

Abstract
Current single-cell mass spectrometry (MS) methods can quantify thousands of peptides per single cell while detecting peptide-like features that may support the quantification of 10-fold more peptides. This 10-fold gain might be attained by innovations in data acquisition and interpretation even while using existing instrumentation. This perspective discusses possible directions for such innovations with the aim to stimulate community efforts for increasing the coverage and quantitative accuracy of single proteomics while simultaneously decreasing missing data. Parallel improvements in instrumentation, sample preparation, and peptide separation will afford additional gains. Together, these synergistic routes for innovation project a rapid growth in the capabilities of MS based single-cell protein analysis. These gains will directly empower applications of single-cell proteomics to biomedical research.
Keywords: single-cell proteomics, data acquisition, data interpretation, peptide identity propagation, ultrasensitive proteomics
Graphical Abstract
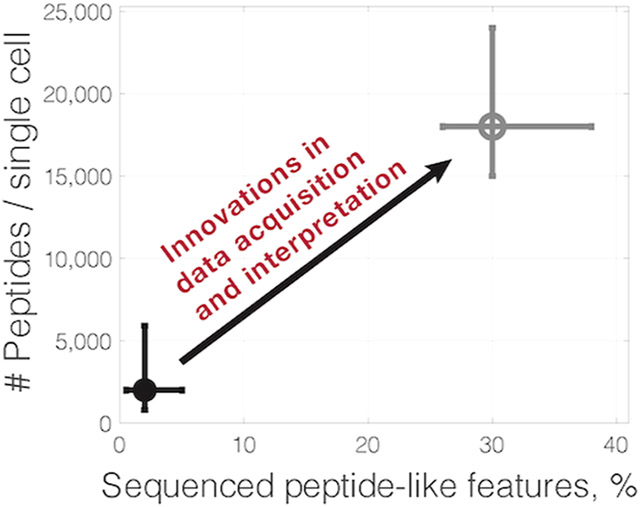
INTRODUCTION
While the sensitivity of mass spectrometry (MS) is often cited as a major limitation for single-cell proteomics, current methods can detect over 60 000 peptide-like features from individual cells analyzed by label-free MS.1,2 Each of these features consists of many data points, an isotopic envelope detected across multiple survey scans. Yet, because of time constraints, shotgun methods can perform MS2 scans on only a fraction of all detected peptide-like features and identify the amino acid sequences of even a smaller fraction, Figure 1. For example, label free analysis of single HeLa cells can identify tens of thousands of peptide-like features but determines the amino acid sequences for only about 1–3% of these features at 1% FDR.1,2 Sequencing even a small fraction of the features has allowed the quantification of over 1000 proteins per single cell and enabled answering biological questions.3-6 Yet, creative new approaches might be able to significantly increase the fraction of sequenced peptide-like features and the consistency of accurately quantified peptides across single cells.
Figure 1.

Number of precursor ions analyzed by shotgun methods decline steeply with increasing ion accumulation times. The graphs show theoretical simulations of the maximum number of MS2 scans that could be performed as a function of MS2 ion accumulation times. The simulations are for a 60 min active gradient, assuming 256 ms MS1 survey scans, full duty cycles, and two TopN methods, either 5 or 20 MS2 scans per duty cycle.
Detected peptide-like features may correspond to modified peptides that are challenging to identify or may not correspond to peptides at all.7 However, based on experience with bulk MS methods, we may expect that the sequences of over 30% of peptide-like features are identifiable.7 Approaching this identification rate with single-cell MS analysis may result in quantifying 10-fold more peptides than what is possible with a 1–3% identification rate. Such improvement of sequence identification will result in sampling more peptide copies per cell than the RNA copies per cell sampled by single-cell RNA sequencing. This alluring potential is not easy to realize, but it does point to a tangible target that seems within the reach of current MS hardware. Thus, investing in realizing this potential will likely produce significant gains for single-cell proteomics that in turn will empower biomedical research.8
Innovations improving sample preparation, peptide separation, ionization, detection, and quantification may advance single-cell proteomics much beyond the 10-fold increase discussed above. These innovations will range from improvements in chromatographic and electrophoretic peptide separation2,9 to hardware advances, such as brighter ion sources, ion mobility technology, and more sensitive MS detectors. Such advancements will further increase the number of detectable peptide features and create new analytical capabilities.10 Instrumentation advancements will likely be driven in a large part by instrument manufacturers, as exemplified by the timsTOF SCP introduced by Bruker and by the FAIMS introduced by ThermoFisher. While low volume sample preparation methods using microfabricated nano wells11 and surface droplets12 have already reduced adsorptive losses and may afford parallel sample preparation of over 2000 single cells,12 further parallelization is needed to increase the number of single cells that can be simultaneously processed with minimal losses and batch effects. Innovations in all of the above steps hold much potential, but they are not the focus of this perspective; rather, the perspective will focus on innovations in data acquisition and interpretation for MS-based single-cell proteomics.
Indeed, independent from hardware advancement, the development of new data acquisition and interpretation methods may enable confident amino acid sequence identification for a large fraction of the currently detectable peptide-like features. Such new methods may also increase the consistency of peptide quantification across cells and the number of peptide copies sampled per cell. Such gains in single-cell protein analysis can be realized using existing equipment, and these gains will likely extend to more advanced equipment when the equipment becomes available. Developing such methods can benefit from the creativity of the mass-spectrometry community; perhaps many of the most fruitful ideas are yet to come. Below are outlined examples of previous advances, challenges to further advances, and suggested future directions.
DATA ACQUISITION METHODS
The challenges in peptide sequence identification have already motivated experimental methods for enhancing it. One such example is the isobaric carrier concept that combines isobarically labeled peptides from small samples (such as single cells) and a larger (carrier) sample.1,3 As a result, the MS2 spectra contain peptide fragments pooled across the small samples and the isobaric carrier, which enhances peptide sequence identification.1 This approach has been successfully applied to many systems by different researchers, as reviewed recently.13 Yet, the isobaric carrier approach enhances the sequence identification of features sent to MS2 scans, while most detected peptide-like features are not sent for MS2 scans during shotgun analysis, Figure 1.
Indeed, a major challenge for analyzing all detected peptide-like features from single cells are the long ion accumulation times needed to sample sufficient copies from each precursor sent for MS2 analysis.1,10 Thus, the rate of peptide quantification by single-cell shotgun analysis is usually limited by long ion accumulation times, not by the scanning speed of the instruments, Figure 1. Single-cell MS methods commonly use accumulation times of a few hundred milliseconds, which allows performing up to about 10 000 MS2 scans per 60 min active gradient.
Employing Intelligent Data Acquisition Methods
The problem of analyzing relatively few precursors by MS2 scans (Figure 1) is exacerbated by the fact that fewer than 50% of these precursors can be confidently identified in typical bulk and single-cell shotgun experiments.5,7 Thus, one direction for increasing proteome coverage is to allocate the limited MS analysis time only to peptide features that result in confidently identified and well quantified peptides. This can be accomplished by performing long MS2 and MS3 scans only for features sequenced by real-time search.14 A second approach to intelligent data acquisition is to perform MS2 scans only on identifiable precursors from an inclusion list. This may be performed efficiently using real-time retention time alignment.15 This approach also provides the opportunity to select peptides of biological interest and to tailor their analysis based on biological considerations. For example, one may increase the ion accumulation times and thus the sampled peptide copies for peptides of relevance to the investigated biological question.
Parallel Analysis of Both Peptides and Single Cells
Another direction for advancing single-cell MS data acquisition is to accumulate ions in parallel as supported by data independent acquisition (DIA). DIA can also enable the identification of coeluting peptide features with similar mass/charge ratios whose hybrid MS2 spectra pose challenges for data dependent acquisition (DDA). Yet, merely acquiring data for precursors and their fragments by DIA is insufficient for identifying peptide sequences. Indeed, current DIA approaches have not significantly exceeded the number of peptides identified by DDA approaches.16
Thus, new data acquisition and interpretation strategies are needed to take advantage of the parallel ion accumulation afforded by DIA. Such innovations appear feasible and ideally will be synergistically combined with multiplexing strategies to support the high throughput needed by most single-cell applications.8,10,13 Indeed, the regular structures in the data introduced by multiplexing, such as precisely known offsets in mass/charge space, may provide additional constraints on peptide identification. Sequence identification may be further advanced by experimental designs that permit pooling peptide fragments across single cells. Such pooling of fragment ions would enhance their detectability and thus amino acid sequence determination in a manner analogous to the gains observed with the isobaric carrier approach. This analogy extends only to the ion pooling for increased sequencing sensitivity while the implementation of the concept must allow for accurate quantification of each peptide, independent of the superposition of reporter ions inherent to DIA analysis of isobarically labeled samples.13
Peptide Sequence Identification
In addition to acquiring data that support increased peptide sequence identification, single-cell proteomics can be advanced by enhancing the sequence identification and propagation from existing MS methods. This potential has been demonstrated by many methods including spectral libraries, feature matching across runs,17-19 and classification models,20,21 as reviewed in ref 13. Many of the existing methods can be seen as arranged along a gradient displayed in Figure 2. The gradient ranges from the most sensitive approaches using only extracted MS1 ion current to the least sensitive and most reliable approaches using MS1 features, MS2 fragments, and additional peptide properties, such as retention time and ion mobility.
Figure 2.
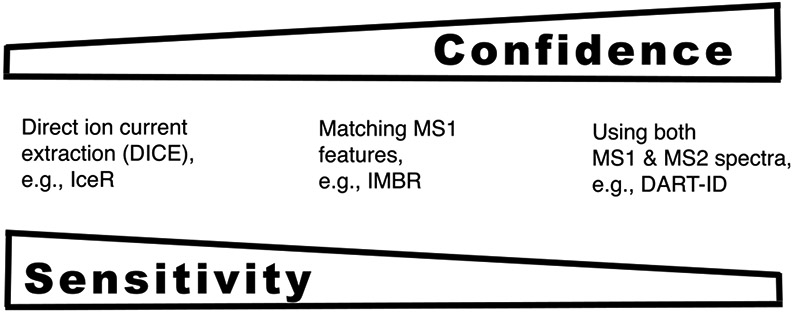
Approaches for peptide sequence identification offer a tradeoff between sensitivity and specificity. Approaches using extracted ion current, such as IceR,22 are likely to offer higher sensitivity of peptide sequence propagation. In contrast, approaches using more features, such as DART-ID,21 are likely to offer higher reliability of peptide sequence identification.
Improving Peptide Identify Propagation
There is a tension between the sensitivity and the reliability of methods for peptide identity propagation (PIP) as shown in Figure 2. The most sensitive methods use extracted ion current, and thus obviate feature identification. Yet, the reliability of these methods may be challenged by isobaric precursors with similar retention times, by variability of peptide separation across experiments, and by contaminant ions. Contaminant ions with charge of +1 can be relatively prevalent in single-cell data, and in the absence of reliable feature identification their extracted ion current might be used for peptide quantification. More generally, methods that rely on accurate retention time and mass/charge ratio of a precursor ion are fundamentally limited by the accuracy of these measurements. These limitations become increasingly restrictive as the proteome coverage increases since more ions must be distinguished from each other.
These limitations may be mitigated by both experimental and computational strategies. Experimental strategies include reducing the variability of peptide separation and decreasing sample contaminants. Contaminants may obscure peptide features and contribute to interferences, which may be mitigated by reducing contaminants by clean and miniaturized sample preparation methods.11,12 Another strategy for enhancing the reliability of amino acid sequence propagation is to advance principled models that use not only MS1-based (precursor) features but also MS2-based (fragment) features to increase the discriminatory power for precursors with very similar masses and separation times, Figure 2. Such methods are best combined with DIA analysis since DIA data provide both MS1 and MS2 features for most detectable peptide ions.
The reliability of new data interpretation methods must be rigorously evaluated not only for reducing missing data but also for quantitative accuracy. The evaluation should use robust benchmarks, such as proteomes of different species mixed in exactly defined ratios. Ultimately, single-cell proteomics needs the most sensitive approaches for peptide identity propagation that result in reliable and robust amino acid sequence identification.
CONCLUSION
The current MS hardware has sufficient sensitivity to support substantial gains in the depth, consistency, and quantitative accuracy of single-cell proteomics. These gains may be realized by innovative data acquisition and interpretation strategies enabling the sequencing of many detectable peptide-like features that are currently unassigned to peptides. These innovations in data acquisition and interpretation will likely interact synergistically with innovations in all other aspects, including sample preparation, peptide separation, and hardware to drive technologies for deeper and quantitatively accurate single-cell proteomics. Investing in such innovations will produce high returns by empowering new biomedical research and discoveries.
ACKNOWLEDGMENTS
This work was funded by an Allen Distinguished Investigator award through The Paul G. Allen Frontiers Group, a New Innovator Award from the NIGMS from the National Institutes of Health under Award Number DP2GM123497, and a Seed Networks Award from CZI, CZF2019-002424.
Footnotes
The author declares no competing financial interest.
REFERENCES
- (1).Specht H; Slavov N Optimizing Accuracy and Depth of Protein Quantification in Experiments Using Isobaric Carriers. J. Proteome Res 2021, 20 (1), 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cong Y; Liang Y; Motamedchaboki K; Huguet R; Truong T; Zhao R; Shen Y; Lopez-Ferrer D; Zhu Y; Kelly RT Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal. Chem 2020, 92 (3), 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Budnik B; Levy E; Harmange G; Slavov N SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity during Cell Differentiation. Genome Biol. 2018, 19 (1), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Schoof EM; Furtwängler B; Üresin N; Rapin N; Savickas S; Gentil C; Lechman E; Keller U. a. D.; Dick JE; Porse BT Quantitative Single-Cell Proteomics as a Tool to Characterize Cellular Hierarchies. Nat. Commun 2021, DOI: 10.1038/s41467-021-23667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Specht H; Emmott E; Petelski AA; Huffman RG; Perlman DH; Serra M; Kharchenko P; Koller A; Slavov N Slavov N Single-Cell Proteomic and Transcriptomic Analysis of Macrophage Heterogeneity Using SCoPE2. Genome Biol. 2021, 22 (1), 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Singh A Towards Resolving Proteomes in Single Cells. Nat. Methods 2021, 18 (8), 856. [DOI] [PubMed] [Google Scholar]
- (7).Chick JM; Kolippakkam D; Nusinow DP; Zhai B; Rad R; Huttlin EL; Gygi SP A Mass-Tolerant Database Search Identifies a Large Proportion of Unassigned Spectra in Shotgun Proteomics as Modified Peptides. Nat. Biotechnol 2015, 33 (7), 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Slavov N Unpicking the Proteome in Single Cells. Science 2020, 367 (6477), 512–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lombard-Banek C; Li J; Portero EP; Onjiko RM; Singer CD; Plotnick DO; Al Shabeeb RQ; Nemes P In Vivo Subcellular Mass Spectrometry Enables Proteo-Metabolomic Single-Cell Systems Biology in a Chordate Embryo Developing to a Normally Behaving Tadpole (X. Laevis)**. Angew. Chem 2021, 133, 12962–12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Specht H; Slavov N Transformative Opportunities for Single-Cell Proteomics. J. Proteome Res 2018, 17 (8), 2565–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian W-J; Kelly RT Nanodroplet Processing Platform for Deep and Quantitative Proteome Profiling of 10–100 Mammalian Cells. Nat. Commun 2018, 9 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Leduc A; Huffman RG; Slavov N Droplet Sample Preparation for Single-Cell Proteomics Applied to the Cell Cycle. bioRxiv, April 26, 2021. DOI: 10.1101/2021.04.24.441211. [DOI] [Google Scholar]
- (13).Slavov N Single-Cell Protein Analysis by Mass Spectrometry. Curr. Opin. Chem. Biol 2021, 60, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schweppe DK; Eng JK; Yu Q; Bailey D; Rad R; Navarrete-Perea J; Huttlin EL; Erickson BK; Paulo JA; Gygi SP Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J. Proteome Res 2020, 19 (5), 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wichmann C; Meier F; Virreira Winter S; Brunner A-D; Cox J; Mann M MaxQuant.Live Enables Global Targeting of More Than 25,000 Peptides. Mol. Cell. Proteomics 2019, 18 (5), 982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cong Y; Motamedchaboki K; Misal SA; Liang Y; Guise AJ; Truong T; Huguet R; Plowey ED; Zhu Y; Lopez-Ferrer D; et al. Ultrasensitive Single-Cell Proteomics Workflow Identifies> 1000 Protein Groups per Mammalian Cell. Chem. Sci 2021, 12 (3), 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yu S-H; Kyriakidou P; Cox J Isobaric Matching between Runs and Novel PSM-Level Normalization in MaxQuant Strongly Improve Reporter Ion-Based Quantification. J. Proteome Res 2020, 19 (10), 3945–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).May D; Fitzgibbon M; Liu Y; Holzman T; Eng J; Kemp CJ; Whiteaker J; Paulovich A; McIntosh M A Platform for Accurate Mass and Time Analyses of Mass Spectrometry Data. J. Proteome Res 2007, 6 (7), 2685–2694. [DOI] [PubMed] [Google Scholar]
- (19).Yu F; Haynes SE; Nesvizhskii AI IonQuant Enables Accurate and Sensitive Label-Free Quantification With FDR-Controlled Match-Between-Runs. Mol. Cell. Proteomics 2021, 20, 100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fondrie WE; Noble WS Machine Learning Strategy That Leverages Large Data Sets to Boost Statistical Power in Small-Scale Experiments. J. Proteome Res 2020, 19 (3), 1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chen AT; Franks A; Slavov N DART-ID Increases Single-Cell Proteome Coverage. PLoS Comput. Biol 2019, 15 (7), e1007082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kalxdorf M; Muller T; Stegle O; Krijgsveld J IceR improves proteome coverage and data completeness in global and single-cell proteomics. Nat. Commun 2021, 12 (1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]