

Abstract
Background:
Chromosome 15q11-q13 duplication syndrome (Dup15q) is a neurogenetic disorder caused by duplications of the maternal copy of this region. In addition to hypotonia, motor deficits, and language impairments, Dup15q patients commonly meet the criteria for autism spectrum disorder (ASD) and have a high prevalence of seizures. It is known from mouse models that synaptic impairments are a strong component of Dup15q pathophysiology, however, cellular phenotypes that relate to seizures are less clear. The development of patient-derived induced pluripotent stem cells (iPSCs) provides a unique opportunity to study human neurons with the exact genetic disruptions that cause Dup15q.
Methods:
Here, we explored electrophysiological phenotypes in iPSC-derived neurons from four Dup15q patients compared to six unaffected controls, one patient with a 15q11-q13 paternal duplication, and three Angelman syndrome patients.
Results:
We identified several properties of Dup15q neurons that could contribute to neuronal hyperexcitability and seizure susceptibility. Compared to controls, Dup15q neurons had increased excitatory synaptic event frequency and amplitude and increased density of dendritic protrusions, along with increased action potential firing and decreased inhibitory synaptic transmission. Dup15q neurons also showed impairments in activity-dependent synaptic plasticity and homeostatic synaptic scaling. Finally, Dup15q neurons showed an increased frequency of spontaneous action potential firing compared to control neurons, in part due to disruption of KCNQ2 potassium channels.
Conclusions:
Together these data point to multiple electrophysiological mechanisms of hyperexcitability that may provide new targets for the treatment of seizures and other phenotypes associated with Dup15q.
Keywords: Autism spectrum disorder, Induced pluripotent stem cells, Dup15q syndrome, electrophysiology
Introduction
Chromosome 15q11-q13 is a region of the genome regulated by genomic imprinting. Most of the imprinted genes in this region are expressed exclusively from the paternally-inherited allele, while only one gene, UBE3A, is expressed exclusively from the maternally-inherited allele in mature neurons(1; 2). Disruption of this genomic region can lead to three clinically distinct neurological disorders: Prader-Willi syndrome (PWS), Angelman syndrome (AS), and chromosome 15q11-q13 duplication syndrome (Dup15q)(3). PWS is caused by the loss of the paternally-expressed genes on chromosome 15q11-q13(4). Although AS is most commonly associated with large maternal deletions of 15q11-q13 encompassing many genes, it is known that loss of function from the maternal allele of UBE3A alone results in the full syndrome(5). Dup15q results from duplications of maternal chromosome 15q11-q13(6). Though the causative gene(s) for Dup15q is less clear, UBE3A is thought to play an important role in Dup15q pathophysiology. This is due to phenotypic similarities between AS and Dup15q, as well as the observation that individuals with maternal, but not paternal, duplications of chromosome 15q11-q13 have Dup15q and UBE3A is the only imprinted gene expressed from the maternally-inherited allele.
In addition to developmental delay, language impairments, and motor impairments, which are also commonly found in AS, Dup15q patients often meet the criteria for autism spectrum disorder (ASD)(7; 8). In fact, the Dup15q chromosomal abnormality is commonly associated with ASD(9). Dup15q patients also have a high prevalence of seizures, which often do not respond well to commonly used medications(10), and have an increased risk for sudden unexplained death in epilepsy (SUDEP)(11). Investigations into cellular phenotypes of Dup15q neurons and their associated mechanistic underpinnings are critically important for the development of targeted therapeutics for seizures and other symptoms in these individuals. Furthermore, understanding these cellular phenotypes may provide insight to the mechanisms underlying idiopathic ASD.
To date, several mouse models of Dup15q have been developed, but it is unclear how well they translate to the human Dup15q syndrome. For instance, one model used chromosomal engineering to duplicate the entire chromosome 15q11-q13 syntenic region. While this mouse model has the best construct validity, disease relevant phenotypes were only observed when duplications were of paternal, rather than maternal, origin(12). Bacterial artificial chromosomes (BAC) transgenics have also been used to express 2x and 3x copies of C-terminal tagged UBE3A to model Dup15q. Although these mice demonstrated behavioral phenotypes reminiscent of ASD, this model does not address potential roles of the other genes that are duplicated in patients(13; 14). Despite these shortcomings, it is clear from these mouse models that synaptic impairments are a strong component of Dup15q pathophysiology(12–16). However, cellular phenotypes in human neurons that can be used for screening novel therapeutics have not been explored for this syndrome. The development of patient-derived induced pluripotent stem cells (iPSCs) provides a unique opportunity to study human neurons with the exact complex genetic disruptions that cause Dup15q(17; 18). Moreover, these neurons provide the ability to study the earliest stages of neuronal development, which is vital in the context of neurodevelopmental disorders such as Dup15q and other ASDs. The goal of our study is to explore a variety of relevant electrophysiological features in human Dup15q neurons to identify cellular phenotypes to be used for eventual cell-based functional screens. Here we present several neuronal cellular phenotypes associated with Dup15q patient iPSC-derived neurons that may be useful for understanding Dup15q pathophysiology and for screening therapeutics, an approach that has already been useful in understanding treatment of bipolar disorder(19).
Methods and Materials
Cell lines/Cell culture.
Studies were carried out using iPSC lines generated from four Dup15q patients, six unaffected controls, three AS patients, and one 15q11-q13 paternal duplication subject. For details regarding cell lines please see supplemental methods.
Electrophysiology.
Recordings and data analysis were carried out as previously described(20). For details regarding recording conditions and protocols see supplemental methods.
Live-cell image acquisition and analysis.
iPSC-derived neurons were imaged on a Zeiss 780 confocal microscope. Neurons with clearly distinguishable processes were imaged as a Z-series (0.2μm interval) and tile scan to capture the entire cell. For details regarding image analysis see supplemental methods.
Immunocytochemistry and flow cytometry.
Immunostaining and flow cytometry were carried out as previously described(20). For details regarding reagents and protocols, see supplemental methods.
Results
Electrophysiological characterization.
To maintain consistency with our previous studies of chromosome 15-associated diseases, iPSC lines were generated and differentiated into neurons using the “embryoid body” approach as previously described(20) from six unaffected control (CTR) subjects, four chromosome 15q duplication (Dup15q) patients, and a single individual with a paternal chromosome 15q11-q13 duplication (Pat Dup) who did not have Dup15q or ASD (Fig. 1a; Supp. Fig. 1a)(21). Given that UBE3A is paternally silenced in neurons, the single Pat Dup line provides a qualitative tool for examining the involvement of overexpression of genes other than UBE3A housed within 15q11–13, including GABAA receptor subunits. Neurons were also generated from three AS patients (Supp. Fig. 1b). Cell cultures consisted of ~80% TBR1+ glutamatergic neurons and a smaller fraction of GAD67+ inhibitory neurons and astrocytes, and these compositions were not different between control and Dup15q (Fig. 1b, Supp. Fig. 1e, Supp. Table 2, Supp. Table 3). These data are similar to our previously published findings in control and AS-derived cultures(20).
Figure 1. Electrophysiological profile of iPSC-derived neurons from control and Dup15q subjects during in vitro development.
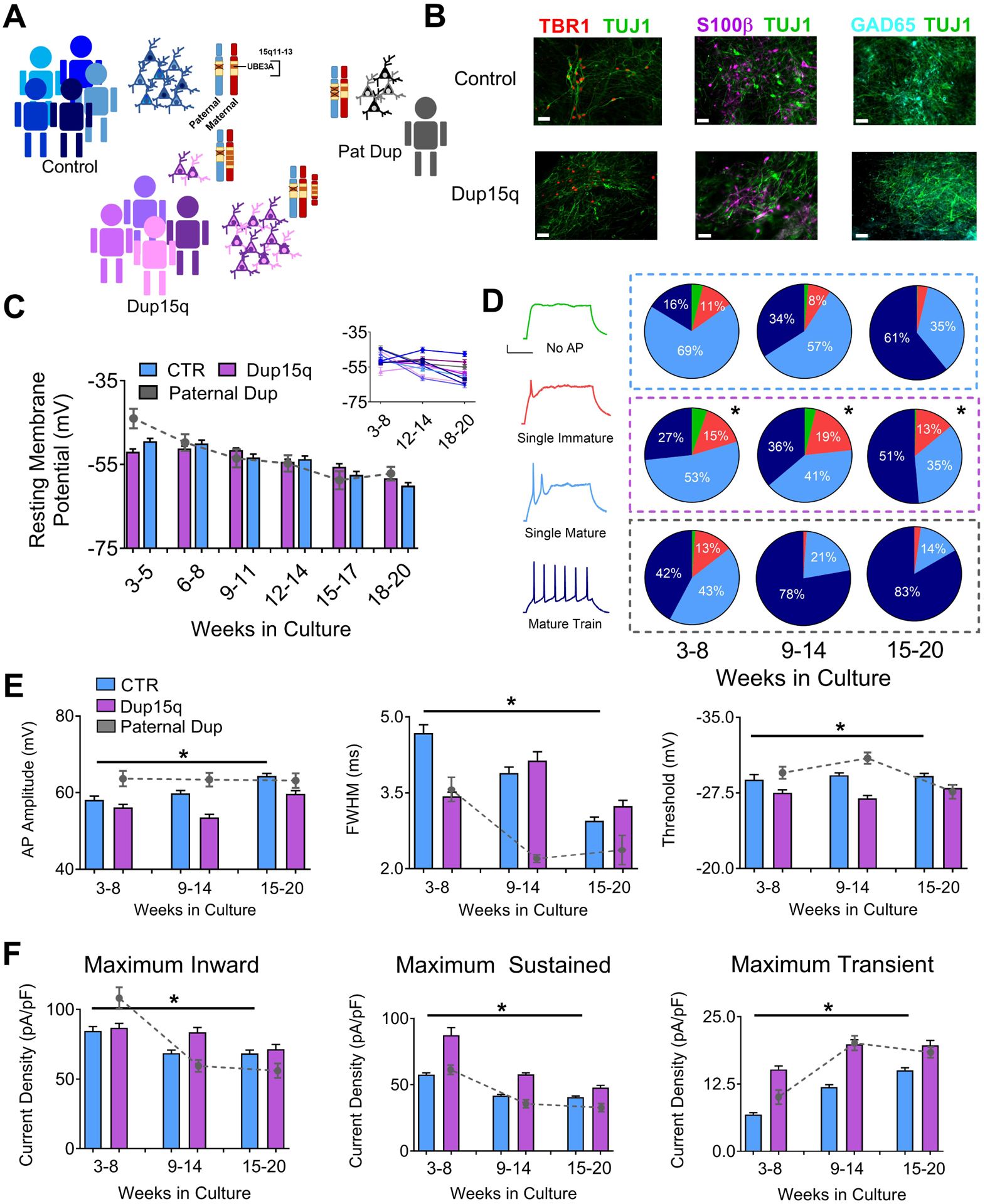
(A) Schematic of patient line genetics of the chromosome 15q11–13 genomic locus. (B) Immunocytochemical staining for TBR1/TUJ1, S100β/TUJI, and GAD65/TUJ1, in control and Dup15q-derived cultures between 20 and 25 weeks in culture. Scale bar, 50 μm (C) Group data for resting membrane potential (RMP) of control (CTR; 5 subjects; n>200 at each time point), Dup15q (4 subjects; n>175 at each time point) and a subject with a paternal duplication of chromosome 15q11-q13 (Paternal Dup; 1 subject; n=45 at each time point) during development. Inset: RMP for all individual lines. For each time bin, n≥30 for all lines. (D) Left: Example traces representing four AP firing patterns used for characterization. Scale bar, 20 mV, 200 ms. Right: Distribution of AP firing patterns for control (blue box; 5 subjects; n>430 at each time point), Dup15q (purple box; 4 subjects; n>430 at each time point), and a 15q11-q13 paternal duplication (grey box; 1 subject; n>60 at each time point) neurons at three developmental time bins. *P<0.0001 for differences between control and Dup15q; χ2 test. #P<0.0001 for differences between control and paternal duplication; χ2 test. (E) AP amplitude (left), full width at half-maximum amplitude (FWHM; middle), and AP threshold (right), for control, Dup15q, and paternal duplication cultures at three time points (n>350 for both control and dup15q at all time points; n>80_for paternal duplication at all time points). *P<0.001 for significant differences between control and Dup15q, #P<0.001 for significant differences between control and paternal duplication (two-way ANOVA). (F) Group data for maximum inward current density (left), maximum sustained outward current density (middle), and maximum transient outward current density (right), for control, Dup15q, and paternal duplication cultures at three time points (n>350 for both control and dup15q at all time points; n>80_for paternal duplication at all time points). *P<0.001 for significant differences between control and Dup15q, #P<0.001 for significant differences between control and paternal duplication (two-way ANOVA).
We recently reported that iPSC-derived neurons from AS patients show impaired maturation of resting membrane potential (RMP), action potential (AP) firing, and synaptic activity. Moreover, these phenotypes were the specific result of loss of UBE3A, as knockdown or knockout of UBE3A recapitulated these developmental deficits(20). Given the close association of AS and Dup15q and the involvement of UBE3A in these syndromes, we examined electrophysiological development of patient-derived Dup15q neurons. Whole-cell patch clamp recordings were performed on ~15 cells/coverslip/week/line starting at plating (week 3) through 20 weeks in culture. Current clamp and voltage clamp protocols were used on all cells to measure RMP, AP firing, inward and outward voltage-gated currents, and synaptic activity. Input and series resistance is listed in Supp. Table 4. Control neurons shifted to more hyperpolarized RMP values across 20 weeks in culture (Fig. 1c), in line with previous observations in iPSC-derived neurons(22). However, unlike AS-derived neurons, Dup15q and Pat Dup neurons showed typical developmental maturation of the RMP, suggesting that this impairment is specific to loss of UBE3A. Results from individual lines are also shown (Fig. 1c).
We next categorized AP firing in neurons during in vitro development. Interestingly, although they do not show the developmental disruption in RMP observed in AS neurons, Dup15q-derived neurons show a delay in AP maturation compared to controls (Fig. 1d; right). Conversely, recordings from the Pat Dup line show an enhanced maturation of AP firing compared to controls, however only a single Pat Dup cell line was investigated. AP development was further characterized by examining amplitude, width, and threshold, as well as voltage-gated inward and outward currents. During in vitro development, control neurons show an increase in AP amplitude, a decrease in FWHM, and no change in AP threshold (Fig. 1e), indicative of AP maturation. These results are consistent with our previously published data on 4 separate control lines(20). Dup15q-derived neurons have a significantly decreased AP amplitude, increased FWHM, and a more depolarized threshold at later stages of development compared to controls. Cell capacitance, which increased during in vitro development, was not different between control and Dup15q neurons (Supp. Fig. 1c). Action potential waveform examples for every cell line measured are depicted in Supp. Fig. 2. Line-by-line quantification of these electrophysiological features is quantified in Supp. Fig. 3. Importantly, cell death, as measured by an LDH assay, was also not significantly different between control and Dup15q cultures (Supp. Fig. 1d).
Synaptic transmission and plasticity.
Both deletion and duplication of UBE3A have been linked to impairments in synaptic transmission and plasticity (13–15; 23; 24). For this reason, we measured the development of spontaneous synaptic activity in control, Dup15q, and Pat Dup cultures. Example traces of spontaneous excitatory activity are depicted in Fig. 2a. Overall, control, Dup15q and Pat Dup cultures showed similar percentages of synaptically-active cells, which increased in all three genotypes during development (Fig. 2b). Both the frequency and amplitude of spontaneous synaptic events were significantly greater in Dup15q neurons compared to controls (Fig. 2c,d). Amplitude of synaptic events was also increased in Pat Dup cells (Fig. 2d). EPSC rise and decay times for each line is listed in Supp. Table 4 and example traces for every cell line measured are shown in Supp. Fig. 3. Spontaneous inhibitory postsynaptic currents (IPSCs) recorded from controls, Dup15q, and AS neurons also showed increased IPSC frequency and amplitude during development (Fig. 2e–g). Interestingly, the frequency of IPSCs was significantly reduced in AS and Dup15q neurons compared to controls at 3–8 and 9–12 weeks in culture (Fig 2f). Subtle but significant differences in IPSC amplitude were also observed (Fig. 2g).
Figure 2. Development of synaptic activity and expression of activity-dependent synaptic plasticity.
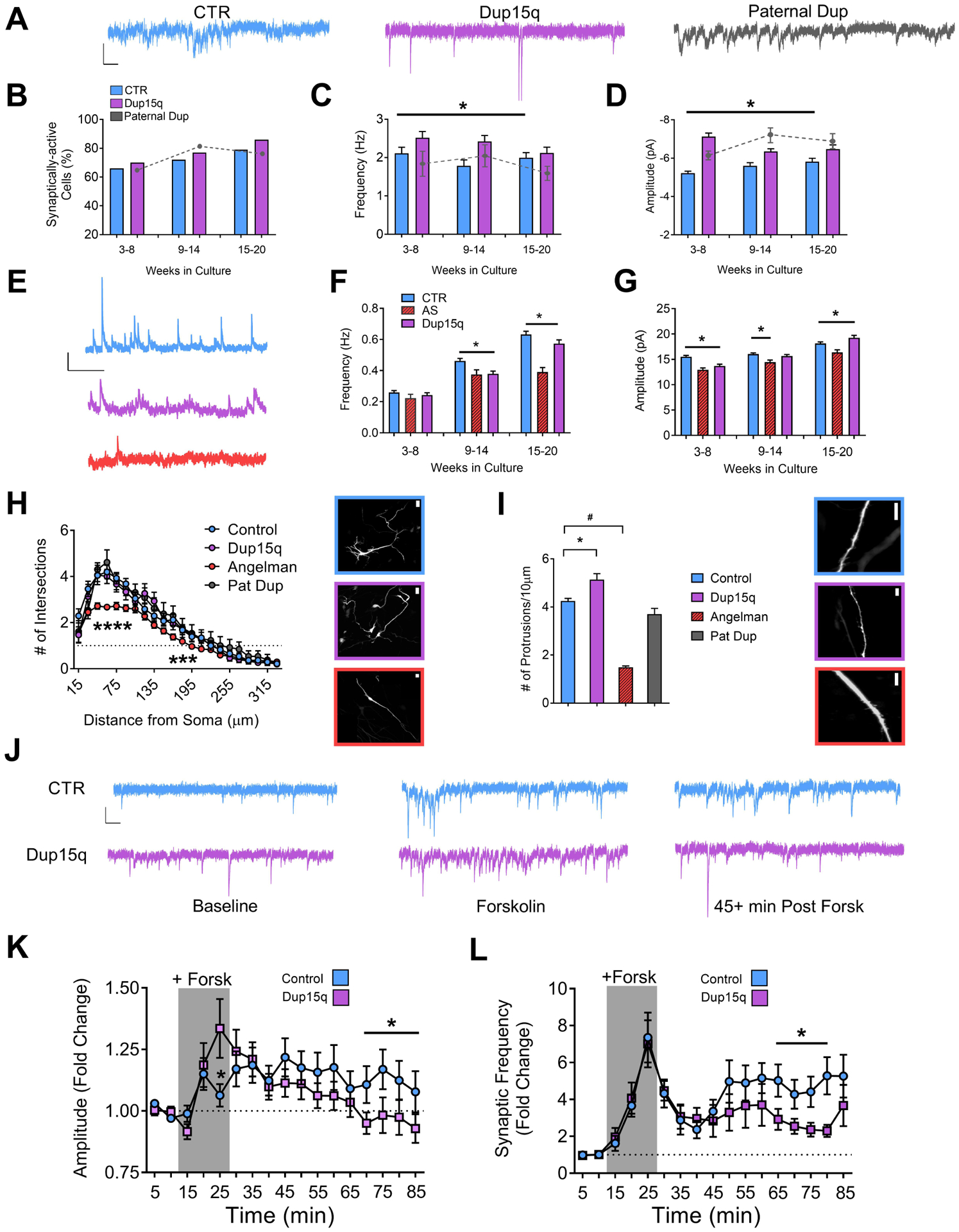
(A) Example traces of spontaneous excitatory synaptic currents from control (CTR), Dup15q, and 15q11–13 paternal duplication (Paternal Dup) neurons at 15–20 weeks in culture. Scale bar: 10 pA, 100 ms. (B) Percent of synaptically-active neurons (event frequency at >0.2 Hz) derived from CTR (5 subjects; n>415 at every time point), Dup15q (4 subjects; n>350 at every time point), and paternal duplication (1 subject; n>80 at every time point). *P<0.05 for significant differences between control and Dup15q (χ2 test). (C) Mean frequency of spontaneous synaptic events for active neurons derived from CTR (5 subjects, n>250 cells at every time point), Dup15q (4 subjects, n>315 cells at every time point) and 15q11–13 paternal duplication (1 line; n>50 cells at every time point). (D) Mean amplitude of spontaneous synaptic events for neurons plotted in (c). *P<0.05 for differences between CTR and AS (two-way ANOVA). (E) Example traces of spontaneous inhibitory synaptic currents from control (CTR; blue), Dup15q (purple), and Angelman syndrome (AS; red) neurons at 15–20 weeks in culture. Scale bar: 20 pA, 1000 ms. (F) Mean frequency of spontaneous inhibitory synaptic events from CTR (6 subjects, n>600 cells at every time point), Dup15q (4 subjects, n>300 cells at every time point) and AS (1 line; n>100 cells at every time point). (G) Mean amplitude of spontaneous inhibitory synaptic events for neurons plotted in (F). *P<0.05 for differences between CTR, AS, and Dup15q as indicated by the bracket above the bars in the graph (Student’s t-test). (H) Left: Grouped Sholl analysis for neurons (28–30 weeks) from control- (n>20), Dup15q- (n=11), AS- (n>40), and 15q11-q13 paternal duplication-derived (n=5) cultures. *P<0.05 indicated significant differences between control and AS (Student’s t-test). Right: example images of transfected neurons used for analysis from control (top; blue), Dup15q (purple; middle), and AS (red; bottom). Scale bar: 25μm. (I) Left: Grouped analysis for number of dendritic protrusions for control (n>45), Dup15q (n=20), AS (n>40), and 15q11–13 paternal duplication cultures (n=9). Data from weeks 15–20 and 28–30 were collapsed into a single bin. *P<0.05 indicates significant differences between control and Dup15q. #P<0.05 indicates significant differences between control and AS (Student’s t-test). Right: example images of transfected neurons used for analysis from control (top; blue), Dup15q (purple; middle), and AS (red; bottom). Scale bar: 10μm. (J) Example traces of spontaneous excitatory synaptic currents from control (CTR; top) and Dup15q (bottom) neurons at (left to right) baseline, during forskolin/rolipram/0 Mg (Forsk), and 45+ min post-Forsk (see Methods for details). Scale bar, 10 pA, 100 ms. (L,K) Group data for CTR (n>60) and Dup15q (n>45) neurons showing (L) mean frequency and (K) mean amplitude of spontaneous synaptic currents during baseline, plasticity induction (indicated by grey box; see Methods for details) and post induction. *P<0.05, indicates differences for CTR (blue) vs Dup15q (purple) (Student’s t-test).
We next examined whether morphological changes were associated with the synaptic phenotype in Dup15q neurons. Neither Dup15q nor Pat Dup neurons showed any differences in dendritic complexity at either 15–20 weeks (Supp Fig. 4a) or 28–30 weeks (Fig. 2h) as compared to control. Unlike Dup15q and Pat Dup neurons, AS neurons had a significant decrease in dendritic complexity compared to controls (Fig. 2h). Example images of transfected neurons are shown in Fig. 2h (right). Additional examples and neuron traces are depicted in Supp. Fig. 4b,c. Sholl analysis for 15–20 week old cultures and for individual lines are also depicted in Supp. Fig. 4a,c. We next counted the number of protrusions along the dendritic tree. Dup15q neurons showed a significant increase, and AS neurons showed a significant decrease, in the number of protrusions, compared to controls (Fig. 2i). Thus, duplications and deletions of 15q11-q13 result in increases and decreases, respectively, in the number of dendritic protrusions and frequency of excitatory synaptic events in iPSC-derived cultures. Example images of dendritic protrusions from transfected control, Dup15q, and AS neurons are depicted in Fig. 2i (right). Number of protrusions for individual control, Dup15q, and AS lines are shown in Supp. Fig. 4d.
We have previously shown that iPSC-derived neurons undergo activity-dependent plasticity following pharmacological elevation of cAMP and enhanced NMDA receptor activation(20; 25). Both control and Dup15q neurons showed an increase in synaptic event amplitude and frequency during the fifteen-minute induction period (Fig. 2k,l; grey box; See Methods). Following washout, control neurons maintained a significant increase in both amplitude (Fig. 2k) and frequency (Fig. 2l) up to one hour post-induction that was diminished in Dup15q neurons (Fig. 2j). We have previously shown that synaptic plasticity changes observed with patch clamp (such as those in Fig. 2j–l) can also be assayed by using calcium imaging as a proxy for AP firing(20). Example raster plots from 4 control experiments and 4 Dup15q experiments using the same plasticity protocol are shown in Supp Fig. 5. The frequency of calcium transients was significantly increased in both control and Dup15q cultures, and significant increases (~2x baseline levels) were still observed 50–60 min post-induction (Supp. Fig. 5b,c). Qualitatively, we observed many experiments where the firing frequency and synchrony were increased in Dup15q neurons compared to controls (Supp. Fig 5a), as reflected in the raster plots and in the trend towards increased frequency observed in Dup15q across baseline, induction, 20–30 post-induction, and 50–60 min post-induction (Supp. Fig. 5b,c). However, no significant differences in plasticity were observed between control and Dup15q neurons. This result differs from that obtained using single-cell patch-clamp measurements of spontaneous excitatory synaptic currents presented in Fig. 2j,k,l. This may indicate that the reduced frequency and amplitude of synaptic events in Fig,2j,k are independent of changes in spontaneous AP firing, and instead reflect specific changes in synaptic function. Future experiments will tease apart these possibilities.
Disrupted synaptic scaling in Dup15q neurons.
In mouse models, deletion or overexpression of UBE3A causes changes in the expression of the protein Arc, though these changes may not be directly related to UBE3A’s role as an ubiquitin ligase(13; 26–29). Arc is a major synaptic protein involved in the trafficking of AMPA receptors at the synapse and modulating synaptic strength(30; 31). Given this correlation and the synaptic deficits related to various models of AS and Dup15q, we next looked at homeostatic synaptic scaling, a well-established phenomenon in which transiently altering activity results in compensatory changes in the number of AMPA receptors at the synapse(32; 33). Importantly, this type of plasticity has been shown in cultured iPSC-derived neurons and is disrupted in a mouse model of AS(27; 34). We therefore measured the amplitude of AMPA receptor-mediated miniature excitatory postsynaptic currents (mEPSCs) subsequent to treatment with the GABAA receptor blocker GABAzine or the sodium channel blocker tetrodotoxin (TTX). Following disinhibition with GABAzine, control neurons showed a leftward shift in the amplitude distribution that was not observed in Dup15q neurons (Fig. 3a). Due to line-to-line variability, data from each line was normalized to its own baseline distribution and then grouped by genotype. Example traces are depicted in Fig.3b. Interestingly, unlike Dup15q neurons, but similar to controls, AS neurons showed a decrease in AMPA event amplitude following GABAzine treatment. To our surprise, all genotypes failed to respond to TTX with a rightward shift in AMPA event amplitude. We next asked whether a low baseline frequency of spontaneous mEPSCs might occlude the effect of TTX. In support of this, we found that 2 control lines with mean baseline activity >3 Hz did in fact display the expected rightward shift in AMPA current amplitudes with TTX treatment (Fig. 3c). Although this distinction between low and high-activity neurons is unexpected, to our knowledge, the study of homeostatic plasticity in human cells is limited (34). None of the Dup15q neurons met the 3 Hz criteria. Additionally, we compared the baseline (vehicle-treated) amplitude distributions of control, Dup15q, and AS neurons. In line with our results in Fig. 2d for Dup15q neurons and our previously published data on AS-derived neurons, both AS and Dup15q baseline amplitudes were significantly increased compared to control (Fig. 3c). These data may be explained in part by the inability of Dup15q neurons to decrease mEPSC amplitudes in response to increases in network activity.
Figure 3. Impaired synaptic scaling in Dup15q-derived neurons.
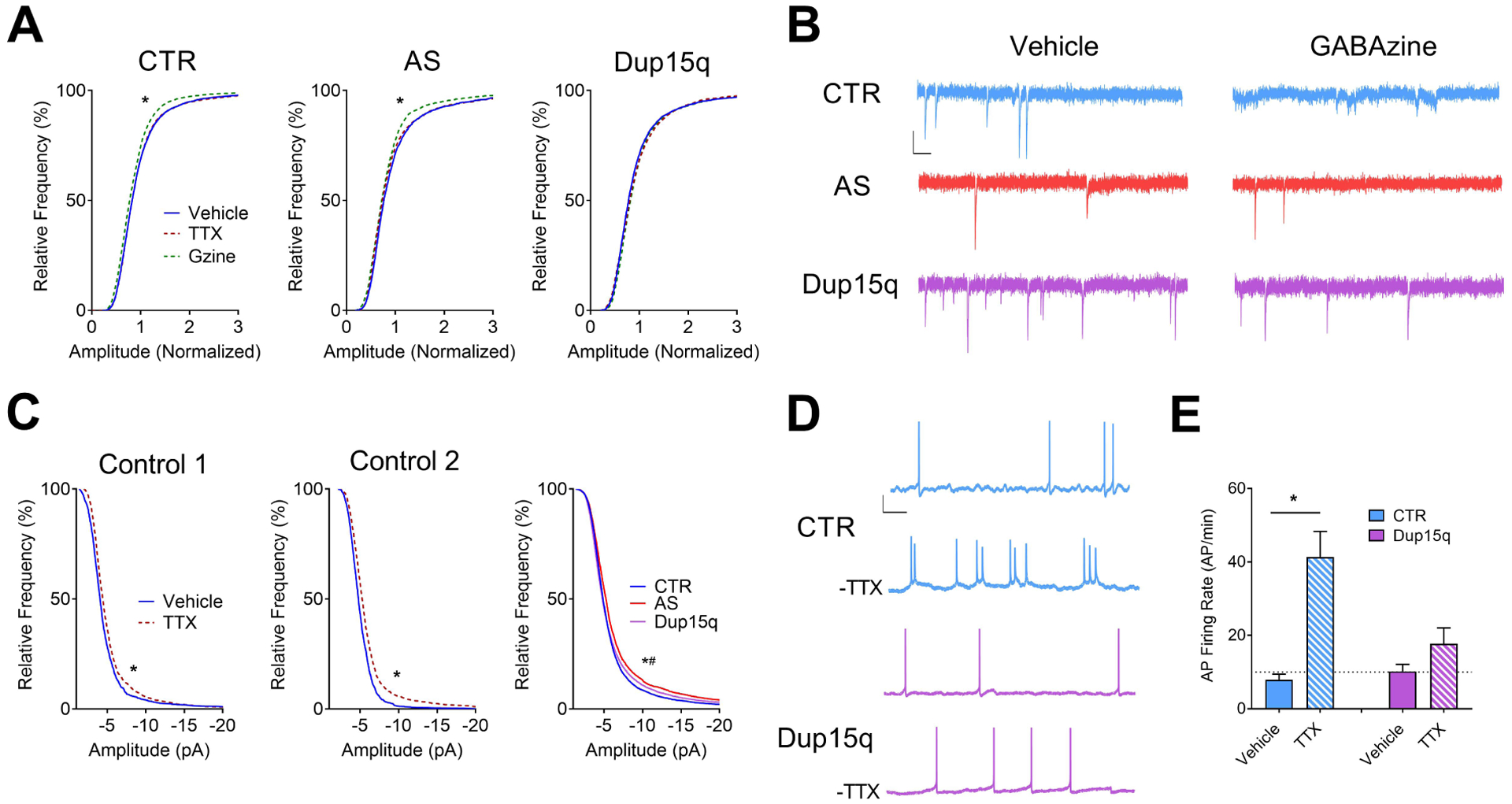
(A) Cumulative frequency histograms of amplitudes (normalized) of miniature spontaneous excitatory synaptic currents for control (CTR; left; 6 subjects; n>140 neurons per treatment), Angelman (AS; middle; 4 subjects; n>80 neurons per treatment), and Dup15q (right; 4 subjects; n>100 neurons per treatment) cultures. *P<0.0001 indicated significant differences between control and GABAzine treatments (Student’s t-test). (B) Example traces of miniature spontaneous excitatory synaptic currents from CTR, AS, and Dup15q neurons following treatment with either vehicle (left) or GABAzine (right). Scale bar: 10 pA, 100ms. (C) Cumulative frequency histograms of raw amplitudes of miniature spontaneous excitatory synaptic currents for 2 control subjects treated with TTX (Control 1; left. Control 2; middle), with baseline synaptic frequencies >3 Hz. Right: Cumulative frequency histograms of raw baseline (vehicle-treated) amplitudes for control, AS, and Dup15q cultures. *P<0.0001 indicates significant differences between control and Dup15q, #P<0.0001 indicates significant differences between control and AS (Kolmogorov-Smirnov test). (D) Example traces of spontaneous action potential firing from control (CTR) and Dup15q neurons from data presented in (E). Scale bar: 20 mV, 1s. (E) Spontaneous action potential (AP) firing rate of vehicle- or TTX-treated neuronal cultures from control and Dup15q subjects following washout of TTX. *P<0.05 indicates significant differences between vehicle- and TTX-treated cultures (Student’s t-test).
It is thought that homeostatic synaptic scaling allows a neuron to maintain a set-point of AP firing in response to changes in network synaptic activity and synaptic strength(35; 36). Thus, the frequency of AP firing should change in the same direction as AMPA currents following treatment with TTX. Example traces of spontaneous APs are depicted in Fig. 3d. As expected, control neurons responded to TTX treatment with a significant increase in spontaneous AP firing (Fig. 3e), whereas Dup15q neurons failed to show a significant increase in AP firing. Overall, these results suggest a disruption in the ability of Dup15q neurons to scale AMPA-mediated synaptic currents in response to changes in network activity.
Spontaneous AP firing.
We next compared changes in spontaneous AP firing. Example traces are shown in Fig. 4a. The frequency of APs recorded from individual lines was variable, but consistent within each genotype (Fig. 4b). On average, control neurons fired less than 0.5 Hz, which was similar to Pat Dup and AS-derived cultures. Dup15q neurons, however, had almost triple the firing rate of the other genotypes (Fig. 4c). The amplitudes of spontaneous APs were not different between genotypes (Fig. 4d, e). We also indirectly monitored AP firing in multiple neurons simultaneously using calcium imaging. Calcium transients were quantified over the course of a 10- minute imaging period and plotted as Raster plots (Fig. 4g). Consistent with patch-clamp recordings, cultures from 2 Dup15q patients showed a higher frequency of calcium transients and, in both cases, Dup15q neurons showed a high degree of synchrony, which was not observed in the control cultures. This was also true for even the most active control neurons from an additional experiment (See example traces in Fig. 4f). Together these data provide an additional aspect of hyperexcitablity that may relate to seizures in Dup15q individuals.
Figure 4. Spontaneous action potential firing.
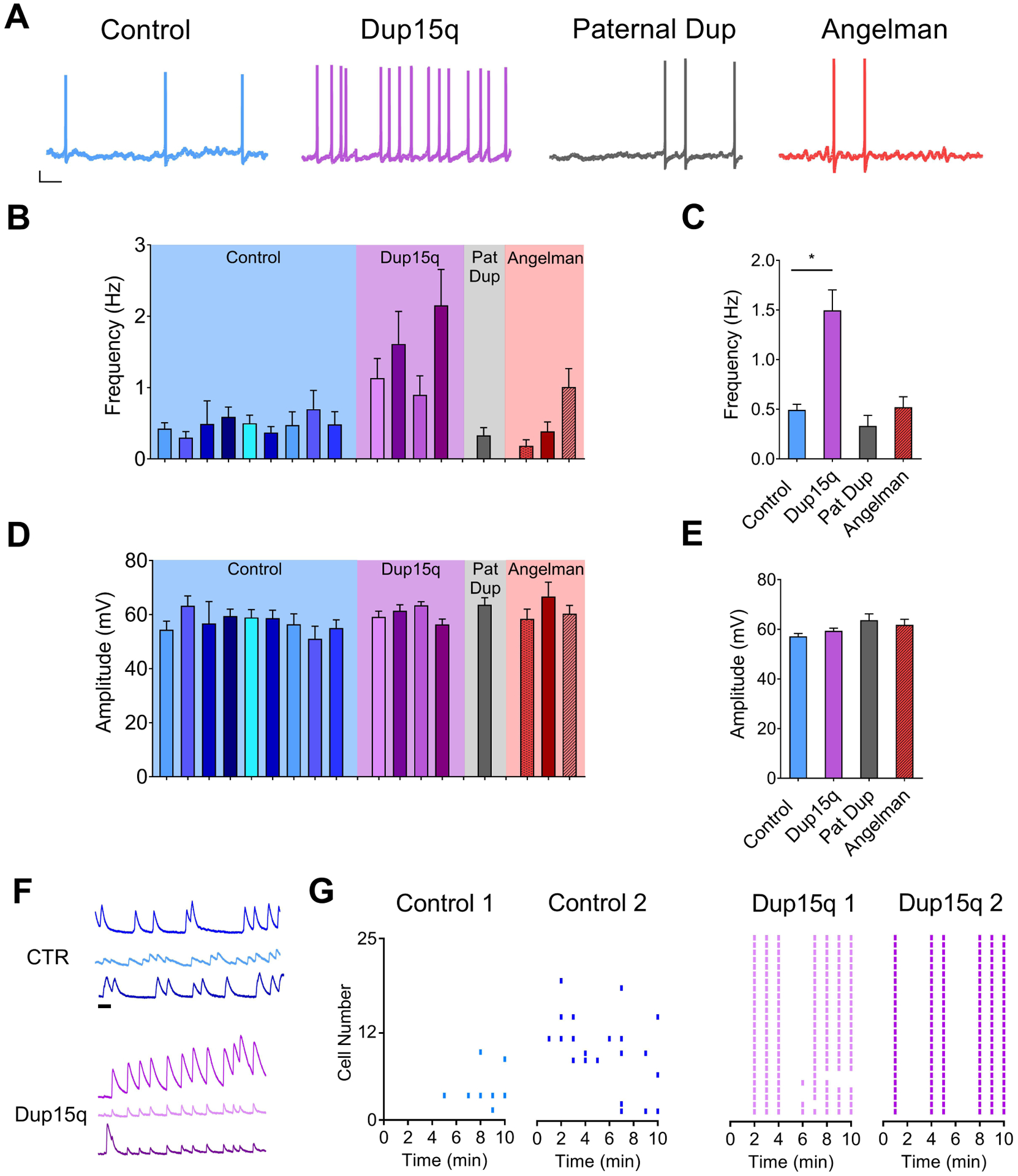
(A) Example traces of spontaneous action potential firing from control (CTR; blue), Dup15q (purple), 15q11–13 paternal duplication (Paternal Dup; grey) and AS (red) neurons at 25+ weeks in culture. Scale bar: 10 mV, 1s. (B,D) Spontaneous action potential frequency (B) and amplitude (D) of neurons derived from individual control lines (blue; 9 subjects; n>15 neurons per bar), Dup15q lines (purple; 4 subjects; n>15 neurons per bar), 15q11-q13 paternal duplication (grey; 1 subject; n>25), and AS (red; 3 subjects; n>25 per bar). (C,E) Grouped data of data presented in (B,D) (n=224, n=110, n=32, n=89 for control, Dup15q, Pat Dup, and AS, respectively). *P<.05 indicates significant differences between control and Dup15q (Student’s t-test). (F) Example traces of spontaneous baseline calcium transients from 3 neurons each from CTR (top; blue) and Dup15q (bottom; purple). Scale bar, 1 min. (G) Raster plots of frequency of calcium transients from cultures derived from control (left; 2 subjects; 25 neurons each) and Dup15q (right; 2 subjects; 25 neurons each).
KCNQ2 channels are disrupted in Dup15q neurons.
Given the role of KCNQ2 potassium channels as a brake on repetitive neuronal AP firing and its involvement in epilepsy early in development(37; 38), we explored potential KCNQ2 deficits in Dup15q-derived neurons. Recordings were made in the presence of either normal ACSF, KCNQ2 blocker XE991, or KCNQ activator flupirtine. Example data of spontaneous AP frequency from 3 individual control (Fig. 5a) and 3 individual Dup15q (Fig. 5b) lines are shown. Additional data from individual lines are also shown in Supp. Fig. 6a,b. Example traces from control and Dup15q neurons during the 3 conditions are depicted in Fig. 5c,d. Under baseline conditions, control neurons fired spontaneous APs at a frequency of ~0.5 Hz (Fig. 5e). As expected, control neurons recorded in the presence of the KCNQ2 activator flupirtine have a significantly reduced frequency of AP firing. In the presence of the KCNQ2 blocker XE991, control neurons have a significantly increased level of firing that is similar to the baseline firing in Dup15q neurons (Fig. 5e). Both Pat Dup and AS neurons responded similarly to controls. Dup15q neurons, however, failed to respond to either drug, suggesting an impairment in KCNQ2 function in these cells. AP amplitude was unaffected across genotype groups and pharmacologic treatment (Fig. 5f, S4b). We also confirmed impairment in KCNQ2 function with recordings of individual neurons before and after acute exposure to the KCNQ2 blocker XE991 (Supp. Fig. 7c). Responses to pharmacological manipulation of Ca2+ channels, SK channels, BK channels, Kv channels, and GABAR were similar between Dup15q and control neurons, suggesting these most likely do not contribute to hyperexcitability in these cells (Supp. Fig. 7). To further explore the lack of response to pharmacological manipulation of KCNQ2 in Dup15q neurons, we stained control and Dup15q cultures for MAP2 and KCNQ2 and then sorted cultures by flow cytometry. We found that >60% of control MAP2+ cells were also positive for KCNQ2 (Fig. 5g), and that this percentage was significantly less in Dup15q cultures (~25%), providing evidence for decreased KCNQ2 expression in Dup15q neurons. Immunocytochemistry for KCNQ yielded a similar finding (Supp. Fig. 6c).
Figure 5. Impaired KCNQ2 channels in Dup15q-derived neurons.
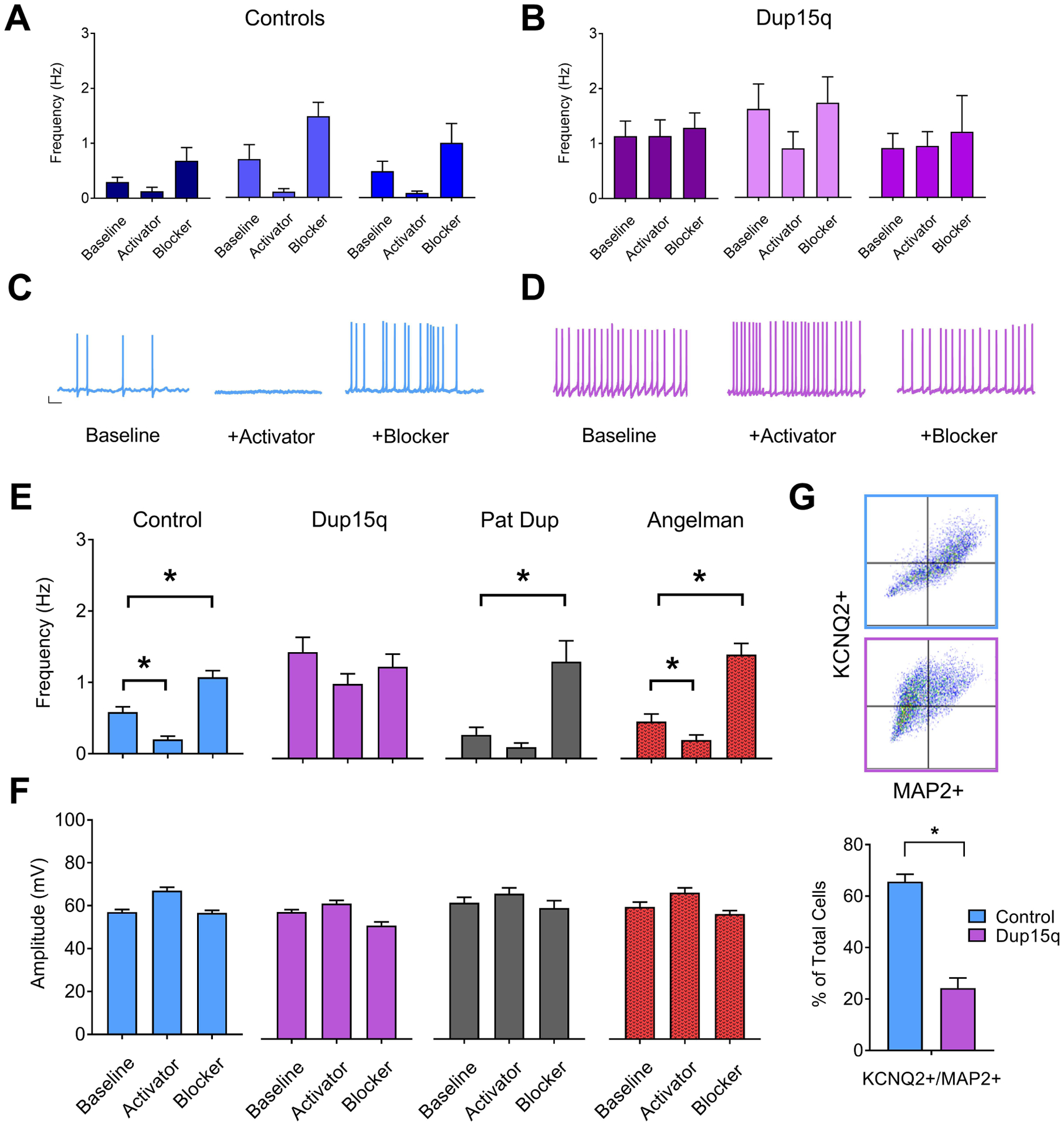
(A,B) Frequency of spontaneous action potential firing for 3 individual control (A) and Dup15q (B) lines at baseline or in the presence of either pharmacological blocker or activator of KCNQ2 channels (n>15 neurons for each bar). (C) Example traces of spontaneous action potentials recorded from control neurons. Scale bar: 10mV, 1s (D) Example traces of spontaneous action potentials recorded from Dup15q neurons (E) Group data for frequency of spontaneous action potential firing for control (left; blue; 9 lines; n=251, 160, 204 neurons for baseline, activator, and blocker, respectively), Dup15q (middle left; purple; 4 lines; n=110, 108, 93 neurons for baseline, activator, and blocker, respectively), 15q11–13 paternal duplication (middle right; grey; 1 line; n=30 neurons for all bars), and Angelman (right; red; 3 lines; n>89 neurons for all bars), recorded at baseline or in the presence of either pharmacological blockers or activators of KCNQ2 channels. *P<0.05 indicates significant differences (student’s t-test). (F) Group data for amplitude of spontaneous action potential firing for data presented in (E). (G) Top: Examples of flow cytometry plots for cultures stained with KCNQ2 and MAP2. Bottom: Flow cytometry quantification of percent of cells positive for both MAP2 and KCNQ2 for control (2 subjects/8 coverslips) and Dup15q (2 subjects/8 coverslips).*P<0.05 indicates significant differences (Student’s t-test).
DISCUSSION
Deletions or duplications of maternal chromosome 15q11-q13 result in AS or Dup15q, respectively, distinct syndromes that share some clinical phenotypes including a high prevalence of seizures(7). In the present study, we investigated the cellular phenotype of Dup15q syndrome patient-derived neurons that have increased expression of UBE3A as well as several other genes in the duplicated region. We used multiple patient lines and a large number of control lines to establish these phenotypes, as the isodicentric chromosome 15q in these patients makes the generation of isogenic cell lines a major challenge for the study of this syndrome. Given that UBE3A is the only paternally-imprinted gene in the duplicated region, we also included a cell line from an individual with a paternal duplication to explore the role of other genes duplicated in the 15q11-q13 region (including a cluster of GABAA receptor subunits).
Mouse models of Dup15q strongly indicate synaptic dysfunction as an important component of cellular pathophysiology(13; 14; 24; 39). Here, we observe altered synaptic function in Dup15q patient-derived neurons. First, Dup15q neurons show significant increases in excitatory synaptic event frequency and amplitude compared to controls, which is maintained over 20 weeks of in vitro development, opposite to phenotypes we have reported in AS-derived neurons(20). IPSC frequency was reduced in both Dup15q and AS neurons, suggesting a relationship between UBE3A and inhibitory transmission. In Dup15q neurons, there was no significant difference in dendritic branching, but there was an increase in dendritic protrusion density, whereas AS neurons showed a decrease in protrusion density. Dup15q neurons also failed to show a sustained increase in the frequency and amplitude of synaptic events in response to pharmacologically-induced plasticity.
Increases and decreases in UBE3A have been linked to altered expression of the synaptic protein Arc, a molecule that regulates internalization of AMPA receptors (13; 27; 28; 40; 41). In line with these data and the observed increase in amplitude of synaptic events across development, Dup15q neurons failed to display homeostatic scaling of synaptic currents and AP firing in response to either disinhibition or Na+-channel block, as seen in both control and AS neurons. This deficit, particularly the inability to down-regulate synaptic currents in response to increased network activity, suggests a hyperexcitable phenotype that could contribute to seizures in Dup15q patients.
Spontaneous AP firing in Dup15q neurons was markedly increased compared to control and AS neurons, which was confirmed with population calcium imaging, suggesting an additional aspect of hyperexcitability specific to Dup15q neurons. Pharmacological manipulations indicate that KCNQ2 channels may be impaired in Dup15q neurons. KCNQ2 channels are potassium channels that act as a brake on repetitive AP firing(42), and mutations in KCNQ2 result in benign familial neonatal convulsions(42). Dup15q neurons failed to respond to pharmacological activation or blockade of KCNQ2, suggesting impaired function and/or a reduction of these channels in Dup15q neurons. Interestingly, blockade of KCNQ2 in control neurons resulted in firing rates that were similar to the baseline firing of Dup15q neurons. In line with these data, KCNQ2 expression was significantly diminished in neurons from Dup15q patients as measured by immunostaining/flow cytometry, suggesting that KCNQ2 may be a potential therapeutic target.
Electrophysiological experiments with the KCNQ2 blocker XE991 yielded opposite responses in Dup15q and AS neurons, which is interesting in light of the opposite direction of change for UBE3A in these patient lines. As Dup15q patient lines have large duplications encompassing a number of genes, it is unclear whether these phenotypes can be directly linked to increases in UBE3A. Recordings from a patient line with paternal duplication to 15q11-q13, in which UBE3A is unchanged, but other genes in the region are duplicated, showed responses to both KCNQ2 block and activation that were identical to control neurons, suggesting a specific role for UBE3A duplication in this phenotype. These experiments lend further support to the idea that KCNQ2 may be downstream of UBE3A changes, though it remains to be determined how and if UBE3A actually regulates KCNQ2 expression and function.
The cellular and molecular causes of seizures in these syndromes remain elusive. It has been reported that increased gene dosage of UBE3A is directly responsible for a seizure phenotype in a mouse model of Dup15q and that network activity generated by this seizure phenotype interacts with the transynaptic protein Cbln1 in the ventral tegmental area to causes changes in sociability in these mice(14). Our study identifies multiple, distinct contributors to neuronal hyperexcitability: 1) Increased frequency and amplitude of excitatory synaptic events, 2) decreases in frequency of IPSCs, 3) impaired synaptic scaling in response to increases in network activity, and 4) increases in spontaneous AP firing as a result of disrupted KCNQ2 channels. It is unclear if and how these mechanisms interact to generate seizures in brain tissue given the lack of proper neuroanatomical microcircuits in cultured neurons. Research using animal models has established that altered excitation-inhibition balance is an important contributor to ASD and ASD-related pathophysiology, including the high prevalence of seizures(43; 44), so it is likely that the increases in spontaneous synaptic event frequency and amplitude and impaired scaling are contributors to a similar pathophysiology in Dup15q. An important next step requires screening mouse models of Dup15q for similar electrophysiological phenotypes, particularly at early developmental time points, to link cellular phenotypes to behavior. Additional insight may also come from studying other genetic subtypes of Dup15q, such as patients with interstitial duplications, where seizures are less prevalent. Overall, this study establishes a robust cellular phenotype in human Dup15q neurons with clinically-relevant genetic disruptions that can be used to screen for compounds aimed at reversing electrophysiological differences as a foundation for treating Dup15q patients.
Supplementary Material
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Antibody | MAP2 | Sigma | ||
| Antibody | KCNQ2 | Sigma | ||
| Antibody | TBR1 | Proteintech | ||
| Antibody | beta III Tubulin | Abcam | ||
| Antibody | S100B | Novus | ||
| Antibody | GAD67 | Abcam | ||
| Antibody | ||||
| Antibody | ||||
| Antibody | ||||
| Bacterial or Viral Strain | ||||
| Biological Sample | ||||
| Cell Line | ||||
| Chemical Compound or Drug | ||||
| Commercial Assay Or Kit | ||||
| Deposited Data; Public Database | ||||
| Genetic Reagent | ||||
| Organism/Strain | ||||
| Peptide, Recombinant Protein | ||||
| Recombinant DNA | ||||
| Sequence-Based Reagent | ||||
| Software; Algorithm | ||||
| Transfected Construct | ||||
| Other |
Acknowledgements
This work was supported by NIH Grants NS078753, NS111986, NS111965, and MH094896 (E.S.L.), the Dup15q Alliance (J.J.F.), and the CT Regenerative Medicine Research Fund (E.S.L., S.J.C., and L.M.L.). We thank Dr. Jon Covault for providing control cell lines and Dr. Evan Jellison for help with flow cytometry. A previous version of this manuscript was posted on BioRxiv.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Chamberlain SJ, Chen P-F, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M (2010): Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci USA 107: 17668–17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, Philpot BD (2013): Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J Comp Neurol 522: 1874–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chamberlain SJ, Lalande M (2010): Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11-q13. Neurobiology of Disease 39: 13–20. [DOI] [PubMed] [Google Scholar]
- 4.Cassidy SB, Driscoll DJ (2008): Prader–Willi syndrome. European Journal of Human Genetics 17: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chamberlain SJ, Lalande M (2010): Angelman syndrome, a genomic imprinting disorder of the brain. J Neurosci 30: 9958–9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Battaglia A (2005): The inv dup(15) or idic(15) syndrome: A clinically recognisable neurogenetic disorder. BRAIN AND DEVELOPMENT 27: 365–369. [DOI] [PubMed] [Google Scholar]
- 7.Kalsner L, Chamberlain SJ (2015): Prader-Willi, Angelman, and 15q11-q13 Duplication Syndromes. Pediatric Clinics of North America 62: 587–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hogart A, Wu D, LaSalle JM, Schanen NC (2008): The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiology of Disease 38: 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. (2008): Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 82: 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conant KD, Finucane B, Cleary N, Martin A, Muss C, Delany M, et al. (2014): A survey of seizures and current treatments in 15q duplication syndrome. Epilepsia 55: 396–402. [DOI] [PubMed] [Google Scholar]
- 11.Friedman D, Thaler A, Thaler J, Rai S, Cook E, Schanen C, Devinsky O (2016): Mortality in isodicentric chromosome 15 syndrome: The role of SUDEP. Epilepsy Behav 61: 1–5. [DOI] [PubMed] [Google Scholar]
- 12.Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, et al. (2009): Abnormal Behavior in a Chromosome- Engineered Mouse Model for Human 15q11–13 Duplication Seen in Autism. Cell 137: Elsevier Inc.1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith SEP, Zhou Y-D, Zhang G, Jin Z, Stoppel DC, Anderson MP (2011): Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci Transl Med 3: 103ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan V, Stoppel DC, Nong Y, Johnson MA, Nadler MJS, Ozkaynak E, et al. (2017): Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature 543: 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isshiki M, Tanaka S, Kuriu T, Tabuchi K, Takumi T, Okabe S (2014): Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nature Communications 5: 4742. [DOI] [PubMed] [Google Scholar]
- 16.Piochon C, Kloth AD, Grasselli G, Titley HK, Nakayama H, Hashimoto K, et al. (2014): Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nature Communications 5: 5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamberlain SJ, Chenb P-F, Ngb KY, Bourgois-Rochab F, Lemtiri-Chlieh F, Levinec ES, Lalande M (2010): Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader–Willi syndromes. PNAS 107: 17668–17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chamberlain SJ, Li X-J, Lalande M (2008): Induced pluripotent stem (iPS) cells as in vitro models of human neurogenetic disorders. Neurogenetics 9: 227–235. [DOI] [PubMed] [Google Scholar]
- 19.Stern S, Sarkar A, Stern T, Mei A, Mendes APD, Stern Y, et al. (2019): Mechanisms Underlying the Hyperexcitability of CA3 and Dentate Gyrus Hippocampal Neurons Derived From Patients With Bipolar Disorder. Biol Psychiatry. doi: 10.1016/j.biopsych.2019.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fink JJ, Robinson TM, Germain ND, Sirois CL, Bolduc KA, Ward AJ, et al. (2017): Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nature Communications 8: 15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urraca N, Cleary J, Brewer V, Pivnick EK, McVicar K, Thibert RL, et al. (2013): The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res 6: 268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fink JJ, Levine ES (2018): Uncovering True Cellular Phenotypes: Using Induced Pluripotent Stem Cell-Derived Neurons to Study Early Insults in Neurodevelopmental Disorders. Frontiers in Neurology. doi: 10.3389/fneur.2018.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallace ML, Burette AC, Weinberg RJ, Philpot BD (2012): Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 74: 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Judson MC, Wallace ML, Sidorov MS, Burette AC, van Woerden GM, King IF, et al. (2016): GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron 90: 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, Lisman J (2003): Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. Journal of Neurophysiology 91: 1955–1962. [DOI] [PubMed] [Google Scholar]
- 26.Greer PL, Greenberg ME (2008): From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59: 846–860. [DOI] [PubMed] [Google Scholar]
- 27.Pastuzyn ED, Shepherd JD (2017): Activity-Dependent Arc Expression and Homeostatic Synaptic Plasticity Are Altered in Neurons from a Mouse Model of Angelman Syndrome. Front Mol Neurosci 10: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kühnle S, Mothes B, Matentzoglu K, Scheffner M (2013): Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc Natl Acad Sci USA 110: 8888–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao C, Rioult-Pedotti MS, Migani P, Yu CJ, Tiwari R, Parang K, et al. (2013): Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol 11: e1001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shepherd JD, Bear MF (2011): New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci 14: 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korb E, Finkbeiner S (2011): Arc in synaptic plasticity: from gene to behavior. Trends in Neurosciences 34: 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turrigiano GG (2008): The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135: 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998): Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892–896. [DOI] [PubMed] [Google Scholar]
- 34.Xu J-C, Fan J, Wang X, Eacker SM, Kam T-I, Chen L, et al. (2016): Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci Transl Med 8: 333ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turrigiano G (2012): Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harbor Perspectives in Biology 4: a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998): Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892–896. [DOI] [PubMed] [Google Scholar]
- 37.Maljevic S, Wuttke TV, Lerche H (2008): Nervous system KV7 disorders: breakdown of a subthreshold brake. The Journal of Physiology 586: 1791–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown DA, Passmore GM (2009): Neural KCNQ (Kv7) channels. British Journal of Pharmacology 156: 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mabb AM, Judson MC, Zylka MJ, Philpot BD (2011): Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends in Neurosciences 34: 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, et al. (2010): The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140: 704–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao C, Rioult-Pedotti MS, Migani P, Yu CJ, Tiwari R, Parang K, et al. (2013): Impairment of TrkB-PSD-95 Signaling in Angelman Syndrome. (Dalva M, editor.)PLoS Biol 11: e1001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jentsch TJ (2001): Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21–30. [DOI] [PubMed] [Google Scholar]
- 43.Zoghbi HY, Bear MF (2012): Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harbor Perspectives in Biology 4:. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson SB, Valakh V (2015): Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 87: 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.