

Abstract
Cocaine addiction is a chronic, relapsing disorder characterized by maladaptation in the brain mesolimbic and nigrostriatal dopamine system. Although changes in the properties of D2-receptor expressing medium spiny neurons (D2-MSNs) and connected striatal circuits following cocaine treatment are known, the contributions of altered D2-receptor (D2R) function in mediating the rewarding properties of cocaine remain unclear. Here we describe how a 7-day exposure to cocaine alters dopamine signaling by selectively reducing the sensitivity, but not expression, of nucleus accumbens D2-MSN D2Rs via an alteration in the relative expression and coupling of G-protein subunits. This cocaine-induced reduction of D2R sensitivity facilitated the development of the rewarding effects of cocaine as blocking the reduction in G-protein expression was sufficient to prevent cocaine-induced behavioral adaptations. These findings identify an initial maladaptive change in sensitivity by which mesolimbic dopamine signals are encoded by D2Rs following cocaine exposure.
eTOC
Gong et al. find that cocaine selectively reduces the sensitivity of accumbens D2-receptors by dynamically regulating G-protein expression in D2-MSNs. These changes were independent of D2R levels and bidirectionally regulated cocaine seeking. This identifies that altered D2R sensitivity is an initial adaptation to cocaine exposure that in turn drives cocaine-seeking behaviors.
Graphical Abstract
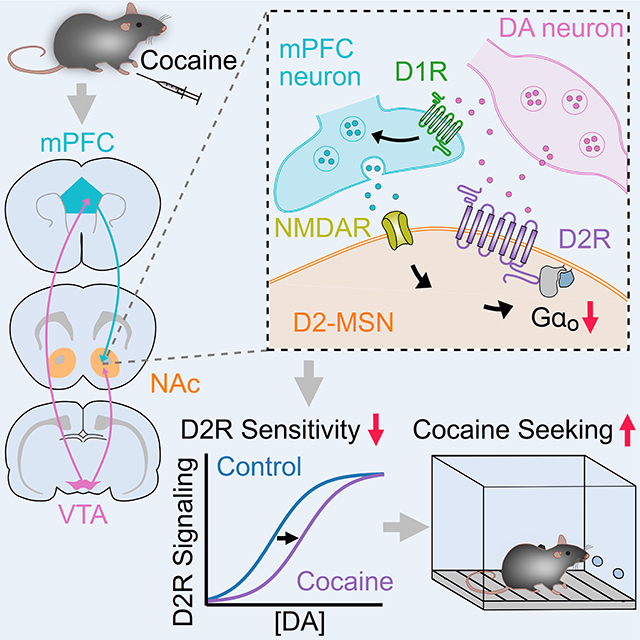
INTRODUCTION
Addictive drugs induce neuroadaptations across a variety of neural circuits, which produce persistent drug seeking and chronic relapse in vulnerable individuals (Koob and Volkow, 2016; Lüscher and Malenka, 2011; Wolf, 2010). A key neural substrate underlying drug addiction involves the midbrain dopamine (DA) system, in particular the mesolimbic and nigrostriatal DA pathways innervating the nucleus accumbens (NAc) and dorsal striatum (DSt). These two striatal regions have distinct afferent and efferent connections, are involved in unique behavioral functions, and are modulated differentially by drugs of abuse. Within the striatum, dopamine mediates reward, reinforcement, and goal-directed action learning via dopamine receptors that, depending on the class, are expressed on output projection medium spiny neurons (MSNs), classes of interneurons and afferent inputs (Gerfen and Surmeier, 2011). As D1-receptors (D1Rs) activate stimulatory Gαs/olf signaling, while D2-receptors (D2Rs) activate inhibitory Gαi/o signaling, dopamine mediates opposing actions on D1- and D2-receptor expressing subpopulations of MSNs. Through their different projections D1-MSNs and D2-MSNs are thought to have opposing actions on behavior, as studies have found that activation of D1-MSNs increases locomotion, motivation and conditioned place preference for cocaine while activation of D2-MSNs decreases those behaviors (Bock et al., 2013; Hikida et al., 2010; Kravitz et al., 2012; Lobo et al., 2010).
Considerable evidence implicates dysregulation of dopaminergic neurotransmission, particularly through inhibitory Gαi/o-coupled D2Rs, as a key contributor to psychostimulant abuse. Manipulation of D2R expression has implicated abnormal D2R levels in the regulation of cocaine related-behaviors (Bello et al., 2011; Dobbs et al., 2016, 2019; Lee et al., 2020; Lewis et al., 2020). In addition, neuroimaging studies have documented that repeated long-term cocaine exposure is associated with reductions in striatal D2R availability, and low striatal D2R availability is inversely correlated with impulsivity and propensity for escalation of cocaine intake as well as compulsive cocaine seeking (Dalley et al., 2007; Morgan et al., 2002; Nader et al., 2006; Volkow et al., 1990, 1993, 1997). Together these findings have indicated that D2Rs are key regulators of motivated behavior and that changes in their levels and availability are instrumental for cocaine-induced behavioral adaptations. However, despite these findings examining chronic changes following long-term cocaine exposure, it has remained unknown how initial or brief exposure to cocaine may alter dopamine D2R signaling, and whether any region-specific changes in D2R function may alter cocaine related motivated behaviors. While the sensitivity of D2R signaling is known to differ across striatal regions (Marcott et al., 2018), the mechanisms and consequences by which cocaine may regulate D2Rs remain unknown. Here we report that repeated cocaine exposure selectively alters dopamine signaling by reducing NAc D2R sensitivity but not the level of D2Rs on D2-MSNs. These cocaine-induced changes were independent of D2R activation and resulted from altered expression and coupling to G-proteins that were driven by NMDA-receptor dependent plasticity at prefrontal cortical to D2-MSN synapses. These changes had direct effects on drug associated behaviors as exacerbating or preventing expression changes in G-protein subunits was sufficient to bidirectionally regulate cocaine-conditioned behaviors. Together this work identifies altered D2R sensitivity in NAc D2-MSNs as an initial adaptation to cocaine exposure that drives cocaine-related behaviors.
RESULTS
Cocaine decreases D2R sensitivity in the NAc
We began by evaluating regional differences in the effect of cocaine exposure on D2R sensitivity in D2-MSNs in the dorsal and ventral striatum. An adeno-associated virus (AAV) encoding G protein-coupled inward rectifying potassium channels (Kir3.2; GIRK2) and a tdTomato fluorophore was injected into the dorsomedial striatum (DSt) and medial shell of the nucleus accumbens (NAc) of wildtype (WT) mice (Fig. 1A and Fig. S1A). As D2Rs efficiently couple to GIRK2 channels in D2-MSNs, the overexpression of GIRK2 provides an electrophysiological read-out of D2R activation in the NAc and DSt allowing for direct measurement of dopamine mediated D2R signaling in D2-MSNs (Marcott et al., 2014, 2018). Expression of GIRK2 had no measurable effect on motor skill learning, locomotor activity, and anxiety- or reward-related behaviors (Fig. S1B–S1E). In the presence of synaptic blockers to isolate D2Rs, whole-cell voltage clamp recordings were made from tdTomato+ MSNs, and a single electrical stimulus was used to evoke a sulpiride-sensitive D2R inhibitory postsynaptic current (D2-IPSC) in D2-MSNs (Fig. 1A). Currents were mediated by D2Rs as they could be blocked by the selective D2R antagonist L-741,626 (200 nM) and not detected in Drd2-floxed mice where D2Rs were genetically ablated following the injection of AAV.Cre (Fig. S1F–S1G). As D2-IPSCs can be evoked in D2-MSNs but not in D1-MSNs (Marcott et al., 2018), D2-MSNs were readily distinguished by the presence of a D2-IPSC following evoked dopamine release. To examine the sensitivity of D2Rs, concentration-response relationships for dopamine were determined by recording from D2-MSNs in the DSt and NAc in the presence of the dopamine reuptake (DAT) inhibitor cocaine (10 μM) while applying different concentrations of dopamine and measuring the resulting D2R-mediated outward currents (Fig. 1B and Fig. S1H). Consistent with previous findings (Marcott et al., 2018), the concentration of dopamine needed to achieve 50% of the maximal effect (EC50) was significantly lower in the NAc than the DSt (Fig. 1C–D), confirming that D2Rs in the NAc have a higher sensitivity for dopamine than in the DSt. As similar levels of GIRK2 were expressed in each region (Fig. S1I), the maximum outward current evoked by dopamine (Emax) was similar across regions (Fig. 1C).
Figure 1. Cocaine sensitization selectively reduces D2R sensitivity in the NAc.
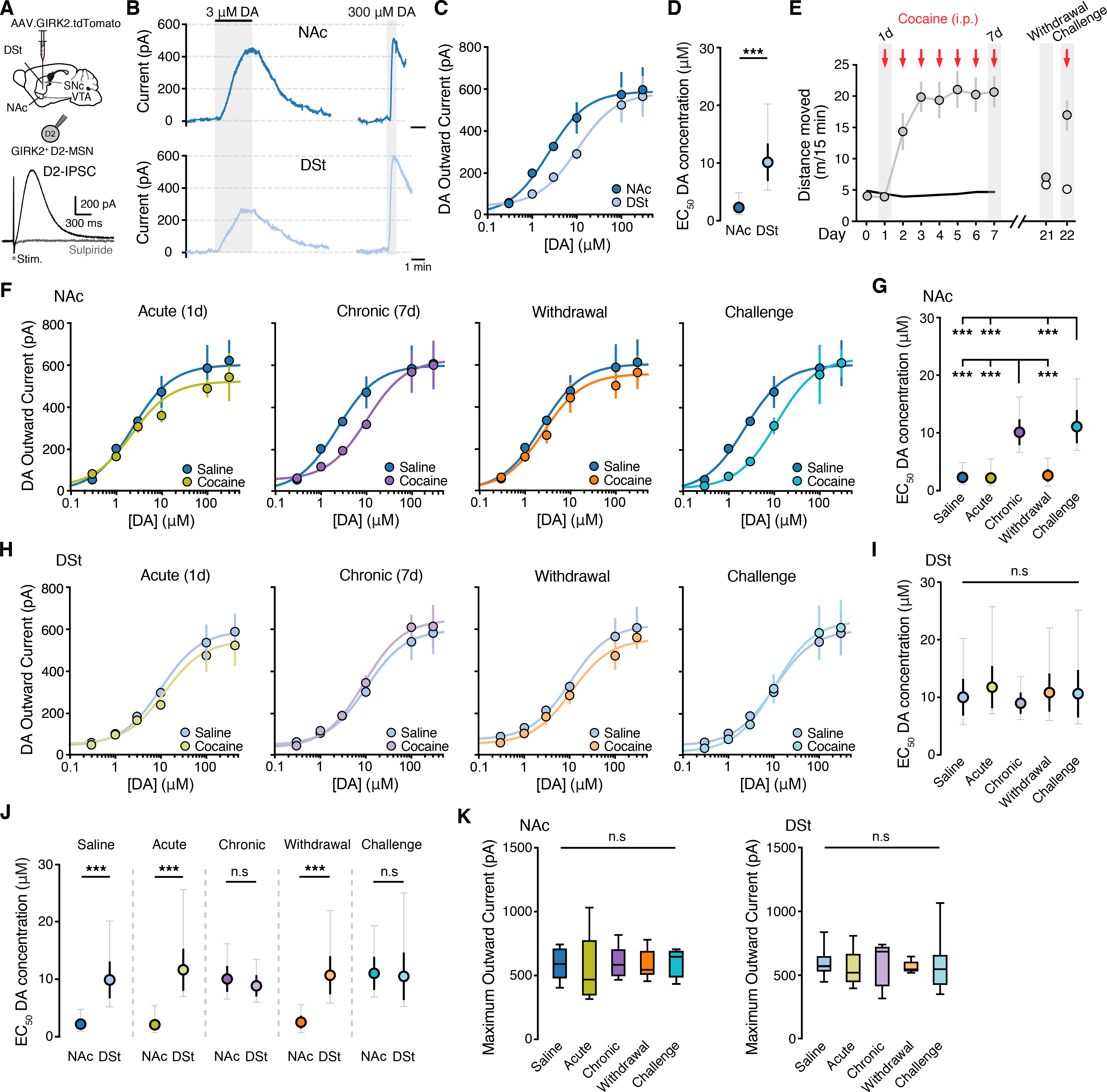
A - Illustration of injection of AAV9.hSyn.tdTomato.T2A.GIRK2 in the NAc (medial shell) and DSt (dorsomedial) and an evoked D2-IPSC from a GIRK2+ D2-MSN blocked by sulpiride (300 nM).
B - Representative traces of D2R mediated outward currents following bath application of dopamine (DA). All recordings were performed in the presence of cocaine (10 μM) to block uptake. Evoked D2-IPSCs blanked for clarity.
C - Dopamine concentration-response relationships for D2R mediated outward GIRK2 currents from D2-MSNs in the DSt and NAc.
D - EC50 values from (C) (two tailed unpaired t-test).
E - Locomotor sensitization comparing saline (open circles) and 7-day administration of cocaine (20 mg/kg; i.p.), 14 days of withdrawal and subsequent administration of cocaine (gray circles).
F - Dopamine D2R dose response curves from NAc D2-MSNs comparing saline treatment to 1d cocaine (Acute), 7d cocaine (Chronic), 14d withdrawal following 7d cocaine (Withdrawal), and a subsequent cocaine challenge (Challenge).
G - EC50 values from (F) (one-way ANOVA, Turkey’s post-test).
H - Dopamine D2R dose response curves from DSt D2-MSNs.
I - EC50 values from (H).
J - EC50 values from (F-I) comparing NAc and DSt across groups (two tailed unpaired t-test).
K - Maximum outward currents evoked by 300 μM dopamine.
Summary data are mean ± SEM (grey traces represent 95% confidence intervals) or boxplots. ns = p > 0.05, *** = p < 0.001. See also Figure S1,S2 and Table S1.
To examine the effect of in vivo cocaine exposure on D2R sensitivity, mice were subjected to a cocaine sensitization regimen (20 mg/kg, intraperitoneally [i.p.] repeated over 7 days) and dopamine concentration-response relationships were generated 24 hours later. Repeated cocaine administration led to the progressive development of enhanced locomotion when compared to saline-treated littermates (Fig. 1E). While acute cocaine treatment (single injection) had no effect on D2R sensitivity in either the NAc or DSt (Fig. 1F–I), chronic exposure for 7 days evoked a rightward shift in the dose-response curve selectively in the NAc (Fig. 1F–I), thus decreasing NAc D2R sensitivity for dopamine to similar levels as in the DSt (Fig. 1J). A similar cocaine-induced shift in D2R sensitivity in the NAc could also be observed in recordings without cocaine present in the bath (Figure S2A–S2B). Repeated exposure to cocaine did not affect the maximum outward current generated by a saturating concentration of dopamine (300 μM) in either region, indicating that cocaine selectively reduces D2R sensitivity in a region-specific fashion (Fig. 1K). Both a lower dose of cocaine (10 mg/kg, i.p., 7 days treatment) and a 7-day exposure to amphetamine (2 mg/kg, i.p.) also reduced the sensitivity of D2Rs in the NAc (Fig. S2D–S2K).
As long-lasting neuroadaptations underlie the persistence of cocaine use and chronic relapse to cocaine seeking in addiction (Bossert et al., 2013; Kalivas, 2004), we hypothesized that the cocaine experience-elicited change in NAc D2R sensitivity might be engaged during the sensitized behavioral response to cocaine during drug re-exposure in cocaine-experienced mice. While we found that the reduced sensitivity of D2Rs in the NAc had returned to basal levels following 14 days of withdrawal from cocaine (Fig. 1F–K), a single challenge injection of cocaine that was sufficient to drive a robust locomotor response (Fig. 1E), was again able to decrease the sensitivity of D2Rs selectively in the NAc (Fig. 1F–K). Taken together these results suggest that cocaine exposure selectively exerts a transient reduction in the sensitivity of D2Rs in the NAc that can be reinstated by re-exposure to cocaine following prolonged withdrawal.
Altering D2R levels does not prevent cocaine-induced changes in sensitivity
Chronic, long-term self-administration of cocaine over prolonged periods is associated with decreased D2R availability and expression (Besson et al., 2013; Conrad et al., 2010; Moore et al., 1998; Nader et al., 2002, 2006; Volkow et al., 2009). To determine whether the decrease in NAc D2R sensitivity following only 7d of cocaine exposure resulted from altered receptor levels, we examined the global expression of D2Rs in the DSt and NAc. Cocaine-treated mice however exhibited similar total levels of expression of D2Rs both in DSt and NAc compared to controls (Fig. 2A–B) as previously observed (Nader et al., 2002), suggesting that the initial effects of cocaine on D2R signaling sensitivity may be independent of changes in expression.
Figure 2. Effect of altering D2R expression on cocaine-induced changes in D2R sensitivity.
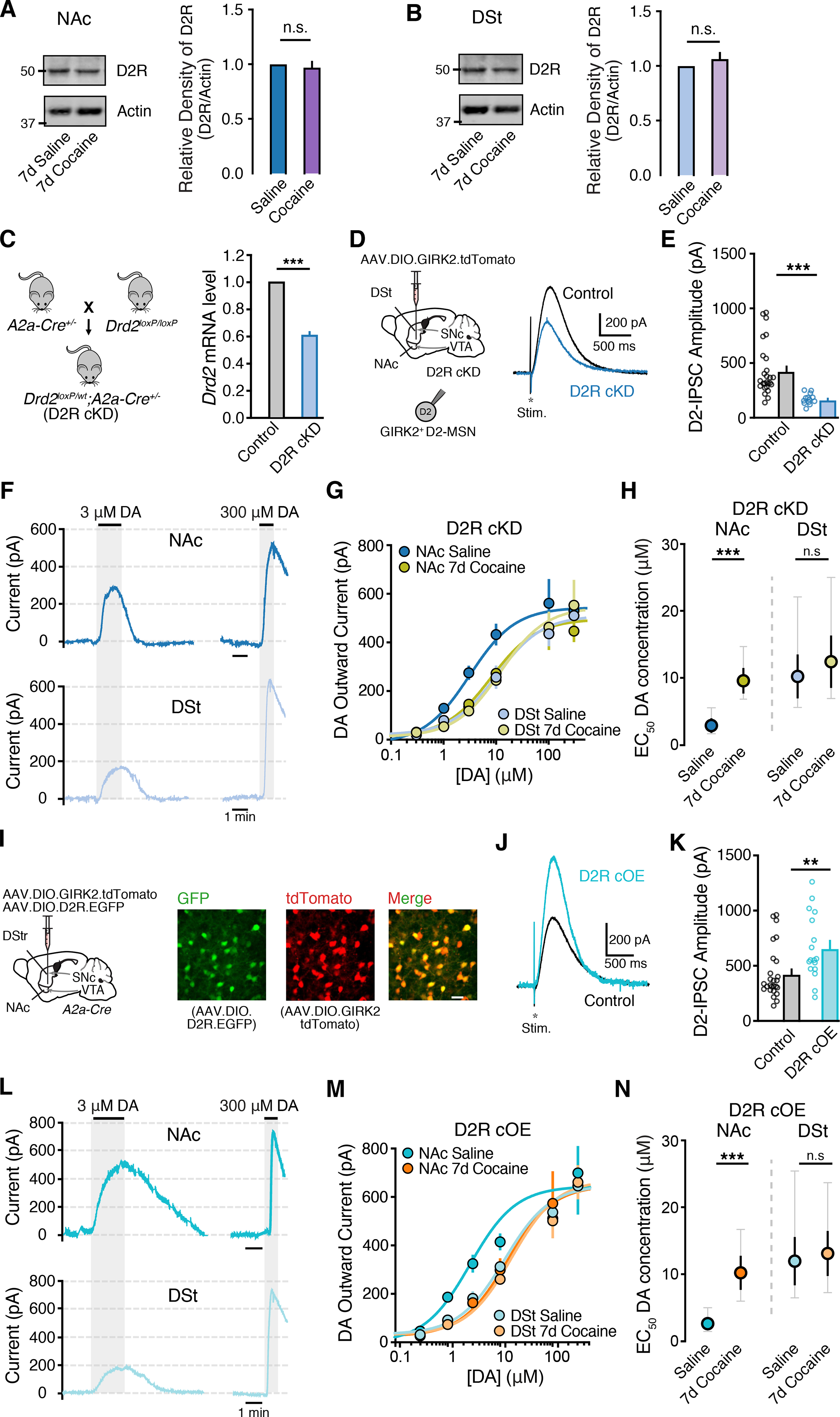
A-B - Representative immunoblot images and quantification of D2R expression levels in the NAc and DSt comparing 7d saline and 7d cocaine (20 mg / kg) treated mice (Mann Whitney test).
C - Generation of D2R conditional knockdown (cKD) mice (Drd2flx/wt; A2a-Cre+/−) and qPCR Drd2 mRNA levels from control (Drd2flx/flx) and D2R cKD groups.
D - Representative D2-IPSCs from GIRK2+ D2-MSNs from control and D2R cKD groups.
E - D2-IPSC amplitudes.
F - Representative traces from D2R cKD D2-MSNs in the NAc and DSt following bath application of dopamine. All recordings were performed in the presence of cocaine (10 μM).
G - Dopamine D2R dose response curves.
H - EC50 values from (G) (two tailed unpaired t-test).
I - Illustration of viral overexpression of AAV9.EF1a.DIO.D2L.P2A.EGFP + AAV.GIRK2.tdTomato to overexpress D2Rs (cOE) and immunofluorescence of eGFP, tdTomato and merged images.
J - Representative D2-IPSCs from control and D2R cOE groups.
K - D2-IPSC amplitudes.
L - Representative traces from D2R cOE D2-MSNs in the NAc and DSt following bath application of dopamine.
M - Dopamine D2R dose response curves.
N - EC50 values from (M).
Summary data are mean ± SEM (grey traces represent 95% confidence intervals). ns = p > 0.05, ** = p < 0.01, *** = p < 0.001. See also Figure S3 and Table S1.
To further examine the potential role of altered D2R expression in mediating the cocaine-induced change in D2R sensitivity, we next proceeded to alter D2R expression levels across the striatum to see if decreasing or increasing receptor levels could occlude the effect of cocaine. We first generated mice where D2R levels were selectively knocked down in D2-MSNs. Adora2a-Cre (A2a-Cre) mice were crossed with D2R floxed mice and the resulting D2R conditional knockdown animals (D2R cKD; A2a-cre+/−; Drd2flx/wt) were injected with a Cre-dependent AAV expressing GIRK2 (AAV.DIO.GIRK2) (Fig. 2C). Striatal immunoblot and qPCR analysis confirmed that D2R protein and mRNA (Drd2) expression was reduced in conditional knockdown animals compared to littermate controls (A2a-cre−/−; Drd2flx/wt) (Fig. 2C and Fig. S3A), as was the amplitude of evoked D2-IPSCs (Fig. 2D–E) and outward currents evoked by a saturating concentration of dopamine (300 μM) (Fig. S3B). D2R cKD mice were then subjected to the cocaine sensitization regimen (20 mg/kg, i.p., 7 d). Concentration-response relationships revealed that despite D2R levels being reduced in these animals, chronic exposure to cocaine was still able to induce a decrease in D2R sensitivity in the NAc while having no effect in the DSt (Fig. 2F–2H). The reduction in sensitivity in the NAc of D2R cKD mice was similar to that observed in the NAc of control WT mice following chronic cocaine.
Next, we examined the effect of increasing D2R expression. We selectively overexpressed D2Rs in striatal D2-MSNs (D2R cOE) by injecting AAV.DIO.D2R.EGFP (Gallo et al., 2015) together with AAV.DIO.GIRK2.tdTomato in A2a-Cre mice. Three weeks following, widespread eGFP and tdTomato fluorescence could be observed in overlapping populations of DSt and NAc neurons (Fig. 2I). Recordings from tdTomato+ and EGFP+ D2-MSNs revealed that the amplitude of D2-IPSCs and the maximum outward current evoked by dopamine (300 μM) were increased in D2-MSNs from D2 cOE mice compared to control animals (Fig. 2J–M and Fig. S3C). However, despite D2R levels being enhanced, we again found that chronic cocaine injections produced a similar selective reduction in D2R sensitivity in the NAc as in control animals (Fig. 2L–N). Together these findings indicate that altering the levels of D2Rs in D2-MSNs does not occlude the cocaine-induced region-specific shift in D2R sensitivity.
Changes in G-protein expression underlie the reduction of D2R sensitivity following cocaine exposure
While the D2R couples to both pertussis toxin sensitive G-protein subunits Gαo and Gαi, dopamine more potently signals via the D2R through Gαo than Gαi (Gazi et al., 2003; Lane et al., 2007; Marcott et al., 2018). Because increased expression and preferential coupling to Gαo underlie the region-specific difference in D2R sensitivity between the DSt and NAc (Marcott et al., 2018) and cocaine exposure regulates the level of Gα proteins within the striatum (Nestler et al., 1990; Striplin and Kalivas, 1993), we next examined whether the underlying mechanism by which repeated cocaine exposure decreases the sensitivity of D2Rs selectively in the NAc is by differentially regulating Gα protein levels in this region. We first measured different Gα protein levels using quantitative western blotting. Gαo protein levels were higher in the NAc than the DSt of saline-treated mice, while Gαi levels were similar between subregions (Fig. 3A–B). Brain punches of the DSt and NAc were then taken 24hr post-injection from saline-treated mice, acute, chronic (7 d), withdrawn (14 d), and cocaine challenged-treated mice (Fig 3C). While acute cocaine did not affect Gαo and Gαi levels in either region, chronic cocaine exposure selectively reduced Gαo protein levels in the NAc, without changing Gαo in the DSt (Fig. 3D–E). Similar to the cocaine-induced changes in D2R sensitivity, we also found that while 14 days of withdrawal from cocaine returned Gαo levels in the NAc to basal control levels, a subsequent challenge of cocaine again selectively reduced Gαo protein levels in the NAc (Fig. 3D). Chronic cocaine exposure, withdrawal and a cocaine challenge had no effect on Gαi protein expression levels in either the DSt or NAc (Fig. 3D–E). Thus, Gαo expression is subject to dynamic regulation in the NAc following cocaine exposure and withdrawal.
Figure 3. Cocaine-induced alterations in G-protein levels.
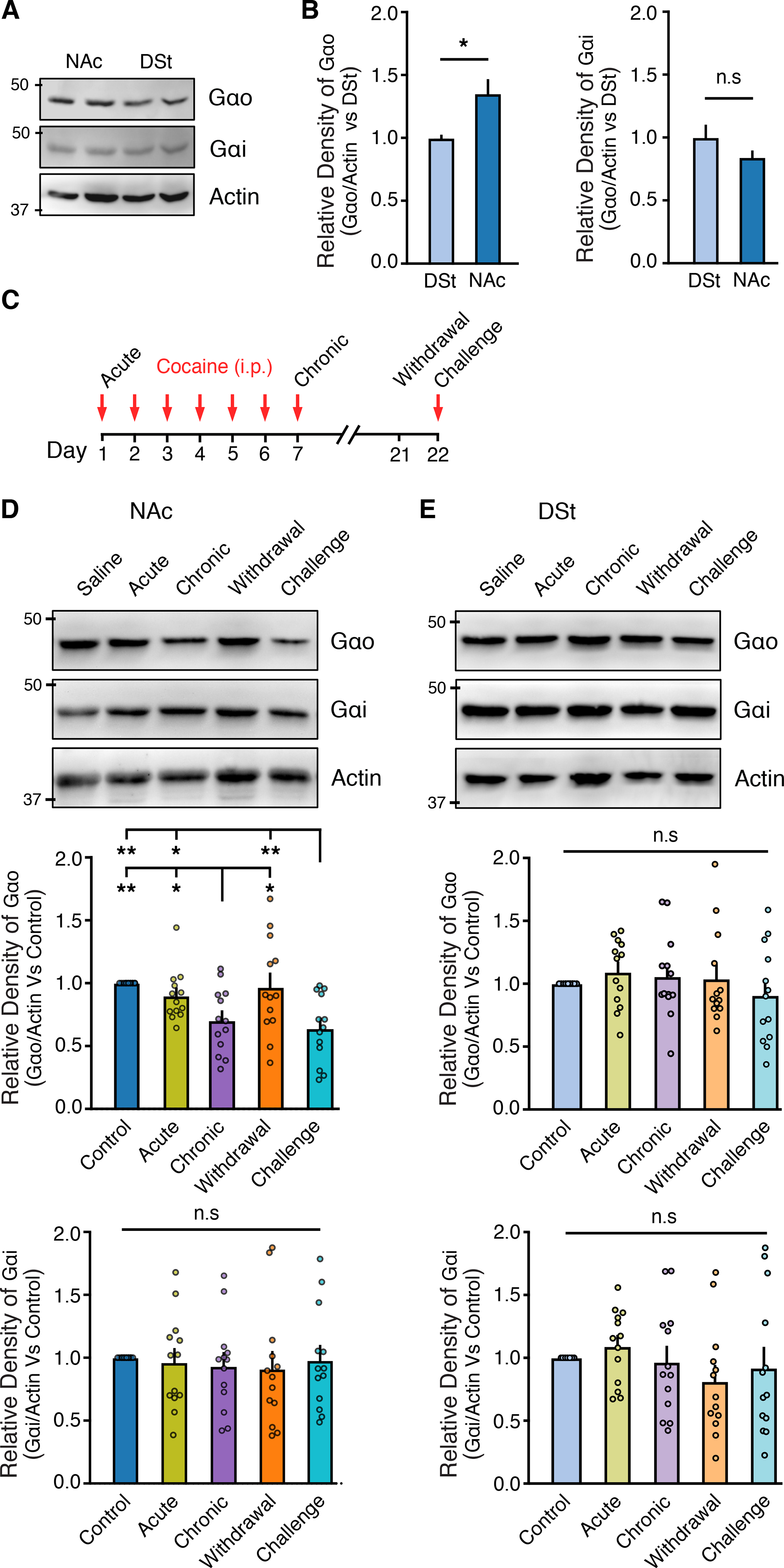
A - Representative western immunoblot from NAc and DSt examining of Gαo and Gαi expression levels.
B - Quantification of Gαo and Gαi expression levels, normalized to saline control in the NAc and DSt (Mann Whitney test).
C - Timeline of experiments. Tissue was taken 24 hrs following the last injection.
D - Representative western immunoblot from NAc and quantification of Gαo and Gαi expression levels, normalized to saline control (RM one-way ANOVA; Tukey’s post-test).
E - Representative western immunoblot from DSt and quantification of Gαo and Gαi expression levels, normalized to saline control.
Summary data are mean ± SEM. ns = p > 0.05, * = p < 0.05, ** = p < 0.01. See also Table S1.
We next tested whether the cocaine-induced alterations in Gαo levels could explain the reduction in D2R sensitivity in cocaine-experienced mice. We first sought to knockdown the expression of Gαo in D2-MSNs. Gαo-floxed mice were crossed with A2a-cre mice and the resulting D2-MSN Gαo conditional knockdown mice (Gαo cKD; Gαoflx/wt; A2a-cre+/−) were injected with AAV.DIO.GIRK2.tdTomato (Fig. 4A–B). Immunoblot examination revealed a ~40 % decrease of Gαo protein level while Gαi levels remained unchanged (Fig. 4A), ruling out compensatory upregulation of Gαi. We found that the reduction of Gαo was sufficient to lower the maximum outward current evoked by dopamine (300 μM) in both the NAc and DSt (Fig. 4C–E and Fig. S4A) and in addition led to a selective reduction in the sensitivity of D2Rs in the NAc, as could be seen by the rightward shift in the concentration-response curve (Fig. 4D). Importantly, we furthermore found that chronic cocaine administration was now no longer able to evoke a subsequent decrease in D2R sensitivity in the NAc in these Gαo cKD mice. Thus, dopamine EC50 values were similar in the NAc between naïve and cocaine-treated Gαo cKD mice (Fig. 4D). Similar results were obtained by virally expressing Cre in the NAc or DSt of Gαo-floxed mice (Fig. S4C–S4G).
Figure 4. Altering the expression of Gαo prevents cocaine-induced changes in D2R sensitivity.
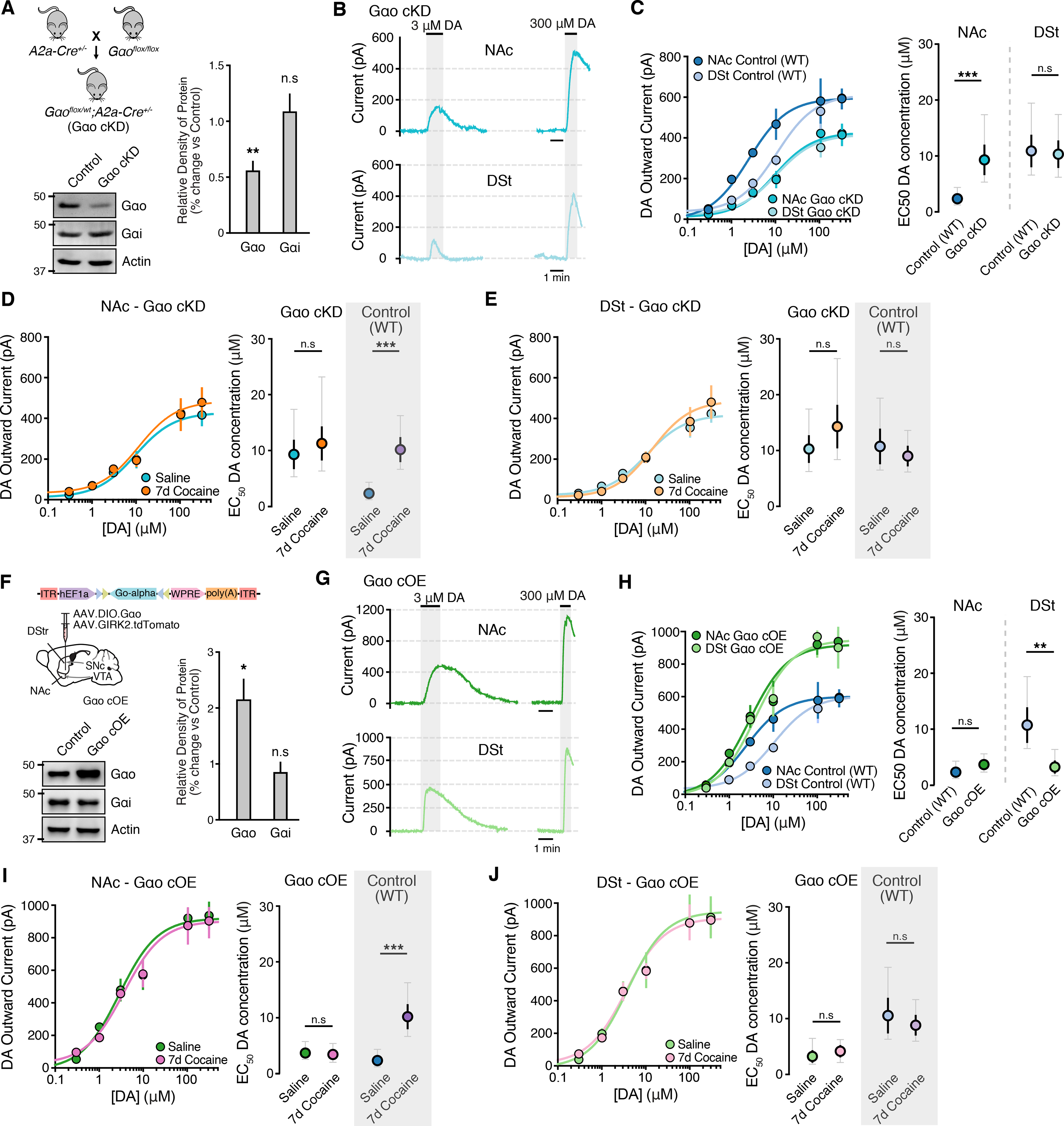
A - Generation of the Gαo conditional knockdown (cKD) group (Gαoflx/wt; A2a-Cre+/−) and representative western immunoblot and quantification of Gαo and Gαi levels (Mann Whitney test).
B - Representative traces from Gαo cKD D2-MSNs in the NAc and DSt following bath application of dopamine. All recordings were performed in the presence of cocaine (10 μM).
C - Dopamine D2R dose response curves and EC50 values from control (WT) and Gαo cKD groups (two tailed unpaired t-test).
D - E - Dopamine D2R dose response curves and EC50 values from NAc and DSt D2-MSNs from saline and 7-day cocaine treated Gαo cKD groups. Control data from wildtype mice following 7-day saline and 7-day cocaine treatment taken from Figure 1 is reshown in boxes in grey.
F - Illustration of injection of AAV9.EF1a.DIO.Gαo + AAV.GIRK2.tdTomato in the NAc and DSt of Gαo cKD animals to overexpress Gαo (cOE) and representative western immunoblot and quantification of Gαo and Gαi levels.
G - Representative traces from Gαo cOE D2-MSNs in the NAc and DSt following bath application of dopamine.
H - Dopamine D2R dose response curves and EC50 values from control (WT) and Gαo cOE groups.
I - J - Dopamine D2R dose response curves and EC50 values from NAc and DSt D2-MSNs from saline and 7-day cocaine treated Gαo OE groups.
Summary data are mean ± SEM (grey traces represent 95% confidence intervals). ns = p > 0.05, ** = p < 0.01, *** = p < 0.001. See also Figure S4 and Table S1.
To confirm the role of Gαo, we next used a viral strategy to overexpress Gαo in Gαo cKD mice to attempt to rescue the loss of Gαo. Three weeks following injection of AAV.DIO.Gαo into the NAc and DSt of Gαo cKD mice western blot analysis revealed ~two-fold increase of Gαo protein levels while Gαi remained unchanged (Fig. 4F). Notably, overexpressing Gαo in the NAc of Gαo cKD mice rescued the reduced D2R sensitivity and further abolished subsequent cocaine-evoked changes (Fig. 4G–I). D2-MSNs over-expressing Gαo in the DSt exhibited an increase in D2R sensitivity, that was still unaffected by cocaine exposure (Fig. 4G–H, J). Maximum outward currents in both regions were also significantly higher than control mice (Fig. S4B). Similar results were obtained by virally overexpressing Gαo in control non-Cre expressing Gαo-floxed mice (Fig. S4H–S4L). Together, with the above results, these findings support the idea that chronic exposure to cocaine initially regulates the sensitivity of D2R signaling by altering the preferential coupling to G-proteins rather than changing the overall level of D2R expression.
Cocaine-induced changes in D2-MSN axon collateral D2R sensitivity
D2-MSNs form functional collateral synapses with neighboring D1-MSNs (Taverna, 2003; Tecuapetla et al., 2009). The inhibition of GABA release from D2-MSNs at these local connections by activation of presynaptic D2Rs (Dobbs et al., 2016; Lalchandani et al., 2013; Marcott et al., 2018; Taverna, 2003; Tecuapetla et al., 2009) contributes to the acute behavioral responses to cocaine (Dobbs et al., 2016). As our results so far examined postsynaptic dendritic D2R signaling in D2-MSNs via exogenously expressed GIRK2 channels, we next wanted to examine if cocaine similarly regulates the sensitivity of D2Rs in D2-MSNs that signal via endogenous pathways to inhibit GABA release. The NAc and DSt of A2a-Cre mice were injected with AAV.DIO.ChR2.eYFP and three weeks later voltage-clamp recordings were made from the neighboring non-fluorescent, putative D1-MSNs (Fig. 5A–B). Photoactivation of D2-MSNs (470 nm, 1 ms) evoked picrotoxin-sensitive (100 μM) GABAA-receptor mediated inhibitory postsynaptic currents (IPSCs) in putative D1-MSNs (Fig. 5B). Optically-evoked GABAA-IPSCs (oIPSCs) were inhibited by bath application of the D2R agonist quinpirole (1 μM) (Fig. S5A, S5C). Dopamine concentration-response curves confirmed that D2R mediated inhibition of local GABA release was substantially more sensitive in the NAc compared to DSt, and similar in value to the EC50 concentrations of dopamine required to activate postsynaptic D2R mediated GIRK2 currents (Fig. 5C–E). Importantly we also found that chronic cocaine exposure produced a similar rightward shift in the NAc, but not DSt, concentration-response curve which corresponded to a decrease in IC50 value selectively in the NAc (Fig. 5C–E). Thus, D2Rs on D2-MSN axon collaterals exhibit region-specific differences in dopamine sensitivity and the sensitivity in the NAc is selectively weakened by chronic cocaine exposure.
Figure 5. Altering the expression of Gαo blocks cocaine-induced changes in sensitivity of axonal D2R regulating collateral transmission.
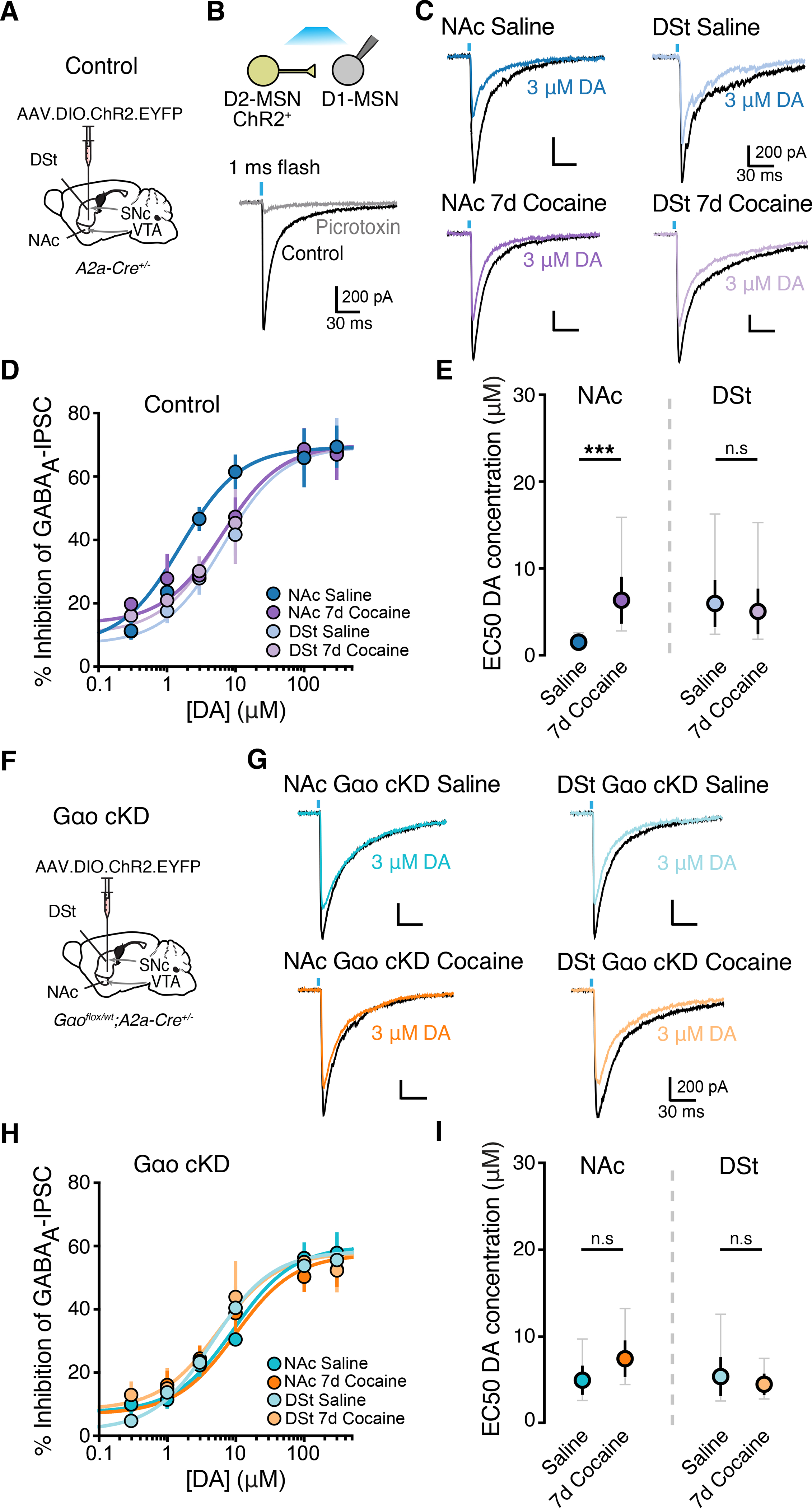
A - Illustration of injection of AAV.EF1a.DIO.hChR2(H134R)-EYFP in A2a-Cre+/− mice.
B - Schematic of experimental condition of expression of ChR2 in D2-MSNs and representative trace of evoked GABAA IPSCs and block by picrotoxin (100 μM) in a D1-MSN following photoactivation of D2-MSNs.
C - Inhibition of D1-MSN GABAA IPSCs recorded in the NAc and DSt of saline or 7-day cocaine treated animals following bath application of dopamine. All recordings were performed in the presence of cocaine (10 μM).
D - Dopamine D2R dose response curves of the inhibition of GABAA IPSCs.
E - IC50 values from (D) (two tailed unpaired t-test).
F - Illustration of injection of AAV5.EF1a.DIO.hChR2(H134R)-EYFP in Gαo cKD mice (Gαoflx/wt; A2a-Cre+/−).
G - Inhibition of D1-MSN GABAA IPSCs from Gαo cKD in the NAc and DSt of saline or 7-day cocaine treated animals following bath application of dopamine.
H - Dopamine D2R dose response curves of the inhibition of GABAA IPSCs in Gαo cKD groups.
I - IC50 values from (H).
Summary data are mean ± SEM (grey traces represent 95% confidence intervals). ns = p > 0.05, *** = p < 0.001. See also Figure S5 and Table S1.
To determine if the cocaine-induced reduction in presynaptic D2R sensitivity on D2-MSN terminals was also mediated by a reduction in coupling to Gαo, we repeated these experiments using Gαo cKD mice (Fig. 5F). While Gαo reduction had no effect on basal oIPSCs, the quinpirole mediated inhibition was ~ 20 % smaller in D1-MSNs from Gαo cKD mice compared to control animals (Fig. S5B–S5C). Consistent with the change in D2R sensitivity caused by Gαo deletion, NAc axonal D2R collateral transmission had lower sensitivity for dopamine in Gαo cKD mice, reflected by an increase in the IC50 (Fig. 5G–I). Furthermore, the cocaine-evoked increase in the IC50 was occluded in NAc slices obtained from cocaine exposed Gαo cKD mice (Fig. 5G–I). These results collectively support the conclusion that cocaine-induced adaptation in D2 receptor signaling throughout D2-MSNs is linked to changes in Gαo expression.
Alterations in D2R sensitivity affect context-driven cocaine seeking
Because cocaine-dependent changes in synaptic Gαo expression in D2-MSNs modulate D2R sensitivity in a manner that might be expected to influence motivation and drug reinforcement, we next investigated whether the Gαo-dependent change in sensitivity would be critical for cocaine-conditioned behaviors. We first assessed cocaine conditioned place preference (CPP), during which mice learn to associate a previously neutral context with the drug (Fig. 6A). Consistent with the cocaine sensitization regimen, cocaine CPP also caused a marked reduction of Gαo in the NAc (Fig. 6B) with the extent of cocaine CPP being correlated with the reduction in Gαo. Compared to control animals (Gαoflx/wt; A2a-cre−/−), Gαo cKD mice (Gαoflx/wt; A2a-cre+/−) spent more time in the cocaine-paired chamber during the CPP test (Fig. 6C–D). Control and Gαo cKD mice exhibited similar basal locomotor activity and similar cocaine induced locomotion following a single acute dose of cocaine (Fig. S6A–S6B). To determine if the reduction of Gαo specifically in NAc D2-MSNs was responsible for the augmentation of conditioned preference, we again used the viral rescue strategy (AAV.DIO.Gαo) to re-express Gαo selectively in NAc D2-MSNs of Gαo cKD mice. This D2-MSN specific rescue manipulation in the NAc abolished cocaine CPP in Gαo cKD mice (Fig. 6C–D). To confirm these results, we also used an alternative viral approach to manipulate Gαo expression levels and similarly found that knocking down or overexpressing Gαo in a non-cell type specific manner in the NAc likewise increased or decreased CPP scores respectfully when compared to control animals injected with AAV.eGFP (Fig. 6E–F). Notably, neither Gαo knockdown nor overexpression in the DSt altered cocaine CPP (Fig. S6C–S6F).
Figure 6. Altering Gαo expression in D2-MSNs affects cocaine reward behaviors.
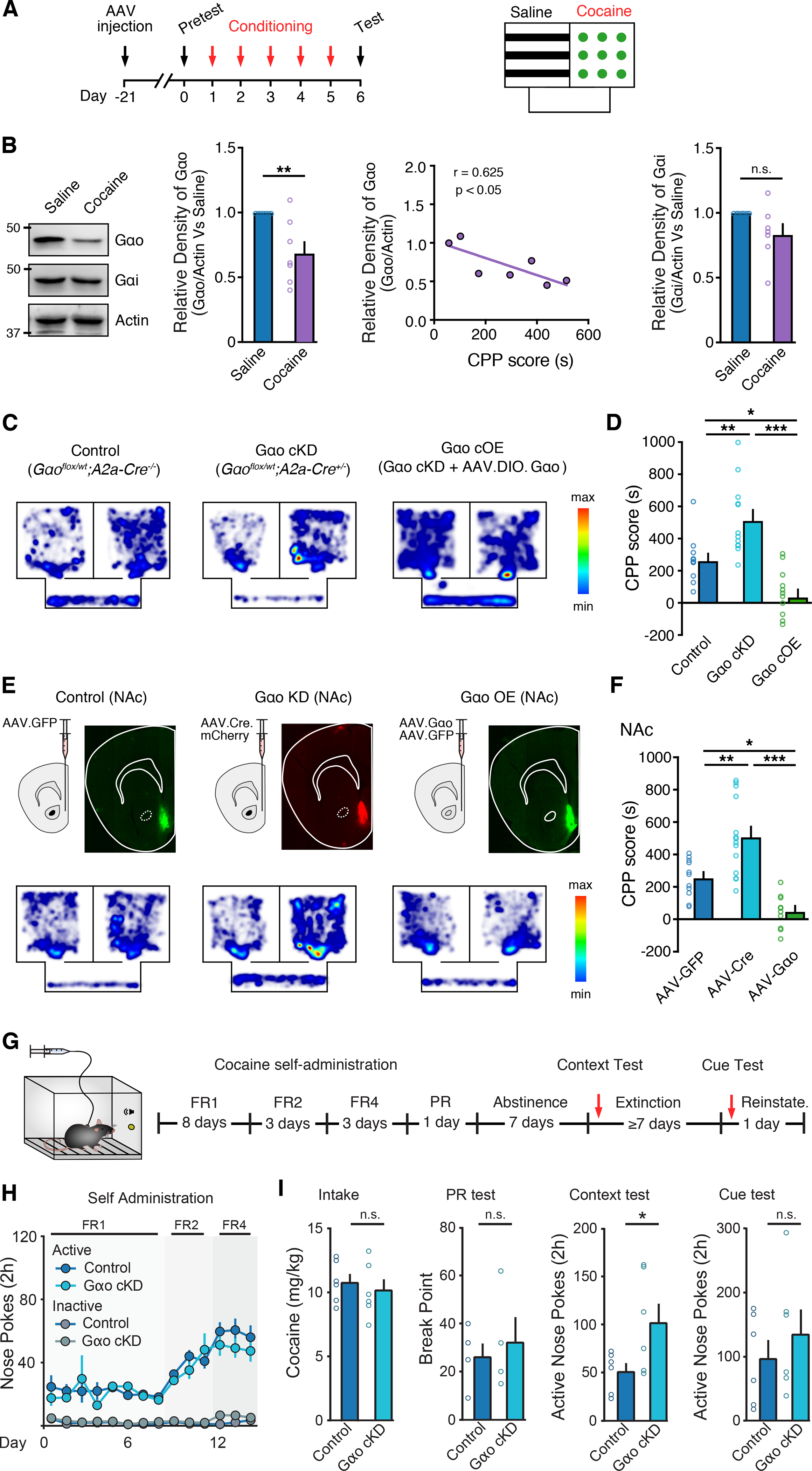
A - Timeline and illustration of cocaine CPP experimental design.
B – Representative western immunoblot, correlation of Gαo and cocaine CPP, and quantification of Gαo and Gαi levels following cocaine CPP from control (Gαoflox/wt;A2a-Cre−/−) mice (Mann-Whitney test).
C - Representative heat map illustrating time spent in chambers during CPP testing for control, Gαo cKD and Gαo cOE groups.
D - Quantification of CPP scores from each group (one-way ANOVA; Tukey’s post-test).
E - Representative GFP and mCherry fluorescence and heat map illustrating time spent in chambers during CPP testing for control, Gαo NAc KD and Gαo NAc OE groups.
F - Quantification of CPP scores from each group.
G - Timeline and illustration of cocaine IV self-administration experimental design.
H - Comparison of nose pokes for cocaine between control and Gαo cKD groups over time (mixed-model ANOVA).
I - Quantification of average daily cocaine intake (two tailed unpaired t-test), break point during PR (two tailed unpaired t-test), and active nose pokes during post-abstinence context and cue tests (two-way ANOVA reinstatement × genotype interaction) comparing between control and Gαo cKD groups.
Summary data are mean ± SEM. ns = p > 0.05, * = p < 0.05, ** = p < 0.01, *** = p < 0.001. See also Figure S6 and Table S1.
Next, we examined whether alterations in D2R sensitivity would interfere with cocaine reinforcement and cocaine seeking in a cocaine self-administration model. Gαo cKD and control mice were initially trained to self-administer cocaine for 8 days on a fixed-ratio (FR) 1 reinforcement schedule in the presence of response-contingent cues (tone/light) delivered during cocaine infusions. Next, mice transitioned to FR2 and FR4 to investigate whether Gαo cKD would affect responding for cocaine at increased effort requirements (Fig. 6G–H and Fig. S6G–S6H). Conditional knockdown of Gαo in D2-MSNs did not affect the acquisition of voluntary cocaine self-administration (Fig. 6H) and no differences were observed in average daily cocaine intake (Fig. 6I and Fig. S6K) or the motivation to take cocaine measured by a progressive ratio test (Fig. 6I). In addition, control and Gαo cKD mice exhibited similar rates of responding for cocaine in a dose-response study (Fig. S6L–S6M). However, given that reduction of Gαo in D2-MSNs in NAc increased cocaine-context mediated behavior in the CPP test, we also investigated the possibility that Gαo downregulation might specifically underlie cocaine-context or cocaine-cue associations. Following a 7-day forced abstinence session in their home cages, cocaine self-administering mice were returned to the cocaine self-administration chambers to perform a cocaine-context relapse test under extinction conditions followed by additional extinction training and a cue test ≥ 7 days later (Fig. 6I). We found that while conditionally knocking down Gαo in D2-MSNs increased drug seeking after abstinence (Fig. 6I), it did not affect cue-induced reinstatement of drug seeking (Fig. 6I and Fig. S6N–S6P). Collectively, these data suggest that the cocaine-induced reduction in NAc Gαo expression levels and associated decrease in the sensitivity of D2Rs regulates learned associations between cocaine reward and cocaine-associated contexts.
Cocaine reduces NAc D2R sensitivity independently of D2Rs
The enhancement of dopamine transmission by cocaine drives circuit and synaptic changes in the NAc, which ultimately contribute to the development of addiction (Kalivas, 2004; Pascoli et al., 2015). We examined whether cocaine-induced reductions in D2R sensitivity resulted from increased D2R activation on D2-MSNs or whether it resulted from circuit adaptations. We first tested whether other manipulations that facilitate extracellular dopamine levels or increase tonic D2R activation could mimic the effect of cocaine. To increase dopamine synthesis, wildtype mice were treated with the dopamine precursor levodopa (L-DOPA; 100 mg/kg) (Fig. 7A–B). However, a 7-day treatment with L-DOPA had no effect on D2R-mediated GIRK2 dopamine concentration-response relationships in either the NAc or DSt (Fig. 7A–C and Fig. S7A–S7D). Likewise, a 7-day treatment with the D2R agonist quinpirole (5 mg/kg, ip, repeated 7d) also failed to alter dopamine D2R dose response curves (Fig. 7c–e and Fig. S7A–S7C). Thus, chronic pharmacological activation of D2Rs was not sufficient to recapitulate the suppression of D2R sensitivity in the NAc induced by cocaine. Furthermore, pretreatment with the D2R antagonist haloperidol (3 mg/kg) 30 min before cocaine injections failed to prevent the reduction of D2R sensitivity in the NAc (Fig. 7A–C). This suggest that alterations in the sensitivity of D2Rs driven by chronic cocaine exposure are unlikely due to cell-autonomous changes resulting from increased D2R activation.
Figure 7. Cocaine-induced decrease of D2R sensitivity in the NAc is dependent on D1R and NMDAR activation.
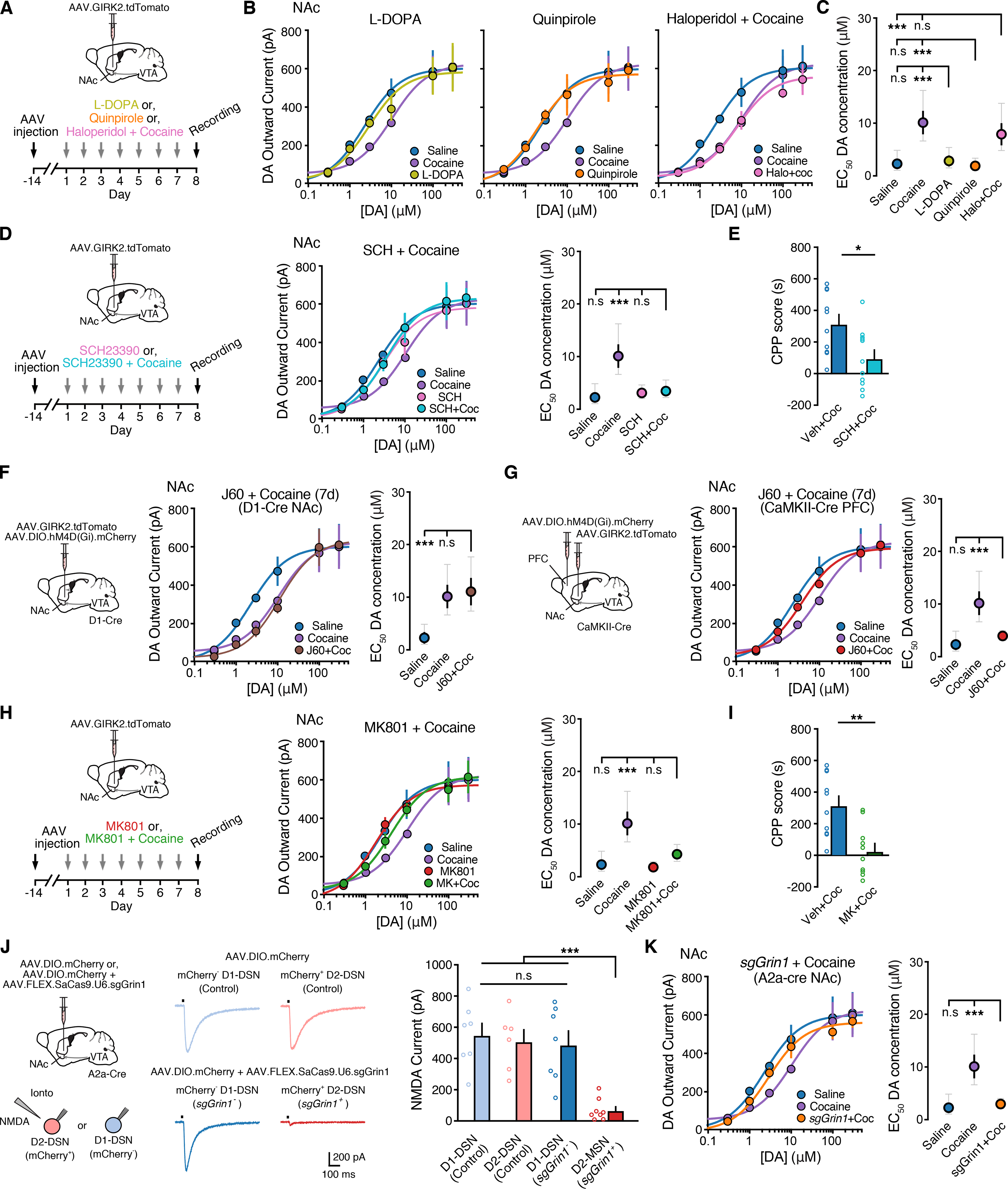
A - Timeline of L-DOPA (100 mg/kg; i.p., 7 days), quinpirole (5 mg/kg; i.p. 7 days) and cocaine (20 mg/kg; i.p.; 7 days) preceded by a haloperidol (3 mg/kg, i.p. 7 days) groups.
B - Dopamine D2R dose response curves from NAc D2-MSNs comparing 7-day treatment of saline and cocaine with L-DOPA (left), quinpirole (middle) and cocaine preceded by a haloperidol (right) pretreatment.
C - EC50 values from (C) (one-way ANOVA; Sidak’s post-test).
D - Dopamine D2R dose response curves and EC50 values from NAc D2-MSNs comparing 7-day treatment of saline and cocaine (20 mg/kg) with cocaine preceded by SCH23390 (0.3 mg/kg, i.p. 7 days) pretreatment.
E - Quantification of cocaine CPP scores from vehicle (saline) and SCH23390 pretreated mice (Mann Whitney test).
F - Illustration of injection of AAV.DIO.hM4D(Gi).mCherry and AAV.GIRK2.tdTomato into the NAc in D1-Cre mice, timeline of cocaine (20 mg/kg; i.p.; 7 days) preceded by JHU37160 (1mg/kg, i.p. 7 days) groups and resulting dopamine D2R dose response curves and EC50 values from NAc D2-MSNs.
G - Illustration of injection of AAV.DIO.hM4D(Gi).mCherry into the PFC and AAV.GIRK2.tdTomato into the NAc in CaMKII-Cre mice, timeline of cocaine preceded by JHU37160 groups and resulting dopamine D2R dose response curves and EC50 values from NAc D2-MSNs.
H - Dopamine D2R dose response curves and EC50 values from NAc D2-MSNs comparing 7-day treatment of saline and cocaine (20 mg/kg; i.p.; 7 days) with cocaine preceded by a MK801 (0.3 mg/kg, i.p. 7 days) pretreatment.
I - Quantification of cocaine CPP scores from vehicle (saline) and MK801 pretreated mice.
J - Left: Illustration of injection of AAV.DIO.mCherry and AAV.DIO.mCherry + AAV.FLEX.SaCas9.U6.sgGrin1 into the NAc of A2a-Cre mice. Right: Representative traces illustration NMDA currents evoked by iontophoretic application of exogenous NMDA (200 mM) from tdTomato− and tdTomato+ putative D1- and D2-MSNs and quantification of NMDA currents (RM one-way ANOVA; Tukey’s post-test).
K - Dopamine D2R dose response curves and EC50 values from NAc D2-MSNs comparing 7-day treatment of saline and cocaine (20 mg/kg; i.p.; 7 days) treated mice with cocaine treated mice expressing sgGrin1.
Summary data are mean ± SEM (grey traces represent 95% confidence intervals). ns = p > 0.05, * = p < 0.05, ** = p < 0.01, *** = p < 0.001. See also Figure S7 and Table S1.
The cocaine-induced reduction in NAc D2Rs sensitivity however could be blocked by pretreatment with the D1-receptor antagonist SCH23390 (0.3 mg/kg) (Fig. 7D and Fig. S7E). As noted previously, SCH23390 also blocked cocaine CPP (Fig. 7E) (Cervo and Samanin, 1995). We set out to determine where within accumbens circuitry these receptors are being engaged. Two major populations of D1-receptor expressing neurons that could regulate D2-MSNs could be D1-MSNs and excitatory inputs from the prefrontal cortex (PFC). A chemogenetic approach was taken to examine the contribution of these two inputs. To inhibit D1-MSNs, the NAc of D1-Cre mice was injected with AAV.DIO.hM4D(Gi).mCherry and three weeks later mice were pre-treated with the DREADD agonist (JHU37160 (J60), 1mg/kg) 30 min prior to cocaine injections (Fig. 7E). Activation of the inhibitory DREADD in D1-MSNs, however had no effect on cocaine-induced D2R sensitivity in the NAc (Fig. 7E). In contrast chemogenetic inhibition of the PFC, following injection of AAV.DIO.hM4D(Gi).mCherry into the PFC of CaMKII-Cre mice and pre-treatment with J60 (1mg/kg), blocked the cocaine-induced reduction in NAc D2R sensitivity in D2-MSNs (Fig. 7F). Thus, activation of D1-receptors on PFC inputs rather than D1-MSNs may mediate the cocaine-induced alteration in D2R sensitivity.
As cocaine induces well known NMDA-receptor dependent glutamatergic excitatory plasticity in the NAc (Kalivas et al., 2005), we found that the cocaine-induced reduction in NAc D2Rs sensitivity could also be blocked by pretreatment with the NMDA-receptor blocker MK-801 (0.3 mg/kg) (Fig. 7G and Fig. S7F). MK-801 also blocked cocaine CPP (Fig. 7I) (Cervo and Samanin, 1995; Kim et al., 1996). To determine if NMDA-receptor dependent activity on D2-MSNs is required for the effect of cocaine, a single Cre-dependent CRISPR/SaCas9 vector was used to genetically inactivate Grin1 (Hunker et al., 2020), the essential NR1 subunit of the NMDA receptor (Fig. 7H). Co-injection of AAV.FLEX.SaCas9.U6.sgGrin1and AAV.DIO.GIRK2.tdTomato in the NAc of A2a-Cre mice dramatically reduced the amplitude of NMDA receptor-mediated inward currents evoked by the exogenous application of NMDA (Fig. 7J) as well as evoked EPSCs in tdTomato+ (sgGrin1+) D2-MSNs (Fig. S7F). Furthermore, we found that inactivating NMDA-receptors in D2-MSNs prevented the cocaine-induced decrease in D2R sensitivity of NAc D2-MSNs (Fig. 7K). Inactivating Grin1 in D1-MSNs by injecting AAV.FLEX.SaCas9.U6.sgGrin1 in D1-Cre mice did not affect D2R sensitivity (Fig. 7K) despite successfully reducing NMDA-currents (Fig. 7J) and NMDA-mediated EPSCs in D1-MSNs (Fig. S7G). Together these results suggest that cocaine-induced changes in D2R sensitivity may be driven by altered glutamatergic transmission and NMDA receptor signaling at prefrontal cortical to D2-MSN synapses.
DISCUSSION
While cocaine-induced alterations in D2-MSN activity and connected circuits are well known (Lobo and Nestler, 2011; Lüscher et al., 2020), direct changes in D2R function and the contribution they may have in regulating drug-associated motivated behaviors have remained largely unclear. The present findings identify that rather than altering expression, an initial adaptation resulting from cocaine exposure is a selective decrease in the sensitivity of D2Rs in the NAc. We found that this decrease in D2R sensitivity facilitated subsequent development of context-driven drug seeking, thus constituting an initial adaptation regulating cocaine-conditioned reward.
Striatal D2Rs have long been implicated in regulating motivation and reinforcement, and alterations in D2R availability and signaling are associated with chronic drug use (Volkow and Morales, 2015). Neuroimaging studies in human, nonhuman primates and rodents have found that low D2R availability is associated with impulsivity and susceptibility to self-administer cocaine (Dalley et al., 2007; Morgan et al., 2002; Nader et al., 2006; Volkow et al., 1990, 1993, 1997). As decreases in D2R expression have been linked to aberrant drug-seeking behaviors (Bello et al., 2011; Bocarsly et al., 2019; Caine et al., 2002; Dobbs et al., 2016, 2019; Nader et al., 2006) and chronic cocaine self-administration leads to a reduction in D2R availability (Dalley et al., 2007; Morgan et al., 2002; Nader et al., 2006; Volkow et al., 1990, 1993, 1997), D2R density, mRNA, and surface expression (Besson et al., 2013; Conrad et al., 2010; Moore et al., 1998; Nader et al., 2002), a decrease in D2R expression is thought to be an important consequence of prolonged cocaine intake that subsequently regulates persistent cocaine use and relapse. However, as we and others found that neither D2R expression nor density (Nader et al., 2002) is altered by brief exposure to cocaine, it has remained unclear what adaptations in D2R function may mediate the initial effects of cocaine reinforcement. Here we found, using both a postsynaptic read-out of dendritic D2Rs activation and a presynaptic measure of axonal D2Rs function, that in vivo cocaine exposure evoked a decrease in sensitivity of dopamine-mediated D2R signaling specifically in the NAc. While this transient decrease did not persist following 14d of abstinence, a single subsequent exposure to cocaine in cocaine-experienced mice was sufficient to rapidly again reduce D2R sensitivity. As the decrease in D2R sensitivity was not mimicked or occluded by conditionally knocking down or overexpressing D2Rs, our results suggest that a reduction in D2R signaling is an initial adaptation preceding changes in expression which is rapidly engaged upon re-exposure to psychostimulants.
Rather than a change in D2R levels, we found that altered expression of G-proteins accounted for the cocaine-induced decrease in NAc D2R sensitivity. Reduced dopamine receptor signaling has been proposed to be an important neuroadaptation in cocaine addiction (Morgan et al., 2002; Park et al., 2013; Volkow et al., 2014), and chronic cocaine administration induces multiple adaptations that can alter the efficacy of dopamine receptor signaling including changes in regulators and activators of G-protein signaling (RGS and AGS proteins), β-arrestin2, G-protein receptor kinases (GRK) and Gα protein expression (Bowers et al., 2004; Daigle et al., 2014; Gaval-Cruz et al., 2016; Labouèbe et al., 2007; Nestler et al., 1990; Padgett et al., 2012; Porter-Stransky et al., 2019; Rahman et al., 2003; Schroeder et al., 2009; Striplin and Kalivas, 1993). We previously uncovered that region-specific differences between the NAc and DSt in the efficacy of dopamine signaling through D2Rs were not due to differences in RGS7/9 nor GRK2/3, but were the result of preferential coupling to Gαo over Gαi in NAc compared to DSt, as D2Rs have a higher sensitivity for dopamine when coupled to Gαo over Gαi and Gαo is more abundant in the NAc (Gazi et al., 2003; Lane et al., 2007; Marcott et al., 2018). As different GPCRs can have distinct profiles by which they engage different G-proteins (Masuho et al., 2015), this region specific difference in D2R sensitivity was not shared by several other GPCRs including opioid or muscarinic receptors (Marcott et al., 2018). We found that the NAc specific changes in D2R sensitivity following cocaine sensitization, abstinence, cocaine challenge and cocaine-induced CPP were paralleled by alterations in the expression of Gαo specifically in the NAc. We further showed that changes in Gαo expression underlie the reduction in D2R sensitivity as selectively knocking down or overexpressing Gαo in D2-MSNs regulated the intrinsic sensitivity of D2Rs and occluded or prevented subsequent cocaine-induced changes in D2R sensitivity. As we found that Gαo levels were ~40% higher in the NAc than DSt and that cocaine decreased Gαo expression in the NAc by ~30%, the results suggest that D2R sensitivity within striatal and accumbens circuits is regulated by a fairly narrow range of Gαo expression levels. This may explain why viral overexpression of Gαo, which more than doubled Gαo levels, decreased the sensitivity of D2Rs in the DSt, but prevented a further decrease in the NAc as well as occluded the effect of cocaine. While our data are in agreement with previous studies showing a transient decrease in Gαo in response to repeated cocaine treatment we did not observe any change in Gαi levels as previously shown (Nestler et al., 1990; Striplin and Kalivas, 1993). Several possible factors likely contribute including differences in the duration of cocaine exposure, species, and sub-region examined.
Within striatal and accumbens circuits D1-MSNs have been classically associated with positive reinforcement and reward, while D2-MSNs are associated with suppression of appetitive motivation or negative reinforcement (Bock et al., 2013; Hikida et al., 2010; Kravitz et al., 2012; Lobo et al., 2010). In line with these observations, optogenetic activation of NAc D2-MSNs has been found to attenuate cocaine CPP and cocaine self-administration, while ablation or chemogenetic inhibition of D2-MSNs can enhance psychostimulant CPP and reinstatement of cocaine-seeking respectively (Bock et al., 2013; Durieux et al., 2009; Heinsbroek et al., 2017; Lobo et al., 2010). To probe the behavioral consequences of cocaine-induced reductions in D2R sensitivity, we used a combination of regional and D2-MSN specific approaches to regulate Gαo expression. We originally predicted that decreases in D2R sensitivity and inhibitory signaling, and potential resulting increase in D2-MSN activity, could have the net effect of decreasing cocaine CPP or the reinstatement of cocaine seeking. However, instead by conditionally manipulating Gαo levels in D2-MSNs, we found surprisingly that decreasing the sensitivity of D2Rs led to increased motivation for cocaine. Altering D2R sensitivity did not affect voluntary cocaine intake during self-administration sessions, which together with previous findings indicating neither increasing nor decreasing D2R expression alters cocaine or alcohol consumption (Bocarsly et al., 2019; Dobbs et al., 2019; Gallo et al., 2015), suggest that alterations in D2 levels or sensitivity likely do not gate the escalation of voluntary drug self-administration. However, as drugs of abuse are thought to produce long-lasting memories that precipitate relapse after abstinence through their association with cues and contexts, the increase in cocaine CPP and context-dependent reinstatement as a result of decreased NAc Gαo levels suggest that cocaine-induced reduced NAc D2R sensitivity may support either reinforcement learning about contextual cues predictive of drug availability or the expression of these memories which drives drug-seeking. Moreover, as recent work has found that genetically reducing D2R levels in D2-MSNs induces an addiction-prone phenotype (Bocarsly et al., 2019), our observations further suggest that cocaine-induced changes in D2R signaling efficacy in D2-MSNs may play more nuanced roles in regulating motivation. Thus, rather than a dichotomous model where D1-MSN and D2-MSN activation drives or inhibits motivation, coordinated activity of microcircuits of populations of neurons may be required. This is in line with recent work that has found that in some cases D2-MSNs may signal both reward and aversion (Soares-Cunha et al., 2016, 2019; Vicente et al., 2016), and that activity in both MSN subpopulations likely underlies goal-directed actions (Cui et al., 2013; Tecuapetla et al., 2016; Vicente et al., 2016).
The mechanism by which decreased D2R sensitivity alters NAc circuits to regulate cocaine-seeking behaviors is likely due to several factors. The reduced ability of dopamine to suppress D2-MSNs via D2Rs would be expected to lead to hyperactivity of D2-MSNs but also a decreased ability of dopamine to regulate collateral inhibition from D2-MSNs to D1-MSNs. As downregulating the expression of D2Rs in D2-MSNs has been found to trigger alterations in D1R signaling in D1-MSNs, behavioral hypersensitivity to D1-agonists and alterations in the strength of striatopallidal projections which facilitate cocaine sensitization (Dobbs et al., 2019), similar mechanisms may contribute to the effects of decreased D2R sensitivity. In addition to these changes within the NAc, decreased sensitivity of D2Rs to dopamine would also suppress the ability of dopamine to regulate striatopallidal projections, as increased enkephalin, likely released from hyperactive D2-MSN terminals, causes a persistent long-term depression at D2-MSN GABAergic synapses to ventral pallidum (VP) and subsequently disinhibits VP neurons to facilitate motivation, cocaine-context association as well as cocaine relapse (Creed et al., 2016; Heinsbroek et al., 2017; Kupchik et al., 2014; Soares-Cunha et al., 2019; Tang et al., 2005). Lastly, given that D2Rs can modulate glutamate synaptic transmission and structural plasticity (Håkansson et al., 2006; Higley and Sabatini, 2010; Iino et al., 2020; Liu et al., 2006), changes in D2R sensitivity likely also regulate the changes in glutamatergic synaptic strength onto accumbens neurons that is associated with cocaine-seeking (Conrad et al., 2008; Roberts-Wolfe et al., 2018).
We found that the cocaine-induced change in D2R sensitivity was not dependent upon the activation of D2Rs, nor was it mimicked by L-DOPA. As cocaine raises dopamine levels more rapidly and transiently than L-DOPA as well as leads to enhanced dopamine spillover as a result of blocking dopamine uptake, the difference between the two manipulations likely results from differences in the extent over which the levels and kinetics over which dopamine is raised. The change in sensitivity however could be prevented by blocking either D1 or NMDA receptors. D1Rs are widely expressed within the NAc and striatum on D1-MSNs, as well as astrocytes and glutamatergic terminals and heterosynaptic plasticity in the NAc is known to involve MSN axon collaterals, interneurons and astrocytes (Corkrum et al., 2020; Dobbs et al., 2016, 2019; Francis et al., 2019). We found that chemogenetically inhibiting the PFC was sufficient to block the effect of cocaine on D2R sensitivity. As inactivating NMDA receptors in D2-MSNs similarly blocked the cocaine-induced decrease in D2R sensitivity, one possibility may be that during cocaine exposure increased D1-receptor activation on PFC inputs to the NAc stimulate glutamatergic transmission to D2-MSNs and the resulting activation of NMDA receptors at these synapses initiates signaling to drive the decrease in Gαo expression which regulates D2R sensitivity.
In conclusion, we show here that cocaine initially induces adaptations in D2R sensitivity rather than changes in D2R levels, which in turn contribute to context-mediated drug seeking and relapse. Future studies will need to elucidate the potential adaptive changes in neural circuits and signaling underlying context-mediated relapse caused by reduced D2R sensitivity, which may have implications for the treatment of relapse to drug-seeking.
STAR METHODS
Lead Contact
Further information and requests for resources and reagents should be direct to and will be fulfilled by the Lead Contact, Christopher Ford (christopher.ford@cuanschutz.edu)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Experimental models
All Animal experiments were carried out according to protocols approved by Institutional Animal Care and Use Committee (IACUC) at the University of Colorado School of Medicine. Both adult males and females were used for all experiments. There was no influence or association of sex on the findings reported. Animals were group-housed in a temperature- and humidity- controlled environment on a 12 h light-dark cycle, with water and food available ad libitum, and experiments were conducted during the light phase. Mice were individually housed for cocaine self-administration experiments. Behavioral and electrophysiology experiments were conducted on Adora2a-cre mice (MMRRC, 036158-UCD), Drd1-Cre mice (MMRRC, 034258), Gαo fl/fl mice (Charles River Laboratory, 129SvGo flx/flx), Drd2loxp/loxp mice(Jackson Laboratory, 020631), Drd2loxP/wt :: A2a-Cre (Drd2loxp/loxp crossed to strain A2a-cre), Gαo fl/wt:: A2a-Cre (Gαo fl/fl crossed to strain A2a-cre), CaMKII-Cre heterozygote mice (generated from WT and homozygous B6.Cg-Tg(Camk2a-cre)T29-1Stl/J, Jackson Laboratory stock# 005359), and wildtype C57BL/6J mice (Jackson Laboratory, 0000664).
Stereotaxic surgery
Mice (3–5 weeks) were anesthetized with isoflurane, transferred to a stereotaxic apparatus (Kopf Instruments) and kept under constant 2% isoflurane anesthesia. The skull surface was revealed via a midline sagittal incision. All AAV viruses were bilaterally injected into the dorsal and ventral part of the striatum using a Nanoject III (Drummond Scientific) at 100 nl/min. Coordinates in millimeters from bregma were as follows: DSt (dorsomedial) (AP +1.2, ML ± 1.85, DV −3.3), NAc (medial shell) (AP +1.5, ML ± 1.15, DV −4.3), PFC (AP +2.0, ML ± 0.4, DV −2.1) The pipette was kept at the site for 5 min and then slowly withdrawn. For experiments in WT mice, 300 nL of AAV9.hSyn.tdTomato.T2A.GIRK2 (University of Pennsylvania Viral Core, V3992) were used. For cell-type specific GIRK expression experiments, 300 nL of AAV9.hSyn.DIO.tdTomato.T2A.GIRK2 (University of Pennsylvania Viral Core, V5688)) was injected into the A2a-Cre or D2R cKD mice. For channelrhodopsin optogenetic studies, A2a-cre or Gαo cKD mice were injected with 500 nL of AAV5.EF1a.DIO.hChR2(H134R)-EYFP (Addgene, 20298). For D2R overexpression, 300 nL AAV9.hSynapsin.DIO.tdTomato.T2A.GIRK2 was combined with 200 nL AAV9.EF1a.DIO.D2L.P2A.eGFP (generous gift from Jonathan A. Javitch, Columbia University). For D2R ablation studies in Drd2loxp/loxp mice, 300 nL AAV9.hSyn.tdTomato.T2A.GIRK2 was combined with 200 nL AAV9.hSyn.Cre.WPRE.hGH (University of Pennsylvania Viral Core, V2676). For Gαo ablation studies, 200 nL of AAV9.hSyn.Cre.WPRE.hGH (University of Pennsylvania Viral Core, V2676) or AAV5.EF1a.mCherry.Cre (University of North Carolina Viral Core, AV6144B) was co-injected with 300 nL of AAV9.hSyn.tdTomato.T2A.GIRK2 into Gαo fl/fl mice. For Gαo overexpression in Gαo fl/fl mice, 200 nL of AAV9.EF1a.Gαo-alpha (Virovek) were co-injected with 300 nL of AAV9.hSyn.tdTomato.T2A.GIRK2. For cell-type specific Gαo expression experiments, AAV9.EF1a.DIO.Gαo-alpha (Virovek) was bilaterally injected with 300 nL of AAV9.hSyn.DIO.tdTomato.T2A.GIRK2. For fluorescent labeling of neurons, 300 nL of AAV5.EF1a.DIO.EYFP (Addgene, 27056) or AAV5.CAG.tdTomato (Addgene, 59462) were used. For chemogenetic studies, 300 nL of AAV2/5.hSyn.DIO.hM4D(Gi).mCherry (Addgene, 44362) was injected into the D1-cre or CaMKII-cre mice. For CRISPR/SaCas9 mediated NMDAR knockout studies, 300 nL of AAV5.FLEX.SaCas9.U6.sgGrin1 (VectorBuilder, addgene, 123852) was co-injected with either 300 nL AAV9.hSyn.DIO.tdTomato.T2A.GIRK2 or 300 nL of AAV9.hSyn.tdTomato.T2A.GIRK2 into the D1-cre or A2a-cre mice. For NMDA iontophoresis studies in A2a-cre mice, 300 nL of AAV5.EF1a.DIO.mCherry was injected with or without 300 nL of AAV5.FLEX.SaCas9.U6.sgGrin1.
Method Details
Slice preparation
Coronal slices containing the striatum were collected at 240 μm. Mice were anesthetized with isoflurane and transcardially perfused with an ice-cold sucrose (95% v/v O2, 5% v/v CO2) cutting solution containing (in mM): 75 NaCl, 2.5 KCl, 6 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, 0.1 CaCl2, 11.1 D-glucose and 1 kyneurenic acid. The brain was subsequently dissected, and sectioned using a vibratome (Leica VT1000 S, Leica Biosystems) in cutting solution. Slices were then transferred to an oxygenated 34 °C chamber filled with aCSF solution consisting of (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, 11.1 D-glucose and 10 μM MK-801 to prevent excitotoxicity. After 1 h, slices were then transferred to the recording chamber where they were constantly perfused with aCSF solutions at a flow rate of 2 mL/min. Solutions also contained SCH23390 (1 μM), scopolamine (200 nM), picrotoxin (100 μM), CGP55845 (300 nM), DNQX (10 μM), and dihydro-β-erthroidine hydrobromide (DHβE, 1μM). MSNs were visually identified using a BXWI51 microscope (Olympus) with custom built infrared gradient contrast optics. Fluorescence was visualized with LEDs (Thorlabs).
Electrophysiology
MSNs were voltage-clamped in whole-cell configuration at −60 mV using Axopatch 200B amplifiers (Molecular Devices), and signals were acquired with Axograph X (Axograph Scientific) at 5 kHz and filtered to 2 kHz or acquired with LabChart (ADInstruments) at 1kHz. Patch pipettes (1.5–2 MΩ, World Precision Instruments) used for MSN recording was filled with intracellular solution containing: 115 mM K-methylsulphate, 20 mM NaCl, 1.5 mM MgCl2, 10 mM HEPES(K), 10 mM BAPTA-tetrapotassium, 1 mg/mL ATP, 0.1 mg/mL GTP, and 1.5 mg/mL sodium phosphocreatine, pH=7.4, 275 mOsm. Dopamine or glutamate release was triggered by electrical stimulation (0.5 ms) using a monopolar glass stimulating electrode filled with aCSF. For concentration-response curve experiments, cocaine (10 μM) was included in the recording solution to block dopamine reuptake. Dopamine was bath applied via perfusion. D2-IPSCs were evoked once per minute and have been blanked in traces for clarity. For GABAA oIPSCs, whole-cell recordings (Vh = − 60 mV) were made from non-fluorescent D1-MSNs and GABA release was evoked by wide-field blue light (470 nm LED, Thorlab, ~2mW/mm2) to the slice every 30 s. The internal solution contained 135 mM CsCl, 0.1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES(K), 0.1 mM BAPTA, 5 mM QX-314, 1 mg/mL ATP, 0.1 mg/mL GTP, and 1.5 mg/mL sodium phosphocreatine, pH=7.4, 275 mOsm. The GABAA oIPSCs were isolated in presence of 1 μM SCH23390, 200 nM scopolamine, 300 nM CGP 55845, 10 μM DNQX, 1μM DHβE and 10 μM cocaine. For AMPA and NMDA receptor-mediated currents, picrotoxin was included in the recording solution to block GABAA currents. The internal solution contained 135 mM CsMeSO3, 0.1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES(K), 0.1 mM EGTA, 1 mg/mL ATP, 0.1 mg/mL GTP, and 1.5 mg/mL sodium phosphocreatine, pH 7.4, 275 mOsm. MSNs were held at −70 mV to measure the AMPA receptor-mediated currents, following by application of DNQX (10 μM). MSNs were then stepped to +40 mV to measure NMDA receptor-mediated currents. AMPA and NMDA traces were digitally subtracted from traces in the presence of DNQX (10 μM) and AP5 (50 μM) to eliminate the stimulus artifact for clarity. All recordings from GIRK2+ MSNs were done within the NAc and DSt in regions showing robust tdTomato reporter fluorescence. For NMDA iontophoresis, an iontophoretic electrode was filled with NMDA (200 mM). NMDA was injected using a negative current (−100 nA) and the leakage was prevented with a retention current (+ 3~10 nA). Recordings were performed in Mg2+ - free aCSF.
qPCR analysis
Striatal tissue punches were collected from coronal sections with vibratome directly into ice-cold Trizol reagent (Invitrogen) and stored at −80 °C until RNA isolation. Total RNA was isolated following the Trizol protocol and the aqueous phase was purified using Qiagen RNAease columns and buggers. RNA quality and quantity were analyzed with a NanoDrop UV-Vis Spectrophotometer (Thermo Scientific). RNA templates were transcribed into cDNA with the Superscript III kit (Invitrogen) and random hexamer primers. cDNA was amplified on a Roche LightCycler 96 system with Fast SYBR Green PCR Master Mix.
Immunoblotting
The DSt and NAc tissue for western analysis was isolated and performed as previously described (Marcott et al., 2018). The samples were homogenized and then denatured with STE buffer (10 mM Tris-Cl, pH 7.5, 1 mM EDTA, pH 8.0, 10% SDS) at 100 °C for 5 min and equivalent amounts of protein were subjected to SDS-PAGE on 10% polyacrylamide gels, and then transferred to methanol activated PVDF membranes (Perkin Elmer). Blots were blocked by 5% milk for 1 hour at 24 °C and then immunolabeled with primary antibody against Gαo protein (1:500, Santa Cruz, sc-13532), Gαi protein (1:500, Santa Cruz, sc-136478), D2R protein (1:500, Millipore, ABN462), GIRK2 protein (1:300, Alamone labs, APC-006) and actin (1:1000, Cell signaling, 4970S), overnight at 4 °C in blocking buffer. Following incubation with species-specific horseradish peroxidase (HRP)-conjugated secondary ECL™ anti-Rabbit (1:2000, GE healthcare, NA934) or anti-Mouse IgG (1:2000, GE healthcare, NA931) antibody for 1 h at 24 °C, blots were detected in Pierce ECL reagent (Thermo Fisher) and visualized with a Cheimager (Alpha Innotech). The densitometry was quantified with ImageJ.
Histology
Mice were anesthetized using isoflurane and perfused transcardially with cold sodium phosphate buffer (PBS) followed by cold 4% (w/v) paraformaldehyde in 0.1 M PBS, pH 7.4. Brains were fixed overnight at 4 °C in 4% PFA and then equilibrated in 30% (w/v) sucrose solution for 2 days. Coronal slices were taken at 30 μm using a Leica CM3050 S Cryostat (Leica Microsystems). Sections were mounted with an anti-fading mounting medium and imaged using a slide scanner microscope (VS120, Olympus) or an inverted LSM 710 Meta confocal microscope (Zeiss).
Behavioral Assessment
Rotarod test
Motor coordination and balance behavior were assessed by accelerated rotarod assay. Mice were trained on an accelerating rotarod apparatus for five trials per day for 3 consecutive days. The rotarod accelerated slowly from 4 to 40 rotations per minute over a 5-min session. The time until falling from the rotating rod or time to reach maximum speed was used to quantify motor performance. Mice were given 10-min breaks between trials to allow for recovery.
Open field test
Locomotion and anxiety-related behavior were assessed using an open field test. Mice were habituated to the room for 1 h before the start of the test. Each mouse was gently placed into the corner of a square arena (42 × 42 cm). The experiment duration was 30 min and locomotion parameters were collected by an overhead camera connected to the video-tracking system (Noldus).
Cocaine behavioral locomotor sensitization
Behavioral sensitization was assessed using the open field arena described above. After habituation to the arena for 15 min, mice were given daily intraperitoneal injections of either saline (0.9 % NaCl) or cocaine (10, 20 mg/kg) and then placed back into the arena for an additional 15 min. After habituation to saline injections (day 1), mice were subdivided into two groups that received either saline or cocaine for 7d. Following a 14d abstinence period, mice were given a challenge dose of cocaine or saline and re-introduced to the arena.
Cocaine CPP
Cocaine CPP was conducted using a standard custom-made three-chamber apparatus, consisting of two conditioning chambers and a corridor, connecting chamber. The two conditioning chambers contained distinct visual stimuli (different color and shape patterns on the walls). Mice were conditioned to cocaine using an unbiased paradigm. On day 1, before the conditioning phase, mice were placed in the corridor chamber and allowed to explore the entire apparatus for 30 min. Mice that showed place bias higher than 30% were excluded from the experiment. Over the next 5 days during the conditioning phase, mice received two paring sessions per day during which they were injected with cocaine (15 mg/kg, i.p.) and placed in the cocaine-paired chamber, or injected with an equivalent volume of saline and placed in the saline-paired chamber, for 20 min each. Cocaine and saline injections were counterbalanced. The two conditioning sessions were separated by at least 4 h. On day 7, during the test phase, mice were placed in the corridor chamber with free access to all chambers, and the time spent in each chamber was quantified over a 30 min period. CPP score was calculated as time spent in the cocaine-paired chamber minus the time spent in the saline-paired chamber.
Cocaine self-administration, extinction, reinstatement and dose-response
Mice were anesthetized with isoflurane (induction 5%, maintenance 1–2% v/v) and implanted with a jugular catheter attached to a back-mounted entry port as described previously (Heinsbroek et al., 2020). Catheters were flushed twice daily with a sterile saline solution containing heparin. After 5–7d of recovery from intravenous catheter surgery, mice were food restricted prior to starting behavior. During cocaine self-administration responses in the active nosepoke port resulted in the intravenous infusion of cocaine (0.75 mg/kg/infusion) over 2s combined with the presentation of a 3s tone/light stimulus followed by a 10s time-out period. Following 8d of acquisition on a FR1 schedule of reinforcement, mice progressed to FR2 and FR4, followed by a PR test. After 7d of forced abstinence, mice returned to the self-administration box for a drug-context relapse test under extinction conditions, followed by at least 7 additional days of extinction training until extinction criterion was reached (last 2d average ≤20 active responses). Afterwards mice underwent a cue test (cue-induced reinstatement) during which drug-paired cues were returned to active nose-pokes. Cocaine dose-response testing was performed as described previously (Dobbs et al., 2019). First, mice were trained 2h/d to acquire cocaine self-administration for 8d at FR1 on a training dose (0.75 mg/kg/infusion). On subsequent days, mice self-administered the training dose during the first 30 min of the session, followed by 1h periods of self-administration of one of 8 doses of cocaine (administered in descending order over 3d; day 1: 3.2 and 1.5 mg/kg, day 2: 1, 0.75 and 0.5 mg/kg, day 3: 0.25, 0.125 and 0.075 mg/kg).
Chemicals
Picrotoxin, MK-801, sulpiride, DNQX, AP5, SCH23390, DHβE, scopolamine, PG01037, L-741626, L-741742, CGP55845 were obtained from Tocris Bioscience. JHU37160 was from Hello Bio. Cocaine and amphetamine were from the NIDA Drug Supply Program. All the other chemicals were from Sigma-Aldrich.
Quantification and Statistical Analysis
Statistical analyses were performed in Prism 7 (GraphPad). Data are shown as mean ± SEM (grey traces represent 95% confidence intervals) or as boxplots (minimum, 1st quartile, median, 3rd quartile and maximum). Statistical significance was defined as p ≥ 0.05 (ns), p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). n represents number of cells; N represents number of animals. Statistical significance was determined using Mann-Whitney U test, Student’s t-test, one-way analysis of variance (ANOVA), Wilcoxon matched-paired signed rank, repeated one-way ANOVA, and mixed-model and two-way ANOVA, as appropriate, with Tukey’s (comparing every mean with every other mean) or Sidak’s (comparing a set of selected means) post hoc analysis and Geisser-Greenhouse correction. Two unpaired groups of data were assumed to be nonparametric. Analysis were two-tailed. The EC50/IC50 of D2-receptor activation in each area was calculated by fitting data points using nonlinear regression (Hill coefficient = 1). EC50/IC50 was assumed to be Gaussian distribution. If the maximum response and inhibition did not differ between regions, the concentration-response curves were constrained to the average maximum values measured in the two regions.
Supplementary Material
Table S1. Statistical analysis and quantification
Statistical analysis and quantification for data presented in Figures 1–7 and Figures S1–S7.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti- Gαo | Santa Cruz | Cat# sc-13532 RRID: AB_2111645 |
| Mouse anti- Gαi | Santa Cruz | Cat# sc-136478 RRID: AB_2722559 |
| Rabbit anti- GIRK2 | Alamone Labs | Cat# APC-006 RRID: AB_2040115 |
| Rabbit anti- D2R | Millipore | Cat# ABN462 RRID: AB_2810225 |
| Rabbit anti- actin | Cell Signaling Technology | Cat# 4970S RRID: AB_2223172 |
| Horseradish peroxidase (HRP)-conjugated secondary ECL™ anti-Rabbit IgG | GE Healthcare | Cat# NA934V RRID: AB_772206 |
| Horseradish peroxidase (HRP)-conjugated secondary ECL™ anti-Mouse IgG | GE Healthcare | Cat# NA931V RRID: AB_772210 |
| Bacterial and Virus Strains | ||
| AAV5.EF1a.DIO.hChR2 (H134R)-EYFP | pAAV-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-HGHpA was a gift from Karl Deisseroth | Addgene plasmid: 20298 |
| AAV9.hSyn.tdTomato.T2A.mGIRK2–1 | Marcott et al., 2014 | Penn Vector Core: V3992 |
| AAV9.hSyn.DIO.tdTomato.T2A.GIRK2 | Marcott et al., 2018 | Penn Vector Core: V5688 |
| AAV5-EF1a-mCherry-Cre | UNC Vector Core | AV6144B |
| AAV9.EF1a.DIO.D2L.P2A.EGFP | Gallo et al., 2015 | N/A |
| AAV9.hSyn.Cre.WPRE.hGH | pENN.AAV.hSyn.Cre.WPRE.hGH was a gift from James M. Wilson | Penn Vector Core: A-9-PV2676 |
| AAV9.EF1a.Gαo-alpha | Marcott et al., 2018 | Virovek |
| AAV9.EF1a.DIO.Gαo-alpha | Virovek | VVK10008799 |
| AAV5.hSyn.DIO.hM4D(Gi).mCherry | Krashes et al., 2011 | Addgene plasmid: 44362 |
| AAV2.hSyn.DIO.hM4D(Gi).mCherry | Krashes et al., 2011 | Addgene plasmid: 44362 |
| AAV5.CAG.tdTomato | pAAV-CAG-tdTomato was a gift from Edward Boyden | Addgene plasmid: 59462 |
| AAV5.EF1a.DIO.EYFP | pAAV-Ef1a-DIO-EYFP was a gift from Karl Deisseroth | Addgene plasmid: 27056 |
| AAV5.FLEX.SaCas9.U6.sgGrin1 | Hunker et al., 2020 | Addgene plasmid: 124852 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Scopolamine | Tocris | Cat# 1414 |
| AP5 | Tocris | Cat# 0106 |
| SCH23390 | Tocris | Cat# 0925 |
| CGP55845 | Tocris | Cat# 1248 |
| DHβE | Tocris | Cat# 2349 |
| DNQX | Tocris | Cat# 0189 |
| Picrotoxin | Tocris | Cat# 1128 |
| MK-801 | Tocris | Cat# 0924 |
| NMDA | Tocris | Cat# 2333 |
| Dopamine | Sigma | Cat# 0214 |
| L-DOPA | Tocris | Cat# 3788 |
| Sulpiride | Tocris | Cat# 0895 |
| JHU37160 dihydrochloride | Hello-Bio | Cat# HB6261 |
| Cocaine hydrochloride | NIDA | N/A |
| Amphetamine hydrochloride | NIDA | N/A |
| Quinpirole | Tocris | Cat# 1061 |
| Haloperidol | Tocris | Cat# 0931 |
| Critical Commercial Assays | ||
| RNeasy Micro Kit | QIAGEN | Cat# 74004 |
| SuperScript cDNA synthesis kit | Thermo Fisher Scientific | Cat# 18091200 |
| TaqMan Fast Advanced master Mix | Thermo Fisher Scientific | Cat# 4444556 |
| Taqman probe: Drd2 | Thermo Fisher Scientific | Mm00438541_m1 |
| Taqman probe: Actb | Thermo Fisher Scientific | Mm00607939_s1 |
| Experimental Models: Organisms/Strains | ||
| Mouse / Adora2a-Cre | MMRRC | 036158-UCD |
| Mouse / Drd1-Cre | MMRRC | 034258 |
| Mouse / Gαo fl/fl | Charles River Laboratory | 129SvGo flx/flx |
| Mouse / CaMKII-Cre | Jackson Laboratory | JAX: 005359 |
| Mouse / Drd2loxp/loxp | Jackson Laboratory | JAX: 020631 |
| Mouse / Drd2loxP/wt :: A2a-Cre | This manuscript | N/A |
| Mouse / Gαo fl/wt:: A2a-Cre | This manuscript | N/A |
| Mouse / C57BL/6J | Jackson Laboratory | JAX: 000664 |
| Software and Algorithms | ||
| Axograph X | Axograph Scientific | RRID: SCR_014284 https://axograph.com |
| LabChart | ADInstruments | RRID: SCR_017551 https://www.adinstruments.com |
| EthoVision XT | Noldus | RRID: SCR_000441 https://www.noldus.com |
| FIJI(ImageJ) | NIH | RRID: SCR_003070 https://imagej.net |
| GraphPad Prism 7 | GraphPad Software | RRID: SCR_002798 https://www.graphpad.com |
Highlight Bullets.
7-day cocaine exposure reduces the sensitivity but not the level of D2Rs in the NAc
Cocaine-induced changes in D2Rs sensitivity result from decreased Gαo expression
D2R sensitivity is regulated by NMDA plasticity at D1R+ PFC inputs to D2-MSNs
Cocaine-induced changes in G protein coupling promote contextual drug seeking
Acknowledgments:
This work was funded by NIH grants R01-DA35821 (CPF), R01-NS95809 (CPF), R21-MH123085 (CPF) and DP5 OD026407 (JAH). We thank Steve Coultrap and Ulli Bayer for assistance with western blot experiments and the University of Colorado Anschutz Behavioral Core, and Savanna Gonzalez for assistance with behavioral experiments. We thank the NIDA drug supply program for providing cocaine and amphetamine.
Footnotes
Declaration of Interests: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited:
- Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, and Rubinstein M (2011). Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat. Neurosci. 14, 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson M, Pelloux Y, Dilleen R, Theobald DE, Lyon A, Belin-Rauscent A, Robbins TW, Dalley JW, Everitt BJ, and Belin D (2013). Cocaine modulation of frontostriatal expression of Zif268, D2, and 5-HT2c receptors in high and low impulsive rats. Neuropsychopharmacology 38, 1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocarsly ME, da Silva E Silva D, Kolb V, Luderman KD, Shashikiran S, Rubinstein M, Sibley DR, Dobbs LK, and Alvarez VA (2019). A Mechanism Linking Two Known Vulnerability Factors for Alcohol Abuse: Heightened Alcohol Stimulation and Low Striatal Dopamine D2 Receptors. Cell Rep 29, 1147–1163.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, Gremel CM, Christensen CH, Adrover MF, and Alvarez VA (2013). Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat. Neurosci. 16, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert JM, Marchant NJ, Calu DJ, and Shaham Y (2013). The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology (Berl.) 229, 453–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, Lanier SM, and Kalivas PW (2004). Activator of G protein signaling 3: a gatekeeper of cocaine sensitization and drug seeking. Neuron 42, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine SB, Negus SS, Mello NK, Patel S, Bristow L, Kulagowski J, Vallone D, Saiardi A, and Borrelli E (2002). Role of dopamine D2-like receptors in cocaine self-administration: studies with D2 receptor mutant mice and novel D2 receptor antagonists. J. Neurosci. 22, 2977–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervo L, and Samanin R (1995). Effects of dopaminergic and glutamatergic receptor antagonists on the acquisition and expression of cocaine conditioning place preference. Brain Res 673, 242–250. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng L-J, Shaham Y, Marinelli M, and Wolf ME (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Ford K, Marinelli M, and Wolf ME (2010). Dopamine receptor expression and distribution dynamically change in the rat nucleus accumbens after withdrawal from cocaine self-administration. Neuroscience 169, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkrum M, Covelo A, Lines J, Bellocchio L, Pisansky M, Loke K, Quintana R, Rothwell PE, Lujan R, Marsicano G, et al. (2020). Dopamine-Evoked Synaptic Regulation in the Nucleus Accumbens Requires Astrocyte Activity. Neuron 105, 1036–1047.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed M, Ntamati NR, Chandra R, Lobo MK, and Lüscher C (2016). Convergence of Reinforcing and Anhedonic Cocaine Effects in the Ventral Pallidum. Neuron 92, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, and Costa RM (2013). Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Ferris MJ, Gainetdinov RR, Sotnikova TD, Urs NM, Jones SR, and Caron MG (2014). Selective deletion of GRK2 alters psychostimulant-induced behaviors and dopamine neurotransmission. Neuropsychopharmacology 39, 2450–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Fryer TD, Brichard L, Robinson ESJ, Theobald DEH, Lääne K, Peña Y, Murphy ER, Shah Y, Probst K, et al. (2007). Nucleus accumbens D2/3 receptors predict trait impulsivity and cocaine reinforcement. Science 315, 1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs LK, Kaplan AR, Lemos JC, Matsui A, Rubinstein M, and Alvarez VA (2016). Dopamine Regulation of Lateral Inhibition between Striatal Neurons Gates the Stimulant Actions of Cocaine. Neuron 90, 1100–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs LK, Kaplan AR, Bock R, Phamluong K, Shin JH, Bocarsly ME, Eberhart L, Ron D, and Alvarez VA (2019). D1 receptor hypersensitivity in mice with low striatal D2 receptors facilitates select cocaine behaviors. Neuropsychopharmacology 44, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux PF, Bearzatto B, Guiducci S, Buch T, Waisman A, Zoli M, Schiffmann SN, and de Kerchove d’Exaerde A (2009). D2R striatopallidal neurons inhibit both locomotor and drug reward processes. Nat. Neurosci. 12, 393–395. [DOI] [PubMed] [Google Scholar]
- Francis TC, Yano H, Demarest TG, Shen H, and Bonci A (2019). High-Frequency Activation of Nucleus Accumbens D1-MSNs Drives Excitatory Potentiation on D2-MSNs. Neuron 103, 432–444.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo EF, Salling MC, Feng B, Morón JA, Harrison NL, Javitch JA, and Kellendonk C (2015). Upregulation of dopamine D2 receptors in the nucleus accumbens indirect pathway increases locomotion but does not reduce alcohol consumption. Neuropsychopharmacology 40, 1609–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaval-Cruz M, Goertz RB, Puttick DJ, Bowles DE, Meyer RC, Hall RA, Ko D, Paladini CA, and Weinshenker D (2016). Chronic loss of noradrenergic tone produces β-arrestin2-mediated cocaine hypersensitivity and alters cellular D2 responses in the nucleus accumbens. Addict Biol 21, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazi L, Nickolls SA, and Strange PG (2003). Functional coupling of the human dopamine D2 receptor with G alpha i1, G alpha i2, G alpha i3 and G alpha o G proteins: evidence for agonist regulation of G protein selectivity. Br. J. Pharmacol. 138, 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, and Surmeier DJ (2011). Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håkansson K, Galdi S, Hendrick J, Snyder G, Greengard P, and Fisone G (2006). Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J. Neurochem. 96, 482–488. [DOI] [PubMed] [Google Scholar]
- Heinsbroek JA, Neuhofer DN, Griffin WC, Siegel GS, Bobadilla A-C, Kupchik YM, and Kalivas PW (2017). Loss of Plasticity in the D2-Accumbens Pallidal Pathway Promotes Cocaine Seeking. J. Neurosci. 37, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinsbroek JA, Bobadilla A-C, Dereschewitz E, Assali A, Chalhoub RM, Cowan CW, and Kalivas PW (2020). Opposing Regulation of Cocaine Seeking by Glutamate and GABA Neurons in the Ventral Pallidum. Cell Rep 30, 2018–2027.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, and Sabatini BL (2010). Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat. Neurosci. 13, 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikida T, Kimura K, Wada N, Funabiki K, and Nakanishi S (2010). Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron 66, 896–907. [DOI] [PubMed] [Google Scholar]
- Hunker AC, Soden ME, Krayushkina D, Heymann G, Awatramani R, and Zweifel LS (2020). Conditional Single Vector CRISPR/SaCas9 Viruses for Efficient Mutagenesis in the Adult Mouse Nervous System. Cell Reports 30, 4303–4316.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino Y, Sawada T, Yamaguchi K, Tajiri M, Ishii S, Kasai H, and Yagishita S (2020). Dopamine D2 receptors in discrimination learning and spine enlargement. Nature 579, 555–560. [DOI] [PubMed] [Google Scholar]
- Kalivas PW (2004). Glutamate systems in cocaine addiction. Curr Opin Pharmacol 4, 23–29. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, and Seamans J (2005). Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron 45, 647–650. [DOI] [PubMed] [Google Scholar]
- Kim HS, Park WK, Jang CG, and Oh S (1996). Inhibition by MK-801 of cocaine-induced sensitization, conditioned place preference, and dopamine-receptor supersensitivity in mice. Brain Res Bull 40, 201–207. [DOI] [PubMed] [Google Scholar]
- Koob GF, and Volkow ND (2016). Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3, 760–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Crusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 121, 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, and Kreitzer AC (2012). Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat. Neurosci. 15, 816–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Scofield MD, Rice KC, Cheng K, Roques BP, and Kalivas PW (2014). Cocaine dysregulates opioid gating of GABA neurotransmission in the ventral pallidum. J. Neurosci. 34, 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouèbe G, Lomazzi M, Cruz HG, Creton C, Luján R, Li M, Yanagawa Y, Obata K, Watanabe M, Wickman K, et al. (2007). RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nature Neuroscience 10, 1559–1568. [DOI] [PubMed] [Google Scholar]
- Lalchandani RR, van der Goes M-S, Partridge JG, and Vicini S (2013). Dopamine D2 receptors regulate collateral inhibition between striatal medium spiny neurons. J. Neurosci. 33, 14075–14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, Powney B, Wise A, Rees S, and Milligan G (2007). Protean agonism at the dopamine D2 receptor: (S)-3-(3-hydroxyphenyl)-N-propylpiperidine is an agonist for activation of Go1 but an antagonist/inverse agonist for Gi1,Gi2, and Gi3. Mol. Pharmacol. 71, 1349–1359. [DOI] [PubMed] [Google Scholar]
- Lee JH, Ribeiro EA, Kim J, Ko B, Kronman H, Jeong YH, Kim JK, Janak PH, Nestler EJ, Koo JW, et al. (2020). Dopaminergic Regulation of Nucleus Accumbens Cholinergic Interneurons Demarcates Susceptibility to Cocaine Addiction. Biol. Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RG, Serra M, Radl D, Gori M, Tran C, Michalak SE, Vanderwal CD, and Borrelli E (2020). Dopaminergic Control of Striatal Cholinergic Interneurons Underlies Cocaine-Induced Psychostimulation. Cell Rep 31, 107527. [DOI] [PubMed] [Google Scholar]
- Liu X-Y, Chu X-P, Mao L-M, Wang M, Lan H-X, Li M-H, Zhang G-C, Parelkar NK, Fibuch EE, Haines M, et al. (2006). Modulation of D2R-NR2B interactions in response to cocaine. Neuron 52, 897–909. [DOI] [PubMed] [Google Scholar]
- Lobo MK, and Nestler EJ (2011). The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, et al. (2010). Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, and Malenka RC (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Robbins TW, and Everitt BJ (2020). The transition to compulsion in addiction. Nat. Rev. Neurosci. 21, 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcott PF, Mamaligas AA, and Ford CP (2014). Phasic dopamine release drives rapid activation of striatal D2-receptors. Neuron 84, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcott PF, Gong S, Donthamsetti P, Grinnell SG, Nelson MN, Newman AH, Birnbaumer L, Martemyanov KA, Javitch JA, and Ford CP (2018). Regional Heterogeneity of D2-Receptor Signaling in the Dorsal Striatum and Nucleus Accumbens. Neuron 98, 575–587.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K, and Martemyanov KA (2015). Distinct profiles of functional discrimination among G proteins determine the actions of G protein–coupled receptors. Sci. Signal. 8, ra123–ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RJ, Vinsant SL, Nader MA, Porrino LJ, and Friedman DP (1998). Effect of cocaine self-administration on dopamine D2 receptors in rhesus monkeys. Synapse 30, 88–96. [DOI] [PubMed] [Google Scholar]
- Morgan D, Grant KA, Gage HD, Mach RH, Kaplan JR, Prioleau O, Nader SH, Buchheimer N, Ehrenkaufer RL, and Nader MA (2002). Social dominance in monkeys: dopamine D2 receptors and cocaine self-administration. Nat Neurosci 5, 169–174. [DOI] [PubMed] [Google Scholar]
- Nader MA, Daunais JB, Moore T, Nader SH, Moore RJ, Smith HR, Friedman DP, and Porrino LJ (2002). Effects of cocaine self-administration on striatal dopamine systems in rhesus monkeys: initial and chronic exposure. Neuropsychopharmacology 27, 35–46. [DOI] [PubMed] [Google Scholar]
- Nader MA, Morgan D, Gage HD, Nader SH, Calhoun TL, Buchheimer N, Ehrenkaufer R, and Mach RH (2006). PET imaging of dopamine D2 receptors during chronic cocaine self-administration in monkeys. Nat. Neurosci. 9, 1050–1056. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Terwilliger RZ, Walker JR, Sevarino KA, and Duman RS (1990). Chronic cocaine treatment decreases levels of the G protein subunits Gi alpha and Go alpha in discrete regions of rat brain. J. Neurochem. 55, 1079–1082. [DOI] [PubMed] [Google Scholar]
- Padgett CL, Lalive AL, Tan KR, Terunuma M, Munoz MB, Pangalos MN, Martínez-Hernández J, Watanabe M, Moss SJ, Luján R, et al. (2012). Methamphetamine-evoked depression of GABA(B) receptor signaling in GABA neurons of the VTA. Neuron 73, 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Volkow ND, Pan Y, and Du C (2013). Chronic Cocaine Dampens Dopamine Signaling during Cocaine Intoxication and Unbalances D1 over D2 Receptor Signaling. J. Neurosci. 33, 15827–15836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Hiver A, and Lüscher C (2015). Sufficiency of Mesolimbic Dopamine Neuron Stimulation for the Progression to Addiction. Neuron 88, 1054–1066. [DOI] [PubMed] [Google Scholar]
- Porter-Stransky KA, Petko AK, Karne SL, Liles LC, Urs NM, Caron MG, Paladini CA, and Weinshenker D (2019). Loss of β-arrestin2 in D2 cells alters neuronal excitability in the nucleus accumbens and behavioral responses to psychostimulants and opioids. Addict Biol e12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman Z, Schwarz J, Gold SJ, Zachariou V, Wein MN, Choi K-H, Kovoor A, Chen C-K, DiLeone RJ, Schwarz SC, et al. (2003). RGS9 modulates dopamine signaling in the basal ganglia. Neuron 38, 941–952. [DOI] [PubMed] [Google Scholar]
- Roberts-Wolfe D, Bobadilla A-C, Heinsbroek JA, Neuhofer D, and Kalivas PW (2018). Drug Refraining and Seeking Potentiate Synapses on Distinct Populations of Accumbens Medium Spiny Neurons. J. Neurosci. 38, 7100–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JA, McCafferty MR, and Unterwald EM (2009). Regulation of dynamin 2 and G protein-coupled receptor kinase 2 in rat nucleus accumbens during acute and repeated cocaine administration. Synapse 63, 863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares-Cunha C, Coimbra B, David-Pereira A, Borges S, Pinto L, Costa P, Sousa N, and Rodrigues AJ (2016). Activation of D2 dopamine receptor-expressing neurons in the nucleus accumbens increases motivation. Nat Commun 7, 11829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares-Cunha C, de Vasconcelos NAP, Coimbra B, Domingues AV, Silva JM, Loureiro-Campos E, Gaspar R, Sotiropoulos I, Sousa N, and Rodrigues AJ (2019). Nucleus accumbens medium spiny neurons subtypes signal both reward and aversion. Mol. Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striplin CD, and Kalivas PW (1993). Robustness of G protein changes in cocaine sensitization shown with immunoblotting. Synapse 14, 10–15. [DOI] [PubMed] [Google Scholar]
- Tang X-C, McFarland K, Cagle S, and Kalivas PW (2005). Cocaine-induced reinstatement requires endogenous stimulation of mu-opioid receptors in the ventral pallidum. J. Neurosci. 25, 4512–4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S (2003). Direct Physiological Evidence for Synaptic Connectivity Between Medium-Sized Spiny Neurons in Rat Nucleus Accumbens In Situ. Journal of Neurophysiology 91, 1111–1121. [DOI] [PubMed] [Google Scholar]
- Tecuapetla F, Koós T, Tepper JM, Kabbani N, and Yeckel MF (2009). Differential Dopaminergic Modulation of Neostriatal Synaptic Connections of Striatopallidal Axon Collaterals. J. Neurosci. 29, 8977–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Jin X, Lima SQ, and Costa RM (2016). Complementary Contributions of Striatal Projection Pathways to Action Initiation and Execution. Cell 166, 703–715. [DOI] [PubMed] [Google Scholar]
- Vicente AM, Galvão-Ferreira P, Tecuapetla F, and Costa RM (2016). Direct and indirect dorsolateral striatum pathways reinforce different action strategies. Curr. Biol. 26, R267–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, and Morales M (2015). The Brain on Drugs: From Reward to Addiction. Cell 162, 712–725. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wolf AP, Schlyer D, Shiue CY, Alpert R, Dewey SL, Logan J, Bendriem B, and Christman D (1990). Effects of chronic cocaine abuse on postsynaptic dopamine receptors. Am J Psychiatry 147, 719–724. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Hitzemann R, Logan J, Schlyer DJ, Dewey SL, and Wolf AP (1993). Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 14, 169–177. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R, Chen AD, Dewey SL, and Pappas N (1997). Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature 386, 830–833. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Baler R, and Telang F (2009). Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology 56 Suppl 1, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Tomasi D, Wang G-J, Logan J, Alexoff DL, Jayne M, Fowler JS, Wong C, Yin P, and Du C (2014). Stimulant-induced dopamine increases are markedly blunted in active cocaine abusers. Mol. Psychiatry 19, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME (2010). The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci. 33, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Statistical analysis and quantification
Statistical analysis and quantification for data presented in Figures 1–7 and Figures S1–S7.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.