| NCT03522246 ATHENA | III | Randomized study evaluating rucaparib and nivolumab as maintenance treatment following response to front-line platinum-based chemotherapy in OC | |
| NCT03992131 SEASTAR | I/II | Study to assess the safety, tolerability, pharmacokinetics, and preliminary efficacy of oral rucaparib in combination with other anticancer agents in OC | |
| NCT03552471 | I | Mirvetuximab soravtansine (IMGN853) and rucaparib for recurrent OC | |
| NCT03840200 | Ib | Evaluating the safety and efficacy of ipatasertib in combination with rucaparib in advanced OC | |
| Ongoing clinical trials combining PARPi with immunotherapies for OC | |||
|---|---|---|---|
| Combinatorial drug | Trial | Phase | Design |
| Olaparib | NCT02477644 PAOLA-1 | III | Randomized olaparib vs. placebo for advanced IIIB–IV HGSOC/EOC with standard first-line platinum-taxane chemotherapy and bevacizumab concurrent and in maintenance, ≥3 cycles of bevacizumab in combination with the 3 last cycles of platinum-based chemotherapy |
| NCT03740165 | III | Randomized study of chemotherapy with or without pembrolizumab followed by maintenance with olaparib or placebo for the first-line treatment of BRCA nonmutated advanced EOC | |
| NCT03737643 DUO-O | III | Randomized multicentre study of durvalumab in combination with chemotherapy and bevacizumab, followed by maintenance durvalumab, bevacizumab and olaparib in newly diagnosed advanced OC | |
| NCT04034927 | II | Randomized olaparib vs. olaparib plus tremelimumab in platinum-sensitive recurrent OC | |
| NCT03699449 AMBITION | II | Randomized olaparib + cediranib, durvalumab + olaparib, durvalumab + chemotherapy, durvalumab + tremelimumab + chemotherapy; a biomarker-driven targeted therapy for HRD platinum-resistant recurrent OC | |
| NCT04361370 | II | Olaparib maintenance with pembrolizumab & bevacizumab in BRCA nonmutated patients with platinum-sensitive recurrent OC | |
| NCT04739800 | II | Randomized triplet therapy trial (a PD-L1 inhibitor (durvalumab) MEDI4736 in combination with olaparib and cediranib) compared to olaparib and cediranib or durvalumab and cediranib or standard of care chemotherapy platinum-resistant recurrent EOC after prior bevacizumab | |
| NCT02571725 | I–II | Nonrandomized, noncomparative olaparib and CTLA-4 blockade tremelimumab for BRCAm recurrent OC | |
| NCT02953457 | I/II | Nonrandomized, noncomparative olaparib together with durvalumab and tremelimumab for gBRCAm recurrent or refractory OC | |
| NCT02734004 MEDIOLA | I/II | Anti–PD-L1 antibody MEDI4736 in combination with olaparib in advanced OC | |
| NCT02484404 | I/II | Anti–PD-L1 antibody MEDI4736 in combination with olaparib and/or cediranib for advanced or recurrent OC | |
| NCT02121990 | I | Dose-escalation study of IP cisplatin, IV/IP paclitaxel, IV bevacizumab, and oral olaparib for newly diagnosed OC | |
| Niraparib | NCT03598270 ANITA | III | Randomized trial of platinum-based chemotherapy with or without atezolizumab followed by niraparib maintenance with or without atezolizumab in recurrent OC |
| NCT04679064 NItCHE-MITO33 | III | Randomized niraparib-TSR-042 (Dostarlimab) vs. physician's choice chemo in recurrent OC | |
| NCT04556071 AVANIRA 3 | II | Study to evaluate the efficacy and safety of niraparib combined with bevacizumab in platinum-refractory/resistant recurrent OC | |
| NCT03326193 | II | Nonrandomized, noncomparative bevacizumab-niraparib for first-line maintenance HGSOC/EOC with CR/PR to front-line platinum-based chemo + bevacizumab ≥1 debulking surgery | |
| NCT03574779 | II | Study to evaluate the safety and efficacy of novel treatment combinations (niraparib + TSR042 + bevacizumab) in recurrent OC | |
| NCT02354131 AVANOVA | I/II | Randomized niraparib vs. bevacizumab-niraparib for platinum-sensitive relapsed HGSOC/EOC | |
| NCT02657889 TOPACIO KEYNOTE 162 | I/II | Niraparib in combination with pembrolizumab (MK-3475) in recurrent OC | |
| NCT03695380 | Ib | Study of cobimetinib and niraparib, with or without atezolizumab in advanced platinum-sensitive OC | |
| NCT04673448 | Ib | Niraparib and neoadjuvant PD-1 inhibitor dostarlimab (TSR-042) in BRCAm OC | |
| Rucaparib | NCT04227522 | III | Rucaparib maintenance after bevacizumab maintenance following carboplatin-based first-line chemotherapy in OC |
| NCT02873962 | II | Safety lead-in of nivolumab in combination with bevacizumab or in combination with bevacizumab and rucaparib for relapsed EOC | |
| NCT03552471 | I | To determine the safety and best dose of mirvetuximab soravtansine and rucaparib camsylate for recurrent OC | |
| Ongoing clinical trials targeting synthetic lethal interactions in OC beyond PARPi | |||
|---|---|---|---|
| Target | Trial | Phase | Design |
| ATR | NCT02487095 | I/II | Trial of topotecan with ATRi VX-970 (M6620) in OC |
| NCT04616534 | I | Gemcitabine combined with the BAY 1895344 ATRi with expansion cohorts in advanced OC | |
| NCT02627443 | I | Dose escalation and expansion cohort of carboplatin and gemcitabine with or without ATRi berzosertib M6620 (VX-970) in first or second recurrence platinum-sensitive EOC | |
| Chk1/2 | NCT02203513 | II | Study of the Chk1/2 inhibitor (LY2606368) In BRCA1/2m HGSOC |
| NCT02797964 | I/II | Chk1 inhibitor (SRA737) administered orally in BRCA1/2m advanced OC | |
| WEE1 | NCT02101775 | II | Randomized trial comparing gemcitabine monotherapy to gemcitabine in combination with WEE1 inhibitor (MK-1775) in recurrent platinum-resistant EOC |
| PI3K | NCT04711161 | I/Ib | Evaluation of the safety, pharmacokinetics and efficacy of GRN-300, a salt-inducible kinase inhibitor, alone and in combination with paclitaxel, in recurrent OC |
| NCT03719326 | I/Ib | Dose-escalation, and dose-expansion study to evaluate the safety, tolerability, pharmacokinetics and clinical activity of etrumadenant (AB928) in combination with pegylated liposomal doxorubicin (PLD) with or without PI3K inhibitor IPI-549 in OC | |
| Akt | NCT04374630 PROFECTA II | II | Study to assess the efficacy and safety of Akt inhibitor afuresertib plus paclitaxel vs. paclitaxel in platinum-resistant OC |
| p53 | NCT03113487 | II | Modified vaccinia virus ankara vaccine expressing p53 and pembrolizumab in recurrent OC |
| NCT02272790 | II | Study of adavosertib plus chemotherapy in p53 mutated platinum-resistant OC | |
| NCT04489706 | N/A | Trial to evaluate the efficacy, safety, and tolerability of arsenic trioxide in recurrent metastatic OC with P53 mutation | |
| ARID1A | NCT04493619 | I/II | Study of BET inhibitor PLX2853 monotherapy in ARID1A-mutated advanced gynecologic malignancies and study of PLX2853/carboplatin combination in platinum-resistant EOC |
Abbreviations: bid, twice daily; HRD, homologous recombination deficiency; IP, intraperitoneal; IV, intravenous; OC, ovarian cancer; po, orally; qd, once daily.
PARPi in First-Line Therapy
On the basis of the success of PARPi maintenance therapy after treatment for recurrent HGSOC, use in the frontline was investigated (52). SOLO1 (NCT01844986) is an international, randomized, double-blind, phase III trial that demonstrated a substantial PFS benefit with the use of olaparib tablets (300 mg, twice daily) in patients with newly diagnosed advanced HGSOC or endometrioid ovarian cancer with germline or somatic BRCA 1/2 mutations who had a complete or partial clinical response after platinum-based chemotherapy (53). After a median follow-up of 41 months, the risk of disease progression or death was 70% lower with olaparib than with placebo [HR = 0.30; (95% CI, 0.23–0.41)], with the median PFS not yet reached (vs. 13.8 months for placebo). A sensitivity analysis performed to assess for attrition bias showed that the median PFS was approximately 36 months longer in the olaparib group compared with the placebo group. Despite the longer duration of treatment, the safety profile of olaparib in the SOLO1 trial was consistent with that observed in the SOLO2 trial in patients with relapsed disease. On the basis of the SOLO1 study, the first FDA approval for PARPi in a first-line setting was granted for olaparib in December 2018. Similarly, PRIMA (NCT02655016) compared niraparib maintenance with placebo following chemotherapy for newly diagnosed ovarian cancer (54). Of the 733 patients who underwent randomization, 373 (50.9%) had HR-deficient tumors. The PFS among women with HRD compared with placebo was 21.9 months versus 10.4 months [HR = 0.43; (95% CI, 0.31–0.59], with a PFS advantage of 13.8 months (niraparib) vs. 8.2 months (placebo) (HR, 0.62; 95% CI, 0.50–0.76) in the overall population. Target enrollment has been completed for the phase III ATHENA trial, which will evaluate rucaparib (Rubraca) as first-line maintenance therapy in patients with newly diagnosed advanced ovarian cancer. Data are expected in 2021.
The first-line setting has also seen addition of another PARPi, veliparib. Veliparib was evaluated in the VELIA phase III trial (NCT02470585) assessing its efficacy when added to first-line chemotherapy followed by veliparib maintenance therapy (55). In this international, phase III, placebo-controlled trial, patients received chemotherapy plus placebo followed by placebo maintenance (control), chemotherapy plus veliparib followed by placebo maintenance (veliparib combination only), or chemotherapy plus veliparib followed by veliparib maintenance (veliparib throughout). A PFS advantage of 34.7 months in the veliparib throughout group versus 22.0 months in the control group (HR, 0.44; 95% CI, 0.28–0.68) was observed in the BRCAm cohort, with a similar PFS advantage seen in the HRD cohort as well. The role of veliparib maintenance therapy alone without its effects during induction therapy has not been established. On the basis of the aforementioned studies, it is likely that the indication for first-line maintenance therapy with a PARPi will be expanded to women with HRD and potentially all patients with newly diagnosed advanced ovarian cancer (Fig. 2).
Figure 2.

Timeline of landmark synthetic lethality advances in ovarian cancer. Several expanded concepts of synthetic lethality have been constantly proposed and studied. After Ashworth and Helleday demonstrated synthetic lethality of PARPi in BRCA1/2-mutated (BRCAm) tumors, several PARPi have been clinically tested for first-line, second-line, and maintenance therapy in ovarian cancer. EMA, European Medicines Agency; CR, complete response; PR, partial response.
PARPi Combinations
There has been interest in combining PARPi maintenance with antiangiogenic maintenance therapy. PAOLA-1 (NCT02477644) evaluated concomitant olaparib and bevacizumab first-line maintenance therapy following platinum-based chemotherapy with concurrent bevacizumab in a randomized, double-blind, international phase III trial (9). Patients received first-line platinum chemotherapy plus bevacizumab and were randomized to receive maintenance placebo or olaparib plus bevacizumab maintenance, regardless of BRCA mutation status. Preliminary results demonstrated a median PFS of 22.1 months in the olaparib/bevacizumab cohort versus 16.6 months in the placebo/bevacizumab cohort (P < 0.0001). However, the benefits of PFS were observed only in patients with BRCA mutations and HRD. Given that no trial cohort assessed PARPi therapy without bevacizumab, and only those with HRD and BRCAm benefited from the addition of olaparib, it is difficult to determine if and how much additional benefit is achieved by adding bevacizumab.
Trials are also underway looking at combining PARPi with immunotherapy (Table 1). This is based on three key observations: (i) BRCA1/2-mutated tumors have higher neoantigen loads, more tumor-infiltrating lymphocytes, and overexpress immune checkpoint modulators, PD-1 and PD-L1 when compared with HR-proficient tumors (56, 57); (ii) PARPi elicits a STING-dependent antitumor immune response independent of its effects on DNA repair (58); and (iii) PARPi causes tumor cells to increase expression of the immune checkpoint molecule PD-L1 (59). DNA repair defects that have resulted (directly or indirectly) from genomic instability are transcribed and translated to encode novel peptide sequences that are unique to the cancer cell. During normal protein degradation, these novel peptides may be bound by MHC proteins that present them on the cell surface as ‘neoantigens’ and this large proportion of mutant neoantigens in mismatch repair–deficient cancers makes them sensitive to immune checkpoint blockade, regardless of the cancer tissue of origin. The MEDIOLA trial (NCT02734004) examined a combination of olaparib and durvalumab in germline BRCA1/2-mutated platinum-sensitive recurrent EOC and found an overall response rate (ORR) of 71.9% (60). The TAPACIO trial (NCT02657889) is looking at niraparib plus pembrolizumab in patients with platinum resistant ovarian cancer irrespective of germline BRCA1/2 status (61). The investigators have reported an ORR of 25% among all platinum-resistant patients and ORR of 45% among patients with somatic BRCA mutations. Two large ongoing clinical trials are also looking at possible PARPi/immunotherapy in the upfront setting. The FIRST trial (NCT03602859) has recently concluded accrual looking at niraparib/dostarlimab followed up niraparib/dostarlimab maintenance, while the ATHENA trial (NCT03522246) is examining rucaparib/nivolumab in the maintenance setting.
Synthetic Lethality Beyond PARPi
Several mechanisms of PARPi resistance have been described and they may be HR-dependent or independent. HR-dependent mechanisms include restoration of BRCA function via secondary or reversion mutations (62) or restoration of HR through other pathways that are independent of BRCA (63). HR-independent mechanisms entail increased replication fork stabilization (64), upregulation of pathways that favor cell survival (65), upregulation of drug efflux pumps (66), and alterations in PARP activity (67). This calls for identifying novel synthetic lethal combinations to overcome PARP resistance mechanisms or sensitize cancer cells to PARP inhibitors, thereby improving clinical effectiveness and patient outcome.
The potential of synthetic lethality to produce genotype-specific cell inhibition has promoted further interest in identifying novel synthetic lethal combinations for ovarian cancer therapy (Table 1). One such approach focuses on loss-of-function mutations in tumor suppressor genes. For example, the tumor suppressor TP53 (p53) is mutated in most cancers, including over 96% of HGSOCs (68). Several lethal approaches to targeting p53-mutant tumor cells have been proposed, including targeting the p38 MAPK MK2 (69), components of the PI3K signaling pathway (70), the DNA damage checkpoint kinase CHK1 (71, 72), and a key determinant of replication fork stability, ATR (ataxia telangiectasia and RAD3-related; refs. 73, 74). Wang and Simon identified potentially druggable genes synthetically lethal for p53 using three microarray datasets for gene expression profiles of the NCI-60 cancer cell lines, one next-generation sequencing (RNA-seq) dataset from the Cancer Genome Atlas (TCGA) project, and one gene expression data from the Cancer Cell Line Encyclopedia (CCLE) project. They demonstrated that prescreening of potential synthetic lethal genes using gene expression profiles is a promising approach for improving the efficiency of synthetic lethal RNAi screening (75). The role and function of p53 as a key driver of tumorigenesis has led to new proposed innovative synthetic lethality strategies against p53-defective cancers (76). Several drugs targeting mutant p53 have entered clinical trials. One strategy is targeting the chaperone protein Hsp90, which is required for proper folding of the mutant p53 protein. For example, GANNET53 was a trial of the Hsp90-inhibitor ganetespib given in combination with paclitaxel. Unfortunately, the phase II trial failed to show an improvement from the combination therapy over paclitaxel alone, but similar strategies are still in development (77).
Another new strategy for synthetic lethality-based therapies is targeting protein-coding genes that are essential for chromatin regulation and reorganization. One promising target includes the SWI/SNF chromatin remodeling complex (78). Helming and colleagues demonstrated that partial loss of ARID1 (AT-Rich Interaction Domain) function via mutation of ARID1A alleles or, less frequently, ARID1B alleles can drive cancer growth but at the same time creates a specific vulnerability compared with nonmutant cells, suggesting ARID1B as a potential target for cancers that contain inactivating ARID1A mutations (79). Dasatinib, an oral dual BCR/ABL and Src family tyrosine kinase inhibitor approved for use in patients with chronic myelogenous leukemia (CML), was found to be consistently lethal in ovarian cancer cells after ARID1A knockdown in ovarian clear cell carcinoma (80). These results implicate ARID1B as a potential therapeutic target for ovarian cancers using synergistic lethality approaches. For example, BET (bromodomain and extra terminal domain) inhibitors cause a reduction in the expression of multiple SWI/SNF members including ARID1B, which produces synthetic lethality with ARID1A loss (81). This strategy is now being tested in a clinical trial of PLX2853, a BET inhibitor, for ARID1A-deficient ovarian cancers (NCT04493619).
Other pathways involved in DNA damage response (DDR) and DNA repair targeting are producing new opportunities for synthetic lethality interventions. The lessons learned from PARPi inhibitors have helped inform DDR inhibitor development. For example, WEE1 protein kinase has been described as an inhibitor of CDK 1 and 2 resulting in temporary cell-cycle arrest and DNA damage repair. In ovarian and endometrial cancers WEE1 inhibitor induce mitotic lethality, rendering p53-deficient cells sensitive to radiation and DNA-damaging agents including PARPi and chemotherapeutic agents (82). The WEE1 inhibitor adavosertib demonstrated preclinical synergy with gemcitabine in early-phase clinical trials (83, 84). Adavosertib efficacy was demonstrated as a monotherapy in a phase I clinical trial with 25 refractory solid tumor patients where two patients with BRCA mutations had partial response (85) and also as a combinatorial modality with chemotherapies which resulted in 53% disease stabilization and 10% partial response (86). A phase II trial with 24 patients with p53-mutated ovarian cancer receiving adavosertib plus carboplatin reported that the overall response rate was 43% including one patient (5%) with a prolonged complete response (87). More recently, in a double-blind, randomized, placebo-controlled, phase II trial, women with measurable recurrent platinum-resistant or platinum-refractory HGSOC, the combination of adavosertib and gemcitabine resulted in an improvement in progression-free survival and overall survival (88). This observed clinical efficacy of a WEE1 inhibitor combined with existing treatments supports ongoing assessment of DNA damage response drugs in HGSOC.
Certainly, additional opportunities for synthetic lethality have yet to be explored. Heinzel and colleagues identified a set of drug combinations currently not tested in late-stage ovarian cancer clinical trials which might produce synthetic lethal interactions and therefore could be worthy of further studies in ovarian cancer (89). This data was mined by mapping a set of synthetic lethal interactions from publicly available data on yeast screens to their respective human orthologs and was complemented by a set of predicted synthetic lethal interactions based on a set of protein meta-data such as molecular pathways. Furthermore, the group established that twelve of the tested drug combinations addressed a synthetic lethal interaction with the anti-VEGF inhibitor bevacizumab in combination with paclitaxel, targeting the synthetic lethal pair between VEGFA and BCL2. They also identified a set of 84 drug combinations with PARPi currently not tested in phase III or IV trials in the context of ovarian cancer which address 102 synthetic lethal interactions.
Conclusions and Future Directions
About 20 years ago, Hartwell and colleagues first proposed that synthetic lethality-based anticancer drug targets can be identified in model organisms, such as yeast (90). Despite the promising nature of synthetic lethality-based therapeutics, the technology faces three major shortcomings that deter translation into the clinic. The first considerable impediment has been the identification of robust, clinically relevant synthetic lethal interactions due to false positive and false negative interactions predicted by high-throughput screening technologies. This calls for follow-up studies to cross-validate potential targets using other experimental strategies such as combinatorial drug testing. Second, these genetic interactions induce lethality, rendering it a challenge to recover and identify mutants. Third, the regulation of signaling pathways and the intracellular environment are heterogeneous due to the genetic and metabolic differences between different cell types making most synthetic lethal interactions condition-dependent and rare. Large numbers of mutant gene-pair combinations are required to understand crosstalk between these pathways and establish potent synthetic lethal interactions. Owing to these reasons, most large-scale synthetic lethal genetic interaction screens have been carried out in budding yeast or fission yeast, as these screens can survey a much larger interaction space and can identify potential cancer-relevant synthetic lethal interactions for direct testing in human cell lines, thereby reducing the number of genetic interaction pairs that need to be tested in human cells. These screens can be constructed by mining genetic networks from existing model organisms for synthetic lethal interactions together with cancer-relevant genes. Furthermore, breakthroughs in RNA interference (RNAi) and CRISPR technology and advances in genome sequencing to rapidly identify genetic and epigenetic changes that differentiate tumor cells from non-tumor cells in a patient have rendered large-scale unbiased synthetic lethality screening directly in human cells feasible.
In conclusion, synthetic lethality provides a means to selectively target tumor cells and spare the patient's nonmalignant cells, resulting in favorable therapeutic indices. In addition, synthetic lethal interactions can facilitate the indirect targeting of nondruggable cancer mutations via identification of a druggable synthetic lethal gene partner, thereby broadening the repertoire of anticancer therapeutic targets. Strategies to exploit synthetic lethality promise to be an area of active research in ovarian cancer for years to come.
Authors' Disclosures
K.M. Elias reports personal fees from Merck KGaA-Pfizer Alliance outside the submitted work. No disclosures were reported by the other authors.
Acknowledgments
This research received support from the grant K12 HD000849, awarded to the Reproductive Scientist Development Program by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (KM Elias). The authors also wish to acknowledge funding support from the GOG Foundation, as part of the Reproductive Scientist Development Program (to K.M. Elias), Robert and Deborah First Family Fund (to A. Chandrasekaran, K.M. Elias), the Saltonstall Research Fund (to A. Chandrasekaran, K.M. Elias), Potter Research Fund (to A. Chandrasekaran, K.M. Elias), the Sperling Family Fund Fellowship (to K.M. Elias), the Bach Underwood Fund (to K.M. Elias), the Minnesota Ovarian Cancer Alliance (to K.M. Elias), the Honorable Tina Brozman Foundation (to K.M. Elias), the Massachusetts Life Sciences Center Bits to Bytes Program (to K.M. Elias), and the Mighty Moose Foundation (to A. Chandrasekaran, K.M. Elias).
References
- 1. American Cancer Society. Cancer facts & figures 2021. Atlanta, GA: American Cancer Society; 2021;13–5. [Google Scholar]
- 2. Kossaï M, Leary A, Scoazec JY, Genestie C. Ovarian cancer: a heterogeneous disease. Pathobiology 2018;85:41–9. [DOI] [PubMed] [Google Scholar]
- 3. Kim J, Park EY, Kim O, Schilder JM, Coffey DM, Cho CH, et al. Cell origins of high-grade serous ovarian cancer. Cancers 2018;10:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kroeger PT, Drapkin R. Pathogenesis and heterogeneity of ovarian cancer. Curr Opin Obstet Gynecol 2017;29:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines®) [Internet]; 2019. Available from: https://www.sciencedirect.com/science/article/abs/pii/S1040842806000643?via%3Dihub.
- 6. Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: evolution of management in the era of precision medicine. CA Cancer J Clin 2019;280–304. [DOI] [PubMed] [Google Scholar]
- 7. Rossi L, Verrico M, Zaccarelli E, Papa A, Colonna M, Strudel M, et al. Bevacizumab in ovarian cancer: a critical review of phase III studies. Oncotarget 2017;8:12389–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med 2019;381:2403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med 2019;381:2416–28. [DOI] [PubMed] [Google Scholar]
- 10. Vergote I, Scambia G, O'Malley D, Van Calster B, Park S-Y, Del Campo JM, et al. Trebananib or placebo plus carboplatin and paclitaxel as first-line treatment for advanced ovarian cancer (TRINOVA-3/ENGOT-ov2/GOG-3001): a randomised, double-blind, phase 3 trial. Lancet Oncol 2019;20:862–76. [DOI] [PubMed] [Google Scholar]
- 11. Hardy-Bessard A-C, Moore K, Mirza M, Asselain B, Redondo A, Pfisterer J, et al. ENGOT-OV44/FIRST study: a randomized, double-blind, adaptive, phase III study of standard of care (SOC) platinum-based therapy ± dostarlimab followed by niraparib ± dostarlimab maintenance as first-line (1L) treatment of stage 3 or 4 ovarian cancer (O). J Clin Oncol 2020. p.TPS6101. [Google Scholar]
- 12. Ménissier De Murcia J, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A 1997;94:7303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murai J, Huang SYN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 2012;72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murai J, Huang SYN, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 2014;13:433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Welcsh PL. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet 2001;10:705–13. [DOI] [PubMed] [Google Scholar]
- 16. Lee EK, Matulonis UA. Emerging drugs for the treatment of ovarian cancer: a focused review of PARP inhibitors. Expert Opin Emerg Drugs 2020;25:165–88. [DOI] [PubMed] [Google Scholar]
- 17. O'Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet 2017;18:613–23. [DOI] [PubMed] [Google Scholar]
- 18. Bridges CB. The origin of variations in sexual and sex-limited characters. Am Soc Nat 1922;56:51–63. [Google Scholar]
- 19. Dobzhansky T. Genetics of natural populations; recombination and variability in populations of drosophila pseudoobscura. Genetics 1946;31:269–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lucchesi JC. Synthetic lethality and semi-lethality among functionally related mutants of drosophila melanfgaster. Genetics 1968;59:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagel R, Semenova EA, Berns A. Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep 2016;17:1516–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Macintyre G, Goranova TE, De Silva D, Ennis D, Piskorz AM, Eldridge M, et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet 2018;50:1262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soong TR, Howitt BE, Miron A, Horowitz NS, Campbell F, Feltmate CM, et al. Evidence for lineage continuity between early serous proliferations (ESPs) in the fallopian tube and disseminated high-grade serous carcinomas. J Pathol 2018;246:344–51. [DOI] [PubMed] [Google Scholar]
- 24. Flaum N, Crosbie EJ, Edmondson RJ, Smith MJ, Evans DG. Epithelial ovarian cancer risk: a review of the current genetic landscape. Clin Genet 2020;97:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loeb LA. A mutator phenotype in cancer. Cancer Res 2001;61:3230–9. [PubMed] [Google Scholar]
- 26. Gourley C, Balmaña J, Ledermann JA, Serra V, Dent R, Loibl S, et al. Moving from poly (ADP-ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J Clin Oncol 2019;37:2257–69. [DOI] [PubMed] [Google Scholar]
- 27. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end joining pathway. Annu Rev Biochem 2011;79:181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morales JC, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, et al. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr 2014;24:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. The Cancer Genome Atlas Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moynahan ME, Chiu JW, Koller BH, Jasint M. Brca1 controls homology-directed DNA repair. Mol Cell 1999;4:511–8. [DOI] [PubMed] [Google Scholar]
- 31. McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006;66:8109–15. [DOI] [PubMed] [Google Scholar]
- 32. Farmer H, McCabe H, Lord CJ, Tutt AHJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21. [DOI] [PubMed] [Google Scholar]
- 33. Bowtell DD. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 2016;15:668–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 2012;366:1382–92. [DOI] [PubMed] [Google Scholar]
- 35. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mateo J, Moreno V, Gupta A, Kaye SB, Dean E, Middleton MR, et al. An adaptive study to determine the optimal dose of the tablet formulation of the PARP inhibitor olaparib. Target Oncol 2016;11:401–15. [DOI] [PubMed] [Google Scholar]
- 37. Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1274–84. [DOI] [PubMed] [Google Scholar]
- 38. Penson RT, Valencia RV, Cibula D, Colombo N, Leath CA, Bidzinski M, et al. Olaparib versus nonplatinum chemotherapy in patients with platinum-sensitive relapsed ovarian cancer and a germline BRCA1/2 mutation (SOLO3): a randomized phase III trial. J Clin Oncol 2020;38:1164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst 2014;106:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lampert EJ, Hays JL, Kohn EC, Annunziata CM, Minasian L, Yu M, et al. Phase I/Ib study of olaparib and carboplatin in heavily pretreated recurrent high-grade serous ovarian cancer at low genetic risk. Oncotarget 2019;10:2855–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geenen JJJ, Dackus G, Schouten PC, Pluim D, Marchetti S, Sonke GS, et al. A phase I dose-escalation study of two cycles carboplatin-olaparib followed by olaparib monotherapy in patients with advanced cancer. J Clin Oncol 2019;37:3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann Oncol 2019;30:551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ison G, Howie LJ, Amiri-Kordestani L, Zhang L, Tang S, Sridhara R, et al. FDA approval summary: niraparib for the maintenance treatment of patients with recurrent ovarian cancer in response to platinum-based chemotherapy. Clin Cancer Res 2018;24:4066–71. [DOI] [PubMed] [Google Scholar]
- 44. Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med 2016;375:2154–64. [DOI] [PubMed] [Google Scholar]
- 45. Del Campo JM, Matulonis UA, Malander S, Provencher D, Mahner S, Follana P, et al. Niraparib maintenance therapy in patients with recurrent ovarian cancer after a partial response to the last platinum-based chemotherapy in the ENGOT-OV16/NOVA trial. J Clin Oncol 2019;37:2968–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu XH, Zhu JQ, Yin RT, Yang JX, Liu JH, Wang J, et al. Niraparib maintenance therapy in patients with platinum-sensitive recurrent ovarian cancer using an individualized starting dose (NORA): a randomized, double-blind, placebo-controlled phase III trial☆. Ann Oncol 2021;32:512–21. [DOI] [PubMed] [Google Scholar]
- 47. Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:1949–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines®) ovarian cancer including fallopian tube cancer and primary peritoneal cancer [Internet]; 2020. Available from: https://www.sciencedirect.com/science/article/abs/pii/S1040842806000643?via%3Dihub.
- 49. Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene 2014;33:3894–907. [DOI] [PubMed] [Google Scholar]
- 50. Kohn EC, Lee Jm, Ivy SP. The HRD decision—which PARP inhibitor to use for whom and when. Clin Cancer Res 2017;23:7155–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takaya H, Nakai H, Takamatsu S, Mandai M, Matsumura N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci Rep 2020;10:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reverdy T, Sajous C, Péron J, Glehen O, Bakrin N, Gertych W, et al. Front-line maintenance therapy in advanced ovarian cancer—current advances and perspectives. Cancers 2020;12:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2018;379:2495–505. [DOI] [PubMed] [Google Scholar]
- 54. González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2019;1–12. [DOI] [PubMed] [Google Scholar]
- 55. Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med 2019;1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016;7:13587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP inhibition elicits STING-dependent antitumor immunity in brca1-deficient ovarian cancer. Cell Rep 2018;25:2972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res 2017;23:3711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Domchek SM, Postel-Vinay S, Im S-A, Hee Park Y, Delord J-P, Italiano A. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol 2020;21:1155–64. [DOI] [PubMed] [Google Scholar]
- 61. Konstantinopoulos PA, Waggoner SE, Vidal GA, Mita MM, Fleming GF, Holloway RW, et al. TOPACIO/Keynote-162 (NCT02657889): a phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. In: J Clin Oncol 2018;106. [Google Scholar]
- 62. Drost R, Dhillon KK, Van Der Gulden H, Van Der Heijden I, Brandsma I, Cruz C, et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J Clin Invest 2016;126:2903–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 2020;11:3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016;535:382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med 2017;9:eaal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, Bray SE, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer 2016;115:431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun 2018;9:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cole AJ, Dwight T, Gill AJ, Dickson KA, Zhu Y, Clarkson A, et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci Rep 2016;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, et al. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response InVivo. Cell Rep 2013;5:868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM, et al. Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-Null tumors. Cell 2013;155:844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Origanti S, Cai SR, Munir AZ, White LS, Piwnica-Worms H. Synthetic lethality of Chk1 inhibition combined with p53 and/or p21 loss during a DNA damage response in normal and tumor cells. Oncogene 2013;32:577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Udai B, Plummer ER, Moreno V, Ang JE, Quinton A. A phase I/II first-in-human trial of oral SRA737 (a Chk1 inhibitor) given in combination with low-dose gemcitabine in subjects with advanced cancer. In: J Clin Oncol 2019;3095. [Google Scholar]
- 73. Reaper PM, Griffiths MR, Long JM, Charrier J-D, MacCormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011;7:428–30. [DOI] [PubMed] [Google Scholar]
- 74. Sangster-Guity N, Conrad BH, Papadopoulos N, Bunz F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene 2011;30:2526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang X, Simon R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med Genomics 2013;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Aning OA, Cheok CF. Drugging in the absence of p53. J Mol Cell Biol 2019;11:255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nicole C, Braicu I, Combe P, Ray-Coquard IL, Joly F, Harter P, et al. Phase II results of GANNET53: a european multicenter phase I/randomized II trial of the Hsp90 inhibitor ganetespib (G) combined with weekly paclitaxel (P) in women with high-grade serous, high-grade endometrioid, or undifferentiated, platinum-resistant ep. In: J Clin Oncol 2018;5567. [Google Scholar]
- 78. Jones S, Wang TL, Shih IM, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010;330:228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med 2014;20:251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Caumanns JJ, Wisman GBA, Berns K, van der Zee AGJ, de Jong S. ARID1A mutant ovarian clear cell carcinoma: a clear target for synthetic lethal strategies. Biochim Biophys Acta Rev Cancer 2018;1870:176–84. [DOI] [PubMed] [Google Scholar]
- 81. Berns K, Caumanns JJ, Hijmans EM, Gennissen AMC, Severson TM, Evers B, et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene 2018;37:4611–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Meng X, Bi J, Li Y, Yang S, Zhang Y, Li M, et al. AZD1775 increases sensitivity to olaparib and gemcitabine in cancer cells with p53 mutations. Cancers 2018;10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, et al. MK-1775, a novel wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res 2011;17:5638–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov 2012;2:524–39. [DOI] [PubMed] [Google Scholar]
- 85. Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I study of single-agent AZD1775 (MK-1775), a wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol 2015;33:3409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Leijen S, Van Geel RMJM, Pavlick AC, Tibes R, Rosen L, Razak ARA, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol 2016;34:4371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Leijen S, Van Geel RMJM, Sonke GS, De Jong D, Rosenberg EH, Marchetti S, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patientswith tp53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol 2016;34:4354–61. [DOI] [PubMed] [Google Scholar]
- 88. Lheureux S, Cristea MC, Bruce JP, Garg S, Cabanero M, Mantia-Smaldone G, et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021;397:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Heinzel A, Marhold M, Mayer P, Schwarz M, Tomasich E, Lukas A, et al. Synthetic lethality guiding selection of drug combinations in ovarian cancer. PLoS One 2019;14:e0210859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997;278:1064–8. [DOI] [PubMed] [Google Scholar]