

Abstract
The emerging clustered regularly interspaced short palindromic repeats (CRISPR)-mediated genome editing technologies have progressed remarkably in recent years, opening up the potential of precise genome editing as a therapeutic approach to treat various diseases. The CRISPR-CRISPR-associated (Cas) system is an attractive platform for the treatment of Duchenne muscular dystrophy (DMD), which is a neuromuscular disease caused by mutations in the DMD gene. CRISPR-Cas can be used to permanently repair the mutated DMD gene, leading to the expression of the encoded protein, dystrophin, in systems ranging from cells derived from DMD patients to animal models of DMD. However, the development of more efficient therapeutic approaches and delivery methods remains a great challenge for DMD. Here, we review various therapeutic strategies that use CRISPR-Cas to correct or bypass DMD mutations and discuss their therapeutic potential, as well as obstacles that lie ahead.
Keywords: CRISPR, Cas, Duchenne muscular dystrophy, genome editing, neuromuscular disorder
Graphical abstract
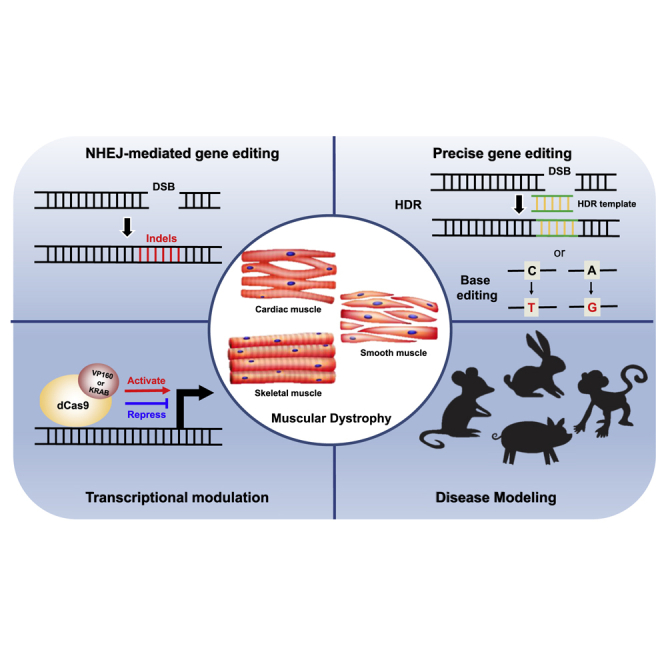
Duchenne muscular dystrophy (DMD) is a neuromuscular disease caused by mutations in the DMD gene. This review summarizes various therapeutic strategies that use CRISPR-Cas to correct or bypass DMD mutations and discuss their therapeutic potential as well as challenges. Finally, the CRISPR-Cas system is suggested as a powerful tool for highly efficient DMD therapy.
Introduction
Duchenne muscular dystrophy (DMD) is a severe, X-linked recessive disease with an average incidence of ∼1 in 5,000 live male births.1 Most DMD patients exhibit progressive muscle degeneration associated with severe muscle weakness, loss of ambulation, cardiac or respiratory complications, and eventually death, in their 20s.2
The DMD gene consists of 79 exons that encode dystrophin, which is a cytoskeletal protein that plays an important role in a complex that connects the cytoskeleton of muscle fibers with the extracellular matrix and is present throughout the cell membrane.3, 4, 5 Different types of mutations in DMD exons and introns cause various forms of dystrophinopathies.3 Approximately 60% of DMD patients harbor a large deletion in the DMD gene, often affecting exons 45–55, a region that represents a mutational hotspot.6 Deletion of a DMD exon can result in a shift in the reading frame and the formation of a premature stop codon, causing either expression of a truncated version of dystrophin that does not function properly or a complete lack of dystrophin expression.
In Becker muscular dystrophy (BMD), a relatively benign type of MD compared to DMD, a semi-functional dystrophin protein is expressed, compensating for the muscle loss.2 Mild BMD symptoms include a relatively slow disease progression, which have little effect on lifespan.7 Thus, alleviating symptoms in DMD patients by expressing a semi-functional protein to mimic a BMD-like disease phenotype could be an efficient strategy for treating DMD. It is notable that a 4% increase in normal dystrophin expression was sufficient to improve muscle function.8, 9, 10
Various pharmacologic therapeutic approaches have focused on converting the DMD phenotype to a BMD-like phenotype by restoring the disrupted DMD reading frame. In 2016, eteplirsen (Exondys 51), an antisense oligonucleotide drug with phosphorodiamidate morpholino oligomer chemistry, became the first medication with such a mechanism to be approved by the US Food and Drug Administration (FDA) for the treatment of DMD. It induces exon 51 skipping in the DMD gene, restoring the expression of semi-functional dystrophin and resulting in BMD-like mild symptoms in eteplirsen-treated DMD patients.11 All of the patients treated with eteplirsen showed an increase in the frequency of dystrophin-positive fibers, by an average of 15.5-fold over untreated controls. In addition, the therapeutic efficacy of golodirsen (Vyondys 53) and viltolarsen (Viltepso), other phosphorodiamidate morpholino oligomer drugs, are under evaluation in clinical trials for treating DMD patients.12,13 Treatment with golodirsen in a Phase I trial resulted in exon 53 skipping and a ∼16-fold increase in dystrophin protein expression over baseline, with 1.02% of normal dystrophin protein expression at week 48. In another study, a 4-week randomized Phase II clinical trial, treatment with viltolarsen caused exon 53 skipping and transcript levels that were 42.4% of normal levels, which in turn led to significant dystrophin production, at 2.8% of normal levels.12, 13, 14 These approaches have reduced disease symptoms, but none have yet eliminated the disease-causing mutation to allow long-term dystrophin expression.
Recently, therapeutic applications of genome editing have been explored for treating various genetic diseases. The clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas) system is a powerful technology for genome editing, especially for correcting disease-causing mutations. Here, we review recent progress in the area of CRISPR-mediated genome editing to treat DMD with various strategies to eliminate pathogenic mutations in the DMD gene. These approaches could also be extensively applied for treating other neuromuscular disorders and other types of genetic diseases.
Genome editing tools: The CRISPR-Cas system
CRISPR-Cas was identified as an adaptive immune system in bacteria and archaea that functions to prevent the invasion of foreign genetic materials.15 Upon viral DNA entry into a bacterium, the cell integrates segments of viral DNA into the CRISPR locus of a bacterial genome. When the same type of virus next invades, RNAs are transcribed from the CRISPR array and cooperate with Cas9 endonuclease to cleave the complementary viral DNA sequence. In the CRISPR system, two RNAs (CRISPR RNA [crRNA] and trans-activating CRISPR RNA [tracrRNA]) are transcribed for target searching. These two components can be linked together to generate a programmable single guide RNA (sgRNA), the form that is now widely used for efficient genome editing. Cas9 derived from the type II CRISPR system of Streptococcus pyogenes (SpCas9) is the most studied and generally used form of the endonuclease; it recognizes a 5′-NGG-3′ or 5′-NAG-3′ protospacer adjacent motif (PAM) and cleaves target DNA 3 bp upstream of the PAM, generating double-strand breaks (DSBs)15, 16, 17 (Figure 1A). Over time, Cas proteins from various other species have been discovered. Among them, Cas12a endonuclease, also called CRISPR from Prevotella and Francisella 1 (Cpf1), is derived from a type V (class II) CRISPR system.18 It has been reported that Cpf1 endonuclease from Acidaminococcus sp. BV3L6 and Lachnospiraceae bacterium ND2006 recognize T-rich PAM motifs (5′-TTTV-3′) and cause staggered end cleavage with equal or greater efficiency than Cas9 orthologs.19 Of special interest, Cpf1 can be used for multiplex genome editing, in which multiple sites are simultaneously edited. Other Cas9 orthologs derived from Staphylococcus aureus (SaCas9)20 or Campylobacter jejuni (CjCas9),21 which are smaller than SpCas9, make it possible to efficiently package the genes encoding these nucleases into small viral vector systems together with its sgRNA. The 2 Cas9 nucleases respectively recognize 5′-NNGRRT-3′ and 5′-NNNVRYAC-3′ PAM sequence before target DNA cleavage.
Figure 1.

CRISPR-Cas genome editing tools
(A) Cas9-sgRNA complexes bind to DNA targets in the genome, generating DSBs 3 bp upstream of the PAM. (B) Cytosine base editors (CBEs), composed of cytidine deaminase fused to nCas9 (D10A) and UGI, enable direct conversion of targeted C:G base pairs to T:A base pairs. (C) Adenine base editors (ABEs), which consist of adenine deaminase fused to nCas9 (D10A), convert targeted A:T base pairs to G:C base pairs. (D) Prime editors consist of nCas9 (H840A) fused to an engineered Moloney murine leukemia virus reverse transcriptase domain. These editors can generate any desired sequence contained in the associated prime editing guide RNA (pegRNA).
More recently, several base editing systems have been developed that allow single base conversion or base editing in cells and organisms in a guide RNA-dependent manner. For targeted base mutagenesis, fusion of a deaminase enzyme, activation-induced cytidine deaminase (AID) or rat APOBEC1, with a catalytically deficient D10A/H840A Cas9 (called dead Cas9 or dCas9) or Cas9 nickase (nCas9) and uracil DNA glycosylase inhibitor (UGI, to prevent base excision repair) enabled direct conversion of a targeted cytidine (C):guanine (G) base pairs to thymine (T):adenine (A) base pairs (Figure 1B).22,23 Furthermore, A base editors (ABEs), which convert A:T base pairs to G:C base pairs, have been constructed using an evolved version of Escherichia coli tRNA adenosine deaminase TadA, TadA∗; they consist of heterodimeric TadA-TadA∗ conjugated with nCas9 (D10A mutation)24 (Figure 1C). These base editing tools convert target bases in a limited editing window located several nucleotide positions upstream of a PAM sequence in the non-target strand. Various approaches are under way to broaden the target window range25, 26, 27, 28 and to increase the efficiency by using various Cas orthologs in this system.29, 30, 31, 32, 33, 34, 35 Although the CRISPR-Cas and base editing systems can precisely install or correct mutations, they have limitations; in particular, Cas9 activity can lead to transversions and random insertions or deletions (indels) at the target site. Moreover, base editors can generate undesired bystander mutations within the base editing window. Most recently, a new genome editing technology, prime editors, was developed with the potential to overcome the limitations of the current genome editing system.36 These editors consist of nCas9 with an inactivated HNH domain (H840A) fused to an engineered Moloney murine leukemia virus reverse transcriptase domain, making it possible to edit the genome to generate any desired sequence36 (Figure 1D). Permanent DNA edits occur when the non-edited strand is replaced by the DNA repair system of the cell using a reverse transcriptase template containing the edit.36 These various CRISPR-Cas systems show great potential for precise genome editing. Here, we summarized the leading strategies for CRISPR-mediated DMD gene editing below (Table 1).
Table 1.
Summary of CRISPR-mediated therapeutic strategies to rescue the DMD phenotype
| Subject | Strategy | Nuclease | DMD mutation | Therapeutic target gene region(s) | Model(s) | Delivery | Reference |
|---|---|---|---|---|---|---|---|
| Therapeutic approach | exon reframing | SpCas9 | DMD exon 48–50 deletions DMD nonsense mutation in exon 51 | DMD exon 51 | human DMD myoblasts | electroporation | 37 |
| SpCas9 | DMD exon 45–52 deletions | DMD exon 53 | human DMD myoblasts | adenovirus | 38 | ||
| SpCas9 | DMD exon 44 deletion | DMD exon 45 | human iPSCs | electroporation | 39 | ||
| CjCas9 | Dmd nonsense mutation in exon 23 | Dmd exon 23 | DMD mice | All-in-one AAV9 | 40 | ||
| LbCpf1 | DMD exon 48–50 deletions DMD nonsense mutation in exon 51 | DMD exon 51 | human iPSCs | nucleofection | 41 | ||
| exon deletion | SpCas9 | Dmd nonsense mutation in exon 23 | Dmd intron 22 and 23 | mdx mice | AAV9 | 42 | |
| SpCas9 | DMD exon 46–51 deletions DMD exon 46–47 deletions | DMD intron 44 and 55 | human DMD myoblasts | nucleofection | 43 | ||
| SaCas9 | Dmd nonsense mutation in exon 23 | Dmd intron 22 and 23 | mdx mice | AAV8 | 44 | ||
| SaCas9 | Dmd nonsense mutation in exon 23 | Dmdexon 23 | Ai9 mdx mice | AAV9 | 45 | ||
| exon skipping | SpCas9 | DMD point mutation in intron 47, exon 51 | DMD exon 47A, exon 51 | human iPSCs | nucleofection | 46 | |
| SpCas9 | DMD exon 44 deletion | DMD splice site of exon 43 or exon 45 | human iPSCs | nucleofection | 47 | ||
| SpCas9 | DMD nonsense mutation in exon 53 | DMD splice acceptor site of exon 53 | human DMD myoblasts | adenovirus | 48 | ||
| SpCas9 | Dmd exon 50 deletion | Dmd splice acceptor site of exon 51 | DMD mice | AAV9 | 49 | ||
| SpCas9 | DMD exon 50 deletion | DMD splice acceptor site of exon 51 | canine model of DMD | AAV9 | 50 | ||
| homology-directed repair | SpCas9 | Dmd nonsense mutation in exon 23 | Dmd exon 23 | mdx mice | injection | 51 | |
| SpCas9 | Dmd nonsense mutation in exon 23 | Dmd exon 23 | mouse muscle stem cells | adenovirus | 52 | ||
| SpCas9 | Dmd nonsense mutation in exon 53 | Dmd exon 53 | mdx4cv mice | AAV6 | 53 | ||
| SpCas9 | DMD exon 44 deletion | DMD exon 44 | human iPSCs | electroporation | 39 | ||
| SpCas9 | DMD exon 7 skipping | DMD splice acceptor site of intron 6 and exon 7 boundary | canine model of DMD | injection | 54 | ||
| LbCpf1 | Dmd nonsense mutation in exon 23 | Dmd exon 23 | mdx mice | injection | 41 | ||
| base editing (exon skipping) | TAM based on SaCas9 (Cytosine base editor) | DMD exon 51 deletion | DMD splice site of exon 50 | human iPSCs | lipotransfection | 55 | |
| base editing (correction) | ABE7.10 | Dmd nonsense mutation in exon 20 | Dmd exon 20 | DMD mice | trans-splicing AAV | 56 | |
| transcriptional modulation | CRISPRa (dCas9-VP160) | DMD exon 45–52 deletions | UTRN A, B promoter | immortalized DMD patient muscle cells | Electroporation | 57 | |
| CRISPRa (dCas9-VP160) | Dmd nonsense mutation in exon 23 | Lama1 promoter | mouse myoblasts, mdx/rag mice | transfection, injection, electroporation | 58 | ||
| CRISPRa (dCas9-VP64) | Dmd nonsense mutation in exon 23 | klotho and Utrn | mdx mice | AAV9 | 59 | ||
| SaCas9 | DMD exon 46–51 deletions | 3′ UTR of UTRN inhibitory miRNA target region | human iPSCs | electroporation | 60 | ||
| CRISPRi (dCas9-KRAB) | epigenetic dysregulation of DUX4 | DUX4 promoter or DUX4 exon 1 | human FSHD myocytes | lentivirus | 61 |
AAV, adeno-associated viral vector; ABE, adenine base editor; CRISPRa, CRISPR activator; CRISPRi, CRISPR interference; FSHD, facioscapulohumeral muscular dystrophy; hiPSC, human induced pluripotent stem cell; TAM, targeted AID mediate mutagenesis.
Therapeutic approach: Exon reframing
Approximately 51% of DMD patients have deleterious frameshifting exon deletion mutations that interrupt the DMD open reading frame (ORF) based on the Leiden DMD mutation database.62 In the case of DMD-causing frameshift mutations, small indels generated by non-homologous end joining (NHEJ)-mediated repair upstream of the premature stop codon have a 1 in 3 probability of reframing the ORF (Figure 2A). Several groups have demonstrated successful DMD exon reframing with this strategy.37, 38, 39, 40, 41 As one example of this approach, in a DMD mouse model, CjCas9 and its Dmd exon 23-specific sgRNA were used to target a site upstream of a premature stop codon caused by a frameshift mutation in Dmd exon 23. After CjCas9-induced cleavage, NHEJ at the cleaved site reframed the ORF. Compared to other Cas9 nucleases, CjCas9 is notable for having the smallest known size to date. In this study, sequences encoding CjCas9 and its sgRNA were packaged into an all-in-one adeno-associated viral (AAV) vector serotype 9, maximizing the delivery efficiency to target muscles.40 This treatment resulted in indel formation at the target site, with a frequency of up to 8%, which in turn led to dystrophin expression in 28%–39% of muscle fibers and improved muscle strength, demonstrating the possibility of applying the CRISPR system to correct the DMD ORF in vivo.40
Figure 2.

Mechanisms of CRISPR-mediated genome editing to correct mutations in the DMD gene or ameliorate the effects of such mutations
(A) Exon reframing induced by NHEJ. Small indels that are generated upstream of the premature stop codon in DMD exon 51 have a 1 in 3 probability of reframing the ORF. (B) Exon deletion using 2 sgRNAs targeting intronic regions flanking the mutated Dmd exon 23. (C) DMD exon 51 skipping induced by disruption of a splice acceptor (SA) site to juxtapose exons 49 and 52 in the mRNA and reframed the ORF. (D) Precise, HDR-mediated mutation correction using Cas9, 2 guide RNAs targeted to sites flanking the mutated Dmd exon 23, and ssODNs. (E) Base editor-mediated exon skipping using a CBE or mutation correction using an ABE. (F) CRISPRa-mediated epigenetic editing to upregulate utrophin expression. (G) CRISPRi-mediated epigenetic editing to inhibit DUX4 expression.
Therapeutic approach: Exon deletion
Deletion of one or more exons can be used to restore disrupted ORF when it is shifted to be out-of-frame by frameshift mutations or the deletion or duplication of exon(s). In addition, the effects of certain point mutations can be rescued by removal of the mutated exon (Figure 2B). As examples of this approach, 2 sgRNAs were designed to remove the mutated exon by targeting intronic regions flanking the mutated Dmd exon 23 in mdx mice, which contain a nonsense mutation.42,44,45 Of note, Ai9 mdx mice treated with AAV9-expressing SaCas9 and appropriate 2 sgRNAs targeting intronic regions flanking the exon 23 exhibited exon 23 deletion at a frequency of 39% in tibialis anterior (TA) muscles, demonstrating the therapeutic potential of this approach in vivo.45 In human genome editing, deletion of a mutation hotspot spanning DMD exons 45 to 55 could treat ∼60% of DMD patients.37 Several groups have demonstrated deletion of this hotspot from the human DMD gene with therapeutic effects. In a study performed in human myoblasts from DMD patients, 2 sgRNAs, one targeting the 5′ end of exon 45 and the other targeting the 3′ end of exon 55, resulted in deletion from the genome of 336 kb that contained exons 45–55. This approach led to dystrophin expression with an edited allele efficiency of 5%–10%.37 Similarly, use of 2 sgRNAs targeting regions flanking exons 45–55 resulted in the deletion of up to 725 kb from both cardiomyocytes and skeletal cells generated from human induced pluripotent stem cells (hiPSCs) derived from a DMD patient.43 NSG-mdx scid mice engrafted with these exon 45- to 55-deleted DMD hiPSCs showed dystrophin expression and colocalization with the dystrophin-associated transmembrane protein, β-dystroglycan at the sarcolemma, suggesting that the deletion of a mutation hotspot would be clinically relevant.
Therapeutic approach: Exon skipping
Exon skipping induced by abolishing conserved RNA splice sites is a powerful strategy for restoring the DMD ORF. The strategy of causing one or more exons to be skipped in the process of RNA splicing could be useful for treating up to 83% of DMD patients.62 The introduction of indels by NHEJ at an RNA splice site or deletion of the splice site of an out-of-frame exon abolishes splice site function, leading to targeted exon skipping (Figure 2C). Several CRISPR-mediated exon skipping approaches, involving splice site targeting, have been demonstrated. In particular, SpCas9-mediated NHEJ at 5′ or 3′ splice sites containing 5′-NAG-3′ or 5′-NGG-3′ PAM motifs can remove essential splice donor or acceptor sequences for skipping of the corresponding target exon. With this strategy, skipping of exons 43, 51, and 53 induced by NHEJ-induced disruption of splice acceptor (SA) sites was demonstrated in hiPSCs and human myoblasts.46, 47, 48 In evaluations of this approach in vivo, skipping of exon 51 was induced, leading to ORF reframing and restoration of dystrophin expression, in mouse49 and canine models of DMD that lack exon 50.50 In these studies, splicing acceptor sites adjacent to exon 51 were modified such that exon 51 was skipped, resulting in the juxtaposition of exon 49 and 52 in the mRNA and ORF reframing.
Therapeutic approach: HDR-mediated gene correction
Homology-directed repair (HDR)-mediated genome editing can restore full-length dystrophin gene expression, whereas NHEJ-mediated exon reframing results in a truncated form of dystrophin. To induce HDR, Cas9, a guide RNA targeting the mutated region, and single-stranded oligodeoxynucleotides (ssODNs) or a donor template with the correct sequence are required (Figure 2D). Several studies have corrected the Dmd gene by knockin strategies targeting Dmd exon 23,41,51,52 exon 53,53 and DMD exon 44.39 As one example of this approach, the nonsense mutation in exon 53 in mdx4cv mice was repaired by intramuscular injection of AAV6 carrying Cas9, sgRNA, and donor template sequences into TA muscles.53 Successful HDR occurred in 0.18% of the total genomes, which led to full-length dystrophin expression that was 1.8%–8.4% of that seen in wild-type (WT) mouse muscles.53 In addition, the nonsense mutation in Dmd exon 23 in the mdx mice was corrected by SpCas9 with a 180-nt ssODN51 or LbCpf1 with a 180-nt ssODNs,41 together with a corresponding gRNA, resulting in correction rates of 17% to 41%51 and 8% to 50%, respectively.41 HDR-mediated genome editing has also been demonstrated in a large animal model. The defect of golden retriever MD dog, which contains splice site mutations that lead to exon 7 skipping, was repaired by CRISPR-induced HDR.54 The ssODNs used in this study included the correct DMD sequence at the intron 6 acceptor splice site.54 With this HDR-mediated repair, examination of muscle biopsies showed that DMD mRNA expression was increased and dystrophin expression was restored to 6% to 16% of normal levels.54
There are several limitations to HDR-based DMD therapy. First, the length of the donor DNA template is limited, so the technique is not applicable to large DMD deletion mutations. Second, HDR is restricted to the S and G2 phases of the cell cycle, when sister chromatids are available to accept the template DNA63; hence, G1-arrested cells (post-mitotic cells) such as mature myofibers and cardiomyocytes are not corrected efficiently by HDR-mediated gene editing.53 Third, unwanted DNA fragments may be integrated into the DMD locus, resulting in an altered dystrophin expression. Lastly, because NHEJ is dominant in mammalian cells, HDR occurs at a much lower frequency than NHEJ. To overcome the low efficiency of HDR in muscles, the recently developed CRISPR-prime editing system has great potential for repairing the target DMD locus with direct reverse transcription of the desired sequence.
Therapeutic approach: Base editing
Therapeutic application of base editing in DMD is a promising strategy because the precise editing of a single base in the targeted site is possible without the generation of DNA DSBs. This method can correct point mutations in the DMD gene, which account for ∼27% of DMD cases.62 CRISPR-Cas-mediated base editing for the treatment of DMD has been demonstrated using two strategies: modulation of splicing or correction of a nonsense mutation (Figure 2E). Using a cytosine base editor (CBE; AID fused to nSpCas9 or nSaCas9), the G in the 5′ splice site of DMD exon 50 was targeted, disrupting the splice site and thereby leading to the skipping of exon 50 during mRNA splicing.55 Approximately 90% of the genome acquired the intended G > A conversion, leading to exon 50 skipping in 99.9% of the DMD transcripts in cardiomyocytes differentiated from hiPSCs.55 Another demonstration of in vivo base editing was the ABE-mediated correction of a nonsense mutation in a DMD mouse model.56 To correct this mutation, located in Dmd exon 20, sequences encoding ABE7.10 (a TadA-TadA∗-nSpCas9 fusion) were delivered to the TA muscles of the DMD mice. The ABE7.10-encoding construct was split into two parts to overcome the packaging limitations of AAV using a trans-splicing AAV (tsAAV) vector system. The two ABE7.10 sequence segments were packaged into independent AAV vectors and then delivered into TA muscles intramuscularly. The AAV vectors were combined via recombination between the two inverted terminal repeat sequences in each AAV vector during AAV concatemerization in a cell. The tsAAV-ABE-treated mouse muscles showed conversion of the stop codon (TAG) into a Gln codon (CAG) with a frequency of 3.3%, leading to increased dystrophin expression (up to 17% of the WT level) and colocalization with the nNOS protein at the sarcolemma, demonstrating the feasibility of ABE-mediated in vivo base editing for DMD.56
Transcriptional modulation using CRISPR-Cas
The CRISPR system has been engineered to regulate gene expression by fusing inactivated dCas9 with a transcriptional activator or repressor, generating tools called CRISPR activator (CRISPRa)64, 65, 66 or CRISPR interference (CRISPRi), respectively.67 Modulation of the expression of a gene related to the disease process, but different from the classic disease-associated gene, could be a new therapeutic approach for DMD and other diseases. Because dystrophin dysfunction has been considered to be the major cause of DMD, compensating for its lack of function with a different protein that functions similarly could be a novel treatment strategy. One advantage of such epigenetic editing is that it could be applicable to any of the DMD-associated mutations; furthermore, no DNA DSB is required to regulate gene expression in this approach. Utrophin, the cytoskeletal protein with a name that is a contraction of “ubiquitous dystrophin” is a homolog of dystrophin that is encoded by the autosomal UTRN gene. This protein is expressed in the myotendinous and neuromuscular junctions of adult skeletal muscles.68,69 Several studies have shown that the upregulation of utrophin could rescue DMD phenotypes.57,59,60,70, 71, 72, 73 Treatment with dCas9-VP160 (dead SpCas9 fused to 10 tandem repeats of the transcriptional transactivator VP16) to target the UTRN A or B promoters, respectively, resulted in 1.7- to 2.7-fold or 3.8- to 6.9-fold increase in utrophin upregulation57 (Figure 2F). Furthermore, SaCas9-mediated deletion of 5 inhibitory microRNA target regions within the UTRN 3′ untranslated region (UTR) resulted in 2-fold higher levels of utrophin in DMD-hiPSCs.60
The laminin protein is another potential compensatory molecule for DMD; the laminin complex links the extracellular matrix to integrin α7β1 in the sarcolemma, and thus could compensate for a loss of dystrophin in dystrophic muscles. Because injection of laminin-111 to mdx mouse muscles stabilized the sarcolemma, dCas9-VP160 was used to target the Lama1 promoter, leading to increased laminin-111 expression.58 In addition, klotho, a transmembrane protein that is epigenetically silenced in muscle cells of mdx mice,74 has been targeted for upregulation by CRISPRa.59 Systemic injection of AAV9 encoding a klotho-targeting sgRNA and dCas9-VP64 to neonatal mdx mice restored klotho expression in muscle tissue and ameliorated DMD phenotypes.59 Expression of DMD inhibitory molecules can be repressed by CRISPRi that inhibits their transcriptional start sites by catalyzing repressive chromatin modifications. In a study of facioscapulohumeral muscular dystrophy (FSHD), which results from epigenetic dysregulation of the DUX4 gene in muscle, CRISPRi was used to downregulate the expression of DUX4 and potential DUX4 activators61 (Figure 2G). The CRISPRi system used here consists of dCas9 fused to a repressive Krüppel associated box (KRAB) domain and an appropriate sgRNA.61 Another potential target for CRISPRi to treat DMD is myostatin (MSTN), a cytokine that is secreted by skeletal muscle cells and is a well-known cause of muscle atrophy with muscle wasting.75 Several studies have reported that CRISPR-induced knockout of Mstn or MSTN increased muscle mass and myotube formation.76,77 However, it has also been reported that the MSTN knockout is only slightly beneficial for inducing excessive muscle growth but causes impaired force generation.78, 79, 80 Therefore, further extensive investigations of this approach are required before it can be applied in humans therapeutically.
Animal modeling of DMD
Generating animal models of DMD makes it possible to study the pathophysiology of the disease and to evaluate the efficacy of biodrugs before clinical trials. With this aim, CRISPR-Cas-mediated DMD animal modeling has been demonstrated in mouse,40,47,49,81, 82, 83, 84, 85 rat,86 rabbit,87 pig,88 and monkey89 (Table 2). A DMD mouse model was generated using CBE, namely Base Editor 3 (BE3): a nonsense mutation (CAG [Gln] to TAG [stop codon]) was introduced in exon 20 of the Dmd gene in the mouse genome (Figure 3A). In this study, BE3 (rat APOBEC1-nCas9 (D10A)-UGI)-encoding mRNA and sgRNAs were introduced into a mouse embryo by microinjection or electroporation, respectively, resulting in 73% and 81% of blastocysts containing the Dmd exon 20 mutation.82 This mouse model was further used to correct nonsense mutations using ABE. as described above in the Therapeutic approach: Base editing section. In addition, the humanized DMD mouse model,90 in which the entire human DMD sequences were integrated into mouse chromosome 5, was used for evaluating the efficiency of CRISPR tools that target the human gene. To place DMD out-of-frame, an exon 45 deletion was induced using Cas9 together with 2 sgRNAs targeting intron 44 and intron 45 of the human DMD gene in a humanized DMD mouse zygote81 (Figure 3B). Treatment of CRISPR targeting intronic regions flanking DMD exons 45–55 in these mice resulted in the rejoining of introns 44 and 55, which in turn led to dystrophin expression at the sarcolemma.81 However, humanized DMD mice are less than ideal for generating knockout models. They contain 2 copies of the DMD transgene, integrated in a tail-to-tail orientation, and both must be modified to result in the desired phenotype.91 A DMD rat model was also generated by targeting both exon 3 and exon 16 to induce a deletion of the region spanning these 2 exons.86 DMD rodent models mimic the defective and pathological features of the disease, yet they do not fully represent the phenotype of DMD patients due to the compensatory effect of utrophin and robust muscle regeneration in these models.92 Thus, therapeutic strategies may benefit from examining more severe mammalian models of DMD that better mimic the pathology of DMD patients.
Table 2.
Summary of animal models of DMD generated by the CRISPR-Cas system
| Subject | Animal | Strategy | Nuclease | DMD mutation | Target gene region(s) | Strain | Delivery | Reference |
|---|---|---|---|---|---|---|---|---|
| Disease modeling | mouse | exon deletion | SpCas9 | DMD exon 45 deletion | DMD intron 44 and 45 | C57BL/10 and DBA/2 | microinjection | 81 |
| SpCas9 | Dmd exon 50 deletion | Dmd intron 49 and 50 | C57BL/6J | not indicated | 49 | |||
| SpCas9 | Dmd exon 44 deletion | Dmd intron 43 and 44 | C57BL/6J | microinjection | 47 | |||
| SpCas9 | Dmd exon 8–34 deletion | Dmd intron 7 and 34 | C57BL/6J xCBA | microinjection | 83 | |||
| SpCas9 | Dmd exon 50 deletion | Dmd intron 49 and 50 | not indicated | not indicated | 84 | |||
| SpCas9 | Dmd exon 43, 45, and 52 deletion |
Dmd intron 42 and 43 Dmd intron 44 and 45 Dmd intron 51 and 52 |
C57BL/6 | microinjection | 85 | |||
| frameshift mutation | CjCas9 | Dmd frameshift mutation in exon 23 | Dmd exon 23 | C57BL/6J | microinjection | 40 | ||
| base editing | BE3 | Dmd nonsense mutation in exon 20 | Dmd exon 20 | C57BL/6J and ICR mice | microinjection and electroporation | 82 | ||
| rat | exon deletion | SpCas9 | Dmd exon 3–16 deletions | Dmd exon 3 and exon 16 | Wistar-Imamichi rats | microinjection | 86 | |
| rabbit | frameshift mutation | SpCas9 | DMD frameshift mutation in exon 51 | DMD exon 51 | New Zealand rabbits | microinjection | 87 | |
| pig | base editing | BE3, hA3A-BE3 | DMD nonsense mutation in exon 13 | DMD exon 13 | Bama miniature pigs and large white pigs | microinjection | 88 | |
| monkey | frameshift mutation | SpCas9 | DMD frameshift mutation in exon 4 and/or exon 46 | DMD exon 4 and/or exon 46 | Macaca mulatta rhesus monkeys | microinjection | 89 |
BE3, base editor 3; hA3A-BE3, hAPOBEC3A base editor 3; hDMD, humanized DMD.
Figure 3.

CRISPR-mediated animal modeling of DMD
(A) DMD mice (left panel) and DMD pigs (right panel) with a nonsense mutation in Dmd exon 20 and DMD exon 13, respectively, generated by the CBE BE3. (B) Humanized DMD mice in which DMD exon 45 has been deleted using SpCas9 and 2 sgRNAs targeting the sites flanking the region to be deleted. (C) DMD monkeys with frameshift mutations in exon 4 and/or exon 46 induced by CRISPR-mediated NHEJ. WT, wild type; MT, mutant type.
Compared to mice, rabbits show more similarities to humans in their physiology, anatomy, and genetics, making them suitable models for cardiac and metabolic diseases. With these advantages, rabbits are an appropriate focus for DMD modeling. CRISPR-Cas was used to induce a desired exon 51 knockout in the rabbit DMD gene; 78.8% of newborn pups carried a single DMD mutation and 84.6% carried biallelic DMD mutations.87 This model sufficiently mimics the histopathological and functional defects of DMD patients, including impaired mobility and defects in muscle regeneration, suggesting it will be a useful model for preclinical studies.87 DMD pig models have also been successfully generated.88 A nonsense mutation was generated in the DMD gene in 75% of embryos injected with BE3 mRNA and an appropriate sgRNA (Figure 3A). A newer version of BE3 based on human APOBEC3A induced mutations in 50% of the embryos.88 Ultimately, 1 live heterozygous piglet, which carried the C-to-T mutation in one DMD allele and an 18-bp deletion in the other was born after mating DMD−/+ female pigs with DMD+/y pigs.88 It is notable that previously generated DMD−/− or DMD−/y pigs generated using CRISPR/Cpf1 could not survive >3 months, whereas the heterozygous female DMD pig mentioned above survived for >1 year.88 Nonhuman primates, represented by rhesus monkeys (Macaca mulatta), are essential for disease modeling given their strong similarities to humans across physiological, developmental, behavioral, immunologic, and genetic levels.93 CRISPR has been successfully used to generate DMD mutant rhesus monkeys by inducing frameshift mutations in exon 4 and/or exon 4689 (Figure 3C). After the injection of Cas9 mRNA and sgRNA, 46.47% of embryos carried different indel mutations in DMD.89 After Cas9-injected embryos were transferred to surrogate rhesus monkey mothers, mosaic frameshifting DMD mutations were observed in 2 stillborn and 9 live monkeys, which represented a gene-targeting rate of 61.1%.89 This monkey showed early muscle atrophy pathology, indicating that a monkey model of DMD was successfully generated.
Further extensive evaluations of pathophysiology in the animal models of DMD are required. Thereafter, these models could provide an alternative option as a new platform for investigating the therapeutic effects of biodrugs before clinical trials.
Challenges: Off-target effects of CRISPR-Cas
Off-target effects of the CRISPR system could lead to indels or base editing in non-targeted regions of the genome, causing unexpected genomic instabilities. Cancer or other diseases may be generated with such off-target nuclease activity, carrying serious risks that would outweigh the benefits of genome editing. Hence, the off-target effects of the CRISPR system must be carefully evaluated. A number of different approaches are being investigated to reduce such off-target activity. To date, several bioinformatic tools, including CRISPR-OFFinder,94 CCTop,95 and CT-Finder,96 have been developed to predict potential off-target sites in the whole genome. In particular, CIRCLE-seq,97 Digenome-seq,98 and GUIDE-seq,99 which are in vitro-based assays, are useful methods for identifying potential off-target sites. These methods account for off-target cleavage sites that may be affected by genetic variation. In addition, efforts to improve the specificity of Cas9 are under way, including the development of enhanced specificity SpCas9 (eSpCas9100), evoCas9,101 HypaCas9,102, and high-fidelity SpCas9 (SpCas9-HF1,103 HeFSpCas9104), and the use of truncated guide RNA.105 In addition, the use of the small anti-CRISPR protein AcrII could regulate Cas expression and thereby reduce indiscriminate cleavage.106
Challenges: Delivery efficiencies, toxicity, and immunogenicity
AAV is the most commonly used vector for DMD gene therapy because of its ability to target muscle tissue with high efficiency.107, 108, 109 AAV vector has several benefits, including low pathogenicity, low immunogenicity, and the ability to provide long-term expression as a gene carrier. However, it is crucial to study the safety of this viral vector for the application of the CRISPR approach before clinical treatment. Several studies have demonstrated that systemic delivery of a high dose of AAV vector (2 × 1014 vg/kg) induces systemic and sensory neuronal toxicity in rhesus monkeys and piglets.110,111 A severe cellular immune response also occurred in the Phase I/II clinical study of recombinant AAV gene therapy for hemophilia.112
In addition, CRISPR components can generate an immune response in vivo because they are derived from foreign bacteria.113 If a patient had been previously exposed to Cas proteins from a given species and then re-exposed during CRISPR-based therapy, a sustained immune response could also lead to Cas endonuclease clearance, which would greatly reduce the effectiveness of treatment and could lead to organ impairment. It has been reported that 58% and 78% of healthy people had anti-Cas9 antibodies against SaCas9 and SpCas9, respectively.114 This result shows the potential for numerous patients to exhibit an immune response following Cas9 treatment, ultimately resulting in reduced gene editing efficiency. It has been reported that such humoral and cellular immune response can be avoided by treating neonatal mice.115 Thus, there is an urgent need to develop highly efficient AAV-CRISPR systems that are effective at low doses and to establish a treatment protocol for the appropriate age group of DMD patients. In addition, modification of the AAV capsid to make it even less immunogenic could maximize the therapeutic effect.
Conclusions
The CRISPR-Cas system provides a powerful genome editing tool for highly efficient DMD therapy. Rapid progress in DMD genome editing is occurring, with the evaluation of the safety and efficacy of therapeutic strategies. When the current limitations of the CRISPR system for the treatment of DMD are overcome, CRISPR-based tools will offer the means to permanently correct DMD mutation or ameliorate their effects. Continued development of the CRISPR system as a means of DMD therapy indicates that it should be an alternative gene therapy technology in the near future.
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea, South Korea (2019R1C1C1005851 to T.K.) and the Technology Innovation Program (20012445 to T.K.) funded by the Ministry of Trade, Industry and Energy, South Korea.
Author contributions
Both of the authors contributed to the writing of this review.
Declaration of interests
The authors declare no competing interests.
References
- 1.Sussman M. Duchenne muscular dystrophy. J. Am. Acad. Orthop. Surg. 2002;10:138–151. doi: 10.5435/00124635-200203000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Cai A., Kong X. Development of CRISPR-Mediated Systems in the Study of Duchenne Muscular Dystrophy. Hum. Gene Ther. Methods. 2019;30:71–80. doi: 10.1089/hgtb.2018.187. [DOI] [PubMed] [Google Scholar]
- 3.Lim K.R.Q., Yoon C., Yokota T. Applications of CRISPR/Cas9 for the treatment of duchenne muscular dystrophy. J. Pers. Med. 2018;8:38. doi: 10.3390/jpm8040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts R.G., Coffey A.J., Bobrow M., Bentley D.R. Exon structure of the human dystrophin gene. Genomics. 1993;16:536–538. doi: 10.1006/geno.1993.1225. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman E.P., Brown R.H., Jr., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 6.Bladen C.L., Salgado D., Monges S., Foncuberta M.E., Kekou K., Kosma K., Dawkins H., Lamont L., Roy A.J., Chamova T. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monaco A.P., Bertelson C.J., Liechti-Gallati S., Moser H., Kunkel L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 8.van Putten M., Hulsker M., Young C., Nadarajah V.D., Heemskerk H., van der Weerd L. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J. 2013;27:2484–2495. doi: 10.1096/fj.12-224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li D., Yue Y., Duan D. Marginal level dystrophin expression improves clinical outcome in a strain of dystrophin/utrophin double knockout mice. PLoS ONE. 2010;5:e15286. doi: 10.1371/journal.pone.0015286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Putten M., van der Pijl E.M., Hulsker M., Verhaart I.E.C., Nadarajah V.D., van der Weerd L. Low dystrophin levels in heart can delay heart failure in mdx mice. J. Mol. Cell. Cardiol. 2014;69:17–23. doi: 10.1016/j.yjmcc.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Heo Y.A. Golodirsen: First Approval. Drugs. 2020;80:329–333. doi: 10.1007/s40265-020-01267-2. [DOI] [PubMed] [Google Scholar]
- 12.Clemens P.R., Rao V.K., Connolly A.M., Harper A.D., Mah J.K., Smith E.C., McDonald C.M., Zaidman C.M., Morgenroth L.P., Osaki H., CINRG DNHS Investigators Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: A phase 2 randomized clinical trial. JAMA Neurol. 2020;77:982–991. doi: 10.1001/jamaneurol.2020.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komaki H., Nagata T., Saito T., Masuda S., Takeshita E., Sasaki M., Tachimori H., Nakamura H., Aoki Y., Takeda S. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci. Transl. Med. 2018;10:eaan0713. doi: 10.1126/scitranslmed.aan0713. [DOI] [PubMed] [Google Scholar]
- 14.Frank D.E., Schnell F.J., Akana C., El-Husayni S.H., Desjardins C.A., Morgan J., Charleston J.S., Sardone V., Domingos J., Dickson G., SKIP-NMD Study Group Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94:e2270–e2282. doi: 10.1212/WNL.0000000000009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Ge X., Yang F., Zhang L., Zheng J., Tan X., Jin Z.B., Qu J., Gu F. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 2014;4:5405. doi: 10.1038/srep05405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tóth E., Weinhardt N., Bencsura P., Huszár K., Kulcsár P.I., Tálas A., Fodor E., Welker E. Cpf1 nucleases demonstrate robust activity to induce DNA modification by exploiting homology directed repair pathways in mammalian cells. Biol. Direct. 2016;11:46. doi: 10.1186/s13062-016-0147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim E., Koo T., Park S.W., Kim D., Kim K., Cho H.Y., Song D.W., Lee K.J., Jung M.H., Kim S. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017;8:14500. doi: 10.1038/ncomms14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y., Zhang J., Yin W., Zhang Z., Song Y., Chang X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods. 2016;13:1029–1035. doi: 10.1038/nmeth.4027. [DOI] [PubMed] [Google Scholar]
- 23.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan J., Zhang F., Karcher D., Bock R. Expanding the genome-targeting scope and the site selectivity of high-precision base editors. Nat. Commun. 2020;11:629. doi: 10.1038/s41467-020-14465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng T.L., Li S., Yuan B., Wang X., Zhou W., Qiu Z. Expanding C-T base editing toolkit with diversified cytidine deaminases. Nat. Commun. 2019;10:3612. doi: 10.1038/s41467-019-11562-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang T.P., Zhao K.T., Miller S.M., Gaudelli N.M., Oakes B.L., Fellmann C., Savage D.F., Liu D.R. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol. 2019;37:626–631. doi: 10.1038/s41587-019-0134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.J., Liquori A.J. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 29.Kim Y.B., Komor A.C., Levy J.M., Packer M.S., Zhao K.T., Liu D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017;35:371–376. doi: 10.1038/nbt.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z., Shan H., Chen S., Chen M., Song Y., Lai L., Li Z. Efficient base editing with expanded targeting scope using an engineered Spy-mac Cas9 variant. Cell Discov. 2019;5:58. doi: 10.1038/s41421-019-0128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Z., Wang S., Zhang C., Gao N., Li M., Wang D., Wang D., Liu D., Liu H., Ong S.G. A compact Cas9 ortholog from Staphylococcus auricularis (SauriCas9) expands the DNA targeting scope. PLoS Biol. 2020;18:e3000686. doi: 10.1371/journal.pbio.3000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X., Wang Y., Liu Y., Yang B., Wang X., Wei J., Lu Z., Zhang Y., Wu J., Huang X. Base editing with a Cpf1-cytidine deaminase fusion. Nat. Biotechnol. 2018;36:324–327. doi: 10.1038/nbt.4102. [DOI] [PubMed] [Google Scholar]
- 35.Kleinstiver B.P., Sousa A.A., Walton R.T., Tak Y.E., Hsu J.Y., Clement K., Welch M.M., Horng J.E., Malagon-Lopez J., Scarfò I. Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 2019;37:276–282. doi: 10.1038/s41587-018-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ousterout D.G., Kabadi A.M., Thakore P.I., Majoros W.H., Reddy T.E., Gersbach C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maggio I., Liu J., Janssen J.M., Chen X., Gonçalves M.A.F.V. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep. 2016;6:37051. doi: 10.1038/srep37051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H.L., Fujimoto N., Sasakawa N., Shirai S., Ohkame T., Sakuma T., Tanaka M., Amano N., Watanabe A., Sakurai H. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2015;4:143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koo T., Lu-Nguyen N.B., Malerba A., Kim E., Kim D., Cappellari O., Cho H.Y., Dickson G., Popplewell L., Kim J.S. Functional Rescue of Dystrophin Deficiency in Mice Caused by Frameshift Mutations Using Campylobacter jejuni Cas9. Mol. Ther. 2018;26:1529–1538. doi: 10.1016/j.ymthe.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y., Long C., Li H., McAnally J.R., Baskin K.K., Shelton J.M., Bassel-Duby R., Olson E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017;3:e1602814. doi: 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young C.S., Hicks M.R., Ermolova N.V., Nakano H., Jan M., Younesi S., Karumbayaram S., Kumagai-Cresse C., Wang D., Zack J.A. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell. 2016;18:533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Long C., Li H., Tiburcy M., Rodriguez-Caycedo C., Kyrychenko V., Zhou H., Zhang Y., Min Y.L., Shelton J.M., Mammen P.P.A. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018;4:eaap9004. doi: 10.1126/sciadv.aap9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Min Y.L., Li H., Rodriguez-Caycedo C., Mireault A.A., Huang J., Shelton J.M., McAnally J.R., Amoasii L., Mammen P.P.A., Bassel-Duby R., Olson E.N. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019;5:eaav4324. doi: 10.1126/sciadv.aav4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maggio I., Stefanucci L., Janssen J.M., Liu J., Chen X., Mouly V., Gonçalves M.A.F.V. Selection-free gene repair after adenoviral vector transduction of designer nucleases: rescue of dystrophin synthesis in DMD muscle cell populations. Nucleic Acids Res. 2016;44:1449–1470. doi: 10.1093/nar/gkv1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amoasii L., Long C., Li H., Mireault A.A., Shelton J.M., Sanchez-Ortiz E., McAnally J.R., Bhattacharyya S., Schmidt F., Grimm D. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017;9:eaan8081. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Long C., McAnally J.R., Shelton J.M., Mireault A.A., Bassel-Duby R., Olson E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu P., Wu F., Mosenson J., Zhang H., He T.C., Wu W.S. CRISPR/Cas9-Mediated Genome Editing Corrects Dystrophin Mutation in Skeletal Muscle Stem Cells in a Mouse Model of Muscle Dystrophy. Mol. Ther. Nucleic Acids. 2017;7:31–41. doi: 10.1016/j.omtn.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mata López S., Balog-Alvarez C., Vitha S., Bettis A.K., Canessa E.H., Kornegay J.N., Nghiem P.P. Challenges associated with homologous directed repair using CRISPR-Cas9 and TALEN to edit the DMD genetic mutation in canine Duchenne muscular dystrophy. PLoS ONE. 2020;15:e0228072. doi: 10.1371/journal.pone.0228072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan J., Ma Y., Huang T., Chen Y., Peng Y., Li B., Li J., Zhang Y., Song B., Sun X. Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol. Cell. 2018;72:380–394.e7. doi: 10.1016/j.molcel.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 56.Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E., Kim J.S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 57.Wojtal D., Kemaladewi D.U., Malam Z., Abdullah S., Wong T.W.Y., Hyatt E., Baghestani Z., Pereira S., Stavropoulos J., Mouly V. Spell Checking Nature: Versatility of CRISPR/Cas9 for Developing Treatments for Inherited Disorders. Am. J. Hum. Genet. 2016;98:90–101. doi: 10.1016/j.ajhg.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perrin A., Rousseau J., Tremblay J.P. Increased Expression of Laminin Subunit Alpha 1 Chain by dCas9-VP160. Mol. Ther. Nucleic Acids. 2017;6:68–79. doi: 10.1016/j.omtn.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liao H.K., Hatanaka F., Araoka T., Reddy P., Wu M.Z., Sui Y., Yamauchi T., Sakurai M., O’Keefe D.D., Núñez-Delicado E. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell. 2017;171:1495–1507.e15. doi: 10.1016/j.cell.2017.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sengupta K., Mishra M.K., Loro E., Spencer M.J., Pyle A.D., Khurana T.S. Genome Editing-Mediated Utrophin Upregulation in Duchenne Muscular Dystrophy Stem Cells. Mol. Ther. Nucleic Acids. 2020;22:500–509. doi: 10.1016/j.omtn.2020.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Himeda C.L., Jones T.I., Jones P.L. CRISPR/dCas9-mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4-fl in FSH muscular dystrophy. Mol. Ther. 2016;24:527–535. doi: 10.1038/mt.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aartsma-Rus A., Fokkema I., Verschuuren J., Ginjaar I., van Deutekom J., van Ommen G.J., den Dunnen J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009;30:293–299. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 63.Symington L.S., Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 64.Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., Iyer E.P.R., Lin S., Kiani S., Guzman C.D., Wiegand D.J. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perez-Pinera P., Kocak D.D., Vockley C.M., Adler A.F., Kabadi A.M., Polstein L.R., Thakore P.I., Glass K.A., Ousterout D.G., Leong K.W. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng A.W., Wang H., Yang H., Shi L., Katz Y., Theunissen T.W., Rangarajan S., Shivalila C.S., Dadon D.B., Jaenisch R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–1171. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Larson M.H., Gilbert L.A., Wang X., Lim W.A., Weissman J.S., Qi L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013;8:2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khurana T.S., Watkins S.C., Chafey P., Chelly J., Tomé F.M.S., Fardeau M., Kaplan J.C., Kunkel L.M. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991;1:185–194. doi: 10.1016/0960-8966(91)90023-l. [DOI] [PubMed] [Google Scholar]
- 69.Ohlendieck K., Ervasti J.M., Matsumura K., Kahl S.D., Leveille C.J., Campbell K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- 70.Loro E., Sengupta K., Bogdanovich S., Whig K., Schultz D.C., Huryn D.M., Khurana T.S. High-throughput identification of post-transcriptional utrophin up-regulators for Duchenne muscle dystrophy (DMD) therapy. Sci. Rep. 2020;10:2132. doi: 10.1038/s41598-020-58737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guiraud S., Edwards B., Babbs A., Squire S.E., Berg A., Moir L., Wood M.J., Davies K.E. The potential of utrophin and dystrophin combination therapies for Duchenne muscular dystrophy. Hum. Mol. Genet. 2019;28:2189–2200. doi: 10.1093/hmg/ddz049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miura P., Jasmin B.J. Utrophin upregulation for treating Duchenne or Becker muscular dystrophy: how close are we? Trends Mol. Med. 2006;12:122–129. doi: 10.1016/j.molmed.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 73.Rafael J.A., Tinsley J.M., Potter A.C., Deconinck A.E., Davies K.E. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat. Genet. 1998;19:79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- 74.Wehling-Henricks M., Li Z., Lindsey C., Wang Y., Welc S.S., Ramos J.N., Khanlou N., Kuro-O M., Tidball J.G. Klotho gene silencing promotes pathology in the mdx mouse model of Duchenne muscular dystrophy. Hum. Mol. Genet. 2016;25:2465–2482. doi: 10.1093/hmg/ddw111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen S., Nathan J.A., Goldberg A.L. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015;14:58–74. doi: 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- 76.Weng S., Gao F., Wang J., Li X., Chu B., Wang J., Yang G. Improvement of muscular atrophy by AAV-SaCas9-mediated myostatin gene editing in aged mice. Cancer Gene Ther. 2020;27:960–975. doi: 10.1038/s41417-020-0178-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y., Wang Y., Yulin B., Tang B., Wang M., Zhang C., Zhang W., Jin J., Li T., Zhao R. CRISPR/Cas9-mediated sheep MSTN gene knockout and promote sSMSCs differentiation. J. Cell. Biochem. 2018;120:1794–1806. doi: 10.1002/jcb.27474. [DOI] [PubMed] [Google Scholar]
- 78.Amthor H., Macharia R., Navarrete R., Schuelke M., Brown S.C., Otto A., Voit T., Muntoni F., Vrbóva G., Partridge T. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Natl. Acad. Sci. USA. 2007;104:1835–1840. doi: 10.1073/pnas.0604893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kramerova I., Marinov M., Owens J., Lee S.J., Becerra D., Spencer M.J. Myostatin inhibition promotes fast fibre hypertrophy but causes loss of AMP-activated protein kinase signalling and poor exercise tolerance in a model of limb-girdle muscular dystrophy R1/2A. J. Physiol. 2020;598:3927–3939. doi: 10.1113/JP279943. [DOI] [PubMed] [Google Scholar]
- 80.Rybalka E., Timpani C.A., Debruin D.A., Bagaric R.M., Campelj D.G., Hayes A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells. 2020;9:2657. doi: 10.3390/cells9122657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Young C.S., Mokhonova E., Quinonez M., Pyle A.D., Spencer M.J. Creation of a Novel Humanized Dystrophic Mouse Model of Duchenne Muscular Dystrophy and Application of a CRISPR/Cas9 Gene Editing Therapy. J. Neuromuscul. Dis. 2017;4:139–145. doi: 10.3233/JND-170218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim K., Ryu S.M., Kim S.T., Baek G., Kim D., Lim K., Chung E., Kim S., Kim J.S. Highly efficient RNA-guided base editing in mouse embryos. Nat. Biotechnol. 2017;35:435–437. doi: 10.1038/nbt.3816. [DOI] [PubMed] [Google Scholar]
- 83.Egorova T.V., Zotova E.D., Reshetov D.A., Polikarpova A.V., Vassilieva S.G., Vlodavets D.V., Gavrilov A.A., Ulianov S.V., Buchman V.L., Deykin A.V. CRISPR/Cas9-generated mouse model of Duchenne muscular dystrophy recapitulating a newly identified large 430 kb deletion in the human DMD gene. Dis. Model. Mech. 2019;12:dmm037655. doi: 10.1242/dmm.037655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Amoasii L., Li H., Zhang Y., Min Y.L., Sanchez-Ortiz E., Shelton J.M., Long C., Mireault A.A., Bhattacharyya S., McAnally J.R. In vivo non-invasive monitoring of dystrophin correction in a new Duchenne muscular dystrophy reporter mouse. Nat. Commun. 2019;10:4537. doi: 10.1038/s41467-019-12335-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Min Y.L., Chemello F., Li H., Rodriguez-Caycedo C., Sanchez-Ortiz E., Mireault A.A., McAnally J.R., Shelton J.M., Zhang Y., Bassel-Duby R., Olson E.N. Correction of Three Prominent Mutations in Mouse and Human Models of Duchenne Muscular Dystrophy by Single-Cut Genome Editing. Mol. Ther. 2020;28:2044–2055. doi: 10.1016/j.ymthe.2020.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakamura K., Fujii W., Tsuboi M., Tanihata J., Teramoto N., Takeuchi S., Naito K., Yamanouchi K., Nishihara M. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci. Rep. 2014;4:5635. doi: 10.1038/srep05635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sui T., Lau Y.S., Liu D., Liu T., Xu L., Gao Y., Lai L., Li Z., Han R. A novel rabbit model of Duchenne muscular dystrophy generated by CRISPR/Cas9. Dis. Model. Mech. 2018;11:dmm032201. doi: 10.1242/dmm.032201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xie J., Ge W., Li N., Liu Q., Chen F., Yang X., Huang X., Ouyang Z., Zhang Q., Zhao Y. Efficient base editing for multiple genes and loci in pigs using base editors. Nat. Commun. 2019;10:2852. doi: 10.1038/s41467-019-10421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen Y., Zheng Y., Kang Y., Yang W., Niu Y., Guo X., Tu Z., Si C., Wang H., Xing R. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 2015;24:3764–3774. doi: 10.1093/hmg/ddv120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.’t Hoen P.A.C., de Meijer E.J., Boer J.M., Vossen R.H.A.M., Turk R., Maatman R.G.H.J., Davies K.E., van Ommen G.-J.B., van Deutekom J.C.T., den Dunnen J.T. Generation and characterization of transgenic mice with the full-length human DMD gene. J. Biol. Chem. 2008;283:5899–5907. doi: 10.1074/jbc.M709410200. [DOI] [PubMed] [Google Scholar]
- 91.Yavas A., Weij R., van Putten M., Kourkouta E., Beekman C., Puoliväli J., Bragge T., Ahtoniemi T., Knijnenburg J., Hoogenboom M.E. Detailed genetic and functional analysis of the hDMDdel52/mdx mouse model. PLoS ONE. 2020;15:e0244215. doi: 10.1371/journal.pone.0244215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deconinck A.E., Rafael J.A., Skinner J.A., Brown S.C., Potter A.C., Metzinger L., Watt D.J., Dickson J.G., Tinsley J.M., Davies K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 93.Vallender E.J., Miller G.M. Nonhuman primate models in the genomic era: a paradigm shift. ILAR J. 2013;54:154–165. doi: 10.1093/ilar/ilt044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao C., Zheng X., Qu W., Li G., Li X., Miao Y.L., Han X., Liu X., Li Z., Ma Y. CRISPR-offinder: a CRISPR guide RNA design and off-target searching tool for user-defined protospacer adjacent motif. Int. J. Biol. Sci. 2017;13:1470–1478. doi: 10.7150/ijbs.21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stemmer M., Thumberger T., Del Sol Keyer M., Wittbrodt J., Mateo J.L. Correction: CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE. 2017;12:e0176619. doi: 10.1371/journal.pone.0176619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu H., Misel L., Graham M., Robinson M.L., Liang C. CT-Finder: A Web Service for CRISPR Optimal Target Prediction and Visualization. Sci. Rep. 2016;6:25516. doi: 10.1038/srep25516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim D., Bae S., Park J., Kim E., Kim S., Yu H.R., Hwang J., Kim J.I., Kim J.S. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods. 2015;12:237–243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 99.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Casini A., Olivieri M., Petris G., Montagna C., Reginato G., Maule G., Lorenzin F., Prandi D., Romanel A., Demichelis F. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018;36:265–271. doi: 10.1038/nbt.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kulcsár P.I., Tálas A., Huszár K., Ligeti Z., Tóth E., Weinhardt N., Fodor E., Welker E. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 2017;18:190. doi: 10.1186/s13059-017-1318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Harrington L.B., Doxzen K.W., Ma E., Liu J.J., Knott G.J., Edraki A., Garcia B., Amrani N., Chen J.S., Cofsky J.C. A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell. 2017;170:1224–1233.e15. doi: 10.1016/j.cell.2017.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y., Li H., Min Y.L., Sanchez-Ortiz E., Huang J., Mireault A.A., Shelton J.M., Kim J., Mammen P.P.A., Bassel-Duby R. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci. Adv. 2020;6:eaay6812. doi: 10.1126/sciadv.aay6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Crudele J.M., Chamberlain J.S. AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 2019;28:R102–R107. doi: 10.1093/hmg/ddz128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Forand A., Muchir A., Mougenot N., Sevoz-Couche C., Peccate C., Lemaitre M., Izabelle C., Wood M., Lorain S., Piétri-Rouxel F. Combined Treatment with Peptide-Conjugated Phosphorodiamidate Morpholino Oligomer-PPMO and AAV-U7 Rescues the Severe DMD Phenotype in Mice. Mol. Ther. - Methods Clin. Dev. 2020;17:695–708. doi: 10.1016/j.omtm.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Flotte T.R., Büning H. Severe Toxicity in Nonhuman Primates and Piglets with Systemic High-Dose Administration of Adeno-Associated Virus Serotype 9-Like Vectors: Putting Patients First. Hum. Gene Ther. 2018;29:283–284. doi: 10.1089/hum.2018.021. [DOI] [PubMed] [Google Scholar]
- 112.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 113.Kim S., Koo T., Jee H.G., Cho H.Y., Lee G., Lim D.G., Shin H.S., Kim J.S. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 2018;28:367–373. doi: 10.1101/gr.231936.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]