

Abstract
A hallmark of pancreatic ductal adenocarcinoma (PDAC) is the presence of a dense, desmoplastic stroma and the consequent altered interactions between cancer cells and their surrounding tumor microenvironment (TME) that promote disease progression, metastasis, and chemoresistance. We have previously shown that IL-6 secreted from pancreatic stellate cells (PSCs) stimulates the activation of STAT3 signaling in tumor cells, an established mechanism of therapeutic resistance in PDAC. We have now identified the tumor cell-derived cytokine interleukin-1α (IL-1α) as an upstream mediator of IL-6 release from PSCs that is involved in STAT3 activation within the TME. Herein, we show that IL-1α is overexpressed in both murine and human PDAC tumors and engages with its cognate receptor IL-1R1 which is strongly expressed on stromal cells. Further, we show that IL-1R1 inhibition using anakinra (recombinant IL-1 receptor antagonist) significantly reduces stromal-derived IL-6, thereby suppressing IL-6-dependent STAT3 activation in human PDAC cell lines. Anakinra treatment results in significant reduction in IL-6 and activated STAT3 levels in pancreatic tumors from Ptf1aCre/+;LSL-KrasG12D/+; Tgfbr2flox/flox (PKT) mice. Additionally, the combination of anakinra with cytotoxic chemotherapy significantly extends overall survival compared with vehicle treatment or anakinra monotherapy in this aggressive genetic mouse model of PDAC. These data highlight the importance of IL-1 in mediating tumor-stromal IL-6/STAT3 crosstalk in the TME and provide preclinical rationale for targeting IL-1 signaling as a therapeutic strategy in PDAC.
Keywords: Pancreatic cancer, interleukin-1, stroma, interleukin-6, inflammation, STAT3
Introduction
Despite significant advances in our understanding of pancreatic ductal adenocarcinoma (PDAC) biology, clinical outcomes in this aggressive malignancy remain dismal due in part to its resistance to most therapeutic strategies (1). A distinguishing feature of PDAC tumors is the presence of a dense, desmoplastic stroma which constitutes a significant portion of tumor mass and plays a critical role in establishing a chemoresistant phenotype (2). Cancer-associated fibroblasts (CAFs) and pancreatic stellate cells (PSCs), major cellular constituents of the stroma, are chiefly responsible for the deposition of extracellular matrix components that impede vascular perfusion and impair drug delivery into tumors (3). Additionally, these cells also coordinate cytokine-mediated inflammatory crosstalk within the tumor microenvironment (TME) that may dictate the response to therapeutic agents (3, 4). Prior work from our laboratory has implicated the pro-inflammatory signal transducer and activator of transcription 3 (STAT3) pathway as a key biomarker of resistance to chemotherapy in PDAC. We have demonstrated that STAT3 inhibition suppresses the growth and invasion of PDAC tumor cells and exerts profound effects on the stroma that improve drug delivery into tumors and enhance the therapeutic response (5–7). Further, we have shown that PSCs engage in crosstalk with tumor cells via the release of IL-6 to activate STAT3 signaling and promote a pro-invasive phenotype (8). However, direct STAT3 inhibition has been unsuccessful in the clinical management of PDAC, highlighting the urgent need to identify and target upstream ligands involved in activation of STAT3 signaling to improve outcomes in PDAC (9, 10).
In the present study, we show the pro-inflammatory cytokine interleukin-1α (IL-1α) is overexpressed in patients with PDAC and acts as a targetable mediator of tumor-stromal crosstalk that promotes the activation of STAT3 in tumors. Blockade of IL-1 receptor 1 (IL-1R1) attenuates the release of IL-6 from PSCs and the subsequent activation of STAT3 within PDAC tumor cells. In addition, IL-1R1 inhibition reduces in vivo levels of IL-6 and pSTAT3 and improves survival when combined with gemcitabine chemotherapy in an aggressive, genetically engineered mouse model of PDAC. Overall, these results provide valuable insight into the use of IL-1R1 blockade to suppress inflammatory and oncogenic signaling in PDAC and expand our understanding of the intricate mechanisms involved in tumor-stromal crosstalk within the TME.
Methods
Cell lines and reagents
Human pancreatic cancer cell lines (BxPC3, SW1990) were obtained from the American Type Culture Collection (ATCC) and maintained according to ATCC guidelines. Immortalized human pancreatic stellate cells (hPSCs) were purchased from (ScienCell Research Laboratories) and plated on poly-L-lysine coated plates (Sigma) as per manufacturer recommendations. Cultured cells are routinely tested in our lab for mycoplasma contamination using PlasmoTest kit (InvivoGen). Cells with low passage numbers (< 10) were used in the study.
Western Blot
In vitro cell lysates were collected following treatment and first washed in 1X PBS followed by rapid lysis on ice using 1X RIPA buffer (Cell Signaling Technology). Lysates were then sonicated and supernatant removed after centrifugation at 4°C. For in vivo studies, tumors were procured and 5–10 mg of tissue was homogenized in 1X RIPA on ice, followed by sonication and centrifugation in an identical manner. For western blotting of nuclear lysates, isolation was performed using Nuclear Extraction Kit (Abcam) per manufacturer’s protocol. Protein quantification was performed using Pierce™ BCA Protein Assay Kit (Thermo Fisher) as per the manufacturer’s protocol. 12.5–40 μg of protein was loaded per lane and separated by SDS-PAGE. Membranes were then transferred to a polyvinylidene difluoride (PVDF) membrane, blocked in 5% milk solution, and incubated with indicated antibodies (Supplementary Table S1) overnight at 4°C. Secondary conjugation was performed using HRP-conjugated anti-rabbit or anti-mouse antibodies (Jackson ImmunoResearch Laboratories) followed by development with chemiluminescent substrate (Thermo Fisher) and imaged using ChemiDoc Imaging System (BioRad). Immunoblots were quantified using ImageJ (NIH, Bethesda, MD) and graphed using Prism Software (GraphPad Software Inc.).
Quantitative Polymerase Chain Reaction (qPCR)
Purified RNA was obtained from in vitro cells or tumor samples using RNeasy Kit (Qiagen) according to manufacturer’s protocol. RNA concentration and quality were verified using NanoDrop spectrophotometer (Thermo Fisher). cDNA was then generated by reverse transcription of RNA product using High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems). qPCR was performed using incubation with gene-specific predesigned primers (RT2 qPCR Primer Assay, Qiagen) to gene targets and iQ™ SYBR® Green Supermix (BioRad). Gene expression was normalized to the housekeeping gene GAPDH using ΔΔCT method and reported as fold change relative to control.
Flow Cytometry
Murine pancreata were enzymatically digested using RPMI solution supplemented with 0.6 mg/ml Collagenase P (Roche), 0.8 mg/ml Collagenase V (Sigma), 0.6 mg/ml soybean trypsin inhibitor (Sigma), and 1800 U/ml DNase I (Roche) for 20–30 minutes at 37°C. Samples were washed and resuspended in cold PEB solution (autoMACS Rinsing Solution, Miltenyi Biotec, supplemented with 0.5% BSA) to quench enzymatic reaction and prevent over-digestion of tissues. The dissociated tissue was strained through 40 μm mesh filter to obtain single cell suspension. Single-cell suspensions were then incubated in Fc receptor blocking reagent (Miltenyi Biotec) and subsequently stained for markers as listed in Supplementary Table S2 per manufacturer’s protocol. Live/dead cell discrimination was performed using Live/Dead Aqua (Thermo Fisher) fixable dye. Fixation of samples was performed using 1% paraformaldehyde solution (Thermo Fisher) and flow data were acquired using the Cytoflex S (Beckman Coulter). Analysis of data was performed using FlowJo software (BD Life Sciences).
Cytokine array
Conditioned media (CM) was first collected from SW1990 tumor cells after incubation in serum free media (SFM) for 48 hours. IL-1α neutralization was performed using 10 μg/ml anti-human IL-1α antibody (Invitrogen) and incubating for 15 minutes at room temperature. Untreated or IL-1α-neutralized CM was then diluted 1:1 (v/v) using SFM and added to hPSCs in biologic triplicate. After 24 hours, hPSC CM was collected and protein concentration measured. Samples were pooled and CM (600 μg total protein) added for analysis by cytokine array (R&D Systems) as per manufacturer’s protocol.
Enzyme-linked immunosorbent assay (ELISA)
Cells were grown in 10% DMEM media until 90% confluent before media was replaced with 0% DMEM for 12–24 hours. CM was harvested and cellular contaminants removed by centrifugation at 4°C. ELISA kits specific for human IL-6, IL-1α, and IL-1β (R&D Systems) were used per manufacturer protocol. For tissue lysates, 5–10 mg of tumor or normal pancreatic tissue was lysed in 1X RIPA buffer and cellular contaminants were removed by centrifugation at 4°C. Lysates were quantified and equal amount of protein loaded per well. Levels of mouse IL-1α (R&D Systems) or mouse IL-6 (R&D Systems) in lysate were determined by manufacturer’s protocol.
In vivo studies
Tumor-bearing Ptf1aCre/+KrasG12D/+Tgfbr2flox/flox (PKT) mice (provided by Dr. Harold Moses, Vanderbilt University Medical Center, Nashville, TN) were generated as previously described (11). Mice were treated with anakinra (50 mg/kg, intraperitoneal [IP] injection twice daily) or equal volume vehicle control (0.9% normal saline, IP injection twice daily) beginning at 4 weeks of age. Swedish Orphan Biovitrum (SOBI) provided anakinra for use in these studies (12). Twice daily dosage for anakinra was selected based on its short half-life and rapid renal excretion (13). For endpoint analysis of vehicle and anakinra treatment, mice were treated until 6.5 weeks of age before sacrifice and downstream processing of pancreatic tissues was performed. For survival studies, mice were treated with vehicle, anakinra, gemcitabine (GEM, 20 mg/kg three times weekly), or combination anakinra and GEM beginning at 4 weeks of age and continued until moribund.
All animal experiments were performed in compliance with the regulations and ethical guidelines for experimental and animal studies of the Institutional Animal Care and Use Committees at The University of Miami (Miami, FL) (Protocol #18-040).
Immunohistochemistry (IHC) and immunofluorescence (IF)
After sacrifice and paraffin embedding of resected specimens, tissue slides were deparaffinized, rehydrated, and permeabilization performed by incubation in 0.1% Triton X-100 solution for 15 minutes. Antigen retrieval was performed using citrate (pH 6.0) or Tris-EDTA buffer as previously described prior to incubation with BlockAid™ Blocking Solution (Thermo Fisher) (7). For IHC, endogenous peroxidase activity was blocked by incubating with 3% H2O2. Tissue sections were stained with primary antibodies at specified concentrations (Supplementary Table S1) overnight at 4°C. IHC slides were developed using 3,3’-diaminobenzidine (DAB) substrate (Vector) followed by counterstain using Meyer’s hematoxylin and imaged using DM750 microscope (Leica Microsystems). For IF images, primary antibody was detected using species-specific Alexa Fluor 594 and/or Alexa Fluor 488 (Thermo Fisher) secondary antibodies incubated on sections for one hour at room temperature. Nuclear staining was performed using Hoechst 33342 dye (Thermo Fisher). Imaging was performed using DMi8 fluorescent microscope (Leica Microsystems). For quantitative analysis of IHC samples from human tissue microarray (TMA), positive staining was quantified using ImageJ image analysis software (NIH, Bethesda, MD) and reported as percentage area of positive staining. Samples were blinded to the analyzing researcher before analysis.
Transcriptional profiling of The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression Projects (GTEx)
The Cancer Genome Atlas (TCGA) database was queried to obtain transcriptional data from patients with a diagnosis of pancreatic adenocarcinoma (14). Patients with missing transcriptomic data from the primary tumor sample were excluded from analysis. Transcriptional data from 178 PDAC patients was downloaded and organized using the R package TCGABiolinks (15). Two separate analyses were performed. As robust sequencing data for normal tissue within the TCGA is often lacking, cross-platform comparison using transcriptional data from the Genotype-Tissue Expression (GTEx) project and TCGA databases was performed using the Recount2 function of the R package TCGABiolinks to compare normal pancreas and PDAC samples, respectively. The Recount2 function is based on workflow by Collado-Torres and colleagues in which transcriptomic data from multiple projects (GTEx, TCGA among others) has been uniformly processed, summarized, and aligned in a single-pipeline using Rail-RNA (16, 17). To account for differences in processing/alignment methodology, GTEx data was imported as a ranged-summarized experiment and scaled to read counts using the scale_counts function from the Recount package (18). Gene count matrices from derived from TCGA and GTEx were then merged and normalization for GC content and quantile filtering completed prior to performing differential gene expression between normal pancreatic tissue and PDAC tumors using limma-voom pipeline as previously described (19). In a subsequent analysis, expression of all genes from TCGA PDAC tumor samples were normalized to fragments per kilobase of transcript per million (FPKM) values and worked as input for the online tool xCell, a gene signatures-based method learned from thousands of pure cell types from various sources, to obtain a “stromal score” for each patient (20). Patients in the upper quartile for stromal score (“stroma enriched”) were compared to those in the lowest quartile (“stroma low”) for subsequent principal component analysis (PCA), heatmap construction, and differential gene expression analysis using the R package DESeq2 (21). Gene set enrichment analysis (GSEA) and generation of enrichment plots were performed with the Molecular Signatures Database v7.2 Hallmark gene sets using GSEA Software and the R package fgsea (22–25).
Statistical Analysis
Statistical analysis was performed using Prism software (Graphpad Software Inc.) and R 3.5.0 (https://www.R-project.org/). Results are shown as values of mean ± SD except where otherwise indicated. Multiple comparisons were performed using ANOVA followed by Tukey’s or Dunnett’s multiple comparisons test where appropriate. Two-tailed Student’s t test was used for two group comparison. Statistical significance was defined using an α cutoff of 0.05. Pairwise comparisons of survival curves for individual treatments were performed using the log-rank (Mantel-Cox) test.
Results
Interleukin-1α is overexpressed in PDAC
To identify targetable mediators of tumor-stromal crosstalk in the PDAC TME, we first profiled transcriptional data from normal pancreatic tissue and PDAC tumors using the GTEx portal (N=197) and TCGA database (N=178), respectively. Compared to normal pancreatic tissue, PDAC tumors were characterized by a significant upregulation in multiple inflammatory signaling nodes, including gene pathways involved in the NF-κB response (Fig. 1A). Inflammatory cytokines and chemokines, particularly activating ligands of NF-κB signaling, have been previously identified as important mediators of stromal activation within the PDAC TME (26–28). Therefore, we further profiled the differential expression of the 200 genes related to the NF-κB signaling pathway within the Hallmark gene set (HALLMARK_TNFA_SIGNALING_VIA_NFKB). Our analysis revealed that PDAC tumor expression of the NF-κB signaling ligand IL1A was among the top 10 most highly upregulated genes from this dataset (Fig. 1B). As cross-platform comparisons between GTEx and TCGA databases can be potentially biased, we then aimed to validate these findings using published expression data (GEO Accession #GSE15471) from Badea and colleagues containing reads from surgically resected PDAC tumors and matched normal tissue from 36 patients (29). In this dataset, expression of IL1A was significantly increased in PDAC tumors compared to normal pancreatic tissue (p<0.0001), further supporting the results of our TCGA/GTEx database comparison analysis (Supplementary Fig. S1A). We next aimed to corroborate these data by examining the staining and localization of IL-1α in human PDAC tumors compared to normal pancreatic tissues. In an analysis of a tissue microarray consisting of human PDAC samples (n=37) and normal adjacent pancreas (n=23), we identified a significant increase in levels of IL-1α within PDAC samples (p<0.001) (Fig. 1C & D). In murine models, IL-1α levels were determined in tumor samples from PKT mice at 4.5- and 6.5-week timepoints as compared to non-tumor bearing Ptf1aCre littermates. IL-1α levels were significantly increased in tumors obtained from 6.5-old PKT mice compared to normal pancreata from non-tumor bearing Ptf1aCre littermates (p=0.005) and 4.5-old PKT mice (p=0.012), demonstrating that a significant increase in IL-1α protein levels accompany the development of invasive PDAC in mice (Fig. 1E & F). Lastly, we aimed to determine associations between IL1A gene expression and survival outcomes in human PDAC patients using publicly available data from TCGA (14). Analysis of TCGA PanCancer data using the cBio Cancer Genomics Portal demonstrated that intratumoral IL1A gene expression has a significant inverse association with overall survival in PDAC (log-rank p=0.028), hepatobiliary cancers (log-rank p<0.0001), and all malignancies within the TCGA database (log rank p<0.0001) (Supplementary Fig. S1B–E) (30, 31). These findings demonstrate that IL-1α levels are significantly increased in human and murine PDAC tumors compared to normal pancreatic tissues and are associated with adverse outcomes in cancer patients.
Figure 1: Interleukin-1α (IL-1) is overexpressed in pancreatic ductal adenocarcinoma (PDAC).
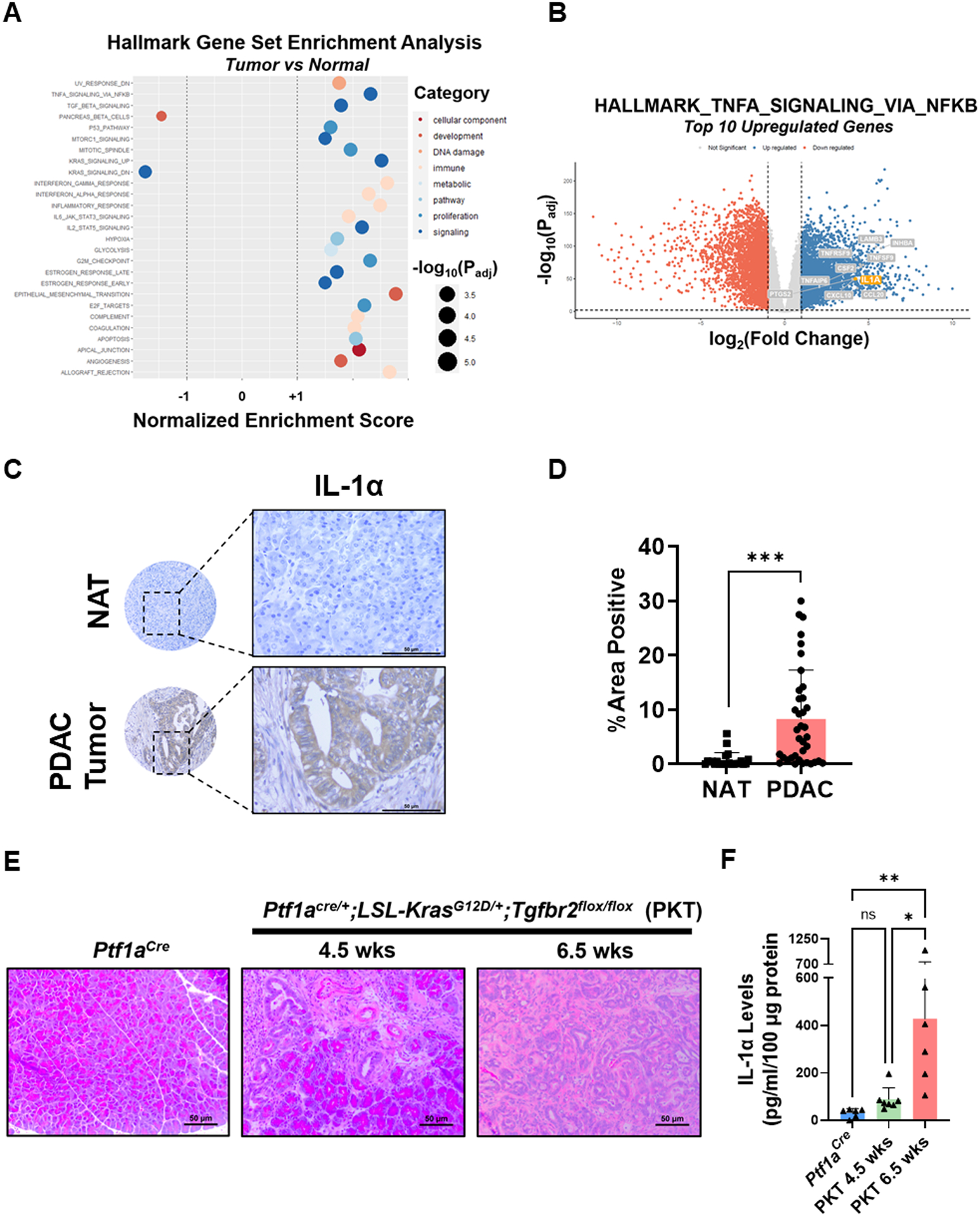
(A) Bubble plot demonstrating significant results of Gene Set Enrichment Analysis (GSEA) for Hallmark gene set in PDAC tumors compared to normal pancreatic tissue. Bubble size indicates the −log10(adjusted p-value) for each pathway. (B) Volcano plot demonstrating the significantly downregulated (red) and upregulated (blue) genes in PDAC tumors compared to normal pancreatic tissues. The top 10 differentially upregulated genes from the MSigDB HALLMARK_TNFA_SIGNALING_VIA_NFKB gene set are labeled in grey, with IL1A highlighted in orange. (C-D) IHC analysis of IL-1α staining was performed on a human tissue microarray, consisting of human PDAC samples (n=37) and normal adjacent tissue (NAT) (n=23) and quantified as percent area of positive staining. (E) Representative H&E staining of histologic sections from non-tumor bearing Ptf1aCre mice (age 6.5–10.5 weeks) and Ptf1aCre/+;LSL-KrasG12D/+; Tgfbr2flox/flox (PKT) mice at harvest at 4.5 weeks and 6.5 weeks of age. (F) Measurement of IL-1α levels by ELISA in non-tumor bearing Ptf1aCre pancreata compared to tumors harvested from 4.5-week-old and 6.5-week-old PKT mice. Data are shown as mean ± SD. Scale bar = 50 μm. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001.
Expression of interleukin-1 receptor type I is enriched in pancreatic stromal cells
Recent evidence suggests that resident mesenchymal cells such as CAFs and PSCs within the PDAC TME may be selectively modulated by paracrine IL-1α derived from tumor cells (27, 32). Therefore, we aimed to determine expression levels of the cognate receptor for IL-1α, IL-1R1, within PKT tumors by flow cytometry. Tumors from PKT mice (aged 6.5 weeks, n=6) showed a significant increase in the population of IL-1R1+ cells in the PDGFR+ population compared to EpCAM+ cells, demonstrating that IL-1R1 is strongly expressed on stromal/CAF populations within the TME (Fig. 2A). Further, immunofluorescent staining of PKT tumors showed strong IL-1R1 expression in PDAC stroma (CDH1 negative, Fig. 2B). To examine this relationship in human PDAC samples, we estimated a “stroma score” using transcriptional data from 187 TCGA PDAC samples with xCell, a cell-type enrichment pipeline (20). Using this platform, we stratified samples into groups with a high (“stroma enriched”) or low (“stroma low”) gene stromal signature, defined as a stroma score in the highest or lowest quartile, respectively. Differential gene expression was then determined between stroma enriched and stroma low groups as outlined in Fig. 2C. Validation analyses revealed distinct increases in canonical fibroblastic markers (ACTA2, VIM, PDGFRA, COL1A1, COL1A2, PDPN) in the “stroma enriched” group (Fig. 2D & Supplementary Fig. S2A–C). Expression of IL1R1 was significantly increased in “stroma enriched” PDAC samples compared to “stroma low” tumors (Fig. 2E, p<0.0001), supporting the results observed in PKT tumors in human PDAC samples. These findings indicate that PDAC fibroblasts are primed to respond to IL-1 signaling within the TME.
Figure 2: Expression of interleukin-1 receptor type 1 (IL-1R1) is enriched in pancreatic stromal cells.
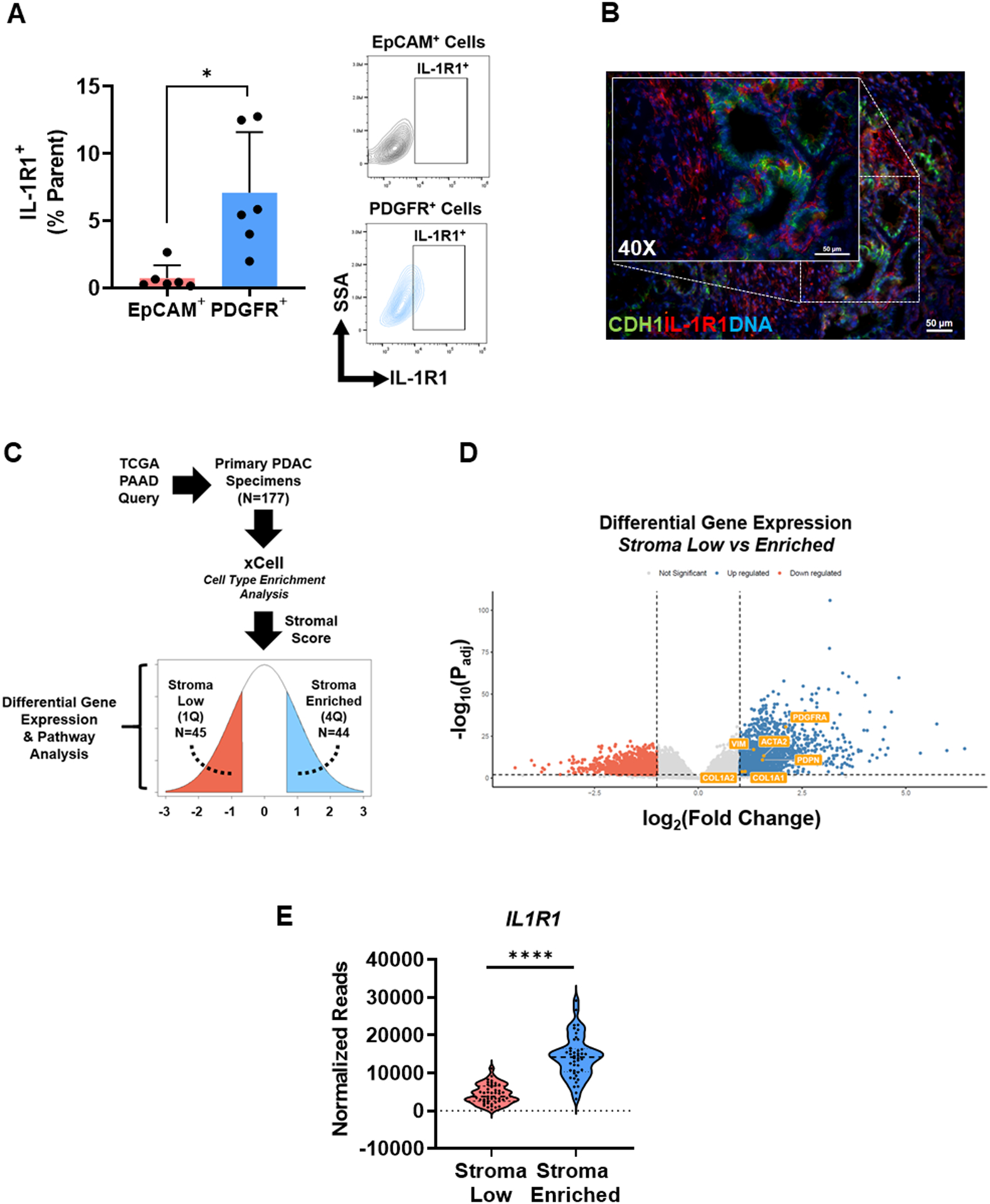
(A) Cell surface expression of IL-1R1 was determined by flow cytometry of tumors from Ptf1aCre/+;LSL-KrasG12D/+; Tgfbr2flox/flox (PKT) mice (aged 6.5 weeks, n=6). Surface staining for co-expression of IL-1R1 on epithelial (EpCAM+) cells or stromal (PDGFR+) cells was quantified. (B) Representative immunofluorescent image demonstrating IL-1R1 (red) and CDH1 (green) co-staining in a PKT tumor section (age 6.5 weeks). (C) Workflow for analysis of The Cancer Genome Atlas (TCGA) human PDAC transcriptional data. xCell was used for the enrichment analysis and subsequent separation of patient samples into stroma enriched (n=44) and stroma low (n=45) quartiles. (D) Volcano plot demonstrating differential expression of characteristic PDAC stromal markers (ACTA2, VIM, PDGFRA, COL1A1, COL1A2, PDPN) between stroma low and stroma enriched groups. (E) Gene expression of IL1R1 in stroma low compared to stroma enriched samples reported as normalized reads. Scale bar = 50 μm. Bar graph data are shown as mean ± SD. *, p<0.05; ****, p<0.0001.
Tumor cell-derived IL-1α induces the release of IL-6 from pancreatic stellate cells
Having established that stromal fibroblasts strongly express IL-1R1, we next aimed to determine changes in the secretory profile of these cells induced by tumor cell-derived IL-1. Human PSCs (hPSCs) were stimulated with conditioned media (CM) from high IL-1α-secreting SW1990 cells (Supplementary Fig. S3A) for 24 hours in the presence or absence of IL-1α neutralizing antibody (10 μg/ml), followed by subsequent analysis of the hPSC CM by cytokine array. Neutralization of IL-1α in condition media resulted in substantial changes to the secretome of hPSCs (Supplementary Fig. S3B), including a significant reduction in the secretion of the pro-tumorigenic cytokine IL-6 (Fig. 3A). These results were validated by ELISA, which demonstrated a significant increase in hPSC secretion of IL-6 upon stimulation with CM from SW1990 or BxPC3 cells that was reduced by IL-1α neutralization in a dose-dependent manner (Fig. 3B & C). Further, we observed that IL-1α neutralization of SW1990 or BxPC3 CM also resulted in a dose-dependent reduction in nuclear localization of the p-p65 subunit of NF-κB, a key activator of IL-6 gene transcription in response to IL-1 (Supplementary Fig. S3C). These findings were further evaluated at the mRNA level, where incubation of hPSCs with SW1990 CM activated IL6 transcription in an IL-1α-dependent manner (Fig. 3D). Similar results were observed using direct IL-1α stimulation (25 ng/ml), which produced a marked increase in IL6 transcription from hPSCs (Fig. 3E). These results demonstrate that tumor-cell-derived IL-1α is a potent inducer of IL-6 release from pancreatic stromal cells.
Figure 3: Tumor cell-derived IL-1α induces the release of IL-6 from pancreatic stellate cells.
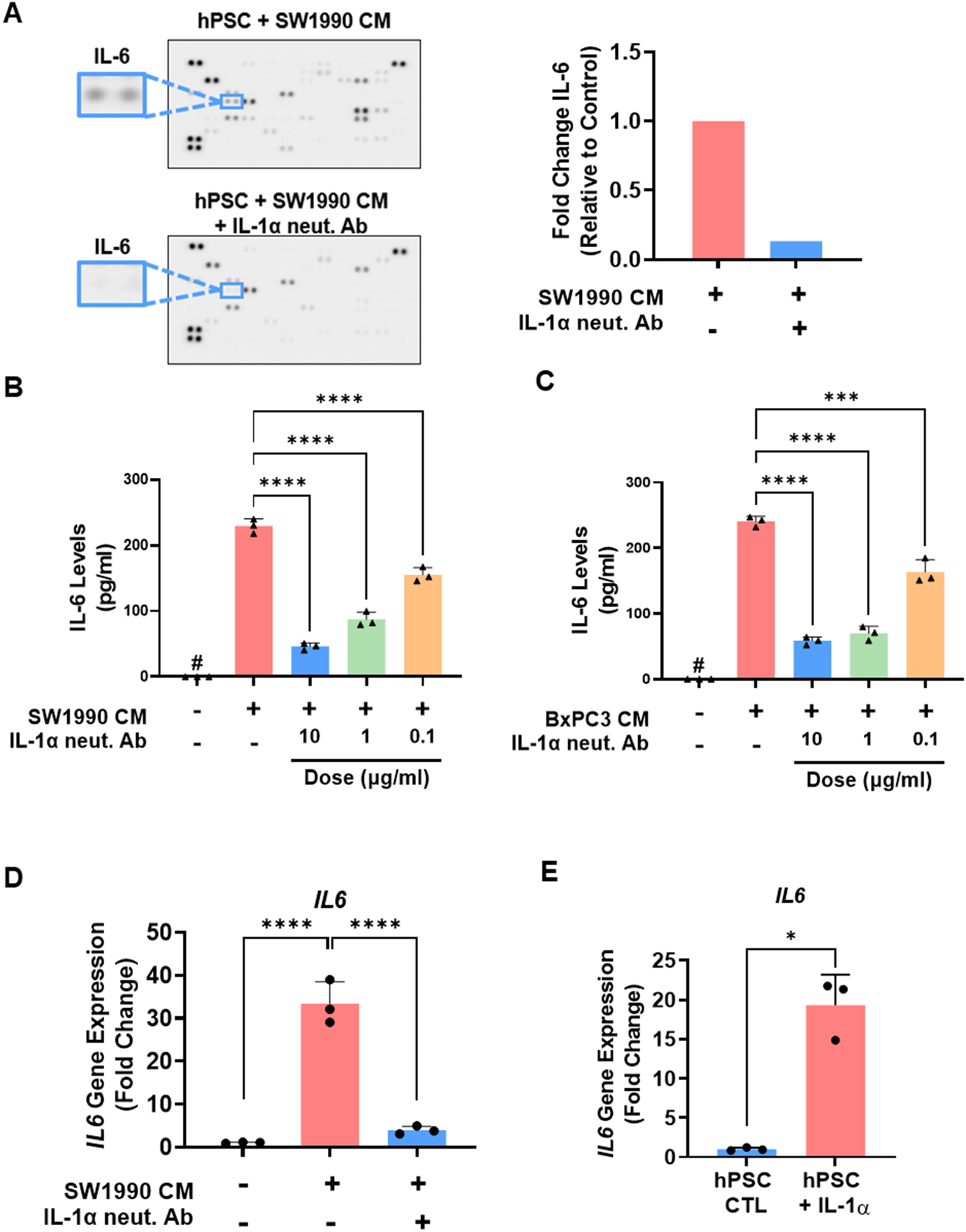
(A) Cytokine array analysis of conditioned media (CM) harvested from human pancreatic stellate cells (hPSCs) after stimulation with SW1990 CM for 24 hours in the presence or absence of IL-1α neutralizing antibody (10 μg/ml), demonstrating a significant reduction in secreted IL-6 levels (right). Measurement of IL-6 levels by ELISA in CM from hPSCs after stimulation with (B) SW1990 CM or (C) BxPC3 CM for 12 hours in the presence or absence of IL-1α neutralizing antibody (0.1, 1, and 10 μg/ml). (D) IL6 gene transcription in human PSCs upon stimulation with SW1990 CM for 1 hour in the presence or absence of IL-1α neutralizing antibody (10 μg/ml). (E) IL6 gene transcription in hPSCs upon stimulation with recombinant IL-1α (25 ng/ml) for 1 hour. Values were normalized to the housekeeping gene GAPDH. Data are shown as mean ± SD. *, p<0.05; ****, p<0.0001. #, not detected.
IL-1α induces IL-6 release from pancreatic stellate cells to activate STAT3 signaling in tumor cells
We and others have shown that IL-6 is a critical cytokine involved in the activation of STAT3 in PDAC (8, 33–35). In parallel with the observed increase in IL-1α during PKT progression (Fig. 1F), we detected a concurrent elevation in IL-6 levels from tumors harvested from PKT mice at 6.5 weeks as compared to non-tumor bearing Ptf1aCre controls (Fig. 4A). Interestingly, this correlated with a concomitant increase in pSTAT3 levels within the tumors of 6.5-week-old PKT mice by both western blot (Fig. 4B & C) and IHC (Fig. 4D). Further, pathway analysis between “stroma enriched” and “stroma low” patients revealed that tumors with a “stroma enriched” gene signature were associated with a significant increase in the expression levels of transcripts involved in the IL-6/STAT3 signaling pathway compared to the “stroma low” quartile (Supplementary Fig. S4A). When examining differential expression of individual components within these gene sets, we observed that “stroma enriched” tumors exhibited significantly higher expression of the IL6 and STAT3 genes (Fig. 4E & F). Together, these results provide associative evidence that IL-1α, IL-6, and pSTAT3 levels all concurrently increase during the development of invasive cancer in mice and that elevated stromal content is correlated with activation of the IL-6/STAT3 pathway in human PDAC tumors.
Figure 4: IL-1α induces IL-6 release from pancreatic stellate cells to activate STAT3 signaling in tumor cells.
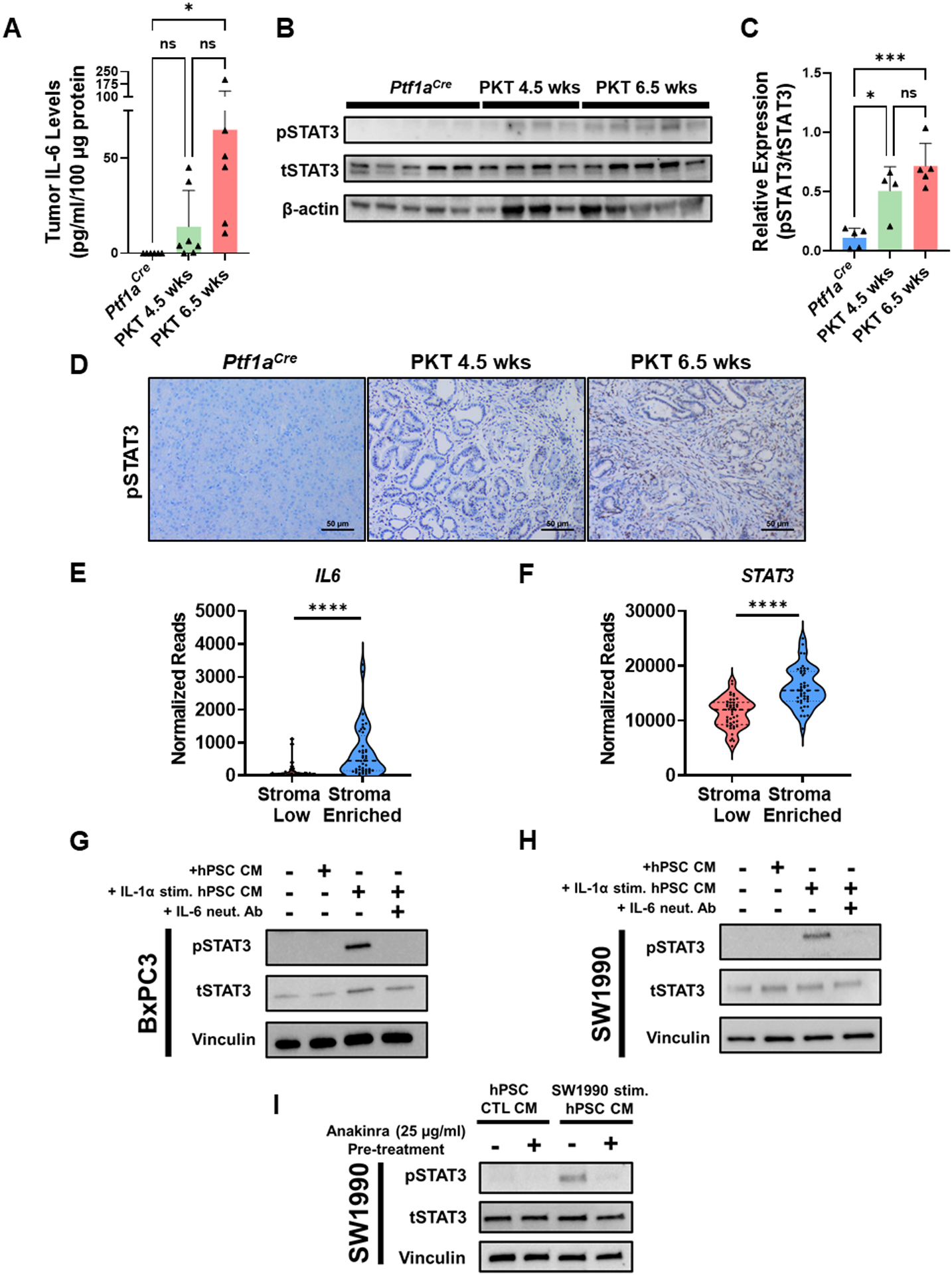
(A) Measurement of IL-6 levels by ELISA in non-tumor bearing Ptf1aCre pancreata compared to tumors harvested from 4.5-week-old and 6.5-week-old Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice. (B–C) Levels of pSTAT3 in Ptf1aCre (n=5), 4.5-week-old PKT (n=4), and 6.5-week-old PKT (n=5) mice as determined by Western blot. Densitometric analysis was performed relative to tSTAT3. (D) Representative IHC images for pSTAT3 levels in non-tumor bearing Ptf1aCre, 4.5-week-old PKT mice, and 6.5-week-old PKT mice. (E) IL6 and STAT3 gene expression between “stroma low” and “stroma enriched” patients from TCGA database analysis. (G–H) Human pancreatic stellate cells (hPSCs) were incubated in either serum-free media (SFM), or SFM supplemented with IL-1α (25 ng/ml) for 12 hours and conditioned media (CM) harvested. Control SFM, unstimulated hPSC CM, or IL-1α-stimulated hPSC CM ± anti-IL-6 antibody (1 μg/ml) were then added to (G) BxPC3 or (H) SW1990 cells and levels of pSTAT3 determined in response by Western blot. (I) In a separate experiment, hPSCs were either cultured in SFM or SW1990 CM ± pre-treatment with anakinra (25 μg/ml) and CM collected after 12 hours. SW1990 cells were then incubated with CM collected from these conditions and levels of pSTAT3 determined in response by Western blot. *, p<0.05; ***, p<0.001; ****, p<0.0001.
As IL-1α potently induces IL-6 release from PSCs and is elevated in parallel with pSTAT3 during PDAC carcinogenesis, we hypothesized that stromal-derived IL-6 produced in response to IL-1α stimulation may activate STAT3 signaling within tumor cells. BxPC3 and SW1990 tumor cells were exposed to CM harvested from control hPSCs or those treated with IL-1α (25 ng/ml) for 12 hours in the presence or absence of IL-6 neutralizing antibody (1 μg/ml, R&D systems) and levels of activated pSTAT3 measured in response (Supplementary Fig. S4B). Tumor cells treated with control media (0% FBS) or CM obtained from unstimulated hPSCs showed low pSTAT3 levels. However, BxPC3 or SW1990 cells treated with CM derived from IL-1α-stimulated hPSCs demonstrated a marked increase in pSTAT3 levels that was significantly reduced by pretreatment with IL-6 neutralizing antibody (Fig. 4G & 4H), indicating that IL-1 stimulation of hPSCs induces STAT3 activation in tumor cells in an IL-6-dependent manner. Of note, neutralization of the STAT3 ligands G-CSF or LIF did not result in a significant reduction in tumor cell pSTAT3 activation under these experimental conditions (Supplementary Fig. S4C).
Next, we aimed to determine if IL-1R1 blockade inhibits STAT3 activation in tumor cells induced by hPSC-derived IL-6. hPSCs were incubated with SW1990 CM for 12 hours in the presence or absence of anakinra (25 μg/ml). CM was then harvested and placed on SW1990 cells and pSTAT3 levels determined in response (Supplementary Fig. S4D). While no appreciable change in pSTAT3 level was observed with incubation of SW1990 cells with unstimulated hPSC CM, a significant increase in pSTAT3 was observed in tumor cells treated with hPSC CM primed by SW1990 supernatant. However, pre-treatment of hPSCs with anakinra was able to abolish this observed increase in pSTAT3 activation in SW1990 cells (Fig. 4I). These results suggest that IL-1R1 blockade inhibits STAT3 activation in PDAC tumor cells through the suppression of IL-6-mediated tumor-stromal crosstalk.
IL-1 receptor blockade reduces IL-6 levels, suppresses STAT3 activation, and improves survival in combination with gemcitabine in Ptf1aCre/+;LSL-KrasG12D/+; Tgfbr2flox/flox (PKT) mice
Having established that IL-1α induces stromal-derived IL-6 and activates STAT3 in PDAC tumor cells, we next aimed to determine if inhibition of IL-1 signaling can suppress the IL-6/STAT3 signaling axis in vivo. For murine studies, we utilized the aggressive PKT mouse model of PDAC. PKT mice develop large, rapidly progressing pancreatic tumors that contain abundant stroma characteristic of human tumors. We have previously demonstrated that this mouse model contains high levels of both IL-6 and activated STAT3 within tumors, making it an optimal model to study this interaction (6, 8, 10). PKT mice were treated with vehicle or anakinra (50 mg/kg twice daily, 5 days per week) via intraperitoneal injection for 2.5 weeks (Fig. 5A). Mice treated with anakinra had a significant reduction of intratumoral Il6 mRNA (p=0.04) and IL-6 protein levels (p=0.002) compared with vehicle-treated mice (Fig. 5B & C). Interestingly, these changes corresponded with a significant reduction in pSTAT3 levels within tumors (Fig. 5D & E).
Figure 5: IL-1 receptor inhibition reduces IL-6 levels, suppresses STAT3 activation, and improves survival in combination with gemcitabine in Ptf1aCre/+;LSL-KrasG12D/+; Tgfbr2flox/flox (PKT) mice.
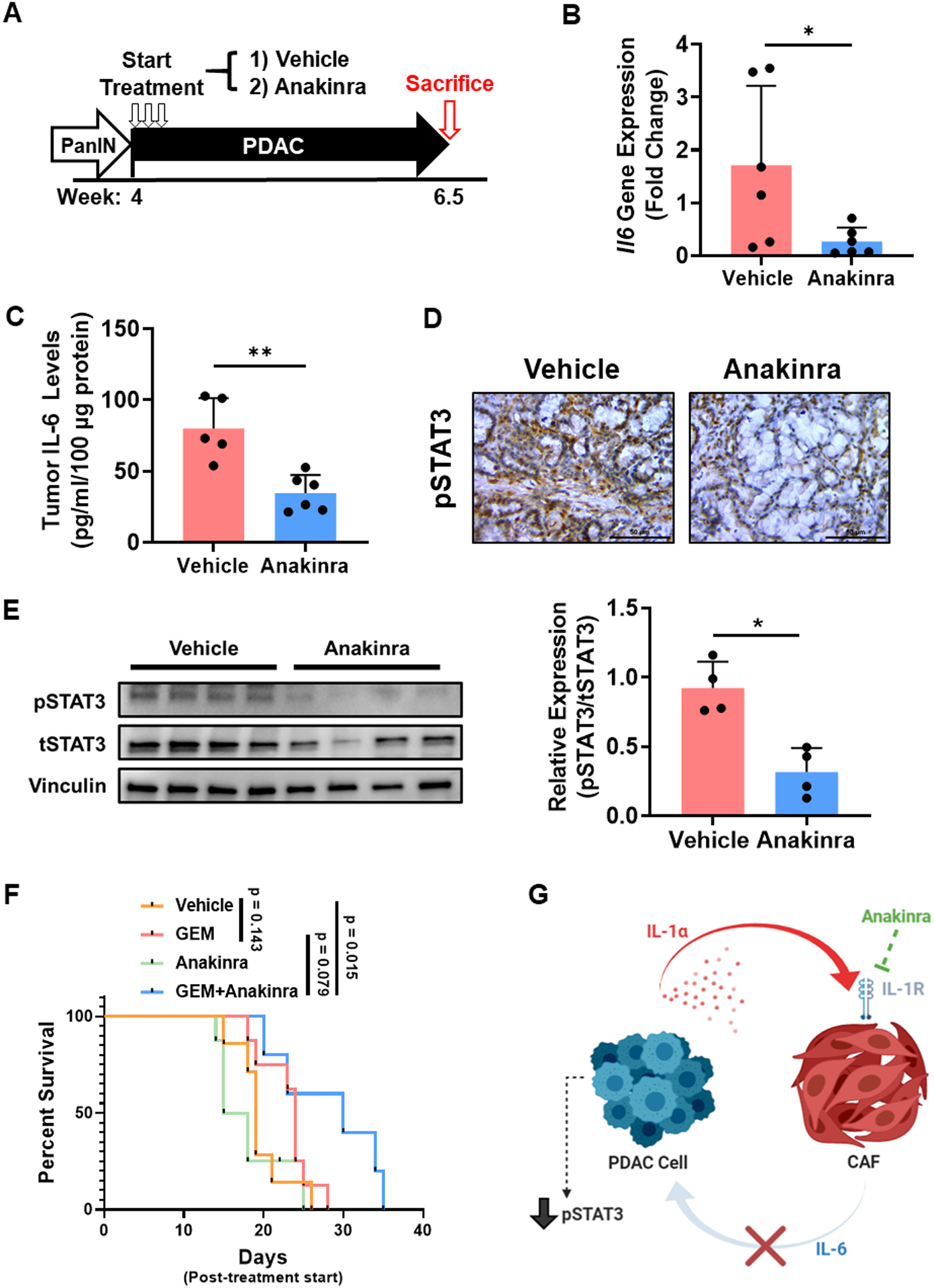
(A) Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice were treated with saline vehicle or anakinra (50 mg/kg twice daily) via intraperitoneal injection beginning at 4 weeks until sacrifice at 6.5 weeks. (B) Il6 gene levels in vehicle and anakinra-treated PKT tumors were determined by qPCR. Values were normalized to the housekeeping gene Gapdh. (C) Intratumoral IL-6 levels in vehicle and anakinra-treated PKT mice were determined by ELISA. (D) Representative images of IHC staining of pSTAT3 in vehicle and anakinra-treated PKT tumor samples. (E) Western blot analysis of pSTAT3 in anakinra-treated and vehicle PKT tumor lysates (n=4/arm). Densitometric analysis was performed relative to tSTAT3. (F) Kaplan-Meier survival curve demonstrating overall survival of vehicle, gemcitabine (20 mg/kg, GEM), anakinra, and combination GEM and anakinra-treated PKT mice (n=5–8 per group). Pairwise comparisons between individual groups were performed using log-rank test. (G) Proposed mechanism of action for anakinra treatment in modulating STAT3 activity in the PDAC tumor microenvironment through the suppression of stromal-derived IL-6. Scale bar = 50 μm. *, p<0.05; **, p<0.01.
As our previous studies have suggested that STAT3 blockade may sensitize PDAC tumors to chemotherapy, we next treated PKT mice with vehicle, gemcitabine (GEM, 20 mg/kg IP injection, 3 times weekly), anakinra (50 mg/kg IP injection twice daily, 5 times weekly) or combination GEM and anakinra beginning at 4 weeks of age and continued until mice were moribund (5–7). Treatment with GEM alone failed to significantly improve survival in PKT mice (median survival 19 days post-treatment initiation vs. 24 days, p = 0.143), in accordance with our previous studies utilizing this transgenic model (6). However, combined treatment with GEM and anakinra significantly extended the survival of PKT mice compared to vehicle treatment (19 vs. 30 days, p=0.015) or anakinra monotherapy (16.5 vs 30 days, p=0.026) and trended towards improved survival compared to GEM alone (24 vs. 30 days, p=0.079) (Fig. 5F). These results demonstrate that IL-1R1 blockade reduces intratumoral IL-6 production, decreases pSTAT3 levels in vivo, and improves survival in PKT mice compared to vehicle treatment when combined with GEM chemotherapy (Fig. 5G).
Discussion
Inflammatory signaling plays an essential role in the initiation and progression of PDAC tumors as evidenced by both the strong association between PDAC and epidemiological risk factors that promote inflammation (smoking, chronic pancreatitis, etc.) as well as the laboratory use of caerulein-induced pancreatitis to drive PDAC carcinogenesis in vivo (36, 37). Oncogenic KRAS mutations, nearly ubiquitous in PDAC tumors, are essential to establishing and maintaining this inflammatory program through the coordinated activation of signaling pathways such as NF-κB and STAT3 that drive tumor growth (38). Among the diverse activators of inflammatory signaling within the TME, IL-1α has been specifically implicated as a driver of Ras-dependent inflammation in PDAC tumors. Activating KRAS mutations have been shown to induce an inflammatory signaling loop mediated by IL-1α/p62, leading to the constitutive activation of IKK2/β and downstream NF-κB signaling that is indispensable for PDAC tumorigenesis in KrasLSL-G12D/+;Ink4a/Arfflox/flox;Pdx1Cre/+ mice (39). We similarly observed a marked increase in IL-1α levels in tumors from PDAC patients and PKT mice compared to normal pancreatic tissue. Levels of IL-1R1 in PKT mouse tumors are increased in PDGFR+ cell populations and transcriptionally elevated in PDAC tumor patient samples with high stromal content, indicating that PSCs and CAFs within the TME are uniquely poised for paracrine activation by IL-1 ligands. These findings corroborate previous studies in KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1Cre/+ (KPC) mice, in which single-cell profiling revealed a marked increase in gene transcription of Il1r1 in intratumoral CAFs, while Il1a ligand expression was comparatively localized to the epithelial cell compartment (27). We show that tumor cells activate the release of pro-inflammatory mediators such glutamic acid-leucine-arginine (ELR) chemokines and IL-6 from stromal cells in an IL-1α-dependent manner. Of these pleiotropic factors, members of the IL-6 superfamily of cytokines have been implicated as potent activators of an inflammatory CAF phenotype and STAT3 signaling within PDAC tumors (8, 27, 40). Herein, we coalesce these findings by demonstrating that tumor cell-derived IL-1α activates IL-6 release from stromal cells leading to intratumoral STAT3 activation, providing further insight into the diverse tumor-stromal interactions that drive oncogenic signaling within the PDAC TME.
STAT3 activation fosters tumor cell growth by promoting uncontrolled proliferation and activating anti-apoptotic mechanisms that significantly reduce the efficacy of cytotoxic chemotherapy (41). Our previous studies have implicated activated STAT3 as a key biomarker of therapeutic resistance in PDAC, a finding that has also been demonstrated in breast, lung, and ovarian cancers (6, 10, 42–44). However, a recent Phase III trial utilizing direct JAK/STAT pathway inhibition with capecitabine in treatment-refractory PDAC patients failed to improve clinical outcomes, highlighting the need to identify alternative strategies to target STAT3 signaling in vivo (9). Although aberrant activation of STAT3 is present in many solid organ cancers, activating mutations in the STAT3 gene itself are infrequent, instead driven predominately by dysregulation of upstream ligands of JAK/STAT signaling (45, 46). This study highlights IL-1 signaling as a targetable, upstream mediator of STAT3 activation via IL-6-dependent tumor-stromal crosstalk mechanisms and provides a rationale for the use of IL-1R1 inhibition to potentially overcome STAT3-mediated treatment resistance in PDAC.
Given the importance of the IL-1 pathway in mediating inflammatory signaling within the PDAC TME, IL-1R1 inhibition likely also yields significant immunologic changes within tumors that influence the response to chemotherapy. IL-1 signaling has been shown to be a critical factor in promoting the characteristic immunosuppressive phenotype of PDAC through the recruitment of diverse immune populations such as regulatory B-cells and myeloid cell subsets that impair the infiltration and function of cytotoxic CD8+ T-cells. Additionally, IL-1 pathway blockade has been shown to improve the response to immune checkpoint inhibition in vivo, further supporting its role as a key cytokine that promotes immune suppression in PDAC and other cancers (47, 48). In array profiling of hPSCs, we identified multiple IL-1-dependent chemotactic factors that are known to modulate macrophage polarization and myeloid-derived suppressor cell (MDSC) trafficking (Supplementary Fig. S3B). Recent studies have demonstrated that resident immune cells, specifically alternatively activated “M2-like” tumor-associated macrophages, play a critical role in the response to chemotherapy in PDAC (49). Likewise, clinical evidence also implicates neutrophil-to-lymphocyte imbalances in patients receiving chemotherapy as a prognostic marker in PDAC, further suggesting modulation of these immune populations may be important in dictating the efficacy of cytotoxic and targeted agents (50). As alterations in these cellular processes undoubtedly influence treatment response, a thorough understanding of the specific immunologic mechanisms of resistance modulated by IL-1R1 inhibition is urgently needed and is the subject of ongoing investigation in our laboratory. Additionally, although IL-1 receptor inhibition significantly reduces pSTAT3 levels in PKT mice, we did observe residual pSTAT3 activity within tumors following treatment (Fig. 5D & E). These findings suggest a potential compensatory role for alternative pro-inflammatory signaling pathways or STAT3 ligands aside from IL-6 in sustaining JAK/STAT signaling during treatment with IL-1R1 inhibitors (27, 40). These redundant signaling mechanisms within the TME highlight the considerable challenges that exist in targeting individual cytokines for the treatment of cancer and likely underlie the modest effect on overall survival that we observed with IL-1R1 treatment in combination with chemotherapy. Further investigation into combinatorial therapeutic approaches that can simultaneously target redundant inflammatory pathways and improve the response to chemotherapy/immunotherapy should therefore be explored in future studies.
Emerging evidence implicates tumor-stromal interactions as a critical determinant of therapeutic resistance in PDAC. This study demonstrates that IL-1α may function as a key regulator of tumor-stromal crosstalk in PDAC that activates oncogenic STAT3 signaling by stimulating the release of IL-6 from resident mesenchymal cells within the TME. These data provide compelling preclinical evidence to explore the use of IL-1R1 blockade in combination with chemotherapy to suppress inflammatory signaling crosstalk that may be explored in future studies in PDAC patients.
Supplementary Material
Acknowledgements
We would like to thank Dr. Oliver Umland for his assistance in flow cytometry experiments and Dr. Deukwoo Kwon of the Sylvester Biostatistics and Bioinformatics Shared Resource (BBSR) for his review of the statistical analyses performed in this manuscript. Figure diagrams were created with BioRender (www.biorender.com). The results published here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 12/31/2020.
Financial Support:
This work was supported by the NIH R01 CA161976 and NIH T32 CA211034 to N. B. Merchant, Society of University Surgeons (SUS) Research Scholar Award GR-013716 and Sylvester Comprehensive Cancer Center (SCCC) Tumor Biology Research Award PG013439 to A.R. Dosch, KL2 career development grant via NIH UL1TR002736 to J.Datta, and NIH NCI R03 CA249401 to N.S. Nagathihalli. A.R. Dosch conducted this work as a fellow on NIH T32 CA211034. Histopathology Core Service was performed through the SCCC support grant (N. B. Merchant and N.S. Nagathihalli). Research reported in this publication was supported by the NCI of the NIH under Award Number P30 CA240139. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest:
N. B. Merchant previously served on the scientific advisory board for Swedish Orphan Biovitrum (SOBI). Research support and investigational compound for this study were in part provided by SOBI.
References
- 1.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25. [DOI] [PubMed] [Google Scholar]
- 2.Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29(3):179–87. [DOI] [PubMed] [Google Scholar]
- 3.Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17(8):487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP, et al. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer. 2019;5(11):724–41. [DOI] [PubMed] [Google Scholar]
- 5.Nagathihalli NS, Washington MK, Merchant NB. Combined blockade of Src kinase and epidermal growth factor receptor with gemcitabine overcomes STAT3-mediated resistance of inhibition of pancreatic tumor growth. Clin Cancer Res. 2011;17(3):483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology. 2015;149(7):1932–43 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dosch AR, Dai X, Reyzer ML, Mehra S, Srinivasan S, Willobee BA, et al. Combined Src/EGFR Inhibition Targets STAT3 Signaling and Induces Stromal Remodeling to Improve Survival in Pancreatic Cancer. Mol Cancer Res. 2020;18(4):623–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagathihalli NS, Castellanos JA, VanSaun MN, Dai X, Ambrose M, Guo Q, et al. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget. 2016;7(40):65982–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurwitz H, Van Cutsem E, Bendell J, Hidalgo M, Li CP, Salvo MG, et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Invest New Drugs. 2018;36(4):683–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagathihalli NS, Castellanos JA, Lamichhane P, Messaggio F, Shi C, Dai X, et al. Inverse Correlation of STAT3 and MEK Signaling Mediates Resistance to RAS Pathway Inhibition in Pancreatic Cancer. Cancer Res. 2018;78(21):6235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32(2):73–5. [DOI] [PubMed] [Google Scholar]
- 12.PubChem Compound Summary for CID 139595263, Anakinra. [Internet]. 2021. [cited April 8, 2021]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Anakinra.
- 13.Powers NE, Swartzwelter B, Marchetti C, de Graaf DM, Lerchner A, Schlapschy M, et al. PASylation of IL-1 receptor antagonist (IL-1Ra) retains IL-1 blockade and extends its duration in mouse urate crystal-induced peritonitis. J Biol Chem. 2020;295(3):868–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32(2):185–203 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nellore A, Collado-Torres L, Jaffe AE, Alquicira-Hernandez J, Wilks C, Pritt J, et al. Rail-RNA: scalable analysis of RNA-seq splicing and coverage. Bioinformatics. 2017;33(24):4033–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collado-Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD, et al. Reproducible RNA-seq analysis using recount2. Nat Biotechnol. 2017;35(4):319–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frazee AC, Langmead B, Leek JT. ReCount: a multi-experiment resource of analysis-ready RNA-seq gene count datasets. BMC Bioinformatics. 2011;12:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mounir M, Lucchetta M, Silva TC, Olsen C, Bontempi G, Chen X, et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019;15(3):e1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73. [DOI] [PubMed] [Google Scholar]
- 24.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garg B, Giri B, Modi S, Sethi V, Castro I, Umland O, et al. NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology. 2018;155(3):880–91 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019;9(2):282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17(2):135–47. [DOI] [PubMed] [Google Scholar]
- 29.Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 2008;55(88):2016–27. [PubMed] [Google Scholar]
- 30.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tjomsland V, Spangeus A, Valila J, Sandstrom P, Borch K, Druid H, et al. Interleukin 1alpha sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia. 2011;13(8):664–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013;73(20):6359–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(4):456–69. [DOI] [PubMed] [Google Scholar]
- 35.Long KB, Tooker G, Tooker E, Luque SL, Lee JW, Pan X, et al. IL6 Receptor Blockade Enhances Chemotherapy Efficacy in Pancreatic Ductal Adenocarcinoma. Mol Cancer Ther. 2017;16(9):1898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382(3):561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lowenfels AB, Maisonneuve P. Risk factors for pancreatic cancer. J Cell Biochem. 2005;95(4):649–56. [DOI] [PubMed] [Google Scholar]
- 38.Kitajima S, Thummalapalli R, Barbie DA. Inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin Cell Dev Biol. 2016;58:127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(1):105–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature. 2019;569(7754):131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Crowe PJ, Goldstein D, Yang JL. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers (review). Int J Oncol. 2012;41(4):1181–91. [DOI] [PubMed] [Google Scholar]
- 42.Qin JJ, Yan L, Zhang J, Zhang WD. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. 2019;38(1):195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Y, Hong Y, Xu Y, Liu P, Guo DH, Chen Y. Inhibition of the JAK/STAT pathway with ruxolitinib overcomes cisplatin resistance in non-small-cell lung cancer NSCLC. Apoptosis. 2014;19(11):1627–36. [DOI] [PubMed] [Google Scholar]
- 44.Pogge von Strandmann E, Reinartz S, Wager U, Muller R. Tumor-Host Cell Interactions in Ovarian Cancer: Pathways to Therapy Failure. Trends Cancer. 2017;3(2):137–48. [DOI] [PubMed] [Google Scholar]
- 45.Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: A review. Int J Cancer. 2016;138(11):2570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wake MS, Watson CJ. STAT3 the oncogene - still eluding therapy? FEBS J. 2015;282(14):2600–11. [DOI] [PubMed] [Google Scholar]
- 47.Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. Tumor Cell-Derived IL1beta Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020;80(5):1088–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A. 2019;116(4):1361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019;29(6):1390–9 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng H, Long F, Jaiswar M, Yang L, Wang C, Zhou Z. Prognostic role of the neutrophil-to-lymphocyte ratio in pancreatic cancer: a meta-analysis. Sci Rep. 2015;5:11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.