

Abstract
Tissue injury induces a long-lasting latent sensitization of spinal nociceptive signaling that is kept in remission by an opposing μ-opioid receptor constitutive activity. To test the hypothesis that supraspinal sites become engaged, we induced hindpaw inflammation, waited three weeks for mechanical hypersensitivity to resolve, and then injected the opioid receptor inhibitors naltrexone, CTOP or β-funaltrexamine subcutaneously and/or into the cerebral ventricles. Intracerebroventricular injection of each inhibitor reinstated hypersensitivity and produced somatic signs of withdrawal, indicative of latent sensitization and endogenous opioid dependence, respectively. In naïve or sham controls, systemic naloxone (3 mg/kg) produced conditioned place aversion and systemic naltrexone (3 mg/kg) increased Fos expression in the central nucleus of the amygdala (CeA). In latent sensitization animals tested three weeks after plantar incision, systemic naltrexone reinstated mechanical hypersensitivity and produced an even greater increase in Fos than in sham controls, particularly in the capsular subdivision of the right CeA. One-third of Fos+ profiles co-expressed protein kinase C delta (PKCδ), and 35% of PKCδ neurons co-expressed tdTomato+ in Oprm1Cre::tdTomato transgenic mice. CeA microinjection of naltrexone (1 μg) reinstated mechanical hypersensitivity only in male mice and did not produce signs of somatic withdrawal. Intra-CeA injection of the MOR-selective inhibitor CTAP (300 ng) reinstated hypersensitivity in both male and female mice. We conclude that mu-opioid receptors in the capsular subdivision of the right central nucleus of the amygdala prevent the transition from acute to chronic postoperative pain.
Keywords: hyperalgesia, dependence, naltrexone, withdrawal, inflammation, aversion
Graphical Abstract
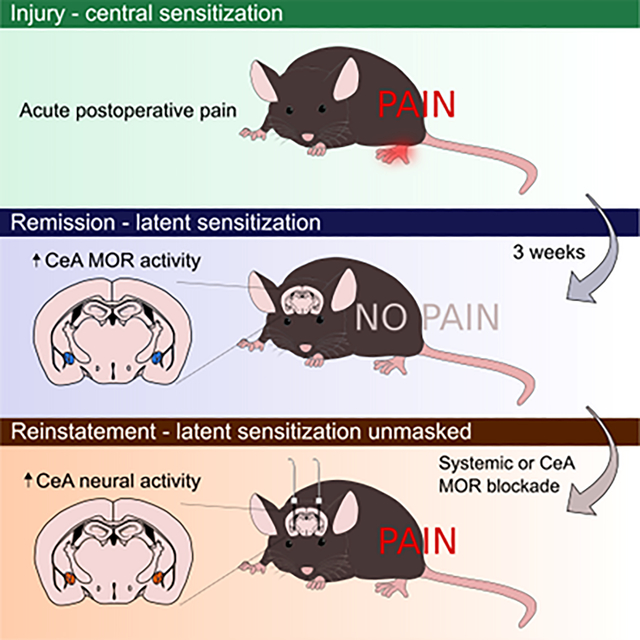
Incision causes neuronal sensitization and acute postoperative pain. After injury resolves, endogenous μ-opioid receptor (MOR) signaling in the central nucleus of the amygdala (CeA) suppresses latent pain sensitization. Inhibiting MOR signaling, either systemically or within the CeA, unmasks latent sensitization, leading to reinstatement of hypersensitivity.
1. Introduction
Inflammation or nerve injury triggers a latent sensitization (LS) of spinal nociceptive signaling that lasts at least several months (Campillo et al., 2011; Corder et al., 2013). LS is maintained in a state of latent remission by the engagement of inhibitory G protein-coupled receptor (GPCR) signaling. In the spinal cord, this includes μ-opioid receptor constitutive activity (MORCA) (Corder et al., 2013; Walwyn et al., 2016), and neuropeptide Y (NPY) Y1 receptor (Fu et al., 2019) and kappa opioid receptor (KOR) (Custodio-Patsey et al., 2020) signaling. Models of LS are initiated by an acutely painful injurious priming event, a waiting period to allow behavioral signs of hyperalgesia to resolve (remission), and then pharmacological challenge with GPCR antagonists or inverse agonists. For example, interruption of MORCA following the injection of an opioid receptor inverse agonist leads to a reinstatement of neuronal activity in the dorsal horn, mechanical hypersensitivity, and affective pain (Corder et al., 2013). This contrasts with models of hyperalgesic priming of primary afferent neurons in which the priming injury is followed by a second noxious event rather than a disruption of inhibitory GPCRs (Aley and Levine, 1999; Araldi et al., 2015, 2017; Kandasamy and Price, 2015).
Much of the previous work investigating mechanisms by which MORCA maintains LS in a state of remission (and thus prevents the transition from acute to chronic pain) has been at the level of the spinal cord dorsal horn (DH) (Corder et al., 2013; Walwyn et al., 2016; Severino et al., 2018). However, tissue injury triggers the development of endogenous opioid receptor dependence; when administered during the remission phase of LS, naltrexone produced numerous behaviors that reflected opioid withdrawal, including jumping behavior, paw tremors, wet-dog shakes, increased locomotion, as well as reinstatement of mechanical hypersensitivity (Corder et al., 2013). Endogenous opioid dependence persisted long after tissue healing, and even increased with the passing of months following the induction of inflammation (Corder et al., 2013). This indicates that physical dependence is not maintained by injury, but rather results from intensifying compensatory opioidergic neuroadaptations (Taylor and Corder, 2014). These likely occur within the brain, since neither intrathecal administration of naltrexone, nor systemic administration of the peripherally restricted naltrexone methobromide, produced withdrawal behaviors.
One potential brain target at the intersection of chronic pain and opioid dependence is the central nucleus of the amygdala (CeA). Neuroplasticity within CeA modulates the mechanical hypersensitivity that develops after cutaneous inflammation or peripheral nerve injury (Carrasquillo and Gereau, 2007; Ji and Neugebauer, 2009; Neugebauer, 2015; Li et al., 2017; Sadler et al., 2017; Cooper et al., 2018), and also contributes to opioid dependence and withdrawal (Stinus et al., 1990; Koob and Le Moal, 2005). To directly test the contribution of opioid receptor activity in the brain to the inhibition of chronic pain and the development of endogenous opioid dependence, we used the intracerebroventricular route of administration to deliver opioid receptor inverse agonists. We also injected drugs directly into the CeA to determine whether this brain area integrates MORCA initiation of both analgesic signaling and a compensatory opponent process that generates endogenous opioid dependence.
2. Methods
2.1. Animals
2.1.1. Animal Husbandry
C57BL/6 mice were purchased from Charles River, USA. Ai14 (Cre-dependent tdTomato expression) mice (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J; stock #007914; RRID: IMSR_JAX:007914) (Madisen et al., 2010) were obtained from The Jackson Laboratory (ME, USA). Homozygous Oprm1Cre:GFP mice were generated as described below and kindly provided by Richard Palmiter (University of Washington). Oprm1Cre:GFP and Ai14 mice were maintained in our in-house colony and first-generation crosses were used for experiments. Male and female mice were aged 6–10 weeks at the beginning of experiments and housed 2–4 per cage and maintained on a 14/10 (Kentucky) or 12/12 (Pittsburgh) hour light/dark cycle in a temperature- (20–22°C) and humidity- (45 ± 10% RH) controlled room, with food and water provided ad libitum. Mice were handled and habituated to testing equipment and experimenter for 20 min/day for 3 consecutive days prior to experimental manipulations and all procedures were performed during lights on. All procedures were approved by the Institutional Animal Care and Use Committees of the University of Kentucky and University of Pittsburgh and carried out in accordance with IASP and ARRIVE guidelines.
2.1.2. Generation of Oprm1Cre:GFP mice
A cassette encoding Cre:GFP was inserted just 5’ of the initiation codon in the first coding exon of the Oprm1 gene. The 5′ and 3′ arms of the Oprm1 gene were amplified from a C57Bl/6 BAC clone by PCR and cloned into polylinkers of a targeting construct that contained mnCre:GFP, a frt-flanked Sv40Neo gene for positive selection, and HSV thymidine kinase and Pgk-diphtheria toxin A chain genes for negative selection. The construct was electroporated into G4 ES cells (C57Bl/6 × 129 Sv hybrid) and correct targeting was determined by restriction enzyme digestion and Southern blot, before injection into blastocysts. Chimeric progeny were bred with Gt(Rosa)26Sor-FLP recombinase mice to remove the SV-Neo gene. Mice were then continuously backcrossed to C57Bl/6 mice. Routine genotyping is performed with 3 primers: 5’ CCT TCC ACT CAG AGA GTG GCG (Oprm1 forward), 5’ CCT TCC ACT CAG AGA GTG GCG (Oprm1 reverse) and 5’ GGC AAA TTT TGG TGT ACG GTC AG (Cre reverse). The wild-type allele gives a band of ~500 bp, while the targeted allele gives a band of ~ 400 bp after 34 cycles with 20-s annealing at 60°C.
2.2. Induction of pain models
2.2.1. CFA model
Mice were lightly restrained and 5 μL of undiluted complete Freund’s adjuvant (Sigma, USA) was injected under the center of the plantar skin of the left hindpaw. Sham-injected mice were injected with an equal volume of saline.
2.2.2. Plantar incision model (PIM)
Over the course of this project, we transitioned from the CFA model to the more clinically relevant plantar incision model of postoperative pain, both of which lead to the development of latent sensitization (Corder et al., 2013). Plantar incision surgery was performed as previously described (Brennan TJ et al., 1996). Briefly, anesthesia was induced with 5% isoflurane (Abbott Laboratories, USA) and then maintained at 2.5%. A midline longitudinal incision was made through the skin and fascia of the proximal edge of the plantar aspect of the left hindpaw. The plantaris muscle was gently separated from underlying tissue and a 4 mm longitudinal incision made through the midline of the muscle using a scalpel blade, without damaging the origin or insertion end of the muscle, and then the skin was closed with two 5–0 PDSII sutures (Ethicon) before covering wound with topical Neosporin ointment (Johnson and Johnson). Sham-operated animals received isoflurane for the same duration as PIM-operated mice.
2.3. Intracerebroventricular and intra-CeA cannula implantation
One week prior to behavioral pharmacology (i.e. 2 weeks after PIM surgery or CFA injection), mice were anesthetized with isoflurane and placed in stereotaxic apparatus (Stoelting, USA). The scalp was shaved, ophthalmic ointment was applied to the eyes, and skin was swabbed with chlorhexidine solution (Chloraprep, BD Healthcare, USA) followed by alcohol. After an incision to expose the cranium, a hole was drilled using a dental burr bit (Stoelting Inc., USA), and then 4.8 mm stainless steel guide cannulas (PlasticsOne, USA) were implanted. To target the cerebrospinal fluid, one cannula was placed into the right lateral ventricle (coordinates relative to bregma: −0.7 mm AP; −1.5. mm ML; −3.3 to 4.0 mm DV). During placement, the cannula was connected to a saline-filled tube, so that correct placement could be confirmed by a drop in the level of the meniscus. To target the CeA, two cannulas were targeted to 1 mm above the center of the left and right CeA (coordinates relative to bregma: −1.45 mm AP; ±2.75 mm ML; −3.75 mm DV), according to Paxinos and Franklin (Paxinos and Franklin, 2013). The guide cannula was affixed to the skull with two bone screws (Stoelting Inc., USA) and dental cement (3M, USA). Skin was then closed around the base of the cannula using 5–0 PDSII suture (Ethicon, USA), and a stylet was inserted into the guide cannula to prevent clogging. Mice were housed in pairs for an additional recovery period of 6–8 d before drug injection and behavioral testing.
2.4. Drug Administration
After behavioral measurements were taken 21 days after CFA or incision, opioid receptor inverse agonists/antagonists were administered at time 0, followed by behavioral observation at timepoint(s) specified in the graphic representation of data.
Subcutaneous.
Mice were gently restrained by scruff at the nape, and drugs were injected at the loose skin over the shoulders in a volume of 3 mL/kg.
Intracranial.
Injections were performed using a 32-gauge injection cannula that extended 1 mm beyond the tip of the guide cannula. The injection cannula was attached to flexible plastic tubing (PlasticsOne, USA), inserted 5 minutes before injection, and then a microliter syringe (Hamilton, USA) was used to deliver naltrexone hydrochloride (NTX; Tocris, UK), D-Pen-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP; Tocris, UK), D-Pen-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP; Tocris, UK) or β-funaltrexamine hydrochloride (β-FNA; Tocris, UK) dissolved in sterile saline. A total volume of 0.2 μL was slowly infused using a syringe pump (Harvard Apparatus, USA) over a period of 6 min (intra-CeA) or 2 min (i.c.v.). The injection cannula was kept in place for an additional 15 min to minimize backflow upon cannula removal. We confirmed successful microinjection by observation of movement of a small air bubble within the tubing, otherwise mice were excluded from further analysis (3 mice in total). Mice receiving i.c.v. injections underwent one injection followed by behavioral testing. Mice receiving an intra-CeA injection on day 21 after induction of hindpaw inflammation received a second injection one week later (either NTX:saline, saline:NTX, saline:CTAP or NTX:CTAP on Day 21:Day 28). At the end of experiments, mice with intra-CeA cannulas were terminally anesthetized and 0.3 μL India ink was injected. Following transcardial perfusion, brains were sectioned and counterstained with Cresyl violet to verify cannula position and injection site. In all mice, cannula placement was <0.5 mm from the outer boundary of the CeA.
2.5. Von Frey assessment of mechanical allodynia
All behavioral measurements were performed by an investigator blinded to experimental treatment by an assistant who randomly assigned treatment groups. Each cohort of experimental animals were tested by a single investigator, including the sex difference studies. Intra-CeA CTAP behavioral measurements were all performed by the same investigator. Hindpaw 50% mechanical withdrawal thresholds were assessed with von Frey filaments (vF; Stoelting, Inc), quantified using an up-down method (Chaplan et al., 1994). The stimulus was localized to the center (CFA) or proximal (PIM) region of the glabrous skin at the plantar surface of the hindpaw. Each hindpaw was stimulated with a series of 8 monofilaments of logarithmically increasing stiffness (0.008 to 6 g). First, an intermediate filament (0.16 g) was applied perpendicular to the skin, causing a slight bending. In case of a positive response (rapid withdrawal or licking of the paw within 3 s), the next smallest filament was tested. In case of a negative response, the next larger filament was tested. Trials continued until 4 measurements beyond the first change in response were taken. von Frey testing was paused during the brief bouts of endogenous opioid withdrawal.
2.6. Assessment of behavioral signs of endogenous opioid withdrawal
Assessment of behavioral signs of physical withdrawal were quantified as previously described (Corder et al., 2013). Briefly, following a 30 min acclimation to a tall but narrow rectangular box, opaque on three sides, each mouse was injected with drug or vehicle and then returned to its box for 60 minutes of video recording. Videos were analyzed by a blinded evaluator to quantify the occurrence of the following behaviors: 1) Jumping, defined as all four paws leaving the testing floor; 2) Paw flutter, defined as a forceful, rapid movement of the forepaw (care was taken to exclude facial grooming motions); and 3) Rearing, defined as both forepaws leaving the testing floor and upward stretching of the body.
2.7. Conditioned place aversion testing
A three-day conditioning protocol using a biased chamber assignment was used for conditioned place aversion (CPA) testing as described previously (Griggs et al., 2015). On the acclimation day (day 1, 21 days after PIM or Sham surgery), mice had free access to explore all chambers of a 3-chamber conditioned place testing apparatus (side chambers: 170 × 150 mm; center chamber: 70 × 150 mm; height: 200 mm; San Diego Instruments, USA) for 30 mins. Mice were able to discriminate between chambers using visual (vertical versus horizontal black-and-white striped walls) and sensory (rough versus smooth textured floor) cues. For pre-conditioning (days 2 and 3), mice were again allowed to freely explore for 15 mins whilst their position was recorded via a 4 × 16 infra-red photobeam array by associated software (San Diego Instruments). For conditioning (days 4–6), each mouse’s non-preferred chamber was paired with saline and preferred chamber with naloxone (NLX). Each morning, mice received a s.c. saline injection, were returned to their home cage for 5 min, then placed in the designated side chamber for 30 min. 4 hours later, mice received s.c. NLX (0.3 or 3 mg/kg; Tocris, UK), returned to their home cage for 5 min, then placed in the NLX-designated chamber for 30 min. On test day (day 7), mice could freely explore whilst their position was recorded, as during pre-conditioning, for 15 min. Difference scores were calculated as the time spent in each chamber on test day minus time spent during pre-conditioning.
2.8. Immunohistochemistry
A separate cohort of adult male and female mice received PIM or sham surgery, and 21 days later were subcutaneously injected with naltrexone hydrochloride (3 mg/kg; Tocris, UK) or saline and mechanical thresholds assessed to confirm reinstatement of mechanical allodynia. 7 days later, animals again received subcutaneous naltrexone or saline, and 2 hours later (i.e. 1 hour after the peak effect of naltrexone-induced hypersensitivity) were terminally anesthetized with pentobarbital and fixed by perfusion with 4% paraformaldehyde (PFA). Brains were post-fixed in 4% PFA overnight, transferred to 30% sucrose for a further 48 hours, then embedded in optimal cutting temperature media (OCT; Tissue Tek, Andwin Scientific, USA) prior to sectioning at 40 μm on a cryostat (Cryostar NX70, Fisher Scientific, USA). Nine non-adjacent sections spanning the range between Bregma −0.9 and −1.7 mm (Paxinos and Franklin, 2013) were randomly selected from each mouse. Free-floating sections from each mouse were washed in PBS, incubated in PBS containing 0.1% Triton X-100 (VWR, USA) for 10 minutes, then blocked in PBS containing 5% normal goat serum (NGS; MP Biomedicals), 1% bovine serum albumin (BSA; Sigma-Aldrich, USA) and 0.1% Triton X-100 for 30 minutes, and then incubated for 18 hours at room temperature with both anti-Fos (1:2000; rabbit anti-phospho-c-Fos (Ser32); Cell Signaling Technology, USA; Cat# 5348; RRID: AB_10557109) and anti-PKCδ (1:1000; BD Biosciences, USA; Cat# 610397; RRID: AB_397780) antibodies that were diluted in 1.5% NGS, 1% BSA and 0.1% Triton X-100. Following washes, sections were incubated with Alexa Fluor-488-conjugated goat anti-rabbit IgG (H+L) (1:1000; Invitrogen, USA; Cat# A11011; RRID: AB_143157) and Alexa Fluor-647-conjugated goat anti-mouse IgG(H+L) (1:100; Invitrogen; Cat# A21235; RRID: AB_2535804) for 1.5 hours. After further washes, sections were mounted on gelatinized slides, rinsed briefly in distilled water, dried, and coverslipped with Vectashield antifade mounting medium with DAPI (Vector Labs, USA).
2.9. Fluorescence in situ hybridization (FISH)
Oprm1Cre:GFP::Ai14 mice were sacrificed with an overdose of pentobarbital, then brains were rapidly collected, embedded in OCT and frozen over dry ice. Brains were sectioned at 12 μm on a cryostat, and sections containing the CeA were collected on Superfrost slides (Fisher Scientific, USA) and immediately frozen and stored at −80°C prior to further processing. Pretreatment, amplification and detection steps were performed using the RNAscope Multiplex Fluorescent Reagent Kit v2 (Advanced Cell Diagnostics, USA; 323100). Slides were immersion-fixed in ice-cold 4% PFA for 15 min then dehydrated in increased concentrations of ethanol (50%, 70% then 100% for 5 min each). Following air drying, slides were pretreated for 15 min with protease per manufacturer’s instructions, and then hybridized with Oprm1 and tdTomato mRNA probes (Advanced Cell Diagnostics 315841-C3 and 317041-C2 respectively) for 2 hr at 40° C in a HybEZ oven. Following amplification and labelling steps per manufacturer instructions, slides were washed in PBS then 0.01 M phosphate buffer without saline, then air-dried and coverslipped with Vectashield antifade mounting medium with DAPI (Vector Labs, USA).
2.10. Microscopy and quantification
Images of sections were captured on a Nikon Ti2 inverted epifluorescence microscope equipped with a 20x, 0.75 NA and a 40x, 0.95 NA objective and a Prime BSI camera (Photometrics, USA). The same exposure time (80 to 500 ms) was used for all images captured in each channel, and each image comprised of 9 individual exposures (3×3) stitched together with 10% overlap. Image capture, adjustments and quantification were performed in NIS Elements (Nikon, Japan). For quantification of immunohistochemistry, the number of immunofluorescence-positive profiles were quantified in 6 to 8 mice (Fos and PKCδ) or 3 mice (MOR-tdTomato and PKCδ) per experimental group. An investigator blinded to treatment first adjusted brightness and contrast in the same manner for each image, and delineations of the CeA and BLA were identified according to Paxinos and Franklin (2013). Then, cell profiles displaying Fos, MOR-tdTomato and/or PKCδ immunofluorescence were counted manually within each region, excluding profiles that were either <4 μm in diameter, largely outside of the plane of view, clearly not representing a soma, or with fluorescence that is readily attributed to artifacts. At least 6 sections per mouse were counted and averaged, with n defined as 1 mouse. Mice were excluded from further analysis where less than 6 sections of sufficient quality to quantify were available at the required rostrocaudal coordinates (2 mice). For analyses and mapping of Fos expression along the rostrocaudal axis of the CeA (nose to tail), the rostrocaudal coordinate of each coronal section was determined as the distance from Bregma to the nearest 0.1 mm, according to Paxinos and Franklin (2013). MOR-tdTomato fluorescence was not readily visible in any section from 26 of the 29 Oprm1Cre::Ai14 mice, and so were excluded from further analysis. This prevented a full quantitative analysis of Fos expression in MOR-tdTomato+ cells and permitted only an analysis of PKCδ and MOR-tdTomato co-localization in the 3 mice where tdTomato fluorescence was present. No mice were excluded from FISH analysis. For quantification of FISH, a cell was deemed to express Oprm1 or tdTomato mRNA when at least 4 fluorescent puncta were observed, with a DAPI-counterstained nucleus visible (Snyder et al., 2018).
2.11. Statistical analysis
All behavioral and immunohistochemical results are presented as mean ± S.E.M, with n defined as a single mouse. Data were analyzed using Prism 8.1 (GraphPad Software Inc., USA) or R 3.5.3 (R Core Team, 2019). Power analyses with GPower 3.1.9.7 determined group sizes (Faul et al., 2007). Data were tested for normality (Shapiro-Wilk tests) prior to analysis by two-way or two-way repeated measures (RM) (examining the interaction of treatment and time) ANOVAs, apart from rostrocaudal distributions of Fos counts which involved mixed effect analyses. Bonferroni or Tukey’s post-hoc tests were performed where appropriate.
3. Results
3.1. Intracerebroventricular administration of opioid receptor inverse agonists induced behavioral signs of endogenous opioid withdrawal and reinstated hypersensitivity
To investigate the potential contribution of supraspinal opioid receptor activity to the suppression of latent pain sensitization, we administered the non-selective opioid receptor inverse agonist naltrexone (NTX; 1 μg) or saline by the intracerebroventricular (i.c.v.) route to male mice, 21 days after intraplantar hindpaw injection of CFA or saline (sham-injured). As illustrated in Fig 1a, NTX but not saline reinstated mechanical hypersensitivity and had no effect in sham-injured mice (2-way RM ANOVA; time x treatment F18,120 = 3.89, P < 0.0001; n = 6). To test the hypothesis that MORs in the brain contributes to the suppression of LS, we challenged CFA mice with i.c.v. injection of the MOR-selective inverse agonists D-Pen-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP) (100 or 300 ng) or β-funaltrexamine (β-FNA) (1 μg). 2-way RM ANOVA revealed a Time x Treatment effect (F19,114 = 2.373, P = 0.017; n = 7–14). As illustrated in Fig 1b, post-hoc analysis revealed that both doses of CTOP and β-FNA elicited mechanical hypersensitivity (Bonferroni post-tests versus saline; P < 0.05 on at least one post-injection timepoint).
Figure 1. Intracerebroventricular (i.c.v.) administration of opioid receptor inverse agonists reinstated mechanical hyperalgesia and induced withdrawal behaviors.

(a) Effect of i.c.v. injection of naltrexone (NTX; 1 μg) or saline on mechanical thresholds in male mice, tested 21 days after intra-plantar injection of CFA or saline (sham) (2-way RM ANOVA with Bonferroni post-tests; n = 6; ** P < 0.01; *** P < 0.001 compared to sham + saline). (b) Effect of i.c.v. injection of the MOR-selective inverse agonists CTOP (100 and 300 ng), β-FNA (1 μg) or saline on mechanical thresholds in mice 21 days after CFA (n = 7 – 14; * P < 0.05, ** P < 0.01, *** P < 0.001 compared to saline). (c to e) Incidence of i.c.v. inverse agonist-induced withdrawal behaviors including (c) jumping, (d) rearing, and (e) paw flutters. ★ P < 0.05 compared to Sham + the respective antagonist. ◆ P < 0.05 compared to CFA-21d + saline. All data shown as mean ± S.E.M.
We then investigated whether endogenous opioid dependence during LS is driven by supraspinal MOR signaling. We found that i.c.v. NTX (1 μg), CTOP (300 ng) or β-FNA (1 μg), but not saline, elicited multiple behavioral signs of somatic withdrawal: jumping (Fig 1c; 2-way ANOVA, injury x drug interaction F3,57 = 66.3, P < 0.001), rearing (Fig 1d; F3,58 = 27.4, P < 0.001), and paw flutters (Fig 1e; F3,60 = 50.3, P < 0.001).
3.2. Systemic opioid receptor inverse agonists increased Fos expression in the right CeA and produced aversion
Several rodents models of persistent pain are associated with an increased activity of CeA neurons, including a pronociceptive population of protein kinase C delta (PKCδ)-expressing GABAergic neurons (Carrasquillo and Gereau, 2007; Ji and Neugebauer, 2009; Wilson et al., 2019). Whether injury produces a much longer-lasting sensitization of CeA neurons that is masked by opioid receptor activity is unclear. To address this question, we conducted PIM or sham surgery in male and female mice, waited 21 days for remission, and then injected NTX (3 mg/kg, s.c.) or saline. After 2 hours, mice were perfused and then the CeA was examined for the expression of Fos, a marker of neuronal activation (Bullitt, 1990), and PKCδ (Fig 2a). PKCδ immunofluorescence was detected largely in the capsular (CeC) and lateral (CeL), not medial (CeM), subdivisions of the CeA, as previously reported (Kim et al., 2017; Wilson et al., 2019).
Figure 2 – Systemic naltrexone produced hypersensitivity and aversion and increased Fos expression in PKCδ positive and negative neurons of the lateral and capsular divisions of the right CeA.

(a) Representative images of Fos (green) and PKCδ (magenta) immunofluorescence in the right CeA 21 days after plantar incision and 2h after systemic naltrexone (NTX; 3 mg/kg) injection. Inset are cropped, enlarged images of the boxed region. Closed arrows denote Fos/PKCδ colocalization, and open arrows denote Fos expression in the absence of PKCδ. CeC, CeL and CeM: capsular, lateral and medial subdivisions of the CeA, respectively. BLA: basolateral amygdala. Scale bars = 100 μm; inset = 10 μm. (b-i) Quantification of Fos+ (green) or Fos+/PKCδ+ (white) cell profiles in the left and right sides of the amygdala. Fos expression in the left CeA (b) and right CeA (c), 21 days after PIM or sham surgery and 2 hours after systemic administration of NTX (3 mg/kg) or saline. NTX increased Fos expression in both PIM- and sham-operated mice (n = 6–8, ***P < 0.001, Tukey’s post-tests after 2-way ANOVA. PIM mice exhibited more Fos-immunoreactive profiles in the right CeA (p<0.001, Injury x Drug interaction), but not left CeA (p = 0.12). Fos+ or Fos/PKCδ+ cell profiles in the left CeC (d) and right CeC (e), the left CeL (f) and right CeL (g), and the left CeM (h) and right CeM (i). NTX increased Fos expression, to a greater extent in PIM mice, in the right CeC and CeL. n = 6–8. *** P < 0.001, Tukey’s post-tests after 2-way ANOVAs. (j) Mechanical thresholds measured 21 days after PIM or sham surgery, and 1h after NTX or saline injection (2-way RM ANOVA with Bonferroni post-tests; n = 6–8; *** P < 0.001). Conditioned place aversion in Sham or PIM mice tested 21 days after surgery, testing saline versus (k) low-dose subcutaneous 0.3 mg/kg naloxone (NLX) (n = 5–8) or (l) high-dose subcutaneous 3 mg/kg NLX (n = 7–8, * P < 0.05, Bonferroni post-tests after 2-way ANOVA).
We examined the effect of NTX on Fos immunoreactivity within the left and right CeA of Sham mice and PIM mice during the remission phase of LS (3 weeks after surgery). Two-way ANOVA revealed that NTX increased the number of Fos-immunoreactive cells in both the left CeA (Fig 2b, Drug (NTX vs saline) F1,23 = 93.32, P < 0.001; n = 6–8 mice, 5–9 sections per mouse) and right CeA (Fig 2c, Drug F1,23 = 214.4, P < 0.001) as compared to saline. No sex differences in Fos-immunoreactive cell counts were found; therefore, data from male and female mice were pooled (3-way ANOVAs; right CeA: Sex x Drug x Injury interaction F1,19 = 1.68, P = 0.21; left CeA: Sex x Drug x Injury interaction F1,19 = 3.89, P = 0.06). Importantly, with regards to the total number of Fos-immunoreactive cells in the right CeA, we observed Drug x Injury (Sham vs PIM) interactions: NTX produced a larger increase in PIM mice as compared to Sham controls (right CeA: Drug x Injury interaction F1,23 = 26.7, P < 0.001; left CeA: Drug x Injury interaction F1,23 = 2.59, P = 0.12). Deeper analysis of just the PKCδ-expressing subgroup of Fos-immunoreactive cells revealed significant NTX-induced increases in cell count in PIM mice (2-way ANOVAs; right CeA: Drug x Injury interaction F1,23 = 24.3, P < 0.001; left CeA: Drug x Injury interaction F1,23 = 5.45, P = 0.03). However, no differences in the percentage of Fos+ cells expressing PKCδ were found between treatment groups (2-way ANOVAs; R CeA: Drug x Injury interaction F1,22 = 0.06, P = 0.81; L CeA: Drug x Injury interaction F1,22 = 0.05, P = 0.83).
We next examined the anatomical segregation of Fos immunoreactivity within capsular, lateral, and medial subregions of the CeA. As illustrated by Fig 2d–i, NTX increased Fos expression primarily in the CeC and in the CeL, but not in the CeM. As illustrated in Table 1, compared to Sham controls, NTX-induced Fos expression during LS was potentiated in the PIM group in the CeC and CeL on the right side (Drug x Injury Interaction, p<0.05) but not on the left side (Drug x Injury Interaction, p>0.05).
Table 1 –
2-way ANOVA degrees of freedom and P values for analyses of the effect of Drug (saline vs naltrexone) and/or Injury (Sham vs plantar incision) in the left or right CeC, CeL or CeM. n = 6–8 mice per group.
| Region | Drug | Injury | Interaction (Drug x Injury) |
|---|---|---|---|
| Right CeC | F1,23 = 83.75, P < 0.0001 | F1,23 = 14.39, P = 0.0009 | F1,23 = 15.82, P = 0.0006 |
| Right CeL | F1,23 = 240.1, P < 0.0001 | F1,23 = 17.48, P = 0.0004 | F1,23 = 12.39, P = 0.0018 |
| Right CeM | F1,23 = 3.51, P = 0.07 | F1,23 = 0.07, P = 0.79 | F1,23 = 0.183. P = 0.673 |
| Left CeC | F1,23 = 35.27, P < 0.0001 | F1,23 = 3.049, P = 0.094 | F1,23 = 2.43, P = 0.13 |
| Left CeL | F1,23 = 95.34, P < 0.0001 | F1,23 = 1.57, P = 0.22 | F1,23 = 0.41, P = 0.53 |
| Left CeM | F1,23 = 5.24, P = 0.032 | F1,23 = 0.41, P = 0.53 | F1,23 = 0.61, P = 0.44 |
NTX reinstated mechanical hypersensitivity following PIM but not Sham surgery (Fig 2j; 2-way RM ANOVA; Time x Treatment interaction F15,115 = 11.09, P < 0.001; n = 6–8 mice). Based on previous reports of aversion induced by opioid receptor inverse agonists (Skoubis et al., 2005; Boulos et al., 2019), we used a conditioned place aversion (CPA) paradigm to test the aversive properties of the opioid receptor inverse agonist naloxone (NLX). The pharmacokinetic half-life of NLX (60–90 minutes) is much shorter than NTX (4 hours) and thus more amenable to our morning vs afternoon testing protocol). We found that NLX dose-dependently induced CPA in both Sham and PIM mice: difference scores (post-minus pre-conditioning) in the saline-paired chamber were not different from the low 0.3 mg/kg NLX-paired chamber paired in either Sham or PIM mice (Fig 2k; Conditioning effect: F1,11 = 2.23, P = 0.16; Conditioning x Injury interaction: F1,11 = 1.48, P = 0.25; n = 5–8); by contrast, differences scores between the saline-paired and 3 mg/kg NLX-paired chambers were significantly different in both Sham and PIM mice (Fig 2l; Conditioning effect: F1,13 = 14.1, P = 0.002; Conditioning x Injury interaction: F1,13 = 0.02, P = 0.88; n = 8).
We also examined the distribution of Fos immunoreactivity along the rostrocaudal axis of the CeA and its subregions. Across the CeA, Fig 3a illustrates that naltrexone-evoked Fos expression did not differ between PIM- and Sham-operated mice in the left CeA (mixed-effects analysis; Treatment effect: F1,10 = 2.03, P = 0.18). In contrast, in the right CeA LS-associated Fos expression (PIM + NTX) was concentrated in the middle to caudal aspect (−1.0 to −1.4 mm relative to Bregma) of the right CeA, with little observable differences versus the Sham + NTX mice at the far rostral and caudal extent of the CeA (mixed-effects analysis with Bonferroni post-tests; Coordinate effect: F2.1, 13.8 = 6.8; P = 0.008; Treatment effect: F1,10 = 23.0, P = 0.0007; n = 6–8 mice, 3–8 sections per rostrocaudal coordinate). More precise anatomical segregation of Fos+ cells in the CeC (Fig 3c–d) and CeL (Fig 3e–f) subregions of the CeA revealed that NTX-induced Fos expression during LS was anatomically segregated to the right CeC (mixed-effects analysis; Coordinate x Treatment interaction: F7,46 = 2.73, P = 0.02; n = 6–8 mice, 3–8 sections per rostrocaudal coordinate), with no significant differences in the left CeC or CeL (mixed-effects analyses; Coordinate x Treatment interactions, L CeC: F7,37 = 0.56, P = 0.78; R CeL: F7,46 = 1.59, P = 0.16; L CeL: F7,37 = 1.39, P = 0.24). The number of Fos+ cells expressing PKCδ along the rostral-caudal axis of the CeA was not different between PIM + NTX and Sham + NTX groups in the left or right CeA (not shown).
Figure 3. Systemic naltrexone during the remission phase of LS increased Fos expression in the capsular subdivision of the right CeA.

(a,b) Rostrocaudal distributions of Fos+ cell profiles in the left (a) or right (b) CeA following naltrexone administration, 21 days after PIM or sham surgery. (c-f) Rostrocaudal distributions of Fos+ cell counts in the left and right CeC and CeL. Fos expression was significantly increased in the right CeC (d) but not the left CeC (c) or either side of the CeL (e,f). n = 6–8 mice, 4–8 sections per rostrocaudal coordinate. *P < 0.05; ** P < 0.01, Bonferroni post-tests after mixed-effect analyses.
MOR expression by PKCδ-expressing neurons has yet to be assessed, and we suspect that pronociceptive PKCδ+ neurons may be directly activated by MOR inverse agonists in the CeA. To address this question, we examined PKCδ expression in the CeA of male and female Oprm1Cre::Ai14 mice (Fig 4). To validate that the Oprm1cre allele is expressed by CeA neurons that express MOR, we performed dual fluorescence in situ hybridization (FISH) for tdTomato and Oprm1 mRNA (Fig 4a). We found that 77.0 ± 3.5% of tdTomato+ neurons expressed Oprm1, and conversely 87.3 ± 5.7% of Oprm1+ neurons expressed tdTomato mRNA (Fig 4b; n = 3 mice, 8–10 sections per mouse), indicating that a large majority of tdTomato+ CeA neurons express Oprm1 mRNA. Upon examination of MOR lineage (MOR-tdTomato positive) neurons and PKCδ-associated immunofluorescence (Fig. 4c), we found that 40.7 ± 4.5% of MOR-tdTomato+ cells were PKCδ+, and 35.7 ± 3.8% of PKCδ+ neurons expressed MOR-tdTomato (Fig 4d; n = 3 mice, 6–8 sections per mouse).
Figure 4 – A large subset of MOR lineage neurons (Opmr1-tdTomato) in the CeA express PKCδ.

(a) Dual FISH of Oprm1 (green) and tdTomato (red) mRNA, plus DAPI counterstaining (white) in the CeA, revealed that a majority of tdTomato+ neurons express Oprm1 mRNA (scale bar = 25 μm). (b) Quantification of a (n = 3 mice, 8–10 sections per mouse). (c) Representative images demonstrating MOR-tdTomato and PKCδ immunofluorescence in the CeA. Insets are cropped, enlarged images of the boxed region. Closed arrows denote MOR-tdTomato/PKCδ colocalization, and open arrows denote MOR-tdTomato in the absence of PKCδ. Scale bars = 100 μm; inset = 10 μm. (d) Quantification of percentage colocalization of MOR-tdTomato and PKCδ (n = 3 mice, 6 to 8 sections per mouse).
3.3. Microinjection of NTX or CTAP into the CeA reinstated hypersensitivity in male mice
The CeA contributes to acute inflammatory pain (Neugebauer, 2015), and systemic administration of NTX increases CeA Fos expression during LS (Fig 2), suggesting an increase in CeA neuron activity. To test the hypothesis that the CeA contributes to chronic postoperative pain (i.e. latent sensitization) that is masked by opioid receptor activity, we bilaterally injected NTX (1 μg/0.2 μL) or saline into the CeA after the resolution of PIM-induced hypersensitivity. As illustrated in Fig 5, NTX did not change mechanical sensitivity in Sham mice of either sex, but reinstated mechanical hypersensitivity in male but not female mice (Fig 5a; 3-way RM ANOVA with Bonferroni post-tests; Time x Injury interaction F5,100 = 5.37, P = 0.002; Sex x Injury interaction F1,20 = 7.88, P = 0.011; Time x Sex x Injury interaction F5,100 = 1.86, P = 0.11; n = 5–7). Intra-CeA injection of saline did not change mechanical sensitivity in female Sham mice or male Sham or PIM, but surprisingly saline produced a robust mechanical hypersensitivity in female PIM mice (Fig 5b; 3-way RM ANOVA with Bonferroni post-tests; Time x Injury interaction F5,95 = 11.81, P < 0.0001; Sex x Injury interaction F1,19 = 0.33, P = 0.57; Time x Sex x Injury interaction F5,95 = 5.19, P = 0.0003; n = 4–7).
Figure 5. Intra-CeA administration of naltrexone reinstated mechanical hyperalgesia in male but not female mice.

(a) Effect of bilateral intra-CeA injection of NTX (1 μg, 0.2 μL) on mechanical thresholds 21 days after plantar incision model (PIM) or sham surgery in male and female mice. Males: n = 6–7; Females: n = 5–6. * P < 0.05, ** P < 0.01, Bonferroni post-tests compared to sham + saline after 2-way ANOVA). (b) Effect of bilateral intra-CeA injection of saline. Males: n = 6–7; Females: n = 4–6. (c, d) Injection sites in males (c;  PIM + NTX;
PIM + NTX;  PIM + saline;
PIM + saline;  Sham + NTX;
Sham + NTX;  Sham + saline) or females (d:
Sham + saline) or females (d:  Sham,
Sham,  PIM) and representative image of guide cannula tracts and India ink injections. (e, f, g) Incidence of intra-CeA NTX-induced withdrawal behaviors in male and female mice, including (e) jumping, (f) rearing and (g) paw flutters.
PIM) and representative image of guide cannula tracts and India ink injections. (e, f, g) Incidence of intra-CeA NTX-induced withdrawal behaviors in male and female mice, including (e) jumping, (f) rearing and (g) paw flutters.
To investigate whether the CeA contributes to endogenous opioid dependence, we quantified withdrawal-like behaviors for 60 minutes following intra-CeA NTX (1 μg/0.2 μL) or saline injection in Sham and PIM mice with LS (Fig 5e–g). Neither saline nor NTX produced withdrawal (2-way ANOVAs; Drug x Injury interactions: Jumping F1, 13 = 1.14, P = 0.31; Rearing F1, 13 = 0.01, P = 0.94; Paw flutters = 0 for all mice; n = 3–4).
To test the hypothesis that MOR in the CeA contributes to the suppression of LS, we challenged PIM mice with bilateral injection of the MOR-selective inverse agonist CTAP (300 ng/0.2 μL) or saline into the CeA during LS (Fig 6). We found that CTAP produced hypersensitivity on both sides in both male and female mice, whilst saline produced hypersensitivity in females but had no effect in males (3-way RM ANOVAs with Bonferroni post-tests; Fig 6a, ipsilateral hindpaw: 3-way RM ANOVA with Bonferroni post-tests; Time x Sex x Treatment interaction F5, 45 = 4.20, P = 0.0032; Fig 6b, contralateral hindpaw: Time x Sex x Treatment interaction F5, 45 = 8.53, P < 0.0001; n = 3–4). Experimental group sizes were determined with a power analysis based on the results of the intra-CeA NTX injection experiments of Fig 5a (α = 0.05, power = 0.8 and Time x Injury effect size of 0.462, projected group size = 3). Post-tests revealed no significant difference in mechanical thresholds at either hindpaw between male CTAP, female CTAP and female saline-treated mice at any timepoint. These data indicate that μ-opioid receptor signaling in the CeA suppresses latent pain sensitization.
Figure 6. Intra-CeA administration of CTAP reinstated bilateral mechanical hyperalgesia in male and female mice.

Effect of bilateral CTAP (300 ng, 0.2 μL) on mechanical thresholds following the resolution of PIM-induced mechanical hypersensitivity at the ipsilateral and contralateral hindpaws. n = 3–4. (a) Ipsilateral, P = 0.0032, Bonferroni post-tests after Time x Sex x Treatment interaction. (b) Contralateral: P < 0.0001. 3-way RM ANOVA.
4. Discussion
Latent sensitization (LS) at the dorsal horn (DH) of the spinal cord is maintained in a state of remission by compensatory Gi-coupled GPCR signaling (Solway et al., 2011; Corder et al., 2013; Walwyn et al., 2016; Severino et al., 2018; Custodio-Patsey et al., 2020). The current study demonstrates that i.c.v. injection of naltrexone, CTOP, or β-FNA reinstates hypersensitivity and precipitates additional signs of opioid withdrawal, indicating that MOR signaling in the brain suppresses LS and maintains a state of endogenous opioid dependence. Furthermore, we found that interruption of opioid receptor signaling activates pronociceptive PKCδ+ neurons in the CeA which may then drive hypersensitivity in male but not female mice.
4.1. MOR signaling in the brain maintains LS in a state of remission
Systemic or intrathecal administration of opioid receptor inverse agonists reinstate hypersensitivity during the remission phase of LS (Campillo et al., 2011; Corder et al., 2013). The current results extend these findings for the first time to the i.c.v. route of administration using not only naltrexone, but also the MOR-selective inverse agonists CTOP and β-FNA; each reinstated mechanical hypersensitivity. We conclude that tissue inflammation induces a long-lasting state of latent pain sensitization that is suppressed by supraspinal MOR signaling.
Our results are consistent with the idea of an injury-induced engagement of brain circuits and/or descending bulbospinal pathways that then maintain LS within a state of remission. Corder et al. (2013) reported that naltrexone evoked a remarkably robust activation of dorsal horn neurons and mechanical hypersensitivity on the side contralateral to injury, leading the authors to suggest the development of a spinal-supraspinal-spinal loop. More recently, local anesthetic block of the cervicothoracic spinal cord, assumed to inhibit descending bulbospinal pain inhibitory pathways during the remission phase of LS, was found to elicit bilateral reinstatement of hyperalgesia at both hindpaws (Chen et al., 2018). Additionally, spinal α2A adrenoreceptor blockade reinstates hypersensitivity during LS (Walwyn et al., 2016) suggesting that injury recruits descending noradrenergic inhibition arising from the locus coeruleus or other brainstem noradrenergic nuclei.
Emerging findings on the supraspinal mechanisms of hyperalgesic priming may provide indications as to the loci that maintain LS. For example, descending dopaminergic input to the DH, arising from the A11 nucleus of the hypothalamus, was reported to be essential in the maintenance of hyperalgesic priming (Megat et al., 2018). Also of interest is the rostral ventral medulla (RVM), a key regulator of chronic inflammatory nociception (Hurley and Hammond, 2001; Fields, 2004; Zhang and Hammond, 2010) that also mediates the manifestation of opiate withdrawal (Vera-Portocarrero et al., 2011); in opiate-dependent animals, lidocaine inactivation of the RVM diminished naloxone-precipitated hyperalgesia. Of particular relevance to the present results, stress and intra-cerebroventricular (i.c.v.) administration of corticotropin-releasing factor reinstate hypersensitivity during the remission phase of LS, and stress-induced reinstatement of hypersensitivity following sumatriptan or repeat opioid priming is driven by κ-opioid receptor (KOR) signaling in the central nucleus of the amygdala (CeA) (Rivat et al., 2007; Xie et al., 2017; Chen et al., 2018; Nation et al., 2018).
4.2. MOR signaling in the brain maintains a state of endogenous dependence
Injury, stress or dietary factors trigger the release of endorphins or enkephalins that then induce physical manifestations of endogenous opioid dependence (Taylor and Corder, 2014). Physical (somatic) or psychological dependence can be revealed upon challenge with an opioid receptor antagonist. Here we report that i.c.v. administration of naltrexone, CTOP, and β-FNA during the remission phase of LS elicited behavioral signs of opioid withdrawal. Our results are consistent with the observation that, when administered during the post-hyperalgesia period after inflammatory insult, systemic, but not intrathecal naltrexone produced numerous behaviors that reflected opioid withdrawal (Corder et al., 2013). Endogenous opioid dependence persists long after tissue healing, and even increased with the passing of months following the induction of inflammation (Corder et al., 2013). This indicates that physical dependence is not maintained by injury, but rather results from intensifying compensatory opioidergic neuroadaptations within the brain. Numerous signs of exogenous opiate physical withdrawal result from receptor blockade in the CNS (Hand et al., 1988). This includes CNS noradrenergic systems such as the locus coeruleus, which contribute to the locomotor hyperactivity associated with opiate withdrawal, such as jumping, rearing and forepaw flutters (Aghajanian, 1978; Llorens et al., 1978; Maldonado et al., 1992). We conclude that tissue injury engages tonic MOR signaling in the brain, leading to endogenous opioid physical dependence. The site of action is not the CeA since local microinjection of naltrexone or CTAP did not produce behavioral signs of withdrawal beyond mechanical hypersensitivity (keeping in mind the caveat that reinstatement of mechanical hypersensitivity likely reflects a cellular dependence to MORCA (Corder et al., 2013)). Additional studies are needed to determine site of action, and whether endogenous physical dependence results from the ligand-dependent tonic release of opioids, or from a ligand-independent transition of opioid receptors to a constitutively active state.
4.3. Systemic naltrexone activates PKCδ-expressing neurons in the CeA to the greatest extent when administered during the remission phase of LS
The CeA has reciprocal connections to both ascending and descending nociceptive pathways, can amplify or attenuate ongoing pain (Neugebauer et al., 2004; Veinante et al., 2013; Wilson et al., 2019), and intrinsic soma, axons and dendrites strongly express MOR (Jaferi and Pickel, 2009; Erbs et al., 2015). The current results indicate that systemic naltrexone induced Fos expression in both the left and right CeA of naïve (i.e. Sham-operated) mice, consistent with the central role that the CeA plays in the modulation of aversion and nociceptive processing (Jin et al., 2005). Importantly, we observed an even greater induction of Fos when naltrexone was administered following the resolution of plantar incision-induced hypersensitivity. We conclude that endogenous opioid signaling tonically inhibits neuronal activity in the CeA during the remission phase of LS.
We found that naltrexone-induced Fos expression during the remission phase of LS was greatest in the right CeA. This is consistent with previous reports indicating that noxious stimulus-evoked increases in neural activity are restricted primarily to the right CeA, regardless of the side of injury (Carrasquillo and Gereau, 2007; Ji and Neugebauer, 2009; Gonçalves and Dickenson, 2012; Sadler et al., 2017). Furthermore, increased neuronal activation in the right CeA is associated with cutaneous inflammatory (Carrasquillo and Gereau, 2007), arthritic (Ji and Neugebauer, 2009), neuropathic (Ikeda et al., 2007; Gonçalves and Dickenson, 2012) and visceral (Sadler et al., 2017) pain, and KOR signaling in the right, not left CeA drives reinstatement of hypersensitivity (Xie et al., 2017). However, given that our injury model was restricted to the left hindlimb, we cannot form any firm conclusions on the lateralization of LS in the CeA.
The lateral (CeL) and capsular (CeC) divisions of the CeA have been termed the “nociceptive amygdala” (Neugebauer et al., 2004), responding predominantly to noxious stimuli and integrating nociceptive-specific signaling via the spinal-parabrachial pathway and polymodal sensory information from the thalamus and cortex, via the basolateral amygdala (BLA). Output from the CeA arises both from the CeC and CeL, as well as the medial division of the CeA (CeM) (Neugebauer et al., 2020). We found that systemic naltrexone during LS induced Fos expression in the CeL and CeC, with the highest expression in the CeC, and little if any induction in the CeM. These results are consistent with Wilson et al. (2019) who reported that noxious stimulation in a neuropathic pain model induced the expression of Fos and phosphorylated extracellular signal-regulated kinase (pERK) in a pattern that was restricted to the CeL and CeC. We also found that the NTX-induced Fos expression was concentrated in the middle-to-caudal aspect of the CeA, again consistent with Wilson et al. (Wilson et al., 2019). As such, our results suggest that the CeL and CeC are activated in a similar pattern in both short-term conventional and long-term LS models of chronic pain states.
Within the CeA, two distinct subpopulations of GABAergic neurons bidirectionally modulate nociception; a heterogenous population of PKCδ-expressing neurons that drive pain hypersensitivity, and a subpopulation of somatostatin-expressing neurons that inhibit pain hypersensitivity (Wilson et al., 2019). These subpopulations comprise the majority of neurons in the CeL and CeC (Kim et al., 2017) and can be considered as functionally homologous to the ON- and OFF-cells of the RVM, respectively (Fields, 2004; Wilson et al., 2019). The current results indicate that during naltrexone-induced reinstatement of hypersensitivity, approximately one third of Fos-expressing neurons in the CeC and CeL express PKCδ (Fig 2); a similar proportion was found by Wilson et al. (Wilson et al., 2019) following Randell-Selitto testing in neuropathic mice. We also found that naltrexone-evoked Fos-expression in both the CeL and CeC occurred in PKCδ-expressing neurons. This again correlates with Wilson et al. (Wilson et al., 2019), and contrasts with reports of anatomical segregation of PKCδ+ neuron function in the CeC and CeL during fear-related behaviors: PKCδ-expressing CeC neurons are activated by threatening stimuli and drive defensive behaviors, whereas PKCδ-expressing neurons in the CeL are activated by states of satiety and drive fear extinction (Kim et al., 2017). Furthermore, our analysis of the rostrocaudal distribution of Fos+ neurons did not fall within the anterior CeA that processes aversion (Kim et al., 2017). Thus, we suggest that during the remission phase of LS, inhibition of MOR signaling in the CeA induces activation of neural circuitry, including but not limited to pronociceptive PKCδ-expressing neurons in the CeC and CeL, in a pattern resembling neuronal activation during overt neuropathic pain states but distinct from fear conditioning.
Previous studies demonstrated that CeA neurons express μ opioid receptors (Jaferi and Pickel, 2009), but whether this extended to PKCδ-expressing neurons of the CeL and CeC was unclear. Our data now demonstrate that a substantial proportion (around 40%) express MOR as evidenced by MOR-tdTomato quantification. We also found that around three quarters of MOR-tdTomato-expressing neurons express Oprm1 mRNA; only a small proportion of MOR-tdTomato signal represents Cre-mediated recombination during pre- or post-natal development and is thus not functional in the adult. Our co-expression data indicates that naltrexone targets opioid receptors on PKCδ-expressing CeA neurons to reinstate hyperalgesia in the setting of LS.
Neurons in the CeL and CeC have been largely segregated within three populations that express PKCδ, somatostatin, or corticotropin-releasing factor (CRF), with some overlap between the somatostatin- and CRF-expressing populations (Kim et al., 2017). Our results indicate that approximately 60% of MOR-tdTomato+ neurons do not express PKCδ. We speculate that the MOR+ / PKCδ- neurons belong to a CRF-expressing subpopulation because: 1) Activation of somatostatin-expressing neurons in CeA is anti-nociceptive (Wilson et al., 2019) and thus unlikely to be inhibited by MOR agonists; and 2) intracerebroventricular administration of CRF reinstates hypersensitivity during LS (Chen et al., 2018). The CeA contains one of the highest densities of CRF-expressing neurons outside of the hypothalamus, with local projections as well as projections to a wide range of brainstem and subcortical nuclei involved in nociceptive processing, including the periaqueductal grey and parabrachial nucleus (Neugebauer et al., 2020). In addition, CRF signaling in the CeA has been implicated in the maintenance of ongoing neuropathic and arthritic pain (Ji and Neugebauer, 2007, 2008, 2019). Future studies would be necessary to determine whether endogenous opioid receptor signaling at CRF-expressing neurons in the CeA contributes to the suppression of LS.
The CeA is an important component of the neural circuitry mediating the aversive experience, and we propose that our observed increases in neuronal activity in the CeA in sham-operated mice are mediated by naltrexone-evoked aversion. Indeed, our naltrexone-induced CPA results agree with and extend previous studies showing that systemic administration of high doses of naltrexone are aversive in uninjured mice (Skoubis et al., 2001, 2005; Shoblock and Maidment, 2006; Kirkpatrick and Bryant, 2015; Boulos et al., 2019). PKCδ-expressing neurons of the CeL encode aversive signals during reinforcement learning (Cui et al., 2017). Furthermore, lesions of the CeA inhibit naloxone-induced conditioned place aversion, albeit in opioid-experienced rats (Watanabe et al., 2002). Importantly, whilst we found that naltrexone induced significantly more Fos-expressing neurons in the right CeA when measured during the remission phase of LS, in sham-operated mice Fos was expressed equally in the right and left CeA. This suggests that lateralization of CeA function is important in pain processing, but not necessarily aversion.
4.4. MOR signaling in the CeA maintains LS in a state of remission
The current results indicate that intra-CeA naltrexone (an inverse agonist at both MOR and KOR) and CTAP (a MOR-specific inverse agonist) produced mechanical hypersensitivity in male mice 21 days after plantar incision but not sham surgery. These findings are consistent with others indicating that the CeA is pronociceptive and becomes engaged in the transition from acute to chronic pain. First, pharmacological activation of CeA metabotropic glutamate receptor 5 (mGluR5) or ERK (Carrasquillo and Gereau, 2007; Kolber et al., 2010) induces mechanical hypersensitivity, and optogenetic activation of CeA neurons increases EMG responses in a visceral pain model (Sadler et al., 2017). Second, inhibition of mGluR1, mGluR5, protein kinase A, ERK or adenylyl cyclase in the right CeA attenuates hypersensitivity in a number of models of persistent pain (Carrasquillo and Gereau, 2008; Ji and Neugebauer, 2009; Kolber et al., 2010; Sadler et al., 2017). Third, following priming with systemic sumatriptan or repeated opioid exposure, stress drives κ-opioid receptor (KOR) signaling in the right CeA to drive mechanical hypersensitivity. Fourth, a reduction in tonic GABAergic CeA signaling to the periaqueductal grey mediates alcohol withdrawal-associated heat hypersensitivity (Avegno et al., 2018).
Our findings with naltrexone and CTAP indicate that endogenous MOR signaling in the CeA maintains mechanical hypersensitivity in a state of remission in male mice and in the case of CTAP, perhaps in female mice as well. These results contrast with studies showing that microinjection of MOR agonists into the CeA did not attenuate inflammatory heat hyperalgesia or neuropathic mechanical hypersensitivity, but did alleviate the affective aspects of pain (Zhang et al., 2013; Navratilova et al., 2020). We suggest that exogenous MOR activation in the CeA inhibits the negative affective component of pain, while endogenous MOR activation in the CeA inhibits the sensory-discriminative component of LS. Perhaps endogenous MOR activation during LS and exogenous MOR activation during ongoing central sensitization have differing consequences on neuronal activity in the CeA. However, it must be kept in mind that bilateral lesions of the CeA blocked systemic morphine antinociception in the rat tail flick and formalin tests (Manning and Mayer, 1995a, 1995b), and further studies are needed to determine whether endogenous MOR activity in the CeA inhibits the affective component of LS.
Systemic or intrathecal administration of MOR inverse agonists reinstated hyperalgesia to a similar extent in male and female mice (Corder et al., 2013), while intrathecal administration of KOR antagonists reinstate hyperalgesia to a greater extent in females (Custodio-Patsey et al., 2020). In the current studies, when CTAP or naltrexone were microinjected into the CeA in male mice, we observed reinstatement of mechanical hypersensitivity. In female mice, CTAP also reinstated hypersensitivity but naltrexone did not. We do not have a clear explanation for this result, nor the paradoxical finding that intra-CeA injection of saline reinstated hyperalgesia in female mice, an effect observed by two independent investigators who observed internally consistent results in two independent experiments: both alongside intra-CeA naltrexone in Figure 5 and alongside intra-CeA injection of CTAP in Figure 6. Perhaps an important clue for these paradoxical findings comes from reports that stress can induce reinstatement of hypersensitivity in models of LS or headache priming (Rivat et al., 2007; Le Roy et al., 2011; Xie et al., 2017; Nation et al., 2018), and the latter has been shown to be driven by KOR signaling in the CeA (Xie et al., 2017; Nation et al., 2018). Indeed, female C57BL/6 mice are more susceptible to stressors than males (Marchette et al., 2018), and this could extend to the stress of intracranial injection. Such stress might explain the reinstatement we observed in the female saline injection groups. Additionally, perhaps KOR-mediated stress underlies opposing actions of the MOR and KOR antagonist properties of naltrexone on nociceptive transmission in the female CeA, leading to one of two possible explanations for our results. First, if intra-CeA naltrexone unmasks endogenous MOR analgesia, then this might be counterbalanced by a reversal of injection stress-induced hyperalgesia by antagonism at KOR, i.e. an inhibition of stress-induced pain sensitization due to the KOR antagonist properties of NTX may have diminished its ability to evoke overt mechanical hypersensitivity in females. Alternatively, perhaps endogenous MOR activity in the CeA does not suppress LS in females, and reinstatement of hypersensitivity in the CTAP group was mediated by injection stress. The lack of hypersensitivity we observed in the naltrexone group could then be explained by KOR antagonism inhibiting stress-induced, KOR-mediated reinstatement of hypersensitivity. Further studies using KOR-selective antagonists are warranted to further investigate sex differences in the neural circuitry and mechanisms involved in the suppression of LS by endogenous MOR and KOR opioid receptor signaling.
4.5. Conclusions and Future Studies
We report that systemic administration of opioid receptor inverse agonists dose-dependently induce aversion in a conditioned place assay. This correlates with increased Fos expression in CeA PKCδ neurons. To rigorously test the hypothesis that opioid receptor inhibitors engage CeA neurons (Watanabe et al., 2002), we predict that intra-CeA infusions will produce aversion, while chemogenetic inhibition of CeA neurons will reduce it.
The current studies indicate that peripheral inflammation induces a long-term state of latent pain sensitization and endogenous opioid dependence that are maintained in remission by supraspinal opioid receptor activity. We conclude that the mechanism of endogenous opioid receptor analgesia involves MOR-expressing neurons in the lateral and capsular subdivisions of the right central nucleus of the amygdala, and suggest that surgical incision induces a latent sensitization in the “nociceptive amygdala” that is suppressed by tonic MOR signaling. If true, then any event that interferes with amygdalar MOR analgesia could unleash a pain episode indicative of chronic pain relapse. Future studies are needed to determine whether this transition from acute to chronic postoperative pain can be alleviated either by facilitating endogenous opioid analgesia in the CeA, thus restricting LS within a state of remission, or by extinguishing LS altogether with a new pharmacotherapy targeted to pronociceptive mechanisms in the CeA.
Significance statement.
Tissue injury can engage endogenous pain control systems in the spinal cord for extended periods of time, but whether this occurs in the brain was unclear. Here, we show that opioid receptor signaling in the brain, specifically to inhibit pain-promoting neurons in the central nucleus of the amygdala, keeps latent pain sensitization in a state of remission. Facilitation of endogenous opioid receptor signaling in the nociceptive amygdala may represent a new approach to prevent the transition from acute to chronic postoperative pain.
Acknowledgements
The authors would like to thank Dr. Richard Palmiter for providing Oprm1Cre:GFP mice, André Martel for analyzing videos and recording (the absence of) withdrawal behaviors, Dr. Tommaso Iannitti for help with blinding and intracerebroventricular infusions, and Dr. Benedict Kolber for useful discussions concerning CeA anatomy and physiology. This work was supported by NIH grants R01DA037621, R01NS45954, R01NS62306 and R01NS112321 to BKT and F31DA032496 to GFC.
Grant support: NIH grants R01DA037621, R01NS45954, R01NS62306 and R01NS112321 to BKT and F31DA032496 to GFC.
Footnotes
Conflict of interest
The authors have no conflict of interest to declare.
Data Accessibility
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Aghajanian GK (1978) Tolerance of locus coeruleus neurones to morphine and suppression of withdrawal response by clonidine. Nature 276:186–188. [DOI] [PubMed] [Google Scholar]
- Aley KO, Levine JD (1999) Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 19:2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araldi D, Ferrari LF, Levine JD (2015) Repeated Mu-Opioid Exposure Induces a Novel Form of the Hyperalgesic Priming Model for Transition to Chronic Pain. J Neurosci 35:12502–12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araldi Di, Ferrari LF, Levine JD (2017) Hyperalgesic priming (type II) induced by repeated opioid exposure: Maintenance mechanisms. Pain 158:1204–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avegno EM, Lobell TD, Itoga CA, Baynes BB, Whitaker AM, Weera MM, Edwards S, Middleton JW, Gilpin NW (2018) Central amygdala circuits mediate hyperalgesia in alcohol-dependent rats. J Neurosci 38:7761–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulos LJ, Ben Hamida S, Bailly J, Maitra M, Ehrlich AT, Gavériaux-Ruff C, Darcq E, Kieffer BL (2019) Mu opioid receptors in the medial habenula contribute to naloxone aversion. Neuropsychopharmacology 45:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TJ, Vandermeulen EP, Gebhart GF (1996) Characterization of a rat model of incisional pain. Pain 64:493–501. [DOI] [PubMed] [Google Scholar]
- Bullitt E (1990) Expression of c-fos-like protein as a marker for neuronal activity following noxious stimulation in the rat. J Comp Neurol 296:517–530. [DOI] [PubMed] [Google Scholar]
- Campillo A, Cabañero D, Romero A, García-Nogales P, Puig MM (2011) Delayed postoperative latent pain sensitization revealed by the systemic administration of opioid antagonists in mice. Eur J Pharmacol 657:89–96. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW (2007) Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci 27:1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW (2008) Hemispheric lateralization of a molecular signal for pain modulation in the amygdala. Mol Pain 4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. [DOI] [PubMed] [Google Scholar]
- Chen W, Taché Y, Marvizón JC (2018) Corticotropin-Releasing Factor in the Brain and Blocking Spinal Descending Signals Induce Hyperalgesia in the Latent Sensitization Model of Chronic Pain. Neuroscience 381:149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AH, Brightwell JJ, Hedden NS, Taylor BK (2018) The left central nucleus of the amygdala contributes to mechanical allodynia and hyperalgesia following right-sided peripheral nerve injury. Neurosci Lett 684:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK (2013) Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science (80- ) 341:1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Lv G, Jin S, Peng J, Yuan J, He X, Gong H, Xu F, Xu T, Li H (2017) A Central Amygdala-Substantia Innominata Neural Circuitry Encodes Aversive Reinforcement Signals. Cell Rep 21:1770–1782. [DOI] [PubMed] [Google Scholar]
- Custodio-Patsey L, Donahue RR, Fu W, Lambert J, Smith BN, Taylor BK (2020) Sex differences in kappa opioid receptor inhibition of latent postoperative pain sensitization in dorsal horn. Neuropharmacology 163:107726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, Vonesch JL, Koch M, Kessler P, Hentsch D, Birling MC, Koutsourakis M, Vasseur L, Veinante P, Kieffer BL, Massotte D (2015) A mu–delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct Funct 220:677–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, Buchner A (2007) G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39:175–191. [DOI] [PubMed] [Google Scholar]
- Fields H (2004) State-dependent opioid control of pain. Nat Rev Neurosci 5:565–575. [DOI] [PubMed] [Google Scholar]
- Fu W, Nelson TS, Santos DF, Doolen S, Gutierrez JJP, Ye N, Zhou J, Taylor BK (2019) An NPY Y1 receptor antagonist unmasks latent sensitization and reveals the contribution of Protein Kinase A and EPAC to chronic inflammatory pain. Pain 160:1754–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves L, Dickenson AH (2012) Asymmetric time-dependent activation of right central amygdala neurones in rats with peripheral neuropathy and pregabalin modulation. Eur J Neurosci 36:3204–3213. [DOI] [PubMed] [Google Scholar]
- Griggs RB, Bardo MT, Taylor BK (2015) Gabapentin alleviates affective pain after traumatic nerve injury. Neuroreport 26:522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TH, Koob GF, Stinus L, Le Moal M (1988) Aversive properties of opiate receptor blockade: evidence for exclusively central mediation in naive and morphine-dependent rats. Brain Res 474:364–368. [DOI] [PubMed] [Google Scholar]
- Hurley RW, Hammond DL (2001) Contribution of endogenous enkephalins to the enhanced analgesic effects of supraspinal μ opioid receptor agonists after inflammatory injury. J Neurosci 21:2536–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda R, Takahashi Y, Inoue K, Kato F (2007) NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain 127:161–172. [DOI] [PubMed] [Google Scholar]
- Jaferi A, Pickel VM (2009) Mu-opioid and corticotropin-releasing-factor receptors show largely postsynaptic co-expression, and separate presynaptic distributions, in the mouse central amygdala and bed nucleus of the stria terminalis. Neuroscience 159:526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V (2007) Differential effects of CRF1 and CRF2 receptor antagonists on pain-related sensitization of neurons in the central nucleus of the amygdala. J Neurophysiol 97:3893–3904. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V (2008) Pro- and anti-nociceptive effects of Corticotropin-Releasing Factor (CRF) in central amygdala neurons are mediated through different receptors. J Neurophysiol 99:1201–1212. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V (2009) Hemispheric lateralization of pain processing by amygdala neurons. J Neurophysiol 102:2253–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V (2019) Contribution of corticotropin-releasing factor receptor 1 (CRF1) to serotonin receptor 5-HT2CR function in amygdala neurons in a neuropathic pain model. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Araki H, Nagata M, Shimosaka R, Shibata K, Suemaru K, Kawasaki H, Gomita Y (2005) Expression of c-Fos in the rat central amygdala accompanies the acquisition but not expression of conditioned place aversion induced by withdrawal from acute morphine dependence. Behav Brain Res 161:107–112. [DOI] [PubMed] [Google Scholar]
- Kandasamy R, Price TJ (2015) The pharmacology of nociceptor priming. Handb Exp Pharmacol 227:15–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Zhang X, Muralidhar S, LeBlanc SA, Tonegawa S, Behaviors A, Kim J, Zhang X, Muralidhar S, LeBlanc SA, Kim J, Zhang X, Muralidhar S, LeBlanc SA, Tonegawa S (2017) Basolateral to Central Amygdala Neural Circuits for Appetitive Behaviors. Neuron 93:1464–1479.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick SL, Bryant CD (2015) Behavioral architecture of opioid reward and aversion in C57BL/6 substrains. Front Behav Neurosci 8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolber BJ, Montana MC, Carrasquillo Y, Xu J, Heinemann SF, Muglia LJ, Gereau RW (2010) Activation of metabotropic glutamate receptor 5 in the amygdala modulates pain-like behavior. J Neurosci 30:8203–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M (2005) Plasticity of reward neurocircuitry and the “dark side” of drug addiction. Nat Neurosci 8:1442–1444. [DOI] [PubMed] [Google Scholar]
- Le Roy C, Laboureyras E, Gavello-Baudy S, Chateauraynaud J, Laulin JP, Simonnet G (2011) Endogenous opioids released during non-nociceptive environmental stress induce latent pain sensitization via a NMDA-dependent process. J Pain 12:1069–1079. [DOI] [PubMed] [Google Scholar]
- Li MJ, Liu LY, Chen L, Cai J, Wan Y, Xing GG (2017) Chronic stress exacerbates neuropathic pain via the integration of stress-Affect-related information with nociceptive information in the central nucleus of the amygdala. Pain 158:717–739. [DOI] [PubMed] [Google Scholar]
- Llorens C, Martres MP, Baudry M, Schwartz JC (1978) Hypersensitivity to noradrenaline in cortex after chronic morphine: relevance to tolerance and dependence. Nature 274:603–605. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R, Stinus L, Gold LH, Koob GF (1992) Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Ther 261:669 LP–677. [PubMed] [Google Scholar]
- Manning BH, Mayer DJ (1995a) The central nucleus of the amygdala contributes to the production of morphine antinociception in the formalin test. Pain 63:141–152. [DOI] [PubMed] [Google Scholar]
- Manning BH, Mayer DJ (1995b) The central nucleus of the amygdala contributes to the production of morphine antinociception in the rat tail-flick test. J Neurosci 15:8199–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchette RCN, Bicca MA, Santos EC da S, de Lima TCM (2018) Distinctive stress sensitivity and anxiety-like behavior in female mice: Strain differences matter. Neurobiol Stress 9:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megat S, Shiers S, Moy JK, Barragan-Iglesias P, Pradhan G, Seal RP, Dussor G, Price TJ (2018) A Critical Role for Dopamine D5 Receptors in Pain Chronicity in Male Mice. J Neurosci 38:379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation KM, De Felice M, Hernandez PI, Dodick DW, Neugebauer V, Navratilova E, Porreca F (2018) Lateralized kappa opioid receptor signaling from the amygdala central nucleus promotes stress-induced functional pain. Pain 159:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova E, Nation K, Remeniuk B, Neugebauer V, Bannister K, Dickenson AH, Porreca F (2020) Selective modulation of tonic aversive qualities of neuropathic pain by morphine in the central nucleus of the amygdala requires endogenous opioid signaling in the anterior cingulate cortex. Pain 161:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V (2015) Amygdala pain mechanisms. Handb Exp Pharmacol 227:261–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS (2004) The amygdala and persistent pain. Neuroscientist 10:221–234. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Mazzitelli M, Cragg B, Ji G, Navratilova E, Porreca F (2020) Amygdala, neuropeptides, and chronic pain-related affective behaviors. Neuropharmacology 170:108052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2013) The Mouse Brain in Stereotaxic Coordinates, 4th Ed. London, UK: Academic Press. [Google Scholar]
- R Core Team (2019) R: A Language and Environment for Statistical Computing. Available at: https://www.r-project.org/.
- Rivat C, Laboureyras E, Laulin JPP, Le Roy C, Richebé P, Simonnet G (2007) Non-nociceptive environmental stress induces hyperalgesia, not analgesia, in pain and opioid-experienced rats. Neuropsychopharmacology 32:2217–2228. [DOI] [PubMed] [Google Scholar]
- Sadler KE, McQuaid NA, Cox AC, Behun MN, Trouten AM, Kolber BJ (2017) Divergent functions of the left and right central amygdala in visceral nociception. Pain 158:747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severino A, Chen W, Hakimian JK, Kieffer BL, Gaveriaux-ruff C, Walwyn W, Marvizón JCG (2018) Mu-opioid receptors in nociceptive afferents produce a sustained suppression of hyperalgesia in chronic pain. Pain 159:1607–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoblock JR, Maidment NT (2006) Constitutively Active Mu Opioid Receptors Mediate the Enhanced Conditioned Aversive Effect of Naloxone in Morphine-Dependent Mice. :171–177. [DOI] [PubMed] [Google Scholar]
- Skoubis PD, Lam HA, Shoblock J, Narayanan S, Maidment NT (2005) Endogenous enkephalins, not endorphins, modulate basal hedonic state in mice. Eur J Neurosci 21:1379–1384. [DOI] [PubMed] [Google Scholar]
- Skoubis PD, Matthes HW, Walwyn WM, Kieffer BL, Maidment NT (2001) Naloxone fails to produce conditioned place aversion in μ-opioid receptor knock-out mice. Neuroscience 106:757–763. [DOI] [PubMed] [Google Scholar]
- Snyder LM et al. (2018) Kappa Opioid Receptor Distribution and Function in Primary Afferents Article Kappa Opioid Receptor Distribution and Function in Primary Afferents. Neuron 99:1274–1288.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solway B, Bose SC, Corder G, Donahue RR, Taylor BK (2011) Tonic inhibition of chronic pain by neuropeptide Y. PNAS 108:7224–7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinus L, Le Moal M, Koob GF (1990) Nucleus accumbens and amygdala are possible substrates for the aversive stimulus effects of opiate withdrawal. Neuroscience 37:767–773. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Corder G (2014) Endogenous Analgesia, Dependence, and Latent Pain Sensitization. Curr Top Behav Neurosci 20:283–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veinante P, Yalcin I, Barrot M (2013) The amygdala between sensation and affect: a role in pain. J Mol Psychiatry 1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Ossipov MH, Lai J, King T, Porreca F (2011) Descending Facilitatory Pathways from the Rostroventromedial Medulla Mediate Naloxone-Precipitated Withdrawal in Morphine-Dependent Rats. J Pain 12:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn WM, Chen W, Kim H, Minasyan A, Ennes HS, McRoberts JA, Marvizón JCG (2016) Sustained Suppression of Hyperalgesia during Latent Sensitization by μ-, δ-, and κ-opioid receptors and α2A Adrenergic Receptors: Role of Constitutive Activity. J Neurosci 36:204–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Yamamoto R, Maeda A, Nakagawa T, Minami M, Satoh M (2002) Effects of excitotoxic lesions of the central or basolateral nucleus of the amygdala on naloxone-precipitated withdrawal-induced conditioned place aversion in morphine-dependent rats. Brain Res 958:423–428. [DOI] [PubMed] [Google Scholar]
- Wilson TD, Valdivia S, Khan A, Ahn H-S, Adke AP, Martinez Gonzalez S, Sugimura YK, Carrasquillo Y (2019) Dual and Opposing Functions of the Central Amygdala in the Modulation of Pain. Cell Rep 29:332–346.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JY, De Felice M, Kopruszinski CM, Eyde N, LaVigne J, Remeniuk B, Hernandez P, Yue X, Goshima N, Ossipov M, King T, Streicher JM, Navratilova E, Dodick D, Rosen H, Roberts E, Porreca F (2017) Kappa opioid receptor antagonists: A possible new class of therapeutics for migraine prevention. Cephalalgia 37:780–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Hammond DL (2010) Cellular basis for opioid potentiation in the rostral ventromedial medulla of rats with persistent inflammatory nociception. Pain 149:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RX, Zhang M, Li A, Pan L, Berman BM, Ren K, Lao L (2013) DAMGO in the central amygdala alleviates the affective dimension of pain in a rat model of inflammatory hyperalgesia. Neuroscience 252:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.