

Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is mainly caused by mutations in genes encoding desmosomal proteins. Variants in plakophilin-2 gene (PKP2) are the most common cause of the disease, associated with conventional ARVC phenotype. The study aims to evaluate the prevalence of PKP2 variants and examine genotype–phenotype correlation in Polish ARVC cohort. All 56 ARVC patients fulfilling the current criteria were screened for genetic variants in PKP2 using denaturing high-performance liquid chromatography or next-generation sequencing. The clinical evaluation involved medical history, electrocardiogram, echocardiography, and follow-up. Ten variants (5 frameshift, 2 nonsense, 2 splicing, and 1 missense) in PKP2 were found in 28 (50%) cases. All truncating variants are classified as pathogenic/likely pathogenic, while the missense variant is classified as variant of uncertain significance. Patients carrying a PKP2 mutation were younger at diagnosis (p = 0.003), more often had negative T waves in V1–V3 (p = 0.01), had higher left ventricular ejection fraction (p = 0.04), and were less likely to present symptoms of heart failure (p = 0.01) and left ventricular damage progression (p = 0.04). Combined endpoint of death or heart transplant was more frequent in subgroup without PKP2 mutation (p = 0.03). Pathogenic variants in PKP2 are responsible for 50% of ARVC cases in the Polish population and are associated with a better prognosis. ARVC patients with PKP2 mutation are less likely to present left ventricular involvement and heart failure symptoms. Combined endpoint of death or heart transplant was less frequent in this group.
Keywords: Arrhythmogenic right ventricular cardiomyopathy, Desmosomal genes, Plakophilin-2, PKP2
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heart muscle disease characterized by fibrofatty replacement of the myocardium leading to electrical instability and ventricular arrhythmias and increasing risk of sudden death. Its prevalence in general population is estimated at 1 in 1000 to 1 in 5000 (Basso et al. 2009) as a result of possible underdiagnosed cases with mild or no symptoms. The diagnosis of ARVC is based on International Task Force Criteria and, since 2010, major diagnostic criteria include the presence of a pathogenic mutation (Marcus et al. 2010). At least 50% of cases are familial and ARVC is currently considered a genetically determined cardiomyopathy, mainly caused by pathogenic variants in genes encoding desmosomal proteins (Basso et al. 2009; Marcus et al. 2010).
The disease usually has an autosomal dominant pattern with age-related, incomplete penetrance and variable expression, leading to an isolated cardiac phenotype (Hoorntje et al. 2017). Pathogenic variants in desmosomal genes are being identified in 33 to 63% of probands (Gandjbakhch et al. 2018). Desmosomes are membrane protein complexes specialized for cell-to-cell adhesion and maintenance of the structural integrity of the ventricular myocardium. Defects in the structure of desmosomes result in cardiac myocyte detachment and death, gap junction remodeling, and dysregulation of the Wnt–beta catenin pathway, leading to fibrofatty tissue substitution and electrical instability (Gandjbakhch et al. 2018).
However, the genetic cause of ARVC remains unknown for 40 to 50% of patients (Gandjbakhch et al. 2018). It is reported that among gene-elusive cases, only about one fifth has a positive family history (Groeneweg et al. 2015). A high percentage of athletes among gene-elusive, non-familial ARVC subjects suggests that high-intensity exercise can play an important role in the disease pathogenesis (Sawant et al. 2014).
Among desmosomal genes, pathogenic variants in plakophilin-2 gene (PKP2) appear to be the most common cause of the disease accounting for 36 to 92% of mutations identified in desmosomal genes (Gandjbakhch et al. 2018). They are associated with an isolated right ventricular involvement and a conventional ARVC phenotype (Xu et al. 2017).
Despite a huge amount of data from many studies, a genotype–phenotype correlation is still lacking and predictive value of genetic testing is sometimes undermined (Sheikh et al. 2018). In the present study, we have evaluated the prevalence of PKP2 variants in an ARVC cohort of Polish patients and investigated the genotype–phenotype correlation.
Methods
Study population
The study cohort comprised 56 patients fulfilling the 2010 International Task Force Criteria (ITFC) for ARVC, who were diagnosed at our institution during the years 1983–2019. As the National Institute of Cardiology is the reference center for ARVC in Poland, the study group consisted of patients from all over the country. All subjects were Caucasian and 48 (86%) were male. Each individual was screened for pathogenic variants in PKP2. The study was approved by the institutional bioethics committee and informed consent was obtained from each patient.
Genetic testing
DNA was extracted from the peripheral blood by phenol extraction or salting-out method. All probands were screened for genetic variants in PKP2, the most frequently affected gene in ARVC patients (https://www.omim.org/entry/602861). Mutation screening was conducted by denaturing high-performance liquid chromatography (DHPLC) (Padua, Italy, tests performed between 2002 and 2005, n = 38) and/or next-generation sequencing (NGS) (Warsaw, Poland, test performed between 2015 and 2016, n = 37) of amplicons designed specifically to cover all PKP2 exons including exon–intron boundaries.
NGS was performed using 454 Genome Sequencer (GS) Junior platform (Roche Diagnostics, Basel, Switzerland) according to the Amplicon Library Preparation Method Manual. All libraries were sequenced with minimum depth of 20 reads. Data were analyzed with GS Amplicon Variant Analyzer (AVA) software (Roche). DHPLC was performed with the use of Wave Nucleic Acid Fragment Analysis System 3500 HT with DNASep HT cartridge technology (Transgenomic Inc, Omaha, NE, USA).
Each suspected DHPLC eluate and/or Junior amplicon found in probands were confirmed with Sanger sequencing using 3500xL Genetic Analyzer (Life Technologies, Carlsbad, CA, USA) and BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies) following the manufacturer’s instructions. Chromatograms were analyzed using Variant Reporter 1.1 (Life Technologies).
Variant frequencies were derived from gnomAD (http://gnomad.broadinstitute.org). VarSome database (https://varsome.com) was used for the bioinformatic prediction scores and evaluating variant’s classification according to the American College of Medical Genetics and Genomics (ACMG) criteria (Richards et al. 2015). The clinical significance of the variants was based on ClinVar (https://www.ncbi.nlm.nih.gov/clinvar). To evaluate genotype–phenotype correlation, we considered PKP2 variants classified as pathogenic and likely pathogenic according to ACMG criteria.
Clinical evaluation
The evaluation of patients involved detailed medical history, including age at diagnosis, family history, history of sports activity, arrhythmia (cardiac arrest, syncope, sustained and nonsustained ventricular tachycardia, premature ventricular beats, atrial fibrillation/flutter, appropriate implantable cardioverter-defibrillator (ICD) interventions, ventricular tachycardia ablation), and heart failure symptoms. ARVC diagnosis was based on the presence of major and minor criteria according to 2010 ITFC. A standard 12-lead electrocardiogram (ECG) was analyzed for negative T waves in precordial leads, QRS duration and dispersion, and presence of epsilon wave and notched S in leads V1–V3. Right ventricular dimensions, and right and left ventricular systolic function were obtained from two-dimensional echocardiographic examinations performed for clinical purposes. Patients were followed for clinical course (right and left ventricular damage progression) and combined endpoint (death or heart transplant).
Statistical analysis
Clinical characteristics of patients were compared between patients with and without identified pathogenic variant in PKP2. For phenotype analysis, three patients with mutations in desmocollin-2, desmoglein-2, and desmoplakin genes (DSC2, DSG2, and DSP, respectively) were classified as PKP2 mutation-negative. Continuous variables were expressed as means with SDs or medians with quartile deviation (QR) depending on symmetrical distribution or not, respectively. The normality of distribution was assessed using the Shapiro–Wilk test. Categorical variables were expressed as frequencies and percentages. Comparative analyses were done using the χ2 test or Fisher’s exact test for categorical data, as appropriate. Continuous variables were assessed with t test or Mann–Whitney U test for non-normally distributed variables. P values < 0.05 were considered statistically significant. Differences in survival probabilities were measured by Kaplan–Meier curve. The statistical significance of the mentioned differences was established by using the log-rank test. All statistical calculations were performed using the Statistica 12 package (Statsoft, Krakow, Poland).
Results
PKP2 variants
In 28/56 cases (50%, 89.3% male), variants in PKP2 were identified. These included five frameshift, two nonsense, two splicing, and one missense variant; five of them were recurring variants (see Table 1 and Fig. 1). All truncating variants (frameshift, nonsense, splicing) are classified as pathogenic or likely pathogenic according to ACMG criteria. The only missense variant at the time, classified as variant of uncertain significance (VUS), has been taken into account on the basis of very low minor allele frequency in general population (gnomAD = 0.000) and the in silico prediction of pathogeneicity (predicted as deleterious by 10 prediction tools).
Table 1.
PKP2 (NM_004572.3) variants analyzed in our cohort
| Variant (dbSNP ID) |
Genomic coordinates (GRCh38) | ACMG verdict | ClinVar clinical significance | gnomAD allele frequency | Number of cases | Described |
|---|---|---|---|---|---|---|
| Frameshift variants | ||||||
|
p.Thr50SerfsTer61 c.148_151delACAG (rs397516997) |
chr12-32,896,581-CTGT- | Pathogenic | Pathogenic | 0 | 1 | Yes |
|
p.His318TrpfsTer10 c.929_951dupTGGATTCCAGCGGGAGGAGAGCG (rs1064792927) |
chr12-32,877,929–CG…CA (23 bp) | Pathogenic | Pathogenic | 0.00000407 | 2 | No |
|
p.Ala568ValfsTer9 c.1703delC (n/a) |
chr12-32,824,148-G- | Likely pathogenic | n/a | 0 | 1 | No |
|
p.His733AlafsTer8 c.2197_2202delCACACCinsG (rs397517021) |
chr12-32,802,500-GGTGTG-C | Pathogenic | Pathogenic | 0 | 1 | Yes |
|
p.Asn809GlufsTer18 c.2423dupA (n/a) |
chr12-32,796,175–T | Likely Pathogenic | n/a | 0 | 1 | Yes |
| Nonsense variants | ||||||
|
p.Arg413Ter c.1237C > T (rs372827156) |
chr12-32,850,907-G-A | Pathogenic | Pathogenic | 0.00001415 | 4 | Yes |
|
p.Gln638Ter c.1912C > T (rs397517012) |
chr12-32,822,526-G-A | Pathogenic | Pathogenic | 0.000007074 | 3 | Yes |
| Splicing variants | ||||||
|
c.2146-1G > C (rs193922674) |
chr12-32,802,557-C-G | Pathogenic | Pathogenic | 0.00003184 | 5 | Yes |
|
c.2489 + 1G > A (rs111517471) |
chr12-32,796,108-C-T | Pathogenic | Pathogenic | 0.00002829 | 9 | Yes |
| Missense variants | ||||||
|
p.Gly673Val c.2018G > T (rs1426480515) |
chr12-32,821,483-C-A | Uncertain Significance | n/a | 0 | 1 | Yes |
Fig. 1.

The distribution of PKP2 variants found in this study
Genotype–phenotype correlation
Table 2 summarizes the medical history, clinical features, arrhythmia, clinical course, and survival outcomes of PKP2 mutation-positive patients compared with PKP2 mutation-negative individuals. In PKP2 mutation-positive group, all 28 patients were unrelated, while in PKP2 mutation-negative group there were 27 probands and one third-degree relative. There was no difference between PKP2 mutation-positive and mutation-negative individuals in sex, family history, history of sports activity, and arrhythmia (meaning cardiac arrest, syncope, ventricular tachycardia (VT), premature ventricular beats, supraventricular arrhythmias, ICD implantation, appropriate ICD interventions, and history of VT ablation) (see Table 2). At diagnosis, PKP2 mutation-positive patients were significantly younger than PKP2 mutation-negative subjects (mean ±SD age 32 ±11 vs. 41 ±12, p = 0.003). Total ITFC 2010 punctation was identical in both groups; however, negative T waves in leads V1–V3 occurred more often in PKP2 mutation-positive patients (75% vs. 43%, p = 0.01), while epsilon wave was more frequent in PKP2 mutation-negative subgroup (75% vs. 21%, p < 0.001). In echocardiography, right ventricular outflow tract dimension and presence of regional right ventricular akinesia or dyskinesia did not differ between those with and without identified PKP2 mutations. Left ventricular ejection fraction (LVEF) was significantly lower in PKP2 mutation-negative subjects (mean ±SD left ventricular ejection fraction 48 ±17 vs. 60 ±7, p = 0.04), which is consistent with the observation that individuals without a PKP2 mutation were more likely to present symptoms of heart failure (54% vs. 21%, p = 0.01). Patients with and without identified PKP2 mutations had similar proportions of right ventricular damage progression (68% vs. 68%; p = 0.83), while left ventricular damage progression was more often observed in PKP2 mutation-negative patients (18% vs. 43%, p = 0.04). Combined endpoint of death or heart transplant occurred more frequently in PKP2 mutation-negative group (11% vs. 39%, p = 0.03). This observation was confirmed by Kaplan–Meier survival analysis (see Fig. 2). However, when analyzing the causes of deaths divided into sudden death and death due to heart failure (this subgroup included also patients who underwent heart transplant), no significant differences were observed.
Table 2.
Clinical characteristics of PKP2 mutation-positive and PKP2 mutation-negative arrhythmogenic right ventricular cardiomyopathy patients
|
PKP2 mutation-positive n = 28 |
PKP2 mutation-negative n = 28 |
p | |
|---|---|---|---|
| Male, n (%) | 25 (89) | 23 (82) | 0.45 |
| Age at diagnosis, years, mean (SD) | 32 (11) | 41 (12) | 0.003 |
| SCD in the young in the family, n (%) | 4 (14) | 4 (14) | 1.0 |
| ARVC in the family, n (%) | 8 (29) | 6 (21) | 0.54 |
| History of sports activity, n (%) | 10 (36) | 10 (36) | 0.92 |
| Cardiac arrest, n (%) | 4 (14) | 5 (18) | 0.72 |
| Syncope, n (%) | 14 (50) | 14 (50) | 1.0 |
| Sustained VT, n (%) | 22 (79) | 20 (71) | 0.54 |
| Nonsustained VT, n (%) | 25 (89) | 24 (86) | 0.69 |
| PVB > 500/day, n (%) | 25 (89) | 27 (96) | 0.30 |
| AF/AFL, n (%) | 8 (29) | 7 (25) | 0.76 |
| ICD, n (%) | 20 (71) | 15 (54) | 0.17 |
| Appropriate ICD interventions, n (%) | 16 (57) | 10 (36) | 0.37 |
| VT ablation, n (%) | 14 (50) | 15 (54) | 0.79 |
| Total ITFC punctation, mean (SD) | 6.5 (1.5) | 6.5 (1.7) | 0.90 |
| Negative T waves in leads V1–V3 (in the absence of RBBB), n (%) | 21 (75) | 12 (43) | 0.01 |
| QRS > 110 ms, n (%) | 14 (50) | 17 (61) | 0.42 |
| QRS dispersion, ms, mean (SD) | 22 (22) | 31 (19) | 0.09 |
| Epsilon wave, n (%) | 6 (21) | 21 (75) | < 0.001 |
| Notched S in leads V1–V3, n (%) | 15 (54) | 13 (46) | 0.31 |
| RVOT PLAX, mm, mean (SD) | 42 (8) | 45 (12) | 0.33 |
| Regional RV akinesia or dyskinesia, n (%) | 25 (89) | 27 (96) | 0.30 |
| LVEF, %, mean (SD) | 60 (7) | 48 (20) | 0.04 |
| Heart failure symptoms, n (%) | 6 (21) | 15 (54) | 0.01 |
| RV damage progression, n (%) | 19 (68) | 19 (68) | 0.83 |
| LV damage progression, n (%) | 5 (18) | 12 (43) | 0.04 |
| Death or HTx, n (%) | 3 (11) | 11 (39) | 0.03 |
| Sudden death, n (%) | 0 (0) | 3 (11) | 0.24 |
| Death of heart failure or HTx, n (%) | 3 (11) | 8 (29) | 0.18 |
| Follow-up duration, years, mean (SD) | 17.4 (9.1) | 14.8 (8.0) | 0.24 |
AF atrial fibrillation, AFL atrial flutter, HTx heart transplant, ICD implantable cardioverter-defibrillator, ITFC International Task Force Criteria, LV left ventricle, LVEF left ventricular ejection fraction, PLAX parasternal long-axis view, PVB premature ventricular beats, RBBB right bundle branch block, RV right ventricle, RVOT right ventricular outflow tract, VT ventricular tachycardia
Fig. 2.
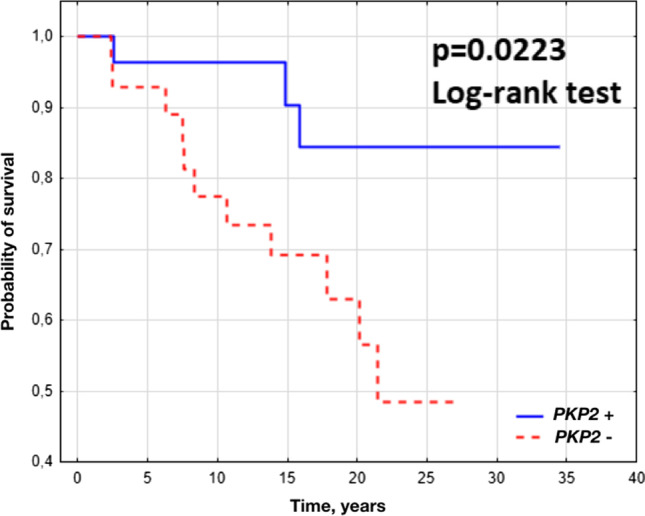
Kaplan–Meier survival curves for combined endpoint of death or heart transplant in PKP2 mutation-positive and PKP2 mutation-negative arrhythmogenic right ventricular cardiomyopathy patients
Discussion
ARVC is mainly caused by pathogenic variants in genes encoding desmosomal proteins, known as desmosomal genes. Among them, mutations in PKP2 are the most common cause of the disease. In our study, PKP2 mutations were identified in 50% of ARVC patients. This observation is consistent with previous studies estimating the frequency of PKP2 mutations for 20–46% (James et al. 2020). Other desmosomal mutations are identified in 3–20% for DSP, 3–20% for DSG2, and 1–15% for DSC2 (James et al. 2020).
PKP2 variants
The PKP2 encodes plakophilin-2, a protein found primarily in cells of the myocardium. This gene belongs to the family of plakophilins characterized by numerous armadillo repeats, localizes to cell desmosomes and nuclei, and participates in linking cadherins to intermediate filaments in the cytoskeleton (Bass-Zubek et al. 2008; Cerrone et al. 2017).
In our cohort, nearly 97% of PKP2 variants were truncating variants which is consistent with other data: according to ClinVar and VarSome databases, truncating variants account for 91.6 and 87.3%, respectively, of pathogenic/likely pathogenic variants in PKP2. The frequency of PKP2 variants found in our cohort is consistent with their frequency in the gnomAD database, with two splice (c.2489 + 1G > A, c.2146-1G > C) and two nonsense (p.Arg413Ter, p.Gln638Ter) variants being the most common. However, one nonsense variant (p.Arg79Ter), which is also very often reported in the literature, was not found in our study group (Tintelen et al. 2006; Lint et al. 2019).
Genotype–phenotype correlation
Despite many studies, the genotype–phenotype relationship in ARVC is not fully understood. It is believed that PKP2 mutations are more likely to cause isolated right ventricular involvement and a conventional ARVC phenotype compared with other desmosomal mutations (Gandjbakhch et al. 2018; Riele et al. 2013). This thesis has been confirmed by our observations regarding the Polish group of ARVC patients.
As previously reported (Xu et al. 2017; Dalal et al. 2006), our results showed a younger age of onset in patients with PKP2 mutation compared with those without PKP2 mutation. As expected, we did not observe any differences between PKP2 mutation-positive and negative individuals in the proportion of males. Although many previous studies indicated that the presence of desmosomal gene mutations as well as PKP2 mutations is associated with a higher incidence of ventricular arrhythmias (Xu et al. 2017; Dalal et al. 2006; Ohno et al. 2013; Bao et al. 2013), this observation was not confirmed by our results. Also, Bhonsale et al. in a large USA/Dutch registry (Bhonsale et al. 2015) did not detect such correlation. However, it is worth noting that our results showed a higher occurrence of T-wave inversion in V1–V3 leads in PKP2 mutation-positive patients than in PKP2 mutation-negative subjects. This has also been demonstrated by the meta-analysis performed by Xu et al. in 2016 (Xu et al. 2017).
Many previous studies suggest that desmosomal mutations other than PKP2 are more often associated with biventricular cardiomyopathy or isolated left ventricular involvement (Gandjbakhch et al. 2018; Bhonsale et al. 2015; Rigato et al. 2013; Castelletti et al. 2017; Fressart et al. 2010; Wong et al. 2019). Moreover, patients with these mutations, especially DSG2 mutation carriers, present a higher risk of developing end-stage heart failure than PKP2 mutation carriers (Gandjbakhch et al. 2018; Hermida et al. 2019). However, not all studies confirm this outcome (Bhonsale et al. 2015). Presence of PKP2 mutation in our population was associated with higher left ventricular ejection fraction compared with mutation-negative individuals. Moreover, we found that ARVC patients with a PKP2 mutation present less frequently left ventricular damage progression and symptoms of heart failure than subjects without PKP2 mutation.
Unlike other researchers, we observed a higher incidence of epsilon wave in PKP2 mutation-negative individuals compared with PKP2 mutation carriers. A possible explanation of this discrepancy could be a more extensive myocardial injury in PKP2 mutation-negative subgroup. It may also be related to the small size of the study group.
Observing our patients for years, we have an impression that PKP2 mutation-positive subgroup presents more frequently classical ARVC phenotype, while PKP2 mutation-negative subgroup includes patients showing clinical features ranging from benign ventricular arrhythmia to severe biventricular involvement leading to end-stage heart failure. Although subjects without PKP2 mutation meet ITFC 2010, the diagnosis is less certain. Moreover, we found that in subjects without PKP2 mutation combined endpoint of death or heart transplant occurred more frequently, which has not been clearly shown in previous studies.
Study limitations
The study was conducted retrospectively; therefore, there was no consistent follow-up protocol. Genetic testing was performed in two laboratories at different times. Some patients were examined a few years ago and then they were lost to follow-up for various reasons. Due to the small size of the study group, we decided to include these patients. As a part of genetic testing, some patients were screened only for mutations in PKP2, while others also had other desmosomal genes tested. In three patients mutations in other ARVC-associated genes (DSC2 /ACMG-VUS/, DSG2 /ACMG-pathogenic/, and DSP /ACMG-VUS/) were found—these patients were classified as PKP2 mutation negative. Furthermore, not all patients underwent myocardial biopsy.
It is worth noting that when analyzing the phenotype of ARVC patients, PKP2 mutation-negative group consists of subjects with other desmosomal or non-desmosomal mutations, as well as patients without any mutation. The possibilities of comparing the phenotype of these subgroups are limited because they are too small for statistical analysis. In this context, it seems reasonable to compare patients with and without PKP2 mutation since the size of these groups is similar.
Conclusions
Pathogenic variants in PKP2 are responsible for 50% of ARVC cases in the Polish population. The presence of these variants is associated with a better prognosis and a lower incidence of combined endpoint, including death or heart transplant. ARVC patients with PKP2 mutation are less likely to have left ventricular involvement and heart failure symptoms.
Author contribution
E.K.B. conceived the concept of the study, was involved in data collection and interpretation of results, and coordinated funding for the project. K.B. was involved in data collection, integration of data, interpretation of results, and preparation of the manuscript. M.F. was involved in data collection, interpretation of results, and preparation of the manuscript. M.S., A.R., O.W., and M.R. contributed to data collection. W.Ś. performed statistical analysis of data. A.L. and P.H. supervised all phases of the project and were involved in data analysis.
Funding
This work was supported by the National Institute of Cardiology in Warsaw, Poland (grant number 2.53/II/15).
Availability of data and material
All data and materials support the published claims and comply with field standards.
Code availability
Not applicable.
Declarations
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the Bioethics Committee of the National Institute of Cardiology, Warsaw, Poland.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Conflicts of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Elżbieta K. Biernacka and Karolina Borowiec equally contributed to the study
References
- Bao J, Wang J, Yao Y, et al. Correlation of ventricular arrhythmias with genotype in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6:552–556. doi: 10.1161/CIRCGENETICS.113.000122. [DOI] [PubMed] [Google Scholar]
- Basso C, Corrado D, Marcus FI, et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- Bass-Zubek AE, Hobbs RP, Amargo EV, et al. Plakophilin 2: a critical scaffold for PKC alpha that regulates intercellular junction assembly. J Cell Biol. 2008;181(4):605–613. doi: 10.1083/jcb.200712133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhonsale A, Groeneweg JA, James CA, et al (2015) Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J ehu509 [DOI] [PubMed]
- Castelletti S, Vischer AS, Syrris P, et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: genotype–phenotype correlation. Int J Cardiol. 2017;249:268–273. doi: 10.1016/j.ijcard.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Cerrone M, Montnach J, Lin X, et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat Commun. 2017;8(1):106. doi: 10.1038/s41467-017-00127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal D, Molin LH, Piccini J, et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation. 2006;113(13):1641–1649. doi: 10.1161/CIRCULATIONAHA.105.568642. [DOI] [PubMed] [Google Scholar]
- Fressart V, Duthoit G, Donal E, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–868. doi: 10.1093/europace/euq104. [DOI] [PubMed] [Google Scholar]
- Gandjbakhch E, Redheuil A, Pousset F, et al. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72(7):784–804. doi: 10.1016/j.jacc.2018.05.065. [DOI] [PubMed] [Google Scholar]
- Groeneweg JA, Bhonsale A, James CA, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015;8:437–446. doi: 10.1161/CIRCGENETICS.114.001003. [DOI] [PubMed] [Google Scholar]
- Hermida A, Fressart V, Hidden-Lucet F, et al. High risk of heart failure in desmoglein-2 mutation carriers in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Europace. 2019;19(suppl_3):iii132. doi: 10.1093/ehjci/eux146. [DOI] [Google Scholar]
- Hoorntje ET, Te Rijdt WP, James CA, et al. Arrhythmogenic cardiomyopathy: pathology, genetics, and concepts in pathogenesis. Cardiovasc Res. 2017;113(12):1521–1531. doi: 10.1093/cvr/cvx150. [DOI] [PubMed] [Google Scholar]
- James CA, Syrris P, van Tintelen JP, Calkins H. The role of genetics in cardiovascular disease: arrhythmogenic cardiomyopathy. Eur Heart J. 2020;41(14):1393–1400. doi: 10.1093/eurheartj/ehaa141. [DOI] [PubMed] [Google Scholar]
- Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31(7):806–814. doi: 10.1093/eurheartj/ehq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S, Nagaoka I, Fukuyama M, et al. Age-dependent clinical and genetic characteristics in Japanese patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ J. 2013;77(6):1534–1542. doi: 10.1253/circj.CJ-12-1446. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigato I, Bauce B, Rampazzo A, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6:533–542. doi: 10.1161/CIRCGENETICS.113.000288. [DOI] [PubMed] [Google Scholar]
- Sawant AC, Bhonsale A, Te Riele ASJM, et al. Exercise has a disproportionate role in the pathogenesis of arrhythmogenic right ventricular dysplasia/cardiomyopathy in patients without desmosomal mutations. J Am Heart Assoc. 2014;3:e001471. doi: 10.1161/JAHA.114.001471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh N, Papadakis M, Wilson M, et al. Diagnostic yield of genetic testing in young athletes with T-wave inversion. Circulation. 2018;138:1184–1194. doi: 10.1161/CIRCULATIONAHA.118.034208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Riele AS, James CA, Philips B, et al. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol. 2013;24:1311–1320. doi: 10.1111/jce.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lint FHM, Murray B, Tichnell C, et al. Arrhythmogenic right ventricular cardiomyopathy-associated desmosomal variants are rarely de novo. Circ Genom Precis Med. 2019;12(8):e002467. doi: 10.1161/CIRCGEN.119.002467. [DOI] [PubMed] [Google Scholar]
- van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- Wong JA, Duff HJ, Yuen T, et al. Phenotypic analysis of arrhythmogenic cardiomyopathy in the Hutterite population: role of electrocardiogram in identifying high-risk desmocollin-2 carriers. J Am Heart Assoc. 2014;3:e001407. doi: 10.1161/JAHA.114.001407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zhu W, Wang C, et al. Genotype–phenotype relationship in patients with arrhythmogenic right ventricular cardiomyopathy caused by desmosomal gene mutations: a systematic review and meta-analysis. Sci Rep. 2017;25(7):41387. doi: 10.1038/srep41387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and materials support the published claims and comply with field standards.
Not applicable.