

Summary
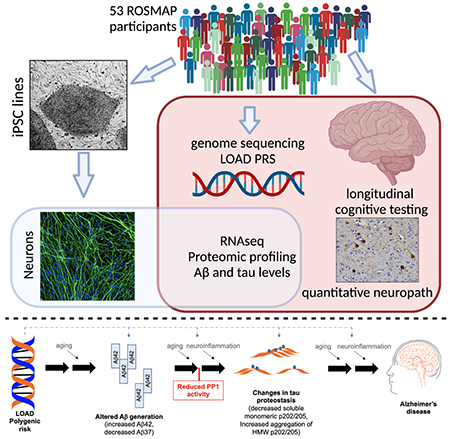
We have generated a controlled and manipulable resource that captures genetic risk for Alzheimer’s disease: iPSC lines from 53 individuals coupled with RNA and proteomic profiling of both iPSC-derived neurons and from brain tissue of the same individuals. Data collected for each person includes genome sequencing, longitudinal cognitive scores, and quantitative neuropathology. The utility of this resource is exemplified here by analyses of neurons derived from these lines, revealing significant associations between specific Aβ and tau species and the levels of plaque and tangle deposition in the brain and, more importantly, with the trajectory of cognitive decline. Proteins and networks are identified that are associated with AD-phenotypes in iPSC-neurons, and relevant associations are validated in brain. The data presented establish this iPSC collection as a resource for investigating person-specific processes in the brain that can aid in identifying and validating molecular pathways underlying AD.
Keywords: iPSC, Alzheimer’s, LOAD, genetics, PRS, cognitive resilience, PP1, PPP1CA, PPP1R1A, MAPT, tau, proteasome, module, Aβ, ROS, MAP, neuron
Graphical Abstract
eTOC blurb
In this study, Lagomarsino et al describe the generation of an iPSC collection from 53 deeply phenotyped individuals. These lines are coupled to RNAseq and proteomic profiling of both iPSC-neurons and brain of the same individuals. Using this system, biological insights are uncovered regarding the molecular mechanisms underlying Alzheimer’s disease.
Introduction
Alzheimer’s disease (AD) affects about 50 million people worldwide, and 2-3 times that number are estimated to be presymptomatic. With limited treatment options available, understanding the molecular causes of AD is imperative. AD is defined by the accumulation of extracellular plaques in the brain composed primarily of amyloid beta peptides (Aβ) and intraneuronal tangles containing excessively phosphorylated tau. The vast majority of AD patients have the “sporadic,” late-onset form (LOAD). Clinicians and pathologists have long recognized that LOAD manifests along a spectrum of cognitive abilities and levels of neuropathology. Although AD is somewhat stereotyped in terms of both cognitive progression with age and the patterns of pathological aggregation of Aβ and tau in the brain, the disease is heterogeneous with respect to rates of progression and the cognitive domains that are primarily affected.
Studies of postmortem brain tissue highlight particular pathways and proteins that are associated with neuropathology, cognitive decline, and clinical AD diagnosis (Zhang et al., 2013a, Seyfried et al., 2017, Mostafavi et al., 2018, Johnson et al., 2020). While a causal chain of events can be hypothesized from such cross-sectional data (Tasaki et al., 2018, Felsky et al., 2019), it cannot be definitively established, as both the alterations which are driving disease progression and those resulting from decades of neurodegeneration are studied at the end stage of disease. Induced pluripotent stem cell (iPSC) technology offers a complementary approach that could fill the scientific gap between genetic and postmortem tissue studies, providing a controlled and manipulable human experimental system that captures intrinsically encoded genetic risk. Most iPSC studies of AD have focused on highly penetrant mutations in APP and PSEN, which cause rare, early-onset cases (Israel et al., 2012, Shi et al., 2012, Kondo et al., 2013, Muratore et al., 2014, Sproul et al., 2014, Moore et al., 2015, Hung and Livesey, 2018, Kwart et al., 2019). While iPSC technology has provided insights into deterministic genes in autosomal dominant AD, it has yet to be shown whether iPSC-derived brain cells are useful in capturing disease-relevant processes in the aging brain and in LOAD.
Here, we have generated iPSC lines from 53 deceased individuals in the Religious Order Study (ROS) or Rush Memory and Aging Project (MAP) that span the clinical and neuropathological spectrum of aging (Bennett et al., 2018). Sixteen of these individuals had a clinical and neuropathological diagnosis of AD/Alzheimer’s dementia. We differentiated these lines into cortical neuronal fate (iNs) and measured RNA and protein profiles as well as quantitative measures of APP cleavage products and tau species. We identify specific proteins and pathways in neuronal cultures that are associated with AD polygenic risk score (PRS), neuropathological burden and/or cognitive trajectory in the donors and validate these findings through analyses of brain tissue. One of the most compelling sets of associations observed were between protein phosphatase 1 (PP1) levels and LOAD PRS, Aβ, and p-tau levels. Experimental modulation of AD-associated Aβ altered PP1 levels, and modulation of PP1 activity in turn altered tau proteostasis. These analyses identified PP1 as both a driver downstream of AD genetic risk and a mechanistic link between AD-associated Aβ and tau, exemplifying the utility of this resource for studying genetic diversity in a complex human disease.
Results
Generation of iPSC lines from ROS and MAP participants
Participants in the ROS/MAP projects range in age from 50 to 100+ and are free of dementia at enrollment (Bennett et al., 2012a, Bennett et al., 2012b, Bennett et al., 2018). Cognitive tests are administered annually that provide quantitative data of multiple cognitive domains (episodic, semantic, and working memory, perceptual speed and visuospatial ability)(Barnes et al., 2001, Wilson et al., 2004, Aggarwal et al., 2005, Wilson et al., 2007). The longitudinal data allows the calculation of person-specific rates of change in cognition over time using a linear mixed model adjusted for age, sex, and education (“slope”). Pathologic AD indices acquired postmortem include quantitative measures of neuritic and diffuse plaques and neurofibrillary tangles determined by silver stain, as well as measures of Aβ load and paired helical filament tau tangles based on immunostaining (Bennett et al., 2004, Bennett et al., 2005, Barnes et al., 2006). Resultant neuropathological scores represent composite data from at least 5 regions: entorhinal cortex, hippocampus, mid-frontal, temporal, and inferior parietal neocortex.
We generated iPSC lines using cryopreserved peripheral blood mononuclear cells (PBMCs) from 53 deceased ROS/MAP participants (Figure 1). Clinical evaluations and postmortem pathological information were used to select participants that spanned the neuropathological and cognitive spectrum of aging (Figure S1). Approximately one third of the subjects selected had neither a pathological nor a clinical diagnosis of AD. Sixteen individuals represent the prototypical profile of LOAD with both a pathological and a clinical diagnosis of AD and Alzheimer’s dementia and no evidence of dementia from other causes. The cohort also contains individuals with high amyloid plaque burden but who were not cognitively impaired. Key demographic data of our iPSC cohort align with the distributions within the ROS/MAP cohorts (Figure S1).
Figure 1. Generation of human iPSC lines from ROS and MAP cohorts.
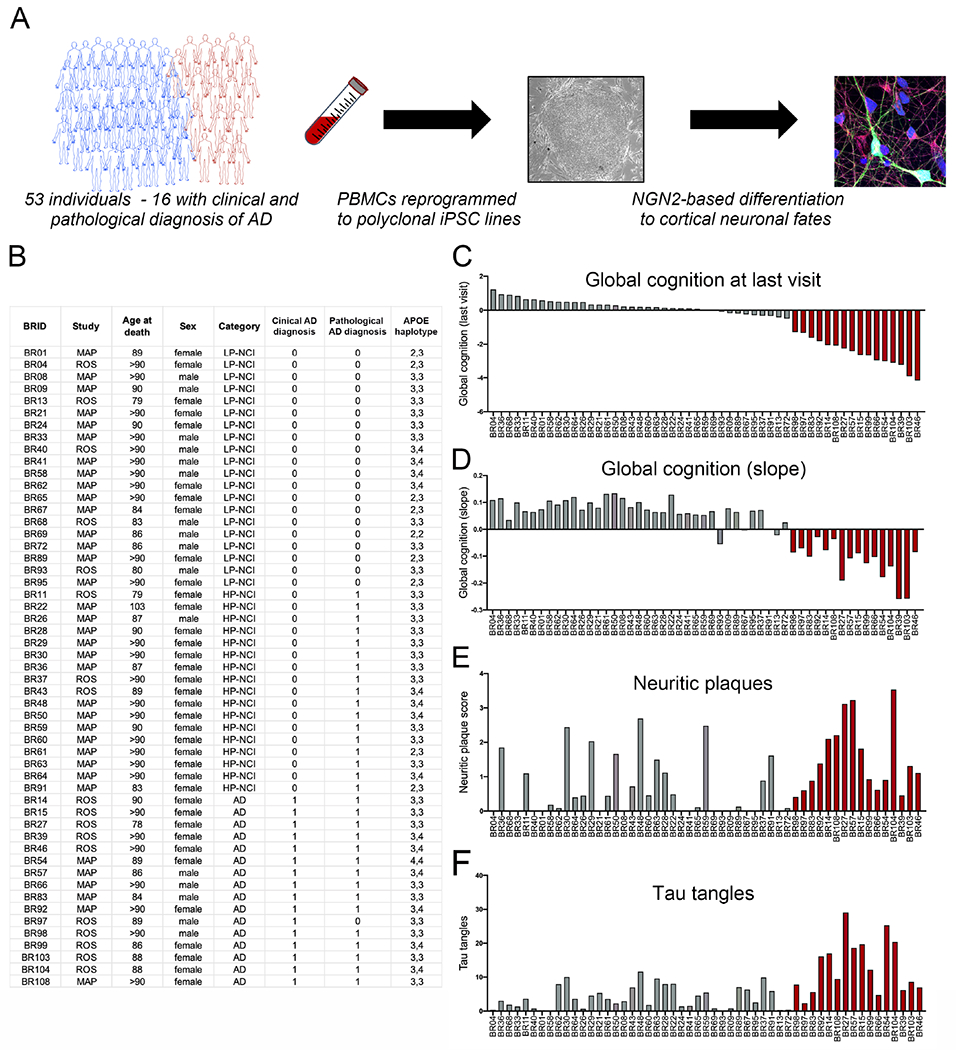
(A) Overview of the study. (B-F) Relevant data on ROS/MAP participants from whom iPSC lines were generated, each line identified using a BWH identifier (BRID). Quantification of plaques in the brain by modified Bielschowsky’s Silver Stain (E), paired helical filament tau by immunostaining using AT8 (F). Red bars display data from those individuals with both a clinical and pathological diagnosis of AD. See also Figures S1, S2.
To generate iPSC lines, PBMCs were transduced with Sendai virus to deliver reprogramming factors, and colonies were consolidated to generate polyclonal lines. Quality control (QC) analytics were performed using NYSCF’s fully automated Global Stem Cell Array technology, minimizing factors that contribute to variability in iPSC behavior and thus reducing line-to-line variability (Paull et al., 2015). Each iPSC line described herein passed several QC tests confirming proper sterility, identity, karyotype, and pluripotency (Figure S2, Figure S3).
Robust and consistent differentiation to neuronal fate across cell lines
The Neurogenin-2 direct induction protocol was employed to differentiate each of the iPSC lines to neuronal fate generating cultures with >98% of cells expressing neuronal markers (Figure 2 and (Zhang et al., 2013b, Srikanth et al., 2018)). D21 induced neurons (iNs) resulting from this protocol exhibited highly consistent morphology across lines (Figures 2A, Figure S3). The cells generated are most similar to upper layer glutamatergic projection neurons of the cerebral cortex based on RNA and protein expression profiling ((Zhang et al., 2013b, Srikanth et al., 2018). Bulk RNA sequencing (RNAseq) and proteomic analyses of these cultures highlighted the consistency of differentiation efficiency across iPSC lines at the level of cell fate (Figure 2B, Figure S3). Single nucleus RNAseq was used to further characterize the cell type composition within iN cultures, benchmarked against iPSC-derived astrocytes and microglia (Figure 2C–G). Over 98% of cells in iN cultures express neuronal markers such as SYP (Figure 2D), but less than 1% express KI67 (a marker of proliferation, Figure 2E), AIF1 (IBA1, a microglial marker, Figure 2F), or GJA1 (an astrocyte marker, Figure 2G).
Figure 2. Consistent differentiation of iPSC lines to neuronal fate.
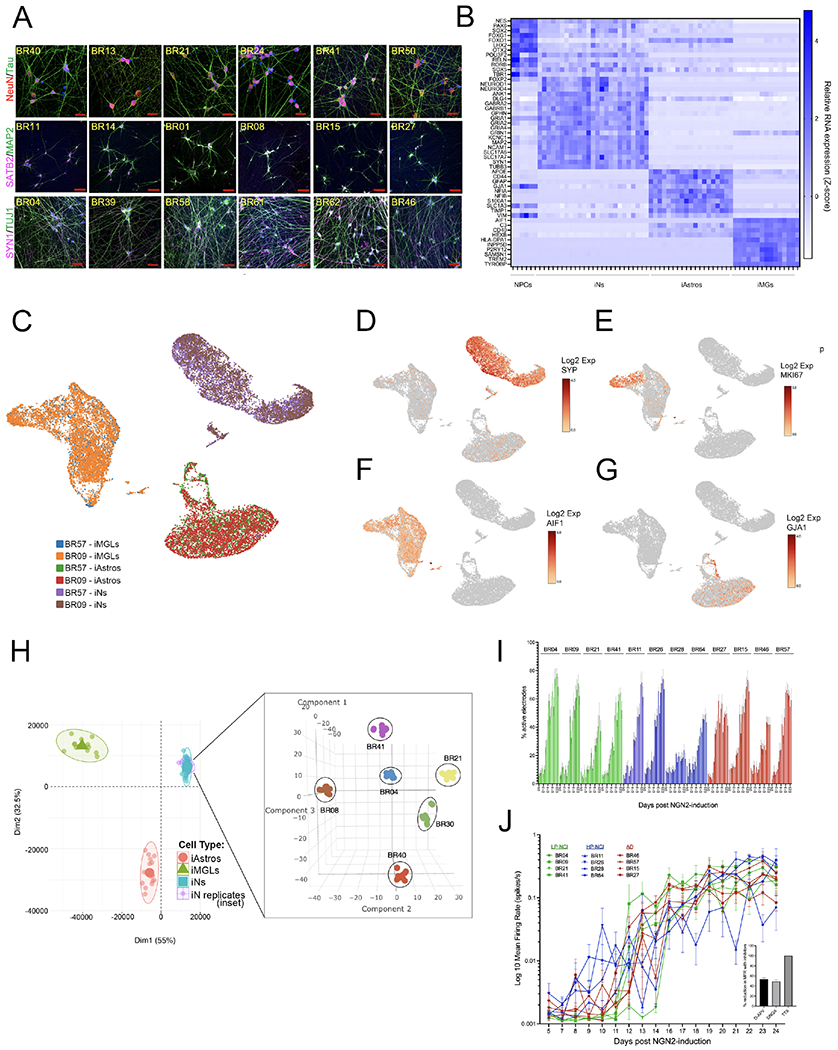
(A) Example images of immunostaining for neuronal markers across iNs from multiple iPSC lines. DNA is stained with DAPI, scale bars = 50μM
(B) Heat map of cell fate markers in iNs across lines, determined by RNAseq and compared to other iPSC-derivatives including neural progenitor cell (NPC), astrocyte (iAstro), and microglialike (iMG) fates. There were no significant differences in cell fate marker expression between NCI and AD individuals (t-test, with Holm-Sidak multiple comparisons test; q-values all >0.75).
(C-G) iPSCs from two donors (BR09, BR57) differentiated to neuronal, astrocyte, and microglial fates in parallel. On the day of harvest iNs were d21, iAstros were d28 and iMGs were d43. Cells from the two donors were pooled and single nucleus RNAseq libraries prepared. UMAP plots show consistency across lines at the single cell level (C), with expression of the neuronal marker SYP (D) in >95% of cells in iN cultures, and <1% of the iN cells expressed the proliferation marker MKI67 (E), the microglial marker AIF1 (IBA1), or the astrocyte marker GJA1 (G). (H) Bulk RNAseq was performed on iNs derived from 6 lines over 6 different batches of differentiation. After controlling for sequencing batch, tSNE analysis showed tight clustering of iN samples by genotype. Data are shown as an inset within another plot showing their location within PCA space relative to other iPSC-derived cell types.
(I,J) Percentage of active electrodes up to d24 of differentiation (I) and well-level mean firing rate over time (J), measured in multi-electrode arrays for 12 lines, n=8 wells/line (16 electrodes/well). After the last recording, wells were treated either with vehicle, TTX, DNQX or AP-5 and activity recorded for 30 mins. Inset shows the electrode-level percent reduction in mean firing rate (MFR) with each compound relative to vehicle, comparing pre-treatment recording to the post-treatment recording. Shown is mean +/−SEM, n>195 electrodes/condition. See also Figure S3.
To further show consistency of differentiation, RNAseq was performed on six iPSC lines across six differentiation rounds and three RNAseq library preparations by two different researchers. Data were processed to eliminate batch effects across library preparation rounds and tested for overall similarity using t-distributed Stochastic Neighbor Embedding (tSNE) analysis. The samples clustered based on genetic background, showing consistency across differentiation rounds and validating the analysis pipeline (Figure 2H).
Multi-electrode array (MEA) analyses of iNs were performed to examine spontaneous neuronal activity over differentiation time. INs from different individuals showed consistent trends in activity over differentiation, defined by the percent of active electrodes and the mean firing rate (Figure 2I,J). Over 98% of activity was inhibited by treatment with tetrodotoxin (TTX), and pharmacological blockade of NMDA receptors (AP-5) or AMPA receptors (DNQX) at the terminal time point each reduced activity by approximately 50% (Figure 2J, inset).
Congruence of gene expression profiles between iPSC-derived neurons and brain tissue
One of the unique strengths of generating iPSC lines from the ROS/MAP cohorts is the rich clinical, pathologic, and -omics data from the donors. This enables a direct comparison between data generated in the cultures and the data acquired from primary tissues of autopsied individuals. Here the level of concordance between RNAseq expression profiles from iNs and the medial prefrontal cortex (PFC; BA9/46) of the same individuals was determined (Figure 3A). We examined genes in the upper half of percentage variance across iN samples to enrich for those with more dynamic expression levels across genetic backgrounds. Pearson correlations (rvals) were calculated for each gene-level RNA detected in both iNs and brain, identifying 110 genes with significant correlation (Figure 3B, Table S1). We rationalized that any genetically encoded congruence between iN and brain would be observed as a skew in the distribution of all rvals towards the positive, as a negative correlation would have suspect biological significance. We observed a positive skew of all brain to iN gene correlations that was significantly higher than what would be expected by chance (Figure 3C,E). These data suggest that this experimental system captures a measurable subset of the intrinsically encoded genetic differences between individuals, and that there is a quantifiable congruence between iNs and the frontal cortex of the individuals from whom they were derived.
Figure 3. Congruence of gene expression profiles between neuronal cultures and brain.
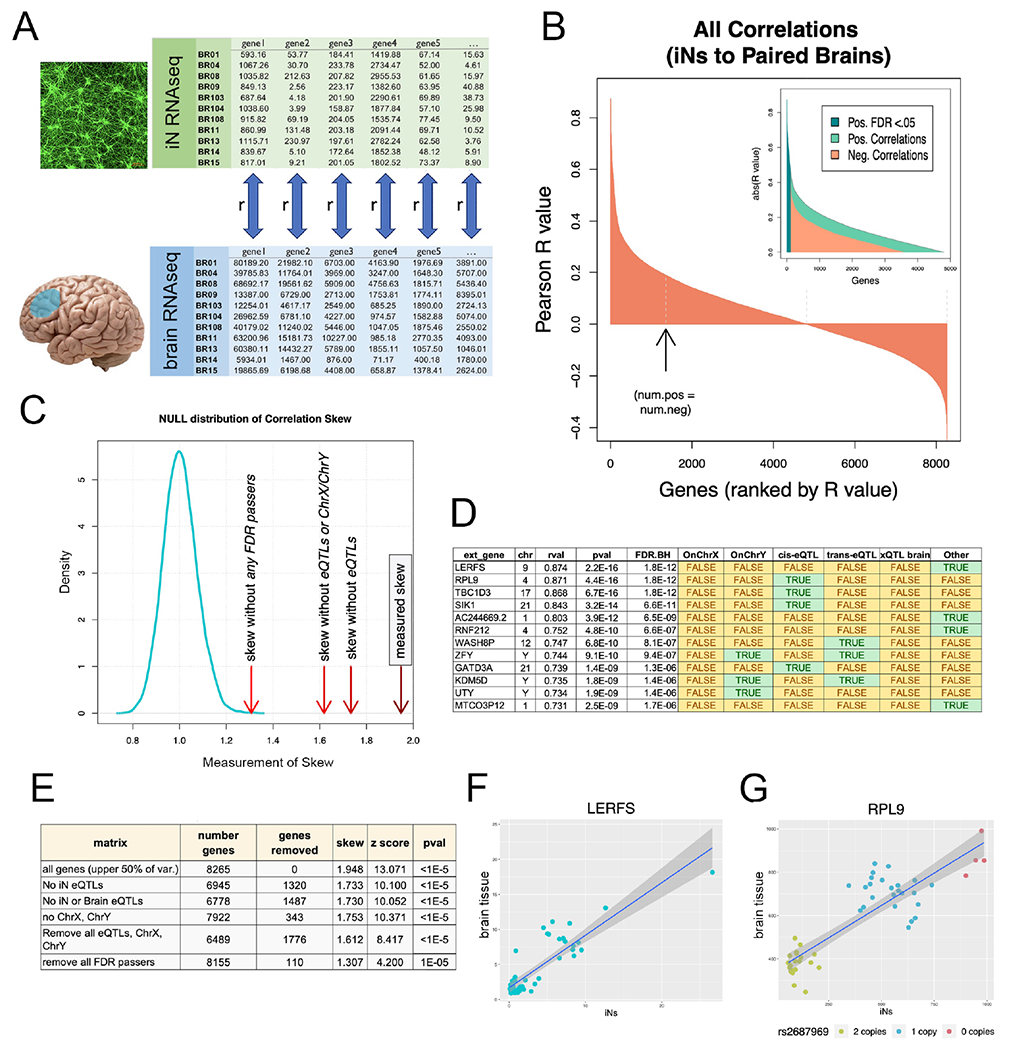
(A) RNAseq profiles from iNs and corresponding brain samples (mPFC) were compared. For each gene in the upper half of percentage variance, a Pearson correlation (rval) was calculated between iNs and brain across 49 human subjects. (B) Waterfall plot of rvals for all 8,265 genes. Arrow denotes point where the number of positive rvals equals the number of negative rvals. 110 genes had correlations that individually passed Benjamini-Hochberg multiple comparisons testing (FDR q<0.05). (C) Positive skew of all rvals as a population was measured by dividing the sum of the highest 100 rvals to the absolute value of the lowest 100 rvals. Density plot of null distribution of skews is shown. The measured skew is significantly higher than what would be expected by chance even after successively removing eQTLs and sex chromosome genes from the dataset (red arrows to the right of null distribution). (D) Top 12 genes with expression correlated between iN and brain. Green shaded cells indicate membership in one of the tested gene sets (sex chromosomes, iN cis and trans eQTLs, brain eQTLs). (E) Table of the number of genes compared after successive gene set removal and effect on skew and significance (z score and pvals) of skew comparison to null distribution. (F) Correlation plot of top correlation (LERFS) between iNs and brain. Each dot indicates a single human subject with expression in iNs on the x-axis and expression in brain on the y-axis. (G) Same plot for RPL9, top cis-eQTL correlation. Dot colors indicate copy number of rs2687969 SNP. See also Figure S4, Table S1.
We expect that this congruence could be due in part to identifiable expression quantitative trait loci (eQTLs) and to differences in sex chromosome complement across human subjects. Removal of eQTLs identified in our iN dataset (see STAR methods) and in ROS/MAP brain (Ng et al., 2017) reduced skew by 27% (Figure 3C–E). Additional removal of genes encoded on the X and Y chromosomes reduced the skew further, but still left a gene set with significant correlation between iNs and brains (Figure 3C,E). Importantly, removing all genes that pass FDR < 0.05 still resulted in a matrix with significant positive skew. This result indicates that the false negatives, inevitable with large comparisons, are sufficient to drive significant congruence between expression sets. Even with the obvious differences between iNs and brain, we observe a readily detectable and significant correlation of genetic regulation. Furthermore, the congruence between iNs and brain can be traced to identified eQTLs, sex chromosome-linked genes, and as yet unidentified genetic variants that regulate gene expression (Figure 3D–G, Table S1).
Concordance between subsets of altered pathways in AD neuronal cultures and AD brain
We determined which genes, proteins and pathways are altered in AD iNs, and whether these same alterations are found in brain from the same individuals. Here, we define categories based upon both clinical and pathological diagnosis of AD, but recognize that individuals within each category are spread across a spectrum of both neuropathological and cognitive measurements. “AD” individuals had both a clinical and pathological diagnosis of AD while “NCI” are not cognitively impaired with no clinical AD diagnosis. Individuals with high pathology (“HP”) had a pathological diagnosis of AD, and low pathology (“LP”) individuals did not.
We performed gene-level differential expression analysis using bulk RNAseq data from d21 iN cultures comparing AD to LP-NCI, and separately comparing AD to HP-NCI. Here, we used iN RNAseq data as a discovery dataset followed by validation analyses using better powered RNAseq data from 579 ROS/MAP brain samples (Mostafavi et al., 2018). In our iN dataset, comparing AD to LP-NCI yielded 667 genes with an uncorrected p<0.05; HP-NCI versus AD yielded 281 genes that met these same criteria. We then examined whether these same genes were differentially expressed in the AD versus LP-NCI in the brain RNAseq data set. In brain, 86 genes had significant differential expression following multiple comparisons testing (FDR<0.05) with the directionality of these gene expression changes concordant between iN and brain data; 11 genes met the same criteria for the AD vs HP-NCI comparison (Table S2).
In a previous study, we identified 47 modules of co-expression using brain RNAseq data from the dorsolateral prefrontal cortex of 476 ROS/MAP participants (Mostafavi et al., 2018). Here, we performed gene set enrichment analysis (GSEA) to examine whether any of these brain-derived modules of co-expression were enriched along the positive or negative tails of the AD vs. LP-NCI expression gradient in iNs. Genes with higher expression in AD iNs were enriched in 8 brain modules (FDR q<0.05). Genes driving enrichment regulate synaptic vesicle localization and transport, axonogenesis and microtubule polymerization (Figure 4A,B, Table S2). Genes with lower expression in AD were enriched in 7 brain modules, and driver genes encoded ribosomal proteins, and proteins involved in transport, focal adhesion, and apoptosis (Figure 4A,C).
Figure 4. Brain-derived modules of RNA and protein co-expression are associated with AD diagnosis in both iNs and prefrontal cortex.

(A-C) Differential gene expression analyses were performed between LP-NCI and AD iNs and between HP-NCI and AD iNs, with GSEA to examine enrichment in brain-derived co-expression modules generated from ROS-MAP DL-PFC brain tissue (Mostafavi et al., 2018). See also Table S2 showing all genes with an uncorrected p-value<0.05 (t-test) in the iN RNAseq dataset that then showed concordant differences (FDR q<0.05) in a better powered brain RNAseq data set (Mostafavi et al., 2018). A) Table of modules significantly enriched in genes upregulated (blue) or down regulated (red) in AD. For iNs n=18 LP-NCI, 15 HP-NCI, 16 AD; t-tests performed. For brain, n=196 LP-NCI, 171 HP-NCI, 212 AD; t-test with multiple comparisons testing: FDR calculation using Benjamini, Krieger, and Yekutieli (BKY).
(B, C) Gene-concept networks of GSEA leading edge genes in m23 and m116.
(D-H) Proteomic analyses of iNs, comparing AD to LP-NCI or HP-NCI. Shown in (D) are all proteins that met an uncorrected p-val<0.05 in iNs, which then showed concordant changes in a better powered brain dataset (FDR q<0.05). For iNs n=16 LP-NCI, 14 HP-NCI, 8 AD; t-tests performed. For brain, n=138 LP-NCI, 131 HP-NCI, 100 AD; t-test with multiple comparisons testing: FDR calculation using BKY.
(E-H) GSEA using protein modules from frontal cortex to compare proteomic results between AD and LP-NCI or HP-NCI iNs. Pearson correlations between each module protein for both brain (E) and iNs (F). Heat maps of correlations show concordant patterns of association between modules and within modules between brain and iNs. Table (G) shows brain-derived modules enriched in AD vs LPNCI and AD vs HPNCI comparisons using GSEA analysis. Rank files for GSEA analyses of all genes were generated by calculating the −log10(pval) from Student’s t (2-sided unpaired, heteroscedastic) comparison and signed to reflect the directionality of differential expression. Leading edge genes for each module with significant enrichment are shown (H). NE = not enriched; NES = normalized enrichment score. See also Figure S5, Table S3.
We next examined protein expression in iNs using liquid chromatography coupled to tandem mass spectrometry. As expected, some changes in RNA are reflected in changes in protein, while in other cases a change in RNA is discordant with protein levels. Transcript abundance and protein levels were significantly associated overall across all genes detected in both datasets (skew=2.98 towards positive correlations relative to null distribution, p = 0.0014). Using the discovery-validation criteria described above, 69 differentially expressed proteins (DEPs) between AD:LP-NCI were identified in iNs, 29% of which met the more stringent cutoff of FDR<0.05 in the brain dataset (Figure 4D). Although similarly powered, the AD:HP-NCI comparison for iNs identified only 26 proteins, 5 of these validating in the brain dataset (Figure 4D).
From 323 ROS/MAP DLPFC samples, 57 modules of protein co-expression were identified using weighted correlation network analysis (Table S3). To examine associations between proteins within each module and the relationship across modules, protein-protein Pearson correlations were calculated. We observe highly similar patterns of correlation both between and within these modules in brain and in iN proteomic datasets (Figure 4E,F, Figure S5). However, as expected, some brain-derived modules (for example those enriched in glial genes) were not well-conserved in iN proteomics data (Figure S5).
Consistent with the transcriptome analyses, protein modules upregulated in AD vs. LP-NCI iNs function in synaptic vesicle regulation (“red”), and protein transport (“brown”), while AD down regulated modules contained ribosomal proteins (“skyblue3”) and regulate RNA splicing (“skyblue”) (Figure 4G,H). Protein modules that were upregulated in AD vs. HP-NCI iNs are involved in nuclear transport, regulation of serine/threonine phosphorylation (“light green”) and synaptic vesicle regulation (“dark green”), while AD downregulated modules contained proteins involved in RNA stabilization and splicing as well as protein transport proteins (“skyblue” and “purple”) (Figure 4G,H). “Skyblue” was the only module enriched in both the “lower in AD vs LP-NCI” and “lower in AD vs HP-NCI” comparisons. Many of the “skyblue” proteins are involved in the regulation of RNA metabolism (splicing, transport, degradation, translation).
Of particular interest are those biological processes implicated in both the RNA and protein analyses that also are associated with AD in brain tissue. M23 (RNA module) and the red protein module each contain factors involved in synaptic vesicle regulation, and both were upregulated in AD iNs (Figure 4A,G,H). Importantly, this finding was validated in brain tissue (FDR q<1x10−5; Figure 4G). Interestingly, while overall numbers of synapses are reduced in the AD brain, a subset of synaptic proteins involved in vesicle trafficking are elevated in AD brain and iNs. Regarding modules downregulated in AD, both m116 (RNA) and the skyblue3 protein module contain ribosomal factors, and both are reduced in AD relative to LP-NCI iNs (Figure 4G,H). This finding also was validated in brain tissue (FDR q<1x10−5; Figure 4G). Taken together, these results demonstrate agreement between a subset of pathways altered in both AD neuronal cultures and AD brain tissue, and highlight specific genes, proteins and processes for further study.
Associations between brain neuropathology and Aβ and tau measures in cultured neurons from the same individuals
We next examined whether Aβ and/or tau levels measured in iNs from individuals across the neuropathological spectrum were correlated to the level of terminal neuritic plaque burden or tau tangle burden in the brain of the same individuals. In fAD, mutations in APP and PSEN either increase Aβ production generally or increase the ratio of Aβ42 to Aβ40 by raising Aβ42 and/or lowering Aβ40 (reviewed in (Bertram et al., 2010)). Different lengths of Aβ are determined by both the site of initial cleavage and the processivity of γ-secretase generating Aβ peptides ranging from 37-49 amino acids. We have developed a robust and specific ELISA to measure Aβ37, and in a parallel study have found that fAD missense mutations elevate Aβ42:40 ratio, but that an even stronger and more consistent effect is observed on the ratio of Aβ42 to Aβ37 (Liu et al., 2021). Further, age of onset for each of the fAD mutations tested was strongly correlated with the 42:37 ratio produced by cells (Liu et al., 2021). Therefore, we measured extracellular levels of Aβ37, Aβ38, Aβ40 and Aβ42 in the media of our iPSC-derived neurons (Figure 5A). Measurements across derivations of independent iPSC lines and across a wide range of cell plating densities were reproducible and consistent (Figure S6). We also measured Aβ peptides in the protein lysates, which captures both intracellular Aβ and cell surface-associated Aβ (referred to here as “intraAβ”).
Figure 5. Significant association between Aβ and tau species in neuronal cultures and postmortem plaque and tangle pathology in the brain.
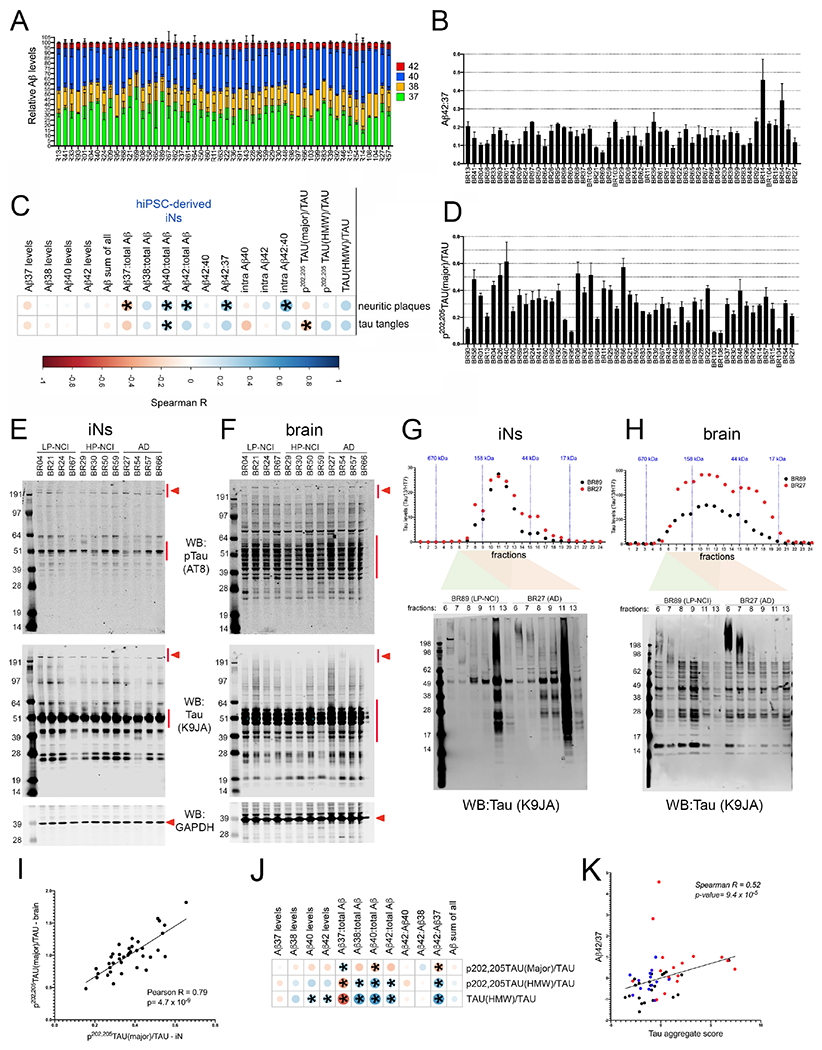
A-F) Aβ37, 38, 40 and 42 were quantified from the media of iNs and Aβ40 and 42 also were measured in cell lysate (“intraAβ”) by ELISA. Tau was quantified in cell lysates via WB. Tau was detected using K9JA (“tau”) and AT8 (p-tau). Measurements were made in at least 3 differentiations (average of 7 differentiations), 2-3 wells per differentiation.
(A) Stacked bar graph showing relative levels of each Aβ peptide measured in the media across lines (mean +/− SEM). (B) Aβ42:37 for each individual across differentiations (mean +/− SEM shown). Lines are listed in order of increasing neuritic plaque burden in the brain.
C) The mean for each metric was calculated across differentiations and a rank-order correlation coefficient calculated between each in vitro measure and neuritic plaque burden or tau tangle burden in the postmortem brain across 51 ROS/MAP individuals. Asterisks mark those associations with p<0.05.
D) Quantification across lines of the level of the major band of ptau relative to tau. Lines are listed in order of increasing tau tangle burden in the brain.
E,F) Representative WBs of iN lysates using AT8, K9JA, and GAPDH for iN (E) and brain tissue (F) lysates. Red arrowheads indicate “HMW” tau and red lines indicate “major” bands quantified.
G,H) iN and brain tissue lysates from two subjects (BR89, LP-NCI and BR27, AD) were fractionated using size exclusion chromatography and tau levels quantified in each fraction via ELISA (Tau13/HT7). Select fractions also were run on reducing gels and immunoblotted for tau (K9JA).
I) P-tau/tau in iNs (x-axis) relative to p-tau/tau in brain tissue (y-axis) quantified by WB (each dot represents data from a single individual).
J) Spearman correlations between Aβ and tau measures in iNs. Asterisks mark those associations with p<0.05.
K) Graph showing the relationship between Aβ42:37 and tau aggregate score in iNs. Tau aggregate scores were calculated by summation of the z-scores of: tau(HMW)/tau, p-tau(HMW)/tau, and −(p-tau(major)/tau). Red dots=AD; blue dots=HP-NCI; black dots=LP-NCI. See also Figures S6–S8.
We hypothesized that neurons derived from individuals who had a high Aβ burden in their brain would produce higher levels of Aβ, and/or higher levels of longer (more amyloidogenic) Aβ peptides relative to shorter peptides. We calculated rank-order correlation coefficients between APP products measured from iNs and neuritic plaque pathology in the brain. The Aβ42:37 ratio in the media, Aβ37, Aβ42 and Aβ40 as a proportion of all Aβ in the media and the Aβ42:40 ratio in the cell lysate (intraAβ42:40) each were significantly associated with neuritic plaque score (Figure 5A–C). The strongest association with neuritic plaque burden observed was with intraAβ42:40 ratio, and this ratio was associated with levels of TMEM35A, SF3B2 and SART3 (Figure S7, Table S4). Further, intraAβ42:40 was highly correlated with Aβ42:40 levels measured in water-insoluble Triton soluble brain extracts (mPFC, BA9/46) from the same individuals for whom we measured intraAβ42:40 in cultured neurons (r=0.44, p=0.0047), as well as Aβ42:40 levels in Triton-insoluble, GuHCl-soluble fractions of the same brain tissue (r=0.47, p=0.0028). These results demonstrate a level of concordance between specific Aβ measures in cultured neurons and from the aged brain of the same individuals.
Tau was measured in iNs via Western blot, and quantified immunoreactive bands include a 50-60 kDa set of “major” bands and >191 kDa high molecular weight (HMW) bands (Figure 5D, example WB shown in Figure 5E). These HMW bands were immunoreactive with both AT8 and K9JA, and also were recognized by antibodies to p217-tau and p181-tau (Figure S8). The proportion of p-tau in the major band had a significant negative association with tau tangle measures in brain (Figure 5C,D). To clarify this seemingly counter-intuitive result, we performed WB of Triton-soluble proteins in brain to determine the relative levels of the major band of AT8+ p-tau (example WB shown in Figure 5F). Tau banding in brain tissue was overlapping but distinct from that observed in iNs. Differences are likely due to variation in both splicing and in the specific patterns of the hundreds of post-translational modifications that have been described for tau. In spite of these differences, the correlation between AT8+ p-tau(major)/tau measured by WB in the brain and the iNs was remarkably high (Figure 5I). Further, levels of p-tau(major)/tau measured by WB in brain tissue were inversely correlated with tangle score (r=−0.41; p=8.3x10−3), as was observed in iNs.
HMW tau present in the human brain has recently been shown to significantly correlate with seeding capacity in an in vitro assay of tau aggregation, and tau seeding ability was associated with aggressiveness of the clinical disease (Dujardin et al., 2020). To confirm the identity of the HMW tau immunoreactive bands, we performed size exclusion chromatography (SEC) on both iN lysates and brain tissue. We quantified tau in each fraction by ELISA using two additional antibodies (Tau13/HT7), and examined tau by WB of the SEC fractions (Figure 5G,H). In both iNs and brain, tau was detected by ELISA in fractions above the 158 kDa SEC molecular weight standard, and a relatively strong HMW anti-tau immunoreactive signal is observed in these fractions via WB in AD samples (Figure 5G,H).
The amyloid hypothesis postulates that phosphorylation of tau occurs downstream of changes in Aβ (reviewed in (Selkoe and Hardy, 2016)). Here, we examined associations between Aβ levels and AD-associated tau levels across these different genetic backgrounds in human neurons. Similar to associations with plaque pathology, the strongest associations between Aβ and tau were between ratiometric measures of Aβ (i.e. Aβ37:total Aβ) rather than with absolute Aβ levels (Figure 5J). A single “tau aggregate score” (summation of signed values of p-tau(major)/tau, p-tau(HMW)/tau, and tau(HMW)/tau for each line) showed significant positive correlation with multiple Aβ measurements, the strongest association being with Aβ42:37 (Figure 5K).
Associations between Aβ and tau in neuronal cultures and measures of cognition
We next calculated rank-order correlations between Aβ peptides or tau and the slope of global cognition. The relative proportions of extracellular Aβ37, 38, 40 and 42 in iN media all were significantly associated with trajectory of cognition (Figure 6A). Examination of correlations between Aβ peptides revealed a striking inverse correlation between the relative levels of Aβ37 and all other Aβ peptides (Figure S7), and indeed higher relative levels of Aβ38, 40 and 42 were associated with worse cognitive outcome while higher relative levels of Aβ37 were associated with better cognitive outcome. The Aβ42:total Aβ and Aβ42:Aβ37 ratios showed the strongest correlations to cognitive trajectory (Figure 6A, r = −0.48, p = 4.2x10−4; and r = −0.44, p = 1.1x1 0−3, respectively) and were significantly correlated with terminal cognition measures. Further, p-tau also showed significant association with cognitive trajectory. Interestingly, p-tau“major”/tau was positively correlated with cognitive slope while the >191 kDa “HMW” p-tau/tau was inversely correlated (Figure 6A, r = 0.52, p = 9.79x10−5; and r = 0.49, p = 3.57x10−4 .respectively). Remarkably, the magnitudes of these associations with cognition in the iNs were equal to or higher than correlations between cognition and plaque or tangle burden in the brain of the same individuals. There were no significant associations between Aβ and baseline visit cognitive scores, suggesting an association only with age-related cognitive decline.
Figure 6. Significant association between Aβ and tau measurements in neuronal cultures and cognitive trajectory in the same individuals.
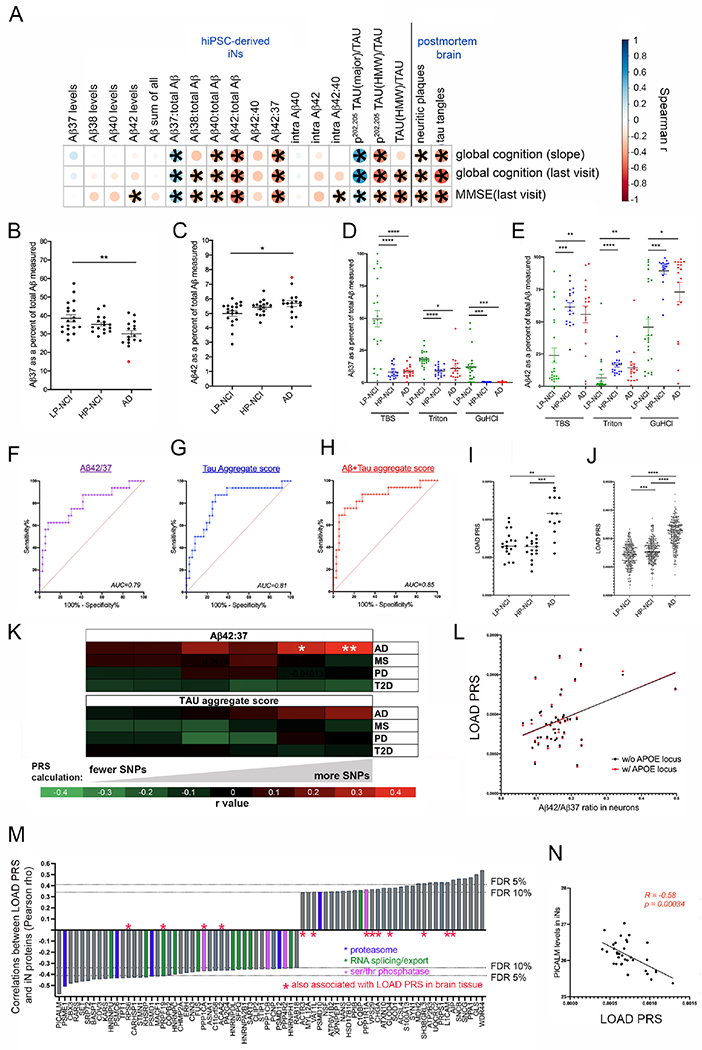
(A) Spearman correlation coefficients were calculated between measures of Aβ and tau in the iNs and slope of global cognition over age, global cognition at last visit and MMSE (n=52 individuals). Also shown are the correlations between brain pathology scores and measures of cognition for the same individuals.
(B,C) Plots of the mean value for each individual across the three diagnostic categories for Aβ37 (B) and Aβ42 (C) as a percent of all Aβ measured. One-way ANOVA with Tukey’s multiple comparisons test comparing across categories. Data points in red highlight an individual that we discovered had a PSEN1 coding variant (see main text).
(D,E) Sequential extractions were performed on postmortem brain (mPFC) of ROS/MAP individuals. Aβ levels were measured in sequential fractions (TBS-soluble, Triton-soluble and GuHCl-soluble) by ELISA. One-way ANOVA with Dunnett’s multiple comparison’s test.
F-H) Area under the curve (AUC) was calculated by plotting the receiver operating characteristic curve between NCI (n=36) and AD (n=16) iNs for Aβ42:37 (F), tau aggregate score (G) and Aβ+tau aggregate score (H). Aβ+tau aggregate score was calculated by summing the z-scores of tau aggregate score and Aβ42:37.
I,J) Polygenic risk scores (PRS) were calculated for each subject in our iPSC cohort (I) and in the entire ROS/MAP cohort (J) by incorporating all SNPs with p<0.1 association with LOAD. One-way ANOVA with Kruskal-Wallis test.
(K) Heat map showing Pearson correlations between iN Aβ42:37 or tau aggregate score and LOAD PRS, calculated using different thresholds of inclusion for SNPs. White asterisks show significant associations (*p=0.026; **p=0.008). Also shown are the absence of associations between Aβ42:37 or tau aggregate score and unrelated diseases: MS=multiple sclerosis; PD=Parkinson’s disease; T2D=type-2 diabetes. (L) Correlation between Aβ42:37 and LOAD PRS with (p=0.008) and without (p=0.009) inclusion of the APOE locus (chr19: 44,000,000-47,000,000).
(M) Waterfall blot of the strongest correlations between LOAD PRS and proteomic measurements acquired from iNs. Protein components related to the proteasome, RNA splicing/export, and serine threonine phosphatases are highlighted by colored bars, as indicated. Red asterisks denote those proteins that also were observed in brain tissue to be associated with LOAD PRS (defined by p<0.05).
(N) Correlation between PICALM protein levels in iNs and LOAD PRS.
For A-J, *p<0.05, **p<0.01, ***p<0.005, ****p<0.001. See also Figure S9.
As a group, AD iN cultures had higher Aβ42 and lower Aβ37 relative to all Aβ peptides. Aβ42:37 was significantly higher in AD than LP-NCI, which was driven by both lower Aβ37 and higher Aβ42 (Figure 6B,C). Using the same ELISA platforms, we see that high pathology brain samples also had a higher proportion of Aβ42 and a lower proportion of Aβ37 in all fractions compared to individuals who had low neuropathological burden, mirroring what was observed in iNs (Figure 6D,E). In line with the above-described observations, Aβ42:37 measured in iN media shows a modest performance in distinguishing NCI from AD (Figure 6F, p=0.0008). Aggregating measures of tau into a single score also shows a modest performance (Figure 6G, p=0.0003). Aggregating the Aβ and tau measures together outperforms either set of measurements alone (Figure 6H, p=5.7x10−5).
Many genetic variants of small effect size contribute to Aβ profile in neurons across individuals
We next examined whether genetic variants or age contribute to variation of Aβ and tau profiles in iNs. No association was observed between age at PBMC collection used for iPSC generation and any Aβ or tau measure (Figure S9). Whole genome sequencing of the individuals for whom iPSC lines were generated revealed rare coding variants in genes previously associated with AD (Figure S9). However, it is not known if these are pathogenic, protective, or benign variants. Intriguingly, the individual in our cohort who had the highest Aβ42 and lowest Aβ37 (BR14) had a PSEN1 coding variant (E318G) that is not a fAD mutation, but that may affect γ-secretase processivity (red dots in Figure 6B,C). APOE haplotype had only weak effects on the iN phenotypes analyzed. Using RNAseq and proteomics datasets, we compared neurons with an e4 allele versus those with no e4 allele. No genes passed multiple comparisons testing at the RNA level. In the proteomics analysis, one protein, AIP (aromatic hydrocarbon receptor interacting protein), was significantly differentially expressed (Figure S9). Of note, analyses of an isogenic set of APOE iNs showed no significant differences in tau measures between 4/4, 3/3 and 2/2 haplotypes (Figure S10). Previous studies have shown that APOE4 exacerbates tau pathology in iPSC experimental systems (Wang et al., 2018, Zhao et al., 2020). Here, the lack of association with APOE genotype may be due to many factors, including a lack of glia in these cultures (Wang et al., 2021).
No individual LOAD SNP (Lambert et al., 2013, Jansen et al., 2019, Kunkle et al., 2019) showed a significant association with Aβ42:37 or tau measures in iNs (Figure S9). Thus, we hypothesized that a combinatorial influence of variants converge to influence the Aβ and tau profiles in our experimental system. Polygenic risk scores (PRS) incorporate the weighted additive effects of common genetic variants genome-wide to quantify heritable disease risk (Dudbridge, 2013). We calculated PRS for LOAD at increasingly stringent p-value thresholds for SNP inclusion, based on available summary statistics from the most recent large-scale genomewide association studies (Felsky et al., 2018). As expected, LOAD PRS was significantly higher in those with an AD diagnosis both in our iPSC cohort and in the larger ROSMAP cohort (Figure 6I,J). PRS for LOAD was associated with iN Aβ42:37 (Figure 6K,L), with the strongest association being with the least stringent cutoff for SNP inclusion. LOAD PRS remained associated with iN Aβ42:37 after exclusion of the APOE locus (Figure 6L). This indicates that, in the absence of very strong perturbations such as fAD mutations, Aβ42:37 is influenced at least in part by numerous genetic variants of small effect size. Tau aggregate score was not associated with LOAD PRS, suggesting that elevated Aβ42:37 may be a more proximal consequence of LOAD PRS than effects on tau in iNs.
We next aimed to identify the pathways and proteins in human neurons most influenced by the genome-wide burden of LOAD genetic risk. Pearson correlations were calculated between all proteins detected in the iNs and LOAD PRS. Interestingly, the top association was a negative association with PICALM (Figure 6M,N), a protein previously linked to LOAD by genetics and function (reviewed in (Van Acker et al., 2019)). Proteins negatively associated with LOAD PRS were enriched in “protein serine/threonine phosphatase complex” (FDR=4.1x10−2), “regulation of mRNA metabolic process” (FDR=3.1x10−8) and “proteasome complex” (FDR=2.4x10−5)(Figure 6M). The observation of a genetic association with these terms suggests that alterations in these pathways are intrinsically encoded in the genome and highlight these as potential causal influencers of LOAD. Proteins also associated with PRS in brain tissue proteomic data with the same directionality are denoted by red asterisks (Figure 6M).
AD-associated Aβ and tau levels are associated with reduced PP1 protein levels in both iNs and in the postmortem human brain
To identify LOAD-associated candidate proteins for experimental validation, we identified proteins that were associated with LOAD PRS, Aβ42:37, and tau measures in both iN cultures and brain tissue (Figure 7A–D). We chose to pursue the relationship between protein phosphatase 1 (PP1) and Aβ and tau for the following reasons: 1) PPP1CA protein levels (a core component of PP1) are associated with LOAD PRS and with Aβ42:37 levels in iNs (Figure 7A,D); 2) PPP1CA protein levels in brain are associated with LOAD PRS and brain Aβ42:37 levels and are significantly lower in AD relative to LP-NCI and HP-NCI (Figure 7B,E,G); 3) Protein levels of PPP1R1A (a negative regulator of PP1 activity) are associated with LOAD PRS, Aβ42:37 and tau aggregate score in iNs (Figure 7A,D), and 4) Brain PPP1R1A protein levels are associated with LOAD PRS and brain Aβ42:37 levels, and are significantly higher in AD relative to LP-NCI and HP-NCI (Figure 7B,F,G).
Figure 7. Elevated Aβ42:37 induces a reduction in PP1 which in turn affects tau proteostasis in human neurons.
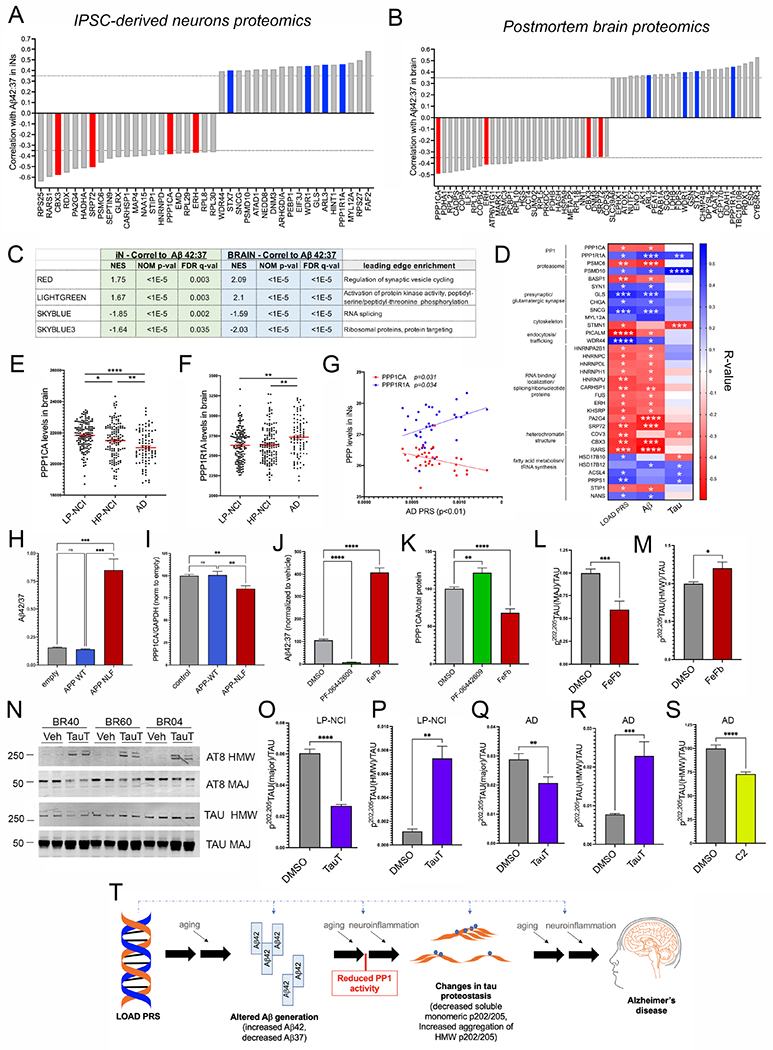
Waterfall plots of top associations (Pearson correlations, p-values ranging from 0.02-1.6x10−5) between Aβ42:37 and proteomic measurements acquired from iNs (A) and between brain Aβ42:37 (Triton-soluble fraction) and proteomic measurements acquired from brain tissue of the same individuals (B). Associations highlighted in red and blue were shared between the iNs and brain, a number greater than would be expected by chance (Chi-square test, p=0.0024).
(C) GSEA analysis of protein correlations in iNs highlight two brain-derived co-expression modules that are enriched with higher and two with lower Aβ42:37. These same protein modules also are associated with Aβ42:37 in the brain tissue. Rank files were generated using the −log10(pValue)*(r value) to factor both the significance as well as the magnitude and directionality of effect.
D) Heat map of all proteins associated with at least 2 of the 3 measures shown (LOAD PRS, Aβ42:37, tau aggregate score).
E,F) Protein levels of PPP1CA are significantly lower and PPP1R1A levels higher in AD brain compared to HP-NCI and LP-NCI. One-way ANOVA with Tukey’s multiple comparisons test.
G) Correlation between PPP1CA and PPP1R1A protein levels in iNs (as quantified by proteomics) and LOAD PRS.
H,I) IPSCs were transduced with lentivirus encoding either wild type APP, APP-NLF, or else with control lentivirus and differentiated to iNs. Aβ was measured in the media and WBs run on lysates to measure PPP1CA levels. Aβ42:37 levels were elevated (H) and PPP1CA levels reduced (I) with APP-NLF expression. Shown are data from three independent differentiations, >11 wells per condition. One-way ANOVA with Tukey’s multiple comparisons test.
J,K) iNs were treated with γ-secretase modulators to reduce (PF-06442609; 0.5 μM) or elevate (FeFb, 100 μM) the generation of longer versus shorter Aβ peptides. iNs were treated for 48-72 hours, and media collected for ELISA for Aβ (J) and cell lysates collected and WB performed for PPP1CA (K). Data show mean +/−SEM from six differentiations, 3 iPSC lines (BR15, BR21, BR89); n=20 wells per condition, each line normalized to vehicle. One-way ANOVA with Tukey’s multiple comparisons test.
L,M) Treatment of LP-NCI iNs with FeFb induces a reduction in p-tau(major)/tau, and an elevation in p-tau(HMW)/tau. Data show mean +/−SEM from three differentiations, 4 iPSC lines (BR21, BR40, BR60, BR89); n=14 wells per condition, each line normalized to vehicle. Welch’s t-test.
N-R) iNs were treated with tautomycin (TauT, 1μM) or vehicle (DMSO) for 24 hrs. Cells were lysed and WBs performed to measure levels of p-tau and total tau. Representative WB of 3 LP-NCI lines (N; see also Figure S11 for AD representative WB). Quantification across LP-NCI iNs (O,P) and AD iNs (Q,R). Data in O-R show mean +/−SEM from 5-7 differentiations, 5 LP-NCI iPSC lines (BR21, BR89, BR04, BR40, BR60), and 5 AD iPSC lines (BR27. BR54, BR14, BR46, BR103), n=18 wells per condition, each line normalized to vehicle. Welch’s t-test.
S) AD iNs (BR27, BR46, BR57, BR83, BR92) were treated with 10 μM C2-ceramide for 48 hours, lysed and tau analyzed by WB. Data shows mean +/−SEM, n=21 wells per condition, each line normalized to mean of vehicle treated wells within each differentiation round. Welch’s t-test.
For D-S, *p<0.05, **p<0.01, ***p<0.005, ****p<0.001. See also Figure S10, S11.
T) Schematic overview outlining cellular phenotypes and hypotheses regarding the cascade of events contributing to AD pathogenesis. Graphic created with BioRender.com.
Elevation of Aβ42:37 induces a reduction in PPP1CA protein levels, and PP1 inhibition subsequently affects tau proteostasis
Lysates of iNs spanning the range of PPP1CA expression were collected in an independent round of differentiation and levels of PPP1CA analyzed by WB. Rank order levels of PPP1CA were identical between WB and proteomic data (Figure S11). To determine whether changes in the Aβ42:37 ratio affect PP1 levels, we expressed APP harboring both the Swedish (KM670/671NL) and Iberian (I716F) fAD mutations (“APP-NLF”), which elevates the Aβ42:37 ratio (Figure 7H). Expression of APP-NLF in iNs significantly decreased protein levels of PPP1CA (Figure 7I). We also used γ-secretase modulators to reduce (PF-06442609 “PF” Pfizer) or to elevate (fenofibrate “FeFb”; Sigma) Aβ42:37 in iNs (Figure 7J). Consistent with the APP-NLF results, pharmacological reduction of Aβ42:37 resulted in an increase in PPP1CA protein levels, while elevation of Aβ42:37 reduced PPP1CA levels (Figure 7K). No changes in PPP1CA RNA levels were detected (Figure S11). These findings suggest that the reduced PPP1CA levels observed in AD neurons are at least in part downstream of alterations in Aβ processing.
FeFb treatment of iNs consistently resulted in an elevation of HMW p-tau/tau and a decrease in major p-tau/tau across four LP-NCI lines (Figure 7L,M). However, PF treatment had inconsistent effects on HMW p-tau/tau across four AD lines: in two lines it had no effect (BR46, BR57), in one line it induced a subtle reduction (BR83), and in the last line (BR27) it significantly reduced HMW p-tau (Figure S11). These results suggest that elevating Aβ42:37 in neurons with “healthy” tau is sufficient to induce changes in tau, but that reducing Aβ42:37 in iNs is not sufficient for lowering already elevated levels of HMW p-tau.
To investigate whether reduction in PP1 activity affects tau phosphorylation and/or Aβ42:37, we treated iNs with the PP1 inhibitor tautomycin (TauT)(Knobloch et al., 2007) for 24hrs followed by quantification of pan phospho-serine/threonine (pSer,Thr) and p-tau. TauT induced a significant elevation in overall pSer-Thr levels but had no effect on Aβ42:37 levels (Figure S11). However, inhibition of PP1 reduced p-tau(major)/tau, and increased p-tau(HMW)/tau in both LP-NCI and AD neurons (Figure 7N–R). Effect sizes of TauT were diminished in AD neurons relative to those observed in LP-NCI iNs, perhaps due to the already reduced levels of PPP1CA and elevated HMW p-tau observed in AD lines at baseline. These effects are consistent with the observed associations between elevated Aβ42:37, reduced PPP1CA, reduced p-tau(major)/tau, and elevated p-tau(HMW)/tau. With TauT treatment, PPP1R1A transcript levels did not change but transcript levels of PPP1CA were elevated, perhaps reflecting a compensatory effort of the cells in response to PP1 inhibition.
These data indicate that elevating PP1 activity in AD neurons can lower HMW p-tau levels. To our knowledge, there are no commercially available small molecules that can specifically elevate PP1 activity. However, C2-ceramide has been described to activate PP1 as well as PP2A (Dobrowsky and Hannun, 1992, Wolff et al., 1994, Chalfant et al., 1999). Here, treatment of AD iNs with C2-ceramide consistently reduced HMW p-tau across five AD lines (Figure 7S, Figure S11). Taken together, these results support the hypothesis that modulation of Aβ can alter levels of PP1, which in turn affects the phosphorylation and aggregation of tau (Figure 7T).
Discussion
We conceptualize two critical steps in the development of AD that are relevant to the design of this study. The first is the accumulation of Aβ oligomers in the brain, and the second is the complex multicellular response to Aβ oligomer accumulation that leads to cognitive decline, in part through effects on tau. By interrogating neurons that span the clinical and neuropathological spectrum, we identified proteins and pathways associated with the consequence of each step: accumulation of plaque and tangle burden in the brain (Figure 5) and cognitive decline (Figure 6,7). Through the analysis of neuron-only cultures, we are undoubtedly not capturing all important aspects of AD pathogenesis. In spite of this we were able to isolate and analyze the neuronal contribution of genetic risk for AD and observe consistent AD-relevant associations between our human culture system and the aged human brain. The unique set of iPSC lines developed and phenotyped here offers a tractable experimental system, ultimately across many CNS cell types, for probing the mechanisms underlying the accumulation of neuropathology and cognitive decline versus resilience across individuals.
Genetic studies of LOAD suggest that a combinatorial influence of variants converge to initiate the deposition of the hallmark amyloid plaques and tau tangles and associated cognitive decline that define the disease. Multiple lines of evidence suggest that soluble Aβ oligomers are upstream of changes in tau phosphorylation (Gotz et al., 2001, Oddo et al., 2004, Jin et al., 2011, Muratore et al., 2014) and that the inductions of AD-related changes in tau are a critical step that determines to what extent one is vulnerable or resistant to the accumulation of Aβ. We have established an experimental system that captures aspects of the cell biological processes relevant to these steps within human neurons (Figure 7T). We find that elevated Aβ42 and decreased Aβ37 in iNs are associated with plaque deposition and cognitive decline in the same individuals. We find that LOAD PRS is significantly associated with Aβ42:37 ratio, suggesting that the effects of multiple LOAD risk variants converge to result in an alteration of γ-secretase mediated cleavage of APP.
Previous studies of deterministic fAD mutations have shown a clear connection between rare genetic mutations in APP or PSEN1/PSEN2 and changes in Aβ profile (Scheuner et al., 1996, Bentahir et al., 2006). While numerous lines of evidence support a role of altered Aβ production in fAD, much less is known about the contribution of Aβ production to LOAD. Our work has revealed for the first time, associations between late-onset AD and measures of neuronal Aβ production. In contrast to fAD, in LOAD the ratio of Aβ42:40 generated in iNs was not associated with cognition. Rather, the strongest association with cognition was with the relative ratio of Aβ42 to the shortest and least amyloidogenic peptide, Aβ37. Remarkably, the associations of cognitive trajectory to Aβ peptide measurements in cultured neurons are stronger than the associations to neuritic plaque burden in the brains of the same cohort (Figure 6A). Moreover, a lower proportion of Aβ37 relative to all other Aβ peptides was observed in LOAD postmortem brain tissue (Figures 6B,C). Thus, the level of the short, non-amyloidogenic Aβ37 peptide is associated with relative protection from amyloid plaque formation and cognitive decline. It is unclear from our existing data if Aβ37 plays a direct protective role in neutralizing longer Aβ peptides, but if further work supports this, there may be important implications for the design of immunotherapeutic agents targeting Aβ.
Through multiple analyses, we consistently observed an elevation of a subset of proteins associated with synaptic vesicle biology in both AD iNs and brain. In the AD brain, reduction in synaptic numbers are the best correlate of cognitive decline (Terry et al., 1991). However, here we observe an upregulation of RNA and protein levels of a subset of pre-synaptic proteins in AD. Additional studies are warranted to experimentally validate these findings and determine if this upregulation is compensatory and/or if the dysregulation perhaps causes synapses to be vulnerable. Proteins involved in RNA splicing and localization, ribosomal proteins, proteasomal components, and trafficking proteins also emerged from multiple analyses. Each of the above-mentioned pathways previously has been implicated in studies of AD using model systems and/or in postmortem brain analyses (e.g., (Bereczki et al., 2016, Garcia-Esparcia et al., 2017, Canchi et al., 2019, Hsieh et al., 2019, Johnson et al., 2020)). Here we show that these same proteins are altered in human neurons that have not been exposed to confounding insults such as decades of degeneration, exogenously introduced mutations or toxic factors. Because these findings are in a well-controlled reductionist system, we raise the possibility that dysregulation of these pathways are initiating events that are tied to genetic risk for LOAD. This collection of iPSC lines provides a readily manipulable system for future studies that can further interrogate these and other hypotheses. As one example of the utility of this resource for probing events relevant to AD, we experimentally validate a mechanistic link between Aβ peptides and p-tau via PP1, a complex repeatedly associated with AD-related phenotypes in this study.
Limitations of the study
This iPSC resource models naturally occurring genetic diversity across humans. As such, we do not expect to observe strong effect sizes like those observed in studies of “knock out vs wild type” or “treatment vs no treatment”. Additionally, with -omics level data, one needs relatively strong effect sizes in order to overcome the burden of multiple comparisons. Our approach to this limitation was two-fold: 1) to strengthen confidence in the biological relevance of a subset of findings through analytic validation in better powered RNAseq and proteomic datasets from human brain and 2) to experimentally validate findings with the strongest evidence, i.e. components of PP1. In this study we predicted that shared biological domains may be disrupted across individuals which leads to the characteristic accumulation of Aβ and tau in the brain, and ultimately to cognitive decline and clinical Alzheimer’s disease. However, the basis of the biological network dysregulation may be subtle disruptions, distributed across many genes. Because of the inherent challenge this poses, GSEA was a useful complementary tool to single gene analyses that allowed us to identify pathways and networks that are associated with AD-relevant measures.
The iPSC system does not capture the accumulation of a lifetime of environmental exposures and aging-relating processes that influence disease-related processes. Additionally, here we focused upon neuronal biology, and processes across a variety of cell types clearly contribute to AD pathogenesis. LOAD is genetically complex, with over 30 loci identified that contribute significantly to disease risk (Lambert et al., 2013, Jansen et al., 2019), and many LOAD genes are expressed and have functions in astrocytes, microglia, and at the blood-brain barrier (BBB). Numerous studies have demonstrated the importance of neuroinflammation and disruption of the BBB in AD pathogenesis, and those findings underscore the utility of future studies in analogous microglial, astrocytic, and endothelial cellular systems to uncover non-neuronal pathways relevant to AD biology.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Tracy Young-Pearse (tpearse@bwh.harvard.edu).
Materials Availability
IPSC lines described here are made available to the scientific community for research purposes via the NYSCF repository following MTA and Data Usage Agreement approval by Rush University Medical Center (https://www.radc.rush.edu/). NYSCF iPSC search tool can be found here (https://nyscf.org/research-institute/repository-stem-cell-search) and more information about their repository by emailing repository@nyscf.org.
Data and Code Availability
RNAseq and proteomic datasets from iPSC-derivatives and brain tissue are available through the AMP-AD Knowledge Portal (http://doi.org/10.7303/syn25169976). ROSMAP cohort data can be requested at https://www.radc.rush.edu.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chick polyclonal Anti-MAP2 | ABCAM | Cat. # AB5392, RRID AB_2138153 |
| Mouse monoclonal Anti-SATB2 (C-terminal) | ABCAM | Cat. # AB51502 RRID AB_882455 |
| Rabbit polyclonal Anti-Synapsin 1 | Calbiochem | Cat. # 574777 |
| Rabbit polyclonal Anti-Tau (MAPT) | Dako | Cat. # A0024 RRID AB_10013724 |
| Mouse monoclonal Anti-Phospho-Tau (Ser202, Thr205) (AT8) | ThermoFisher | Cat. # MN1020 RRID AB_223647 |
| Rabbit monoclonal Anti-β-Tubulin III (clone 5H16) | Millipore | Cat. # MAB1637 RRID AB_2210524 |
| Mouse monoclonal Anti-NeuN | Millipore | Cat. # MAB377 RRID AB_2298772 |
| Rabbit monoclonal Anti-PPP1R1A (clone EP902Y) | Abcam | Cat. # Ab40877 RRID AB_775773 |
| Mouse monoclonal Anti-PP1, PPP1CA (clone E-9) | Santa Cruz | Cat. # sc-7482 RRID AB_628177 |
| Rabbit monoclonal anti-β-Amyloid (1-37) (D2A6H) | Cell Signaling Technology | Cat. # 12467S RRID AB_2797928 |
| Mouse monoclonal Anti-pT181 Tau (AT270) | ThermoFisher | Cat. # MN1050 RRID AB_223651 |
| Rabbit polyclonal Anti-pT217 Tau | GeneTex | Cat. # GTX24851 RRID AB_373931 |
| Bacterial and Virus Strains | ||
| FUdeltaGW-rtTA | Zhang et al., 2013 | Addgene plasmid # 19780 |
| Tet-O-FUW-EGFP | Zhang et al., 2013 | Addgene plasmid # 30130 |
| pTet-O-Ngn2-puro | Zhang et al., 2013 | Addgene plasmid # 52047 |
| Biological Samples | ||
| HUMAN BRAIN SAMPLES MPFC | RUSH RADC BRAIN BANK | https://www.radc.rush.edu/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PP1 inhibitor: Tautomycin | Sigma | CAS 109946-35-2 |
| iGSM Fenofibrate | Santa Cruz Biotechnologies | cat. # F6020 |
| GSM PF-06442609 | Pfizer | cat. # PF-06442609 |
| DNQX disodium salt, Tocris Bioscience | Fisher Scientific | cat. # 23-121-0 |
| D-AP5, Tocris Bioscience | Fisher Scientific | cat. # 01-061 |
| Streptavidin Sulfo-TAG | Meso Scale Discovery | cat. # R32AD-5 |
| C2 Ceramide | MedChemExpress | cat. # HY-101180 |
| Critical Commercial Assays | ||
| MSD V-PLEX Plus Aβ Peptide Panel 1 (6E10) Kit | MesoScale Discoveries | cat. # K15200G-1 |
| MSD V-PLEX sAPPα/sAPPβ Kit | MesoScale Discoveries | cat. #K15120E-1 |
| Aβx-37 ELISA | Liu et al., 2021. | N/A |
| Deposited Data | ||
| RNAseq data from human iNs | This paper | http://doi.org/10.7303/syn25169976 |
| Proteomics data from human iNs | This paper | http://doi.org/10.7303/syn25169976 |
| Experimental Models: Cell Lines | ||
| HUMAN IPSC LINE: BR01 | This paper | BR01, AJ0006 |
| HUMAN IPSC LINE: BR04 | This paper | BR04, AJ0056 |
| HUMAN IPSC LINE: BR08 | This paper | BR08, AJ0044 |
| HUMAN IPSC LINE: BR09 | This paper | BR09, AJ0038 |
| HUMAN IPSC LINE: BR103 | This paper | BR103, AJ0121 |
| HUMAN IPSC LINE: BR104 | This paper | BR104, AJ0113 |
| HUMAN IPSC LINE: BR108 | This paper | BR108, AJ0117 |
| HUMAN IPSC LINE: BR11 | This paper | BR11, AJ0002 |
| HUMAN IPSC LINE: BR13 | This paper | BR13, AJ0039 |
| HUMAN IPSC LINE: BR14 | This paper | BR14, AJ0003 |
| HUMAN IPSC LINE: BR15 | This paper | BR15, AJ0008 |
| HUMAN IPSC LINE: BR21 | This paper | BR21, AJ0046 |
| HUMAN IPSC LINE: BR22 | This paper | BR22, AJ0021 |
| HUMAN IPSC LINE: BR24 | This paper | BR24, AJ0040 |
| HUMAN IPSC LINE: BR26 | This paper | BR26, AJ0043 |
| HUMAN IPSC LINE: BR27 | This paper | BR26, AJ0001 |
| HUMAN IPSC LINE: BR28 | This paper | BR28, AJ0029 |
| HUMAN IPSC LINE: BR29 | This paper | BR29, AJ0030 |
| HUMAN IPSC LINE: BR30 | This paper | BR30, AJ0042 |
| HUMAN IPSC LINE: BR33 | This paper | BR33, AJ0047 |
| HUMAN IPSC LINE: BR36 | This paper | BR36, AJ0022 |
| HUMAN IPSC LINE: BR37 | This paper | BR37, AJ0031 |
| HUMAN IPSC LINE: BR39 | This paper | BR39, AJ0005 |
| HUMAN IPSC LINE: BR40 | This paper | BR40, AJ0045 |
| HUMAN IPSC LINE: BR41 | This paper | BR41, AJ0024 |
| HUMAN IPSC LINE: BR43 | This paper | BR43, AJ0020 |
| HUMAN IPSC LINE: BR46 | This paper | BR46, AJ0004 |
| HUMAN IPSC LINE: BR48 | This paper | BR48, AJ0028 |
| HUMAN IPSC LINE: BR50 | This paper | BR50, AJ0041 |
| HUMAN IPSC LINE: BR54 | This paper | BR54, AJ0048 |
| HUMAN IPSC LINE: BR57 | This paper | BR57, AJ0073 |
| HUMAN IPSC LINE: BR58 | This paper | BR58, AJ0068 |
| HUMAN IPSC LINE: BR59 | This paper | BR59, AJ0072 |
| HUMAN IPSC LINE: BR60 | This paper | BR60, AJ0067 |
| HUMAN IPSC LINE: BR61 | This paper | BR61, AJ0076 |
| HUMAN IPSC LINE: BR62 | This paper | BR62, AJ0069 |
| HUMAN IPSC LINE: BR63 | This paper | BR63, AJ0070 |
| HUMAN IPSC LINE: BR64 | This paper | BR64, AJ0066 |
| HUMAN IPSC LINE: BR65 | This paper | BR65, AJ0080 |
| HUMAN IPSC LINE: BR66 | This paper | BR66, AJ0094 |
| HUMAN IPSC LINE: BR67 | This paper | BR67, AJ0099 |
| HUMAN IPSC LINE: BR68 | This paper | BR68, AJ0095 |
| HUMAN IPSC LINE: BR69 | This paper | BR69, AJ0103 |
| HUMAN IPSC LINE: BR72 | This paper | BR72, AJ0090 |
| HUMAN IPSC LINE: BR83 | This paper | BR83, AJ0083 |
| HUMAN IPSC LINE: BR89 | This paper | BR89, AJ0089 |
| HUMAN IPSC LINE: BR91 | This paper | BR91, AJ0115 |
| HUMAN IPSC LINE: BR92 | This paper | BR92, AJ0116 |
| HUMAN IPSC LINE: BR93 | This paper | BR93, AJ0114 |
| HUMAN IPSC LINE: BR95 | This paper | BR95, AJ0119 |
| HUMAN IPSC LINE: BR97 | This paper | BR97, AJ0123 |
| HUMAN IPSC LINE: BR98 | This paper | BR98, AJ0109 |
| HUMAN IPSC LINE: BR99 | This paper | BR99, AJ0107 |
| Human iPS Cell Line (Episomal, CD34+, ApoE2) | Alstem cell advancement | iPS46 |
| Human iPS Cell Line (Episomal, CD34+, ApoE Knockout) | Alstem cell advancement | iPS36 |
| Human iPS Cell Line (Episomal, CD34+, ApoE3) | Alstem cell advancement | iPS26 |
| Human iPS Cell Line (Episomal, CD34+) | Alstem cell advancement | iPS16 |
| Software and Algorithms | ||
| R Studio, v3.6.1 of R; v1.2.5019 of R Studio | R Core Team, 2020 | https://www.rstudio.com |
| pairwiseCorrSkew.R for generating Figure 3 correlations and skew | This paper; zenodo | https://doi.org/10.5281/zenodo.5161656 |
| FIJI | Schindelin et al., 2012 | https://imagej.net/Fiji/Downloads |
| Kallisto pseudoalignment quantification program, v0.43.1 | Bray et al., 2016. | https://github.com/pachterlab/kallisto |
| Sleuth, v0.30.0 | Pimentel et al., 2017 | https://www.rdocumentation.org/packages/sleuth/versions/0.30.0 |
| Gene Set Enrichment Analyses (GSEA) | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ComBat algorithm in the SVA (v3.34.0) package in R | Johnson et al., 2007 | https://www.bioconductor.org/packages/release/bioc/vignettes/sva/inst/doc/sva.pdf |
| MaxQuant v1.6.3.4 | Cox et al., 2008 | https://github.com/galaxyproteomics/tools-galaxyp/ |
| WGCNA | Langfelder et al., 2008 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| TAMPOR | Johnson et al., 2020 | https://github.com/edammer/TAMPOR/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Induced Pluripotent Stem Cell Generation and lines
ROS and MAP studies were approved by an Institutional Review Board of Rush University Medical Center. All participants signed an informed consent, an Anatomical Gift Act, and a repository consent to allow their data and biospecimens to be repurposed. IPSC lines were generated following IRB review and approval through Partners/BWH IRB (#2015P001676). See also Figure S1 for details on human subjects cognition, pathology and distribution.
Peripheral blood mononuclear cell (PBMC) samples from the ROS and MAP cohorts were first expanded in StemPro-34 SFM Complete Medium (Thermo Fisher, 10639-011) supplemented with cytokines SCF (100ng/mL, R&D Systems, 255-SC-050), Flt3 (100ng/mL, R&D Systems, 308-FK-025/CF), IL3 (20ng/mL, Cell Signaling, 8918SF), and IL6 (20ng/mL, Cell Signaling, 8904SC) as previously described (Zhou et al., 2015) for 4 days to enrich for myeloid and erythroblast lineages and to generate samples for sterility testing by Chocolate Agar or SteriTEC sterility assays, cell line ID fingerprinting with SNPtrace genotyping panel (Fluidigm, PN100-6280) as described (Paull et al., 2015), and to prime cells for reprogramming. Primed PBMC samples passing sterility tests were seeded at either 30k or 50k cells/well in a 96-well Cultrex (HESC qualified Cultrex, Trevigen, 3434-001-02) coated plate for reprogramming using CytoTune™-iPS Sendai Reprogramming v2.0 Kit (Thermo Fisher, A16517) per the manufacturer’s recommendations modified for cell number and plate format. In some cases, primed PBMCs were first frozen into barcoded Matrix tubes (Thermo Fisher, 50823819) in Synth-a-freeze Cryopreservation Media (Thermo Fisher, A125542) before reprogramming. After infection, cells were gradually transitioned to Freedom media (DMEM-F12 with Freedom-1 Supplement, Life Technologies, Custom) for 5 days post-infection with partial daily media changes on the NYSCF Global Stem Cell Array® platform (Paull et al., 2015). Live cell surface staining using the iPSC marker Tra-1-60 (Tra-1-60 Antibody, Life Technologies) was performed 12-14 days post-transfection to identify reprogrammed cells. Successfully reprogrammed cell lines were expanded from 96-well plates to 24- and then to 6-well Cultrex coated plates to isolate colonies. Colonies were expanded via automation on the NYSCF Global Stem Cell Array® platform for further quality control assays and then frozen into barcoded Matrix tubes in Synth-a-freeze Cryopreservation Media at ≥500k cells/vial. All iPSC lines made by the NYSCF Global Stem Cell Array® undergo a rigorous quality procedure that includes a sterility check, mycoplasma testing, karyotyping, and a pluripotency assays (Figure S2). iPSCs were maintained using StemFlex Medium (Thermo Fisher Scientific). APP mutation lines were previously described (Muratore et al., 2014, Muratore et al., 2017). Sex of all iPS cell lines reported in Figure 1. See also Figure S2, S3 for detailed QC data.
Induced neuron differentiation
Induced neurons were generated as described with minor modifications described below. Several previously published studies have described the use of NGN2 to induce differentiation to neuronal fate (Pang et al., 2011, Zhang et al., 2013b, Ho et al., 2016, Nehme et al., 2018). iPSCs were plated in mTeSR1 media at a density of 95K cells/cm2 on Matrigel-coated plates for viral transduction. Media was changed from StemFlex to mTeSR1 as we found better transduction viability with mTeSR1 Viral plasmids were obtained from Addgene (plasmids #19780, 52047, 30130). FUdeltaGW-rtTA was a gift from Konrad Hochedlinger (Addgene plasmid # 19780). Tet-O-FUW-EGFP was a gift from Marius Wernig (Addgene plasmid # 30130). pTet-O-Ngn2-puro was a gift from Marius Wernig (Addgene plasmid # 52047). Lentiviruses were obtained from Alstem with ultrahigh titers (109) and used at the following concentrations: pTet-O-NGN2-puro: 0.1 ul/ 50K cells; Tet-O-FUW-eGFP: 0.05ul/ 50K cells; Fudelta GW-rtTA: 0.11ul/50K cells. Transduced cells were dissociated with Accutase and plated onto Matrigel-coated plates at 50,000 cells/cm2 in mTeSR1 (day 0). On day 1, media was changed to KSR media with doxycycline (2 ug/ml, Sigma). Doxycyline was maintained in the media for the remainder of the differentiation. On day 2, media was changed to 1:1 KSR: N2B media with puromycin (5 ug/ml, Gibco). Puromycin was maintained in the media throughout the differentiation. On day 3, media was changed to N2B media + 1:100 B27 supplement (Life Technologies), and puromycin (10 ug/ml). From day 4 on, cells were cultured in NBM media + 1:50 B27 + BDNF, GDNF, CNTF (10 ng/ml, Peprotech). All analyses presented here were at day 21 (d21) post induction of neuronal differentiation unless otherwise noted. Frozen stocks were generated at day 4 for all lines to facilitate the synchronized differentiation of dozens of lines in parallel.
Induced neuron protocol media:
•KSR media: Knockout DMEM, 15% KOSR, 1x MEM-NEAA, 55 uM beta-mercaptoethanol, 1x GlutaMAX (Life Technologies).
•N2B media: DMEM/F12, 1x GlutaMAX (Life Technologies), 1x N2 supplement B (Stemcell Technologies), 0.3% dextrose (D-(+)-glucose, Sigma).
•NBM media: Neurobasal medium, 0.5x MEM-NEAA, 1x GlutaMAX (Life Technologies), 0.3% dextrose (D-(+)-glucose, Sigma).
Induced astrocyte differentiation
Induced astrocytes (iAstros) were generated following a previously published paper (Canals et al., 2018) with minor modifications. IPSCs were plated at 95k cells/cm2 on a growth factor reduced matrigel (Corning #354230) coated plate, then were transduced with three lentiviruses – TetO Sox9 puro (Addgene plasmid #117269), TetO Nfib Hygro (Addgene plasmid #117271), and FUdeltaGW-rtTA (Addgene plasmid #19780). The cells were then dissociated with Accutase, plated at 200,000 cells/cm2 using Stemflex and ROCK inhibitor (10uM) (D0). From D1 to D6, the media was gradually switched from Expansion media (DMEM/F12, 10% FBS, 1% N2 supplement, 1% Glutamax) to FGF media (Neurobasal media, 2% B27, 1% NEAA, 1% Glutamax, 1% FBS, 8ng/ml FGF, 5ng/ml CNTF, 10ng/ml BMP4). On day 8, cells are replated at 84k cells/cm2 and from D8 to the end of differentiation D21, cells were cultured with Maturation media (1:1 DMEM/F12 and Neurobasal, 1% N2, 1% Glutamax, 1% Sodium Pyruvate, 5ug/ml N-acetyl cysteine, 5ng/ml heparin-binding EGF-like GF, 10ng/ml CNTF, 10ng/ml BMP4, 500ug/ml dbcAMP) and fed every 2-3 days.
Generation of iMGs from iPSCs
For the differentiation of iPSC-derived microglia-like cells (iMGs), iPSCs were differentiated using a previously-established protocol (Abud et al., 2017, McQuade et al., 2018) that was further optimized, as described here. HPCs were generated from iPSCs using the StemDiff Hematopoietic Kit. Cells were replated at day 12 at 100,000 cells per 35 mm well (1 well of a 6 well plate). Cells were plated in 2 mL of iMG media per well: DMEM/ F12, 2X insulin-transferrinselenite, 2X B27, 0.5X N2, 1X glutamax, 1X non-essential amino acids, 400μM monothioglycerol, 5 μg/mL insulin. Microglia media is supplemented with fresh cytokines before each use: 100 ng/mL IL-34, 50 ng/mL TGFβ1, and 25 ng/mL M-CSF. On days 14, 16, 18, 20, and 22, each well was supplemented with 1 mL of iMG media with freshly added cytokines. On day 24, all media is removed from the wells and the cells are dissociated by incubating with 5 minutes of PBS at room temperature. Both the media and the PBS is centrifuged at 300 x G for 5 minutes to pellet adherent and non-adherent cells. Cells are combined and replated at 100,000 cells per 15.6 mm well (1 well of a 24 well plate) in 1 mL of a 1:1 mixture of old media and fresh iMG media with tricytokine cocktail. On days 26, 28, 30, 32, 34, 36, each well is supplemented with 0.5 mL of iMG media with freshly added 3 cytokines. On day 37, all but 0.5 mL of media was removed from each well. Media was centrifuged for 5 minutes at 300 x G to pellet non-adherent cells. Cells are resuspended in iMG media supplemented with 100 ng/mL IL-34, 50 ng/mL TGFβ1, 25 ng/mL M-CSF, 100 ng/mL CD200 and 100 ng/mL CX3CL1. On days 39 and 41, cells are fed with microglia media with five cytokine cocktail (0.5 mL per well).
METHOD DETAILS
STR Profiling
Genomic DNA (gDNA) was prepared using QuickExtract DNA extraction solution (Epicentre, QE09050). iPS-NGN2 transduced cell lines were routinely sent to Genetica DNA Laboratories to confirm identity through STR profiling. gDNA from neuronal cultures were also STR profiled to confirm identity prior to RNAseq and proteomic analyses. Any samples failing identity verification were eliminated from all analyses.
Bulk RNA sequencing
For iNs, at least 250 ngs of total RNA input was oligo(dT) purified, then double-stranded cDNA was synthesized using Superscript III Reverse Transcriptase with random hexamers. Sequencing libraries were generated by processing 1 ng of generated cDNA through the Illumina Nextera Tagmentation library protocol. RNA and cDNA were quantified on a TapeStation 4200. Multiplexed libraries were sequenced on an Illumina NextSeq 500 to an average depth of 24 million mapped paired-end reads (150 bases) per sample. RNAseq reads were quality trimmed, then quantified using the Kallisto pseudoalignment quantification program (v0.43.1) (Bray et al., 2016) running 100 bootstraps against a Kallisto index generated from GRCh38. Kallisto quantified samples were compared using Sleuth (Pimentel et al., 2017)(v0.30.0) in R Studio (v3.6.1 of R; v1.2.5019 of R Studio). Differentially expressed genes were identified using a Wald test after controlling for library preparation batch. Expression values were exported from the Sleuth object as normalized TPM values. The RNAseq library preparation of all ROSMAP libraries took several rounds to complete. To generate a master expression matrix the exported expression vectors were combined, quantile normalized, then batch effects were regressed using the ComBat algorithm in the SVA (v3.34.0) package in R. Any gene with very low average expression (0.6TPM for gene level analysis, and 0.2TPM for transcript level analysis) was excluded from the matrix. Samples with abnormally high levels of either Oct2/3 or LEFTY2 (>20TPM) were suspected to have a high percentage of undifferentiated cells and were removed from the analysis. One additional outlier was identified in PCA analysis and was removed from the matrix. Of the remaining samples, 18 of the lines had replicate entries and their expression vectors were averaged in the final expression matrix. The final RNAseq master expression matrix has 49 samples and quantifies expression of 104,090 transcripts from 22,799 genes.
Single nucleus RNA sequencing
At the end of iN and iA differentiation, all media was removed from the wells and cells were washed once in ice-cold PBS with 0.04% BSA. Cells were lysed on ice for 15 minutes using lysis buffer: 10 mM Tris, 0.49% CHAPS, 0.1% BSA, 21 mM magnesium chloride, 1 mM calcium chloride, 146 mM sodium chloride. The nuclei were pelleted through centrifugation (5 minutes at 500 x G) and resuspended in PBS + 1% BSA for counting prior to loading into the 10X. At the end of iMGL cultures, cells were dissociated by incubating for 5 minutes in PBS at room temperature. Cells were centrifuged for 5 minutes at 300 x G and the pellets were combined and washed once in ice-cold PBS with 0.04% BSA. The suspension was centrifuged for 5 minutes at 500 x G to pellet cells and then lysed on ice for 15 minutes with lysis buffer. Nuclei were pelleted similar to previously described. Filtered single nucleus suspensions at 1k cells/ul were used to generate gel emulsion bead suspensions using the 10xGenomics Chromium controller and NextGEM 3’ reagents v3.1 (10X Genomics, San Francisco, CA) with a targeted recovery of 10000 cells. Single cell libraries were generated following the “Chromium NextGEM” protocol (CG000204 Rev D). A TapeStation 4200 was used to assay library quantity and size distribution. Libraries were sequenced at the New York Genome Center using a NovaSeq at a depth of 4E8 reads per sample (4E4 reads per cell). Fastq files were processed using the 10xGenomics CellRanger pipeline and standard CellRanger outputs were visualized using the Loupe Browser. Because samples from different donors were multiplexed together, individual nuclei were assigned to donors with the demuxlet software package (PMID:29227470), using genotype files derived from whole genome sequence data on these donors. Only nuclei that were called “singlets” and unambiguously identified as belonging to one donor were used for downstream visualization.
Gene Set Enrichment Analyses (GSEA)
GSEA were performed using both the desktop GSEA application (https://www.gsea-msigdb.org/gsea/index.jsp), and with the updated fgsea package for R, both giving highly comparable results (Mootha et al., 2003, Subramanian et al., 2005). Rank files for GSEA analyses of differentially expressed genes were generated by calculating the −log10(pValue) from Student’s t (2-sided unpaired, heteroscedastic) comparison and signed to reflect the directionality of differential expression. For GSEA of correlations we generated rank files using the -log10(pValue)*(r value) to factor both the significance as well as the magnitude and directionality of effect. Gene sets (*.gmt files) were generated using the gene set modules described in (Mostafavi et al., 2018) and separately using WGCNA (see “Generation of Co-expression Modules from ROS/MAP Brain Tissue Proteomics”). Normalized Enrichment Scores reflect the amount of enrichment of each gene set (module) in the top (+) or bottom (−) of the gene rank files (from DEG or correlation analysis), normalized to account for gene set sizes.
qPCR
In brief, samples were prepared using the Power SYBR™ Green Cells-to-Ct kit (Invitrogen #4402953) according to manufacturer’s instructions. Each biological sample had three technical replicates, and the qPCR was performed using ViiA7 System (Applied Biosystems), analyzed with Comparative CT method (ΔΔCT) normalized to GAPDH. Primers were ordered from IDT. Primers:
| Forward | Reverse | |
|---|---|---|
| GAPDH | GGGAGCCAAAAGGGTCATC | TGGTTCACACCCATGACGAA |
| PPP1CA | ACTACGACCTTCTGCGACTAT | AGTTCTCGGGGTACTTGATCTT |
| PPP1R1A | CCACGGCAACGGAAGAAGAT | CCCCAGGTGATGTTCAACCA |
LC-MS/MS proteomics
For each cell line, 1x106 cells were plated per well of a 6 well plate and were cultured for 21 days. On D21 iNs were lysed in NP40 lysis buffer (500 uL NP40, 1.5 mL 5M NaCl, 2.5 mL 1M Tris pH 7.6, 1.0 mL 0.5 M EDTA, 1 Complete protease inhibitor tablet, and brought up to 50 mL with MilliQ water). Lysates were stored at −80 degrees and then used for label-free LC-MS/MS proteomics through the Emory School of Medicine Proteomics Core, as described (Seyfried et al., 2017).
Protein concentrations were determined by the bicinchoninic acid (BCA) method, and 100ug of total protein was aliquoted for each sample. Protein homogenates were treated with 1 mM (final concentration) dithiothreitol (DTT) at 25°C for 30 minutes, followed by 5 mM (final concentration) iodoacetimide (IAA) at 25°C for 30 minutes in the dark and digested with 1:100 (w/w) lysyl endopeptidase (Wako) at 25°C overnight. The samples were then diluted with 2-fold with 50 mM NH4HCO3 and further digested overnight with 1:50 (w/w) trypsin (Promega) at 25°C. Detergent was removed using detergent removal spin columns (Thermo Scientific Pierce) according to manufacturer’s protocol. The samples were then acidified and desalted with a Sep-Pak C18 column (Waters). All samples were dried under vacuum.
The dried peptides were resuspended in peptide loading buffer (0.1% formic acid, 0.03% trifluoroacetic acid, 1% acetonitrile). Peptide mixtures (1ug) were separated on a self-packed C18 (1.9 um Dr. Maisch, Germany) fused silica column (25 cm x 75 uM internal diameter; New Objective, Woburn, MA) by Thermo Easy-nLc 1200 and monitored on a Q-Exactive HFX mass spectrometer (ThermoFisher Scientific , San Jose, CA) (Seyfried et al., 2017). Elution was performed over a 60 minute gradient at a rate of 300nl/min with buffer B ranging from 3% to 50% (buffer A: 0.1% formic acid in water, buffer B: 0.1 % formic in 80% acetonitrile). The mass spectrometer cycle was programmed to collect one full MS scan followed by 20 data dependent MS/MS scans. The MS scans (400-1600 m/z range, 1,000,000 AGC, 100 ms maximum ion time) were collected at a resolution of 60,000 at m/z 200 in profile mode and the MS/MS spectra (2 m/z isolation width with an isolation offset of 0.5 m/z, 32% collision energy, 50,000 AGC target, 50 ms maximum ion time) were acquired at a resolution of 15,000 at m/z 200. Dynamic exclusion was set to exclude previous sequenced precursor ions for 10 seconds within a 10 ppm window. Precursor ions with +1, and +6 or higher charge states were excluded from sequencing.
RAW data for all samples were analyzed using MaxQuant v1.6.3.4 with Thermo Foundation 2.0 for RAW file reading capability. The search engine Andromeda, integrated into MaxQuant, was used to build and search a concatenated target-decoy IPI/Uniprot human reference protein database (retrieved April 20, 2015; 90,411 target sequences), plus 245 contaminant proteins from the common repository of adventitious proteins (cRAP) built into MaxQuant. Methionine oxidation (+15.9949 Da), asparagine and glutamine deamidation (+0.9840 Da), and protein N-terminal acetylation (+42.0106 Da) were variable modifications (up to 5 allowed per peptide); cysteine was assigned a fixed carbamidomethyl modification (+57.0215 Da). Only fully tryptic peptides were considered with up to 2 miscleavages in the database search. A precursor mass tolerance of ±20 ppm was applied prior to mass accuracy calibration and ±4.5 ppm after internal MaxQuant calibration. Other search settings included a maximum peptide mass of 6,000 Da, a minimum peptide length of 6 residues, 0.05 Da tolerance for high resolution MS/MS scans. Co-fragmented peptide search was enabled to deconvolute multiplex spectra. The false discovery rate (FDR) for peptide spectral matches, proteins, and site decoy fraction were all set to 1 percent. Quantification settings were as follows: re-quantify with a second peak finding attempt after protein identification has completed; match MS1 peaks between runs; a 0.7 min retention time match window was used after an alignment function was found with a 20 minute RT search space. The quantitation method only considered razor plus unique peptides for protein level quantitation. Quantitation of proteins was performed using LFQ intensities given by MaxQuant. The full list of parameters used for MaxQuant are available as parameters.txt accompanying the public release.
Proteomic data were log2 transformed, filtered to remove any proteins with 7 or more missing values (out of 40 samples), and imputed with MinProb, q = 0.1. The ComBat algorithm was used to remove variance induced by differentiation round.
Generation of Co-expression Modules from ROS/MAP Brain Tissue Proteomics
ROS/MAP DLPFC Proteome abundance data was batch-normalized using a median polish algorithm as previously described ((Seyfried et al., 2017, Johnson et al., 2020), code available on https://www.github.com/edammer/TAMPOR/), 60/400 case samples not meeting AD, AsymAD, or NCI-low pathology status were excluded, 17 additional outliers based on a preliminary sample connectivity network (|z.k|>3) were removed, and then data were bootstrap regressed to remove age, sex, and PMI covariance, as previously published (Seyfried et al., 2017, Johnson et al., 2020). The WGCNA package for R v3.5.3, blockwise Modules function was used to cluster co-expression modules with the following parameters: scale free topology power beta=7; deepSplit=4; minimum module size 20; mergeCutHeight=0.07; TOMDenominator-’mean”; corType-’bicor”; “signed” network type; pamStage=TRUE; pamRespectsDendrogram=TRUE; and a p value reassignment threshold of 0.05. 56 modules met a minimum module size of 20, and additional smaller module member proteins were returned to grey (not in a module). Following calculation of signed kMEs using signedKME function with parameter corFnc-’bicor”, module members with kMEintramodule < 0.30 were first returned to grey, and then all proteins with a kME>0.30 were assigned to the module with the highest correlation, signed kMEs were recalculated, and the process was iterated until all assignments met the above criteria. Data for the 323 regressed and 400 unregressed ROSMAP proteomes can be found on https://www.synapse.org/Consensus.
For analyses of individual proteins, data were filtered to include tissue from only those participants that met the criteria for our three categories: LP-NCI (no clinical diagnosis of AD “pm_AD”=0, no pathological diagnosis of AD “pathoAD”=0); HP-NCI (no clinical diagnosis of AD “pm_AD”=0, pathological diagnosis of AD “pathoAD”=1); AD (clinical diagnosis of AD “pm_AD”=1, pathological diagnosis of AD “pathoAD”=1). This resulted in analysis of 366 brain samples. Following filtering for no missing values, 4,291 proteins were quantified.
Immunocytochemistry and microscopy
Cells were fixed with 4% paraformaldehyde (Sigma) for 10 minutes, followed by membrane permeabilization and blocking with 0.25% Triton X-100 (Sigma) and 2% donkey serum (Jackson Immunoresearch) in PBS for 1 hour at room temperature. Cells were then incubated with primary antibodies overnight at 4°C, secondary antibodies for 1 hour at room temperature, and 1:1000 DAPI (Life Technologies) for 10 minutes, with multiple washes between each step. Samples were imaged using Zeiss LSM710 or LSM880 confocal microscopes and Zen black software. Zen black and FIJI were used to pseudo-color images and add scale bars. Antibodies are listed below.
Western blotting
Lysates were prepared in a buffer containing 1% NP40, 10mM EDTA, 150mM NaCl, 50mM Tris, complete Protease Inhibitors (PI) and phosSTOP (Roche). BCA protein assays were performed on all samples to normalize for protein content (Pierce). Equal protein amounts 1.5-10 ug, depending on target) for each sample was loaded onto 4–12% Bis-Tris NuPAGE gels (Life Technologies) and transferred to nitrocellulose membranes. Prior to blocking with LI-COR TBS blocking buffer, blots were treated with REVERT Total Protein Stain (LI-COR). Blots were imaged in the 700 channel of the Odyssey-Clx system (LI-COR). REVERT Total Protein Stain was then removed with REVERT Destaining Solution (LI-COR) and then blocked in LI-COR TBS blocking buffer. Blots were then probed with primary antibodies (see “Antibodies for Immunocytochemistry and Western blotting” and KRT) in LI-COR TBS blocking buffer overnight. Following washes in TBST, blots were incubated with fluorophore-conjugated secondary antibodies and imaged on the Odyssey system (LI-COR).
Antibodies for Immunocytochemistry and Western blotting:
| Antigen | Host | ICC/WB | Dilution | Vendor | Catalog # |
|---|---|---|---|---|---|
| MAP2 | Chicken | ICC | 1/2000 | Abcam | ab5392 |
| SATB2 | Mouse | ICC | 1/500 | Abcam | ab51502 |
| SYN1 | Rabbit | ICC | 1/1000 | Calbiochem | 574777 |
| Tau (MAPT) | Rabbit | ICC | 1/200 | Dako | A0024 |
| Tau (MAPT) | Rabbit | WB | 1/2000 | Dako | A0024 |
| P202,205 AT8 (MAPT) | Mouse | WB | 1/250 | ThermoFisher | MN1020 |
| TUJ1 | Mouse | ICC | 1/1000 | Millipore | MAB1637 |
| NeuN | Mouse | ICC | 1/1000 | Millipore | MAB377 |
| PPP1CA | Mouse | WB | 1/200 | Santa Cruz | sc-7482 |
| PPP1R1A | Rabbit | WB | 1/2000 | Abcam | ab40877 |
Sequential human brain extraction for Αβ and tau analyses
For analyses of Aβ levels in the brain, grey matter from medial prefrontal cortex (BA9/46) was dissected and homogenized in Tris-buffered saline (TBS) + PI +phosSTOP and centrifuged at 100,000 g for 30 minutes. Supernatant was collected (“TBS-soluble”) and the pellet homogenized in 1% Triton X-100 in TBS + PI +phosSTOP and centrifuged at 100,000 g. Supernatant was collected (“Triton-soluble”) and the pellet homogenized in 6M guanidine hydrochloride (GuHCl) using a sonicator and centrifuged at 100,000g. The supernatant was then collected (“GuHC-soluble”).
Size-exclusion chromatography
Brain or iN TBS homogenate (350 μL total volume) were injected onto a Superdex 200 increase (24 mL bed volume) and run on a fast protein liquid chromatography (FPLC) system (AKTA; GE Healthcare) in PBS, pH 7.4. 500-μL fractions were collected after void volume for downstream experiments. Columns were calibrated with Gel Filtration Standard (Bio-Rad), which ranges from 1,350 to 670,000 Da.
ELISAs
48 hours before harvest (D19), neuronal media was changed and 100ul fresh NBM was dispensed into each well. On D21, media was collected and stored in −80C. Cells were then washed with cold PBS and lysed with 1% NP40 lysis buffer for 30 minutes on ice, as described above in LC-MS/Proteomics methods. Extracellular Aβ 38, 40, and 42 levels were measured using MSD V-PLEX Plus Aβ Peptide Panel 1 (6E10) Kit (cat. # K15200G-1) with controls diluted in given buffer. Intracellular Aβ 38, 40, and 42 were measured with the same kit, with controls diluted in lysis buffer. Each cell line had at least 3 experimental replicates (differentiation rounds) and 2-3 technical replicates (wells) for both intra-and extracellular ELISAs. sAPPα and sAPPβ levels were examined with the MSD sAAPα/sAAPβ Kit (cat. #K15120E-1).
For Aβx-37 assay, each well of an uncoated 96-well multi-array plate (Meso Scale Discovery, #L15XA-3) was coated with a PBS solution containing 3 μg/mL of 266 capture antibody (Elan) and incubated at room temperature overnight. A detection antibody solution was prepared with biotinylated monoclonal antibody recognizing C-terminus residues (D2A6H, Cell Signaling Tech), 100 ng/mL Streptavidin Sulfo-TAG (Meso Scale Discovery, #R32AD-5), and 1% BSA diluted in wash buffer. Following overnight incubation, 50 μL/well of the sample, followed by 25 μL/ well of detection antibody solution were incubated for 2 hr at room temperature with shaking at >300 rpm, washing wells with wash buffer between incubations. The plate was read and analyzed according to manufacturer protocol.
Aβ values were batched normalized to control for lot-to-lot variability in the ELISA kits and standards. Outliers were excluded (ROUT 10%) and a mean value calculated for each Aβ peptide (37, 38, 40, 42). The means were then summed to generate a “total” Aβ measurement, and this value used to calculate a “percent of total” measurement for each Aβ peptide. Mean values were used to examine associations with clinical and pathological data from the same individuals from whom the iPSC lines were derived.
For our home-made MSD electrochemiluminescence platform for the tau ELISA, each well of an uncoated 96-well multi-array plate (Meso Scale Discovery, #L15XA-3) was coated with 30 μL of a PBS solution containing capture antibody (3 μg/mL Tau-13, Biolegend, for both human Tau ELISAs) and incubated at room temperature (RT) overnight followed by blocking with 5% BSA in wash buffer for 1h at RT with shaking at >300 rpm. A detection antibody solution was prepared with biotinylated detection antibody, 100 ng/mL Streptavidin Sulfo-TAG (Meso Scale Discovery, #R32AD-5), and 1% BSA diluted in wash buffer. Following the blocking step, 50 μL/well of the sample, followed by 25 μL/well of detection antibody solution were incubated for 2 h at room temperature with shaking at >300 rpm, washing wells with wash buffer between incubations. The plate was read and analyzed according to the manufacturer’s protocol. The antibodies used to detect specific antigens were for for N-terminal tau (biotinylated HT-7, Thermofisher) and phospho tau (biotinylated AT8, Thermofisher). N-terminal tau concentration was calculated through calibration by recombinant Tau 441 (rPeptide).
IncuCyte assay
Day 4 iNs were dissociated and plated on Matrigel coated 96 well plates (Greiner Bio-One, 655090) at ~15,000 cells/cm2. Plates were cultured and imaged in an Essen live cell analysis IncuCyte system for up to 120 hours (DIV 9). Each cell line was plated in 3 wells per 96wp and 4 images were taken per well. Neurite outgrowth and cell body cluster analysis was performed using IncuCyte ZOOM software, neurite outgrowth pipeline.
Multi-Electrode Array (MEA)
The night before MEA culturing, each well of a 96 well plate (Axion Biosystems) was coated with Poly-D-Lysine (Sigma, P6407) and laminin (Invitrogen, 23017-015) overnight. The next day, laminin was washed with PBS and coated with Matrigel for 1 hour. D4 iN stocks were then thawed and plated in NBM media at a density of 25K cells per well (78K/cm2) (n=8 wells per line). During differentiation, iNs were fed with NBM media every 3-4 days. Doxycycline and puromycin treatment were stopped after D14. MEA plates were recorded every 1-2 days for 30 minutes for up to 20 days after plating (D24). For the inhibitor treatment, plates were first recorded for baseline neuronal activity and then 20 μM DNQX (Fisher Scientific, 23-121-0), 50 μM D-AP5 (Fisher Scientific, 01-061-0), or 0.1 μM TTX (Biotium, 00060) were applied for the final 30 minute recording. Background noise in spike data per electrode was calculated using TTX-treated wells (>95% of noise removed by eliminating all electrodes with 0 or 1 spikes/recording period). Mean firing rates over time (spikes/second) were calculated (Negri et al., 2020). Percent reduction in firing after inhibitor treatments were calculated per electrode after removing outliers (ROUT 10%, Prism).
QUANTIFICATION AND STATISTICAL ANALYSIS
Information regarding statistical analyses can be found in the figure legends. All statistical tests were computed using GraphPad Prism or R Studio. More information on the software and codes used for computational analyses can also be found in the Method Detail section and the KRT. All data is graphed with +/− standard error of the Mean (SEM) or else with standard deviation (SD), as noted in the figure legends. For correlation analyses Pearson correlations were employed when measurements were proximal or linear relationships were anticipated, while rank-order (Spearman) correlations were used when comparing more distant measures whose linearity could not be assumed. Analyses were executed using algorithms in the Harrell (Hmisc) biostatistical package and visualized using corrplot in R.
Supplementary Material
Table S1. Table of all genes showing significant concordance (FDR<0.05) between iN and brain RNAseq expression levels, related to Figure 3. RNAseq expression profiles from iN cultures and corresponding postmortem brain samples (medial prefrontal cortex) were compared. For each gene in the upper half of percentage variance, a Pearson correlation coefficient (rval) was calculated between iN and brain expression levels across 49 human subjects. 110 genes had correlations that individually passed BH multiple comparisons testing. Columns indicate chromosome (chr), rval, significance of correlation (pval), adjusted pval (FDR.BH). Green shaded cells indicate membership in one of the tested gene sets (sex chromosomes, eQTLs) or membership in none of the known sets (other).
Table S2. RNAseq analysis of iNs and brain tissue by category, related to Figure 4. Shown are genes that showed a trend of differential expression by category (p<0.05) in iNs, that then were validated in brain tissue to be differentially expressed for the same comparison following multiple comparisons testing (FDR q<0.05).
Table S3. Protein assignment to modules derived from ROSMAP brain tissue, related to Figure 4. WGCNA was used to generate co-expression modules using proteomics data from ROS and MAP brain tissue, as described in STAR methods.
Table S4. Gene ontology enrichment of proteomic correlations with intraAβ42:40 in iNs, related to Figure 4. GO analysis of proteins positively and negatively correlated with intraAβ42:40, significance determined by Fisher’s exact test with FDR correction.
Highlights.
Generation and characterization of iPSC lines from 53 deeply phenotyped aged humans
Degree of congruence in expression profiles between iNs and brain of the same person
Association of cognitive decline and measures of Aβ and tau in iPSC-derived neurons
Identification and validation of PP1 as a molecular link between LOAD PRS, Aβ and tau
Acknowledgements
This work was part of the AD Deep Phenotyping program supported by a gift from Rick and Nancy Moskovitz (TYP, DJS). This work also was supported by NYSCF, the Head Foundation, the Ellison Foundation, the Orchard Foundation, NIH grants R01AG061894, U01AG046170, U01AG061356, RF1AG057440, R01AG061798, R01NS117446, R01AG055909, R21AG056011, and R21MH118576. The results published here are in part based on human brain tissue data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org/). Study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. The authors thank Charles Jennings for critical reading of the manuscript. Data collection for ROS and MAP was supported through funding by NIA grants P30AG10161, R01AG15819, R01AG017917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, the Illinois Department of Public Health, and the Translational Genomics Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
DJS is a director and consultant of Prothena Biosciences.
References
- ABUD EM, RAMIREZ RN, MARTINEZ ES, HEALY LM, NGUYEN CHH, NEWMAN SA, YEROMIN AV, SCARFONE VM, MARSH SE, FIMBRES C, CARAWAY CA, FOTE GM, MADANY AM, AGRAWAL A, KAYED R, GYLYS KH, CAHALAN MD, CUMMINGS BJ, ANTEL JP, MORTAZAVI A, CARSON MJ, POON WW & BLURTON-JONES M 2017. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron, 94, 278–293 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AGGARWAL NT, WILSON RS, BECK TL, BIENIAS JL & BENNETT DA 2005. Mild cognitive impairment in different functional domains and incident Alzheimer’s disease. J Neurol Neurosurg Psychiatry, 76, 1479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNES LL, NELSON JK & REUTER-LORENZ PA 2001. Object-based attention and object working memory: overlapping processes revealed by selective interference effects in humans. Prog Brain Res, 134, 471–81. [DOI] [PubMed] [Google Scholar]
- BARNES LL, SCHNEIDER JA, BOYLE PA, BIENIAS JL & BENNETT DA 2006. Memory complaints are related to Alzheimer disease pathology in older persons. Neurology, 67, 1581–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENNETT DA, BUCHMAN AS, BOYLE PA, BARNES LL, WILSON RS & SCHNEIDER JA 2018. Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis, 64, S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENNETT DA, SCHNEIDER JA, ARVANITAKIS Z & WILSON RS 2012a. Overview and findings from the religious orders study. Curr Alzheimer Res, 9, 628–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENNETT DA, SCHNEIDER JA, BUCHMAN AS, BARNES LL, BOYLE PA & WILSON RS 2012b. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res, 9, 646–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENNETT DA, SCHNEIDER JA, WILSON RS, BIENIAS JL & ARNOLD SE 2004. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol, 61, 378–84. [DOI] [PubMed] [Google Scholar]
- BENNETT DA, SCHNEIDER JA, WILSON RS, BIENIAS JL & ARNOLD SE 2005. Education modifies the association of amyloid but not tangles with cognitive function. Neurology, 65, 953–5. [DOI] [PubMed] [Google Scholar]
- BENTAHIR M, NYABI O, VERHAMME J, TOLIA A, HORRE K, WILTFANG J, ESSELMANN H & DE STROOPER B 2006. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem, 96, 732–42. [DOI] [PubMed] [Google Scholar]
- BERECZKI E, FRANCIS PT, HOWLETT D, PEREIRA JB, HOGLUND K, BOGSTEDT A, CEDAZO-MINGUEZ A, BAEK JH, HORTOBAGYI T, ATTEMS J, BALLARD C & AARSLAND D 2016. Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Aizheimers Dement, 12, 1149–1158. [DOI] [PubMed] [Google Scholar]
- BERTRAM L, LILL CM & TANZI RE 2010. The genetics of Alzheimer disease: back to the future. Neuron, 68, 270–81. [DOI] [PubMed] [Google Scholar]
- BRAY NL, PIMENTEL H, MELSTED P & PACHTER L 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–7. [DOI] [PubMed] [Google Scholar]
- CANALS I, GINISTY A, QUIST E, TIMMERMAN R, FRITZE J, MISKINYTE G, MONNI E, HANSEN MG, HIDALGO I, BRYDER D, BENGZON J & AHLENIUS H 2018. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat Methods, 15, 693–696. [DOI] [PubMed] [Google Scholar]
- CANCHI S, RAAO B, MASLIAH D, ROSENTHAL SB, SASIK R, FISCH KM, DE JAGER PL, BENNETT DA & RISSMAN RA 2019. Integrating Gene and Protein Expression Reveals Perturbed Functional Networks in Alzheimer’s Disease. Cell Rep, 28, 1103–1116 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHALFANT CE, KISHIKAWA K, MUMBY MC, KAMIBAYASHI C, BIELAWSKA A & HANNUN YA 1999. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem, 274, 20313–7. [DOI] [PubMed] [Google Scholar]
- DOBROWSKY RT & HANNUN YA 1992. Ceramide stimulates a cytosolic protein phosphatase. J Biol Chem, 267, 5048–51. [PubMed] [Google Scholar]
- DUDBRIDGE F 2013. Power and predictive accuracy of polygenic risk scores. PLoS Genet, 9, e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUJARDIN S, COMMINS C, LATHUILIERE A, BEEREPOOT P, FERNANDES AR, KAMATH TV, DE LOS SANTOS MB, KLICKSTEIN N, CORJUC DL, CORJUC BT, DOOLEY PM, VIODE A, OAKLEY DH, MOORE BD, MULLIN K, JEAN-GILLES D, CLARK R, ATCHISON K, MOORE R, CHIBNIK LB, TANZI RE, FROSCH MP, SERRANO-POZO A, ELWOOD F, STEEN JA, KENNEDY ME & HYMAN BT 2020. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med, 26, 1256–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELSKY D, PATRICK E, SCHNEIDER JA, MOSTAFAVI S, GAITERI C, PATSOPOULOS N, BENNETT DA & DE JAGER PL 2018. Polygenic analysis of inflammatory disease variants and effects on microglia in the aging brain. Mol Neurodegener, 13, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELSKY D, ROOSTAEI T, NHO K, RISACHER SL, BRADSHAW EM, PETYUK V, SCHNEIDER JA, SAYKIN A, BENNETT DA & DE JAGER PL 2019. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat Commun, 10, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARCIA-ESPARCIA P, SIDERIS-LAMPRETSAS G, HERNANDEZ-ORTEGA K, GRAU-RIVERA O, SKLAVIADIS T, GELPI E & FERRER I 2017. Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am J Neurodegener Dis, 6, 15–25. [PMC free article] [PubMed] [Google Scholar]
- GOTZ J, CHEN F, VAN DORPE J & NITSCH RM 2001. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science, 293, 1491–5. [DOI] [PubMed] [Google Scholar]
- HO SM, HARTLEY BJ, TCW J, BEAUMONT M, STAFFORD K, SLESINGER PA & BRENNAND KJ 2016. Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods, 101, 113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HSIEH YC, GUO C, YALAMANCHILI HK, ABREHA M, AL-OURAN R, LI Y, DAMMER EB, LAH JJ, LEVEY AI, BENNETT DA, DE JAGER PL, SEYFRIED NT, LIU Z & SHULMAN JM 2019. Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep, 29, 301–316 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNG COY & LIVESEY FJ 2018. Altered gamma-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer’s Disease. Cell Rep, 25, 3647–3660 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISRAEL MA, YUAN SH, BARDY C, REYNA SM, MU Y, HERRERA C, HEFFERAN MP, VAN GORP S, NAZOR KL, BOSCOLO FS, CARSON CT, LAURENT LC, MARSALA M, GAGE FH, REMES AM, KOO EH & GOLDSTEIN LS 2012. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature, 482, 216–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANSEN IE, SAVAGE JE, WATANABE K, BRYOIS J, WILLIAMS DM, STEINBERG S, SEALOCK J, KARLSSON IK, HAGG S, ATHANASIU L, VOYLE N, PROITSI P, WITOELAR A, STRINGER S, AARSLAND D, ALMDAHL IS, ANDERSEN F, BERGH S, BETTELLA F, BJORNSSON S, BRAEKHUS A, BRATHEN G, DE LEEUW C, DESIKAN RS, DJUROVIC S, DUMITRESCU L, FLADBY T, HOHMAN TJ, JONSSON PV, KIDDLE SJ, RONGVE A, SALTVEDT I, SANDO SB, SELBAEK G, SHOAI M, SKENE NG, SNAEDAL J, STORDAL E, ULSTEIN ID, WANG Y, WHITE LR, HARDY J, HJERLING-LEFFLER J, SULLIVAN PF, VAN DER FLIER WM, DOBSON R, DAVIS LK, STEFANSSON H, STEFANSSON K, PEDERSEN NL, RIPKE S, ANDREASSEN OA & POSTHUMA D 2019. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet, 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIN M, SHEPARDSON N, YANG T, CHEN G, WALSH D & SELKOE DJ 2011. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A, 108, 5819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON ECB, DAMMER EB, DUONG DM, PING L, ZHOU M, YIN L, HIGGINBOTHAM LA, GUAJARDO A, WHITE B, TRONCOSO JC, THAMBISETTY M, MONTINE TJ, LEE EB, TROJANOWSKI JQ, BEACH TG, REIMAN EM, HAROUTUNIAN V, WANG M, SCHADT E, ZHANG B, DICKSON DW, ERTEKIN-TANER N, GOLDE TE, PETYUK VA, DE JAGER PL, BENNETT DA, WINGO TS, RANGARAJU S, HAJJAR I, SHULMAN JM, LAH JJ, LEVEY AI & SEYFRIED NT 2020. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG HM, SUBRAMANIAM M, TARG S, NGUYEN M, MALISKOVA L, MCCARTHY E, WAN E, WONG S, BYRNES L, LANATA CM, GATE RE, MOSTAFAVI S, MARSON A, ZAITLEN N, CRISWELL LA & YE CJ 2018. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol, 36, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOBLOCH M, FARINELLI M, KONIETZKO U, NITSCH RM & MANSUY IM 2007. Abeta oligomer-mediated long-term potentiation impairment involves protein phosphatase 1-dependent mechanisms. J Neurosci, 27, 7648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONDO T, ASAI M, TSUKITA K, KUTOKU Y, OHSAWA Y, SUNADA Y, IMAMURA K, EGAWA N, YAHATA N, OKITA K, TAKAHASHI K, ASAKA I, AOI T, WATANABE A, WATANABE K, KADOYA C, NAKANO R, WATANABE D, MARUYAMA K, HORI O, HIBINO S, CHOSHI T, NAKAHATA T, HIOKI H, KANEKO T, NAITOH M, YOSHIKAWA K, YAMAWAKI S, SUZUKI S, HATA R, UENO S, SEKI T, KOBAYASHI K, TODA T, MURAKAMI K, IRIE K, KLEIN WL, MORI H, ASADA T, TAKAHASHI R, IWATA N, YAMANAKA S & INOUE H 2013. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell, 12, 487–96. [DOI] [PubMed] [Google Scholar]
- KUNKLE BW, GRENIER-BOLEY B, SIMS R, BIS JC, DAMOTTE V, NAJ AC, BOLAND A, VRONSKAYA M, VAN DER LEE SJ, AMLIE-WOLF A, BELLENGUEZ C, FRIZATTI A, CHOURAKI V, MARTIN ER, SLEEGERS K, BADARINARAYAN N, JAKOBSDOTTIR J, HAMILTON-NELSON KL, MORENO-GRAU S, OLASO R, RAYBOULD R, CHEN Y, KUZMA AB, HILTUNEN M, MORGAN T, AHMAD S, VARDARAJAN BN, EPELBAUM J, HOFFMANN P, BOADA M, BEECHAM GW, GARNIER JG, HAROLD D, FITZPATRICK AL, VALLADARES O, MOUTET ML, GERRISH A, SMITH AV, QU L, BACQ D, DENNING N, JIAN X, ZHAO Y, DEL ZOMPO M, FOX NC, CHOI SH, MATEO I, HUGHES JT, ADAMS HH, MALAMON J, SANCHEZ-GARCIA F, PATEL Y, BRODY JA, DOMBROSKI BA, NARANJO MCD, DANIILIDOU M, EIRIKSDOTTIR G, MUKHERJEE S, WALLON D, UPHILL J, ASPELUND T, CANTWELL LB, GARZIA F, GALIMBERTI D, HOFER E, BUTKIEWICZ M, FIN B, SCARPINI E, SARNOWSKI C, BUSH WS, MESLAGE S, KORNHUBER J, WHITE CC, SONG Y, BARBER RC, ENGELBORGHS S, SORDON S, VOIJNOVIC D, ADAMS PM, VANDENBERGHE R, MAYHAUS M, CUPPLES LA, ALBERT MS, DE DEYN PP, GU W, HIMALI JJ, BEEKLY D, SQUASSINA A, HARTMANN AM, ORELLANA A, BLACKER D, RODRIGUEZ-RODRIGUEZ E, LOVESTONE S, GARCIA ME, DOODY RS, MUNOZ-FERNADEZ C, SUSSAMS R, LIN H, FAIRCHILD TJ, BENITO YA, et al. 2019. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet, 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWART D, GREGG A, SCHECKEL C, MURPHY EA, PAQUET D, DUFFIELD M, FAK J, OLSEN O, DARNELL RB & TESSIER-LAVIGNE M 2019. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP beta-CTFs, Not Abeta. Neuron, 104, 256–270 e5. [DOI] [PubMed] [Google Scholar]
- LAMBERT JC, IBRAHIM-VERBAAS CA, HAROLD D, NAJ AC, SIMS R, BELLENGUEZ C, DESTAFANO AL, BIS JC, BEECHAM GW, GRENIER-BOLEY B, RUSSO G, THORTON-WELLS TA, JONES N, SMITH AV, CHOURAKI V, THOMAS C, IKRAM MA, ZELENIKA D, VARDARAJAN BN, KAMATANI Y, LIN CF, GERRISH A, SCHMIDT H, KUNKLE B, DUNSTAN ML, RUIZ A, BIHOREAU MT, CHOI SH, REITZ C, PASQUIER F, CRUCHAGA C, CRAIG D, AMIN N, BERR C, LOPEZ OL, DE JAGER PL, DERAMECOURT V, JOHNSTON JA, EVANS D, LOVESTONE S, LETENNEUR L, MORON FJ, RUBINSZTEIN DC, EIRIKSDOTTIR G, SLEEGERS K, GOATE AM, FIEVET N, HUENTELMAN MW, GILL M, BROWN K, KAMBOH MI, KELLER L, BARBERGER-GATEAU P, MCGUINESS B, LARSON EB, GREEN R, MYERS AJ, DUFOUIL C, TODD S, WALLON D, LOVE S, ROGAEVA E, GALLACHER J, ST GEORGE-HYSLOP P, CLARIMON J, LLEO A, BAYER A, TSUANG DW, YU L, TSOLAKI M, BOSSU P, SPALLETTA G, PROITSI P, COLLINGE J, SORBI S, SANCHEZ-GARCIA F, FOX NC, HARDY J, DENIZ NARANJO MC, BOSCO P, CLARKE R, BRAYNE C, GALIMBERTI D, MANCUSO M, MATTHEWS F, EUROPEAN ALZHEIMER’S DISEASE, I., GENETIC, ENVIRONMENTAL RISK IN ALZHEIMER’S, D., ALZHEIMER’S DISEASE GENETIC, C., COHORTS FOR, H., AGING RESEARCH IN GENOMIC, E., MOEBUS S, MECOCCI P, DEL ZOMPO M, MAIER W, HAMPEL H, PILOTTO A, BULLIDO M, PANZA F, CAFFARRA P, et al. 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet, 45, 1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU L, LAURO BM, WOLFE MS & SELKOE DJ 2021. Hydrophilic loop 1 of Presenilin-1 and the APP GxxxG transmembrane motif regulate gamma-secretase function in generating Alzheimer-causing Abeta peptides. J Biol Chem, 100393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCQUADE A, COBURN M, TU CH, HASSELMANN J, DAVTYAN H & BLURTON-JONES M 2018. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol Neurodegener, 13, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOORE S, EVANS LD, ANDERSSON T, PORTELIUS E, SMITH J, DIAS TB, SAURAT N, MCGLADE A, KIRWAN P, BLENNOW K, HARDY J, ZETTERBERG H & LIVESEY FJ 2015. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep, 11, 689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOOTHA VK, LINDGREN CM, ERIKSSON KF, SUBRAMANIAN A, SIHAG S, LEHAR J, PUIGSERVER P, CARLSSON E, RIDDERSTRALE M, LAURILA E, HOUSTIS N, DALY MJ, PATTERSON N, MESIROV JP, GOLUB TR, TAMAYO P, SPIEGELMAN B, LANDER ES, HIRSCHHORN JN, ALTSHULER D & GROOP LC 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet, 34, 267–73. [DOI] [PubMed] [Google Scholar]
- MOSTAFAVI S, GAITERI C, SULLIVAN SE, WHITE CC, TASAKI S, XU J, TAGA M, KLEIN HU, PATRICK E, KOMASHKO V, MCCABE C, SMITH R, BRADSHAW EM, ROOT DE, REGEV A, YU L, CHIBNIK LB, SCHNEIDER JA, YOUNG-PEARSE TL, BENNETT DA & DE JAGER PL 2018. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci, 21, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURATORE CR, RICE HC, SRIKANTH P, CALLAHAN DG, SHIN T, BENJAMIN L, WALSH DM, SELKOE DJ & YOUNG-PEARSE TL 2014. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet, 23, 3523–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURATORE CR, ZHOU C, LIAO M, FERNANDEZ MA, TAYLOR WM, LAGOMARSINO VN, PEARSE RV 2ND, RICE HC, NEGRI JM, HE A, SRIKANTH P, CALLAHAN DG, SHIN T, ZHOU M, BENNETT DA, NOGGLE S, LOVE JC, SELKOE DJ & YOUNG-PEARSE TL 2017. Cell-type Dependent Alzheimer’s Disease Phenotypes: Probing the Biology of Selective Neuronal Vulnerability. Stem Cell Reports, 9, 1868–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEGRI J, MENON V & YOUNG-PEARSE TL 2020. Assessment of Spontaneous Neuronal Activity In Vitro Using Multi-Well Multi-Electrode Arrays: Implications for Assay Development. eNeuro, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEHME R, ZUCCARO E, GHOSH SD, LI C, SHERWOOD JL, PIETILAINEN O, BARRETT LE, LIMONE F, WORRINGER KA, KOMMINENI S, ZANG Y, CACCHIARELLI D, MEISSNER A, ADOLFSSON R, HAGGARTY S, MADISON J, MULLER M, ARLOTTA P, FU Z, FENG G & EGGAN K 2018. Combining NGN2 Programming with Developmental Patterning Generates Human Excitatory Neurons with NMDAR-Mediated Synaptic Transmission. Cell Rep, 23, 2509–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NG B, WHITE CC, KLEIN HU, SIEBERTS SK, MCCABE C, PATRICK E, XU J, YU L, GAITERI C, BENNETT DA, MOSTAFAVI S & DE JAGER PL 2017. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci, 20, 1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ODDO S, BILLINGS L, KESSLAK JP, CRIBBS DH & LAFERLA FM 2004. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron, 43, 321–32. [DOI] [PubMed] [Google Scholar]
- PANG ZP, YANG N, VIERBUCHEN T, OSTERMEIER A, FUENTES DR, YANG TQ, CITRI A, SEBASTIANO V, MARRO S, SUDHOF TC & WERNIG M 2011. Induction of human neuronal cells by defined transcription factors. Nature, 476, 220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAULL D, SEVILLA A, ZHOU H, HAHN AK, KIM H, NAPOLITANO C, TSANKOV A, SHANG L, KRUMHOLZ K, JAGADEESAN P, WOODARD CM, SUN B, VILBOUX T, ZIMMER M, FORERO E, MOROZIEWICZ DN, MARTINEZ H, MALICDAN MC, WEISS KA, VENSAND LB, DUSENBERRY CR, POLUS H, SY KT, KAHLER DJ, GAHL WA, SOLOMON SL, CHANG S, MEISSNER A, EGGAN K & NOGGLE SA Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods. [DOI] [PubMed] [Google Scholar]
- PAULL D, SEVILLA A, ZHOU H, HAHN AK, KIM H, NAPOLITANO C, TSANKOV A, SHANG L, KRUMHOLZ K, JAGADEESAN P, WOODARD CM, SUN B, VILBOUX T, ZIMMER M, FORERO E, MOROZIEWICZ DN, MARTINEZ H, MALICDAN MC, WEISS KA, VENSAND LB, DUSENBERRY CR, POLUS H, SY KT, KAHLER DJ, GAHL WA, SOLOMON SL, CHANG S, MEISSNER A, EGGAN K & NOGGLE SA 2015. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods, 12, 885–92. [DOI] [PubMed] [Google Scholar]
- PIMENTEL H, BRAY NL, PUENTE S, MELSTED P & PACHTER L 2017. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods, 14, 687–690. [DOI] [PubMed] [Google Scholar]
- SCHEUNER D, ECKMAN C, JENSEN M, SONG X, CITRON M, SUZUKI N, BIRD TD, HARDY J, HUTTON M, KUKULL W, LARSON E, LEVY-LAHAD E, VIITANEN M, PESKIND E, POORKAJ P, SCHELLENBERG G, TANZI R, WASCO W, LANNFELT L, SELKOE D & YOUNKIN S 1996. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med, 2, 864–70. [DOI] [PubMed] [Google Scholar]
- SELKOE DJ & HARDY J 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEYFRIED NT, DAMMER EB, SWARUP V, NANDAKUMAR D, DUONG DM, YIN L, DENG Q, NGUYEN T, HALES CM, WINGO T, GLASS J, GEARING M, THAMBISETTY M, TRONCOSO JC, GESCHWIND DH, LAH JJ & LEVEY AI 2017. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst, 4, 60–72 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHI Y, KIRWAN P, SMITH J, MACLEAN G, ORKIN SH & LIVESEY FJ 2012. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med, 4, 124ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPROUL AA, JACOB S, PRE D, KIM SH, NESTOR MW, NAVARRO-SOBRINO M, SANTA-MARIA I, ZIMMER M, AUBRY S, STEELE JW, KAHLER DJ, DRANOVSKY A, ARANCIO O, CRARY JF, GANDY S & NOGGLE SA 2014. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE, 9, e84547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SRIKANTH P, LAGOMARSINO VN, PEARSE RV 2ND, LIAO M, GHOSH S, NEHME R, SEYFRIED N, EGGAN K & YOUNG-PEARSE TL 2018. Convergence of independent DISC1 mutations on impaired neurite growth via decreased UNC5D expression. Transl Psychiatry, 8, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUBRAMANIAN A, TAMAYO P, MOOTHA VK, MUKHERJEE S, EBERT BL, GILLETTE MA, PAULOVICH A, POMEROY SL, GOLUB TR, LANDER ES & MESIROV JP 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A, 102, 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TASAKI S, GAITERI C, MOSTAFAVI S, YU L, WANG Y, DE JAGER PL & BENNETT DA 2018. Multi-omic Directed Networks Describe Features of Gene Regulation in Aged Brains and Expand the Set of Genes Driving Cognitive Decline. Front Genet, 9, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERRY RD, MASLIAH E, SALMON DP, BUTTERS N, DETERESA R, HILL R, HANSEN LA & KATZMAN R 1991. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol, 30, 572–80. [DOI] [PubMed] [Google Scholar]
- VAN ACKER ZP, BRETOU M & ANNAERT W 2019. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol Neurodegener, 14, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG C, NAJM R, XU Q, JEONG DE, WALKER D, BALESTRA ME, YOON SY, YUAN H, LI G, MILLER ZA, MILLER BL, MALLOY MJ & HUANG Y 2018. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med, 24, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG C, XIONG M, GRATUZE M, BAO X, SHI Y, ANDHEY PS, MANIS M, SCHROEDER C, YIN Z, MADORE C, BUTOVSKY O, ARTYOMOV M, ULRICH JD & HOLTZMAN DM 2021. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron, 109, 1657–1674 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSON RS, BECK TL, BIENIAS JL & BENNETT DA 2007. Terminal cognitive decline: accelerated loss of cognition in the last years of life. Psychosom Med, 69, 131–7. [DOI] [PubMed] [Google Scholar]
- WILSON RS, FLEISCHMAN DA, MYERS RA, BENNETT DA, BIENIAS JL, GILLEY DW & EVANS DA 2004. Premorbid proneness to distress and episodic memory impairment in Alzheimer’s disease. J Neurol Neurosurg Psychiatry, 75, 191–5. [PMC free article] [PubMed] [Google Scholar]
- WOLFF RA, DOBROWSKY RT, BIELAWSKA A, OBEID LM & HANNUN YA 1994. Role of ceramide-activated protein phosphatase in ceramide-mediated signal transduction. J Biol Chem, 269, 19605–9. [PubMed] [Google Scholar]
- ZHANG B, GAITERI C, BODEA LG, WANG Z, MCELWEE J, PODTELEZHNIKOV AA, ZHANG C, XIE T, TRAN L, DOBRIN R, FLUDER E, CLURMAN B, MELQUIST S, NARAYANAN M, SUVER C, SHAH H, MAHAJAN M, GILLIS T, MYSORE J, MACDONALD ME, LAMB JR, BENNETT DA, MOLONY C, STONE DJ, GUDNASON V, MYERS AJ, SCHADT EE, NEUMANN H, ZHU J & EMILSSON V 2013a. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell, 153, 707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, PAK C, HAN Y, AHLENIUS H, ZHANG Z, CHANDA S, MARRO S, PATZKE C, ACUNA C, COVY J, XU W, YANG N, DANKO T, CHEN L, WERNIG M & SUDHOF TC 2013b. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron, 78, 785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO J, FU Y, YAMAZAKI Y, REN Y, DAVIS MD, LIU CC, LU W, WANG X, CHEN K, CHERUKURI Y, JIA L, MARTENS YA, JOB L, SHUE F, NGUYEN TT, YOUNKIN SG, GRAFF-RADFORD NR, WSZOLEK ZK, BRAFMAN DA, ASMANN YW, ERTEKIN-TANER N, KANEKIYO T & BU G 2020. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat Commun, 11, 5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU H, MARTINEZ H, SUN B, LI A, ZIMMER M, KATSANIS N, DAVIS EE, KURTZBERG J, LIPNICK S, NOGGLE S, RAO M & CHANG S 2015. Rapid and Efficient Generation of Transgene-Free iPSC from a Small Volume of Cryopreserved Blood. Stem Cell Rev, 11, 652–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Table of all genes showing significant concordance (FDR<0.05) between iN and brain RNAseq expression levels, related to Figure 3. RNAseq expression profiles from iN cultures and corresponding postmortem brain samples (medial prefrontal cortex) were compared. For each gene in the upper half of percentage variance, a Pearson correlation coefficient (rval) was calculated between iN and brain expression levels across 49 human subjects. 110 genes had correlations that individually passed BH multiple comparisons testing. Columns indicate chromosome (chr), rval, significance of correlation (pval), adjusted pval (FDR.BH). Green shaded cells indicate membership in one of the tested gene sets (sex chromosomes, eQTLs) or membership in none of the known sets (other).
Table S2. RNAseq analysis of iNs and brain tissue by category, related to Figure 4. Shown are genes that showed a trend of differential expression by category (p<0.05) in iNs, that then were validated in brain tissue to be differentially expressed for the same comparison following multiple comparisons testing (FDR q<0.05).
Table S3. Protein assignment to modules derived from ROSMAP brain tissue, related to Figure 4. WGCNA was used to generate co-expression modules using proteomics data from ROS and MAP brain tissue, as described in STAR methods.
Table S4. Gene ontology enrichment of proteomic correlations with intraAβ42:40 in iNs, related to Figure 4. GO analysis of proteins positively and negatively correlated with intraAβ42:40, significance determined by Fisher’s exact test with FDR correction.
Data Availability Statement
RNAseq and proteomic datasets from iPSC-derivatives and brain tissue are available through the AMP-AD Knowledge Portal (http://doi.org/10.7303/syn25169976). ROSMAP cohort data can be requested at https://www.radc.rush.edu.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chick polyclonal Anti-MAP2 | ABCAM | Cat. # AB5392, RRID AB_2138153 |
| Mouse monoclonal Anti-SATB2 (C-terminal) | ABCAM | Cat. # AB51502 RRID AB_882455 |
| Rabbit polyclonal Anti-Synapsin 1 | Calbiochem | Cat. # 574777 |
| Rabbit polyclonal Anti-Tau (MAPT) | Dako | Cat. # A0024 RRID AB_10013724 |
| Mouse monoclonal Anti-Phospho-Tau (Ser202, Thr205) (AT8) | ThermoFisher | Cat. # MN1020 RRID AB_223647 |
| Rabbit monoclonal Anti-β-Tubulin III (clone 5H16) | Millipore | Cat. # MAB1637 RRID AB_2210524 |
| Mouse monoclonal Anti-NeuN | Millipore | Cat. # MAB377 RRID AB_2298772 |
| Rabbit monoclonal Anti-PPP1R1A (clone EP902Y) | Abcam | Cat. # Ab40877 RRID AB_775773 |
| Mouse monoclonal Anti-PP1, PPP1CA (clone E-9) | Santa Cruz | Cat. # sc-7482 RRID AB_628177 |
| Rabbit monoclonal anti-β-Amyloid (1-37) (D2A6H) | Cell Signaling Technology | Cat. # 12467S RRID AB_2797928 |
| Mouse monoclonal Anti-pT181 Tau (AT270) | ThermoFisher | Cat. # MN1050 RRID AB_223651 |
| Rabbit polyclonal Anti-pT217 Tau | GeneTex | Cat. # GTX24851 RRID AB_373931 |
| Bacterial and Virus Strains | ||
| FUdeltaGW-rtTA | Zhang et al., 2013 | Addgene plasmid # 19780 |
| Tet-O-FUW-EGFP | Zhang et al., 2013 | Addgene plasmid # 30130 |
| pTet-O-Ngn2-puro | Zhang et al., 2013 | Addgene plasmid # 52047 |
| Biological Samples | ||
| HUMAN BRAIN SAMPLES MPFC | RUSH RADC BRAIN BANK | https://www.radc.rush.edu/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PP1 inhibitor: Tautomycin | Sigma | CAS 109946-35-2 |
| iGSM Fenofibrate | Santa Cruz Biotechnologies | cat. # F6020 |
| GSM PF-06442609 | Pfizer | cat. # PF-06442609 |
| DNQX disodium salt, Tocris Bioscience | Fisher Scientific | cat. # 23-121-0 |
| D-AP5, Tocris Bioscience | Fisher Scientific | cat. # 01-061 |
| Streptavidin Sulfo-TAG | Meso Scale Discovery | cat. # R32AD-5 |
| C2 Ceramide | MedChemExpress | cat. # HY-101180 |
| Critical Commercial Assays | ||
| MSD V-PLEX Plus Aβ Peptide Panel 1 (6E10) Kit | MesoScale Discoveries | cat. # K15200G-1 |
| MSD V-PLEX sAPPα/sAPPβ Kit | MesoScale Discoveries | cat. #K15120E-1 |
| Aβx-37 ELISA | Liu et al., 2021. | N/A |
| Deposited Data | ||
| RNAseq data from human iNs | This paper | http://doi.org/10.7303/syn25169976 |
| Proteomics data from human iNs | This paper | http://doi.org/10.7303/syn25169976 |
| Experimental Models: Cell Lines | ||
| HUMAN IPSC LINE: BR01 | This paper | BR01, AJ0006 |
| HUMAN IPSC LINE: BR04 | This paper | BR04, AJ0056 |
| HUMAN IPSC LINE: BR08 | This paper | BR08, AJ0044 |
| HUMAN IPSC LINE: BR09 | This paper | BR09, AJ0038 |
| HUMAN IPSC LINE: BR103 | This paper | BR103, AJ0121 |
| HUMAN IPSC LINE: BR104 | This paper | BR104, AJ0113 |
| HUMAN IPSC LINE: BR108 | This paper | BR108, AJ0117 |
| HUMAN IPSC LINE: BR11 | This paper | BR11, AJ0002 |
| HUMAN IPSC LINE: BR13 | This paper | BR13, AJ0039 |
| HUMAN IPSC LINE: BR14 | This paper | BR14, AJ0003 |
| HUMAN IPSC LINE: BR15 | This paper | BR15, AJ0008 |
| HUMAN IPSC LINE: BR21 | This paper | BR21, AJ0046 |
| HUMAN IPSC LINE: BR22 | This paper | BR22, AJ0021 |
| HUMAN IPSC LINE: BR24 | This paper | BR24, AJ0040 |
| HUMAN IPSC LINE: BR26 | This paper | BR26, AJ0043 |
| HUMAN IPSC LINE: BR27 | This paper | BR26, AJ0001 |
| HUMAN IPSC LINE: BR28 | This paper | BR28, AJ0029 |
| HUMAN IPSC LINE: BR29 | This paper | BR29, AJ0030 |
| HUMAN IPSC LINE: BR30 | This paper | BR30, AJ0042 |
| HUMAN IPSC LINE: BR33 | This paper | BR33, AJ0047 |
| HUMAN IPSC LINE: BR36 | This paper | BR36, AJ0022 |
| HUMAN IPSC LINE: BR37 | This paper | BR37, AJ0031 |
| HUMAN IPSC LINE: BR39 | This paper | BR39, AJ0005 |
| HUMAN IPSC LINE: BR40 | This paper | BR40, AJ0045 |
| HUMAN IPSC LINE: BR41 | This paper | BR41, AJ0024 |
| HUMAN IPSC LINE: BR43 | This paper | BR43, AJ0020 |
| HUMAN IPSC LINE: BR46 | This paper | BR46, AJ0004 |
| HUMAN IPSC LINE: BR48 | This paper | BR48, AJ0028 |
| HUMAN IPSC LINE: BR50 | This paper | BR50, AJ0041 |
| HUMAN IPSC LINE: BR54 | This paper | BR54, AJ0048 |
| HUMAN IPSC LINE: BR57 | This paper | BR57, AJ0073 |
| HUMAN IPSC LINE: BR58 | This paper | BR58, AJ0068 |
| HUMAN IPSC LINE: BR59 | This paper | BR59, AJ0072 |
| HUMAN IPSC LINE: BR60 | This paper | BR60, AJ0067 |
| HUMAN IPSC LINE: BR61 | This paper | BR61, AJ0076 |
| HUMAN IPSC LINE: BR62 | This paper | BR62, AJ0069 |
| HUMAN IPSC LINE: BR63 | This paper | BR63, AJ0070 |
| HUMAN IPSC LINE: BR64 | This paper | BR64, AJ0066 |
| HUMAN IPSC LINE: BR65 | This paper | BR65, AJ0080 |
| HUMAN IPSC LINE: BR66 | This paper | BR66, AJ0094 |
| HUMAN IPSC LINE: BR67 | This paper | BR67, AJ0099 |
| HUMAN IPSC LINE: BR68 | This paper | BR68, AJ0095 |
| HUMAN IPSC LINE: BR69 | This paper | BR69, AJ0103 |
| HUMAN IPSC LINE: BR72 | This paper | BR72, AJ0090 |
| HUMAN IPSC LINE: BR83 | This paper | BR83, AJ0083 |
| HUMAN IPSC LINE: BR89 | This paper | BR89, AJ0089 |
| HUMAN IPSC LINE: BR91 | This paper | BR91, AJ0115 |
| HUMAN IPSC LINE: BR92 | This paper | BR92, AJ0116 |
| HUMAN IPSC LINE: BR93 | This paper | BR93, AJ0114 |
| HUMAN IPSC LINE: BR95 | This paper | BR95, AJ0119 |
| HUMAN IPSC LINE: BR97 | This paper | BR97, AJ0123 |
| HUMAN IPSC LINE: BR98 | This paper | BR98, AJ0109 |
| HUMAN IPSC LINE: BR99 | This paper | BR99, AJ0107 |
| Human iPS Cell Line (Episomal, CD34+, ApoE2) | Alstem cell advancement | iPS46 |
| Human iPS Cell Line (Episomal, CD34+, ApoE Knockout) | Alstem cell advancement | iPS36 |
| Human iPS Cell Line (Episomal, CD34+, ApoE3) | Alstem cell advancement | iPS26 |
| Human iPS Cell Line (Episomal, CD34+) | Alstem cell advancement | iPS16 |
| Software and Algorithms | ||
| R Studio, v3.6.1 of R; v1.2.5019 of R Studio | R Core Team, 2020 | https://www.rstudio.com |
| pairwiseCorrSkew.R for generating Figure 3 correlations and skew | This paper; zenodo | https://doi.org/10.5281/zenodo.5161656 |
| FIJI | Schindelin et al., 2012 | https://imagej.net/Fiji/Downloads |
| Kallisto pseudoalignment quantification program, v0.43.1 | Bray et al., 2016. | https://github.com/pachterlab/kallisto |
| Sleuth, v0.30.0 | Pimentel et al., 2017 | https://www.rdocumentation.org/packages/sleuth/versions/0.30.0 |
| Gene Set Enrichment Analyses (GSEA) | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ComBat algorithm in the SVA (v3.34.0) package in R | Johnson et al., 2007 | https://www.bioconductor.org/packages/release/bioc/vignettes/sva/inst/doc/sva.pdf |
| MaxQuant v1.6.3.4 | Cox et al., 2008 | https://github.com/galaxyproteomics/tools-galaxyp/ |
| WGCNA | Langfelder et al., 2008 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| TAMPOR | Johnson et al., 2020 | https://github.com/edammer/TAMPOR/ |