

Abstract
Cell therapies based on reprogrammed adaptive immune cells have great potential as “living drugs.” As first demonstrated clinically for engineered chimeric antigen receptor (CAR) T cells, the ability of such cells to undergo clonal expansion in response to an antigen promotes both self-renewal and self-regulation in vivo. B cells also have the potential to be developed as immune cell therapies, but engineering their specificity and functionality is more challenging than for T cells. In part, this is due to the complexity of the immunoglobulin (Ig) locus, as well as the requirement for regulated expression of both cell surface B cell receptor and secreted antibody isoforms, in order to fully recapitulate the features of natural antibody production. Recent advances in genome editing are now allowing reprogramming of B cells by site-specific engineering of the Ig locus with preformed antibodies. In this review, we discuss the potential of engineered B cells as a cell therapy, the challenges involved in editing the Ig locus and the advances that are making this possible, and envision future directions for this emerging field of immune cell engineering.
Keywords: B cells, genome editing, Cas9, cell therapy, immunotherapy, HIV
Graphical abstract
B cells have recently emerged as a novel immune cell therapy due to innovations in genome editing at the native immunoglobulin locus. Rogers and Cannon highlight the potential of engineered B cells as a therapeutic, review recent work toward these goals, and discuss future directions for the field.
Introduction
Advances in genome editing are driving innovations in existing gene and cell therapies and catalyzing the development of entirely new treatments.1 This includes chimeric antigen receptor (CAR) T cell therapies, which have seen clinical success against certain hematological cancers2 and are now being explored against solid tumors3 and infectious diseases such as HIV.4,5 Here, T cells are reprogrammed using artificial receptors that link recognition of a targeted antigen to the signaling and effector functions of the T cell.6 Genome editing is being used to further enhance receptor functionality,7 ablate immune checkpoints that limit T cell functionality,8 and facilitate off-the-shelf allogeneic cell therapies that would not require manipulation of a patient’s own cells.9, 10, 11, 12
These recent successes of CAR T cell therapies mean that immune cell reprogramming is now also being considered for a population that has received relatively less attention to date—the B cell. These cells comprise the humoral arm of the adaptive immune system and are responsible for the production of antibodies. The therapeutic potential of monoclonal antibodies is already well established and has delivered highly successful treatments for cancer, autoimmune diseases, and infectious diseases.13 In particular, monoclonal antibodies have proven invaluable in applications where such antibodies cannot be induced naturally, for example when targeting self-antigens as treatments for cancer or autoimmune diseases.14,15 Monoclonal antibodies are also necessary when the antibodies cannot be elicited by vaccination, as is currently the case with broadly neutralizing antibodies (bnAbs) against HIV.16,17 However, recombinant antibody therapies cost on average $100,000 per year,18 and the need for frequent re-administration is an additional challenge when prolonged treatments are required.
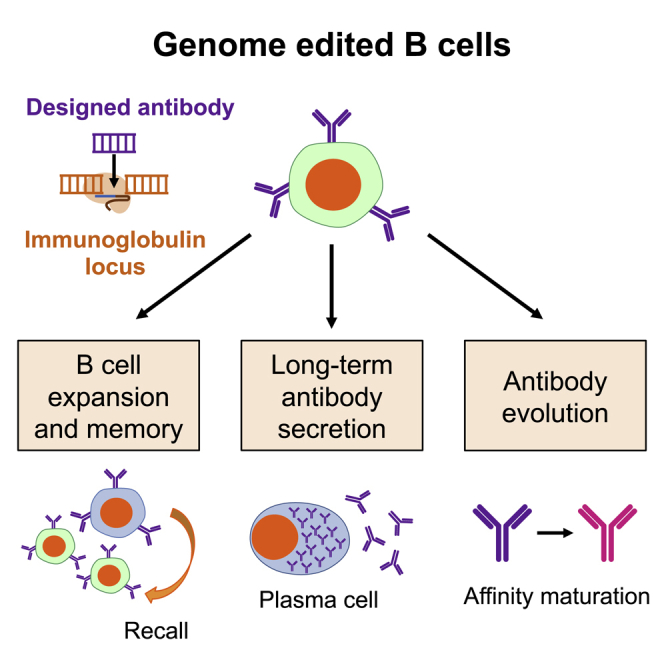
These limitations of monoclonal antibody therapies have led to interest in using gene therapy to achieve sustained antibody production in vivo. These include the use of adeno-associated virus (AAV) vectors to deliver antibody expression cassettes to tissues such as skeletal muscle or liver.19 However, such approaches can be limited by challenges in achieving desired antibody expression levels, or the emergence of anti-drug antibodies against the antibody transgene product.20, 21, 22 Additionally, expression of antibody cassettes from such ectopic cells does not recapitulate the characteristic and advantageous features of natural antibody production from B cells. These include the capacity for prolonged antibody secretion, memory recall responses that can be boosted by vaccination, the evolution of antibodies over time through affinity maturation, and communication with other cells of the immune system (Figure 1). For these reasons, we and others are interested in harnessing genome editing to reprogram B cells to express antibodies with pre-selected specificities.
Figure 1.

Potential features of engineered B cells as an immune cell therapy
The functional potential of engineered B cells and antibodies
Antibodies play many roles that can be exploited for therapeutic applications. They are perhaps best known for their ability to bind to soluble targets, neutralizing the function of secreted proteins, or inhibiting infection by extracellular viruses and bacteria. However, many functions of antibodies also require that they interact with other immune system components through their Fc domains. This allows them to recruit natural killer (NK) cells for cell killing through antibody-dependent cellular cytotoxicity (ADCC); direct macrophages, neutrophils, and dendritic cells to take up and degrade immune complexes or infected cells through antibody-dependent cellular phagocytosis (ADCP); and harness the complement system to lyse viruses, bacteria, and cells through antibody-dependent complement deposition (ADCD).23 These functions can also be further enhanced by modifications to the antibody’s Fc stalk.24
At the same time, B cells communicate with the rest of the immune system through both general (cytokine secretion) and specific (antigen presentation) processes. Antigen-specific interactions can occur either directly, when a B cell endocytoses an antigen and presents it to CD4+ T cells on major histocompatibility complex (MHC) class II,25 or indirectly, when an antibody opsonizes the antigen, leading to presentation by dendritic cells to both CD4+ and CD8+ T cells.26 Indeed, local infiltration of pro-inflammatory B cells has been associated with improved outcomes in a number of different cancers,27, 28, 29, 30, 31 in particular during checkpoint inhibitor blockade, suggesting a previously underappreciated role for B cells in anti-tumor immunity. Increasingly, the importance of holistic immune responses involving both arms of the adaptive immune response is being appreciated, so that the ability of an engineered B cell to maintain these natural communications with other immune cells would be an advantage in an immunotherapy setting.
Finally, B cells have the potential to provide lifelong immune surveillance. This has been seen in studies of immune memory to pathogens experienced many years ago. For example, although smallpox-specific CD4+ T cells were reported to decline with a half-life of roughly 14 years,32 stable secretion of protective antibodies and robust anamnestic B cell responses could still be observed in individuals for at least 48 years after infection.32,33 This suggests that humoral immunity driven by B cells may be more capable of prolonged surveillance than is the cellular immune response.
Exploiting the range of B cell phenotypes
B cells have a relatively complex life cycle and adopt a variety of different phenotypes and functions at different stages of differentiation (Figure 2). Naive B cells undergo VDJ recombination to produce a unique antibody specificity during development in the bone marrow, then circulate through the blood and secondary lymphoid organs until they encounter their cognate antigen. After antigen encounter, the range of differentiation possibilities opens up through clonal expansion. Some B cells immediately become short-lived plasmablasts, producing immunoglobulin (Ig)M subtype antibodies to mount an immediate response. Others may enter a germinal center (GC), which allows them to hone the functionality of their antibody through somatic hypermutation (SHM), as well as to undergo class-switch recombination (CSR) to acquire the functionalities of different antibody isotypes and subclasses, for example IgG. Of note, while CSR has classically been considered a hallmark of the GC, recent evidence has suggested that CSR may actually occur prior to GC entry and independently of SHM.34 Upon emergence from the GC, some cells may once again become short-lived plasmablasts, whereas others differentiate into long-lived plasma cells (LLPCs) that can survive for decades in the absence of further proliferation.35 Still other cells adopt a memory B cell phenotype capable of prolonged survival and primed to re-expand and boost the immune response upon antigen re-encounter. Alternatively, a subset of memory B cells emerges in a GC-independent manner early in the immune response, prior to CSR or SHM.36 The mechanisms that govern these various fate decisions are complex and have been discussed in more detail elsewhere.37, 38, 39
Figure 2.

Simplified life cycle of B cells
After immune activation, some B cells can differentiate directly into antibody-secreting plasmablasts and early memory B cells. Others will enter a germinal center (GC) reaction, where the diversification and specialization pathways of somatic hypermutation (SHM) and class-switch recombination (CSR) enhance antibody specificity and functionality. Antibody-secreting plasmablasts and long-lived plasma cells (LLPCs), as well as memory B cells, emerge from the GC. Recall responses from memory B cells (pink arrows) can then re-enter the GC or directly differentiate into both LLPCs and short-lived plasmablasts.
The many different functions of B cells provide a variety of possibilities for engineered immune cell therapies. For example, naive B cells can be modified to become tolerogenic antigen-presenting cells by expression of an antigen fused to the heavy chain (HC) of IgG, which thereby promotes immunological tolerance in autoimmune diseases or monogenetic disorders.40, 41, 42, 43 Alternatively, B cells can be turned into long-lived in vivo factories for expression of therapeutic proteins such as factor IX44 or bnAbs against HIV and hepatitis C virus (HCV).45, 46, 47, 48 Such cellular factories can be derived from ex vivo-modified B cells44,47 or hematopoietic stem and progenitor cells (HSPCs), where B cell-specific promoters are used to confer B cell specificity.45,46,48 While such modifications are commonly done using lentiviral vectors to deliver the expression cassette, site-specific genome editing has also been used to insert expression cassettes at precise loci in the genome of the B cells and to express factor IX, BAFF, or anti-PD-1 antibodies.49,50
In this review, we focus on perhaps the most compelling potential of engineered B cells: using gene editing to reprogram antigen specificity with a preformed antibody in ways that enable access to the full suite of functions of a B cell throughout its life cycle.
The challenges of engineering antibody expression in B cells
A major goal for engineering B cells is to produce a cell that will not only secrete the engineered antibody but will also respond to the presence of the cognate antigen. This is expected to allow a certain amount of in vivo titration, including boosting of the antibody dose through vaccination, and enable long-term surveillance functions through memory recall responses. To do this, it is necessary to recapitulate the two states of an endogenous antibody, that is, the secreted antibody isoform and the cell surface B cell receptor (BCR) (Figure 3A).
Figure 3.

Challenges of engineering the Ig locus
(A) Expression of the membrane-bound BCR that coordinates clonal expansion, or the secreted antibody that mediates effector functions, is regulated by alternative splicing as the B cell differentiates. (B) The sequences of the IgH locus are unique in each B cell, with variation generated by three processes: VDJ recombination; CSR, which changes the associated constant region sequences; and SHM. (C) The light chain (LC) can be generated from one of two distinct loci, whose unique sequences are generated by VJ recombination and, for Igλ, choice of constant region. (D) Co-expression of both endogenous and engineered heavy chains (HCs) and LCs in the same cell can lead to HC heterodimers that result in bi-specific antibodies, HC-LC cross-pairings that could generate novel and potentially deleterious antigen-binding domains, or combinations of both.
Naive and memory B cells display their antibody as a BCR that regulates clonal expansion, whereas antibody-secreting plasmablasts and plasma cells instead secrete antibodies that provide the effector functions of humoral immunity.51 This is normally achieved through regulation of alternative splicing at the 3′ end of the antibody transcript during B cell differentiation.52, 53, 54 Attempts have been made to mimic this process using lentiviral vectors. For example, Yu et al.55 used 2A ribosome skipping motifs of varying efficiency to generate defined ratios of BCR and antibodies in human B cells. However, this approach generates a defined ratio of products that is unable to change as the B cell differentiates. More physiologically, Fusil et al.47 included the natural intron responsible for alternate antibody splicing in a lentiviral vector to regulate expression of a hepatitis C virus-specific antibody. This appeared to allow physiological regulation of antibody splicing, but the large size of this sequence (∼3.8 kb for IgG1), when combined with the rest of an antibody sequence, could prove challenging for vector packaging and delivery. Consequently, a genome editing approach that instead used the regulatory features of the endogenous Ig locus to achieve splicing regulation would have distinct advantages.
As an additional consideration, expression from the authentic Ig locus may be necessary to harness the full range of antibody functionality. Antibodies expressed in B cells can undergo functional specialization in two ways: SHM of the antigen-binding sequence drives a competitive evolution process known as affinity maturation, while CSR allows specialization of antibody effector functions that can improve the overall response against a specific pathogen (Figure 3B). Both of these processes are mediated by an enzyme called activation-induced cytidine deaminase (AID). The targeting of AID activity is highly complex and not fully elucidated,56 but some studies have implicated the topologically associated domain (TAD) and attendant regulatory features of the Ig locus in restricting AID activity to this region of the genome.57, 58, 59 Since the region containing the Ig constant region genes encompasses roughly 0.3 Mb, engineering this functionality elsewhere in the genome would be extremely challenging, highlighting again the value of editing the native Ig locus.
However, genome editing of the Ig locus also poses a number of challenges. First, as described above, the editing strategy must support the regulated expression of both the membrane BCR and the secreted antibody (Figure 3A). Next, antibody production is made complicated by the fact that the two distinct polypeptide chains that form the molecule are encoded by three possible loci: the HC locus, IgH, and two alternate light chain (LC) loci, Igκ and Igλ (Figures 3B and 3C). In addition, antibodies can promiscuously pair HC and LC sequences, so that co-expressing an engineered antibody in a B cell that expresses an endogenous antibody could lead to mismatched cross-pairings between the different HCs and LCs (Figure 3D). This has the potential to generate deleterious new specificities, including possible self-reactive B cells. Finally, the unique variable sequences generated in each B cell by VDJ recombination (Figure 3B) complicate strategies based on variable sequence replacements, which would effectively require customized gene editing tools for each B cell clone. In the following sections, we discuss the approaches being taken to surmount these challenges and successfully reprogram the antigen specificity of B cells through genome editing.
Strategies for genome editing in B cells
Genome editing can be used to precisely insert new genetic material at a defined site, such as the Ig locus. It takes advantage of the ability of targeted nucleases, for example CRISPR-Cas9, to create site-specific double-stranded DNA breaks (DSBs).60 Repair of this DNA can then be directed by a co-introduced DNA homology donor, which contains the sequence to be inserted flanked by regions with homology to the site of the DSB.60, 61, 62 Building on earlier work in T cells63 and HSPCs,64,65 B cell editing has been developed using electroporation of Cas9 ribonucleoproteins (RNPs) that pre-assemble the nuclease with a target-specific guide RNA (gRNA),66 and combined with transduction by AAV vectors to deliver the homology donor.49
Screening a panel of common AAV serotypes identified AAV6 as the most efficient capsid for human B cell transduction and genome editing,49 as previously shown for other human hematopoietic cells. However, we and others have found that AAV6 transduction of human B cells is less efficient compared to other hematopoietic cell types, and it can require AAV multiplicities of infection (MOIs) of 105–106 vector genomes (vg)/cell.67, 68, 69, 70 This is >300-fold more than has been used to engineer HSPCs.64 To address this issue, we recently optimized protocols for AAV6 transduction of human B cells that allow AAV6 MOIs to be reduced 100-fold without any loss of genome editing efficiencies.70
Several different approaches can be envisioned to engineer the Ig locus (Table 1). Initial attempts focused on replacing the unique rearranged variable sequence of the HC in a mature B cell. For example, Voss et al.71 developed a universal dual gRNA strategy (Figure 4A), to drop out and replace the IgH variable region (VH), by targeting gRNAs to the farthest upstream V segments (VH7–81 or VH3–74) and the farthest downstream J segment. At the same time, a plasmid homology donor was used to knock in the re-arranged VH region from the anti-HIV bnAb PG9. A key aspect of this approach is that the PG9 HC retains Env recognition when paired with different LCs, thereby tolerating the retention of a non-engineered endogenous LC.71
Table 1.
B cell engineering strategies
| Study |
gRNA targets |
Insert |
Approach to prevent HC-HC cross-pairing |
Approach to prevent HC-LC cross-pairing |
% Edited B cells |
Persistence in vivo |
Serum antibody concentration |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Strategy: Replacement of variable regions | |||||||||||
| Voss et al.71 | VH7–81 or VH3–74 and JH6 | VH | drop out and replace | none (endogenous LC tolerated) | ∼0.05% | N/D | N/D | ||||
| Greiner et al.72 | VH3–23 and Vκ3–20 | VH: engineered; VH + CH Vκ: engineered VL + CL | replace with a defined cassette | replace with a defined cassette | ∼8.6% | N/D | N/D | ||||
| Strategy: Universal IgH engineering | |||||||||||
| Moffett et al.73 | JH-Eμ intron | VL-CL-linker-VH | tested biallelic IgH editing | physical linker | ∼8%–24% | >3 weeks | ∼10–40 μg/mL | ||||
| Hartweger et al.74 | JH-Eμ intron | pA-VL-CL-2A-VH | pA signal to stop endogenous transcription | Igκ knockout | ∼0.1%–0.5% | >3 weeks | ∼300 ng/mL | ||||
| Huang et al.75 | JH-Eμ intron | VL-CL-2A-VH | none | clonal deletion | ∼10% | >41 weeks | up to ∼2.4 mg/mL | ||||
| Nahmad et al.76 | JH-Eμ intron | pA-VL-CL-2A-VH | pA signal to stop endogenous transcription | tested both physical linker and Igκ knockout | ∼40%–80% | >4 weeks | ∼1 μg/mL | ||||
VH, HC variable region (or V segment, as indicated); JH, HC J segment; Vκ variable κ LC; CH, HC constant region; VL, LC variable region; CL, LC constant region; Eμ, HC intronic enhancer; IgH, immunoglobulin HC; pA, polyadenylation signal; 2A, ribosome-skipping 2A peptide motif Igκ, immunoglobulin kappa; N/D, no data.
Figure 4.

Strategies for genome editing of the Ig locus
(A) A simplified schematic of the IgH and Igκ loci is shown. Arrows indicate gRNA targets used to generate DNA DSBs and are color coded by publication: Voss et al.71 (teal), Greiner et al.72 (brown), Moffett et al.73 (purple), Hartweger et al.74 (pink), Huang et al.75 (green), Nahmad et al. 76 (gold), and Rogers et al. (unpublished data) (orange). Homology donor constructs are illustrated and labeled by source publication. The approaches from Voss et al. and Greiner et al. are more specific, either only introducing an engineered HC (Voss et al.) or only able to engineer a subset of cells using common VH and VL segments and cannot undergo CSR (Greiner et al.). In contrast, the box highlights homology donors for universal IgH engineering that target the intron upstream of the Eμ enhancer. The approach from Rogers et al. targets immunoglobulin constant regions downstream of the CH1 exon to introduce a promoter-driven HC antibody cassette. (B) Diagram of approaches to ablate cross-pairing between the endogenous LC and engineered HC. (Left) Genetic knockout of the endogenous LC to prevent expression. (Middle) Introduction of a long flexible linker between the CL and VH domains favors the desired HC-LC pairing. (Right) camelid-like HC antibodies are unable to pair with LCs because they lack the CH1 exon necessary for HC-LC heterodimerization. VH, HC variable region. CH, Ig HC constant region. VL, LC variable region. CL, Ig LC constant region; P, promoter; L, linker; pA, poly(A) signal; 2A, ribosome-skipping 2A peptide motif; VHH: camelid HC variable region; sd, splice donor.
Primary human B cells engineered in this way acquired the ability to bind to HIV Env antigen and could undergo CSR from IgM to IgG isotypes.71 Additionally, evidence of SHM was observed in both the engineered HC and the endogenous LC sequences in edited cell lines and primary cells. Interestingly, in vitro selection for Env binding in engineered Ramos human B cells resulted in mutations in LC that enhanced antibody specificity, suggesting that this approach could allow adaptations of the endogenous LC partners to enhance binding to HIV Env in concert with the introduced PG9 HC.
Greiner et al.72 used a similar approach to express engineered antibodies, although they engineered both of the variable regions of the Ig locus. While they also explored dual gRNA dropout and replacement strategies, the authors preferred an approach that used single gRNAs targeting the commonly used VH (VH3–23) and LC variable region (VL) (Vκ3–20) sequences (Figure 4A; Table 1). Double-stranded DNA homology templates were used to insert the desired antibody sequences at these sites. In one iteration, an HC-only single-domain antibody77 was created by insertion of a VH domain at the VH3–23 site alone.72 In addition, a conventional two-chain antibody was engineered by also including constant region sequences with the specific VH and VL domains, and inserting these full-chain cassettes at their respective loci.
While these two variable region replacement strategies were the first to successfully genome edit the Ig locus, neither approach managed to fully reprogram B cells. Voss et al.71 only replaced the HC sequence, and the insertion of HC and LC cassettes at the sites chosen by Greiner et al.72 would not produce matched BCR isoforms. Further iterations would be necessary to develop a strategy that supported all aspects of B cell function after insertion of preformed antibody sequences.
A universal strategy to reprogram B cells
As a strategy for editing the Ig locus that could be used in all B cells, several recent studies have coalesced around the idea of inserting antibody expression cassettes between the farthest downstream JH segment and the Eμ enhancer in IgH (Figure 4A). Since this region is downstream of the segments involved in VDJ recombination, the targeted sequences are conserved across all B cells, simplifying a CRISPR-Cas9 editing approach.
Moffett et al.73 were the first to report this strategy, using CRISPR-Cas9 and AAV6 homology donors to engineer both human and mouse B cells. They inserted a cassette comprising a minimal IgH promoter, a full-length LC including both VL and CL regions, an elongated flexible peptide linker, a partial HC comprising the antigen-recognizing VH domain, and a splice donor (Figure 4A; Table 1). Using several different anti-viral antibodies and AAV6 MOIs of 105–106, a wide range of successful editing frequencies (5%–59%) was observed.
In contrast to the previous approaches, this strategy inserts both engineered LC and HC sequences at a single site in IgH.73 The inserted cassette is placed so that the engineered VH exon will be spliced to the downstream constant regions of IgH, completing the formation of the HC and allowing the natural regulatory mechanisms in this region to produce both BCR and secreted isoforms of the engineered antibody. In addition, the physical linkage of the introduced LC and HC sequences was designed to minimize mismatched cross-pairings with endogenous antibody chains (Figure 4B). Adoptive transfer experiments in mice demonstrated that murine B cells engineered in this way with an antibody specific for the respiratory syncytial virus (RSV) F protein could expand, secrete antibodies, adopt phenotypes consistent with LLPCs and class-switched memory B cells, and protect the mice from RSV challenge. This suggests that this editing approach can faithfully recapitulate many of the characteristics of a natural antibody response.
Even with this elegant engineering strategy, some challenges remain. For example, it is clear that editing can occur at both rearranged and non-rearranged IgH alleles in a cell, since biallelic editing resulted in the simultaneous expression of two different BCR specificities in edited B cells.73 This also means that monoallelic editing at just a non-rearranged allele is possible, and that this could thereby result in expression of both endogenous and introduced antibodies. This, in turn, could create bispecific antibodies through HC-HC heterodimerization (Figure 3D). To avoid such heterodimers, the authors proposed that full biallelic editing would be needed, or, alternatively, monoallelic editing at only the productively rearranged allele to eliminate endogenous antibody expression. It is hypothesized that insertion of an engineered antibody cassette at the selected editing position should “override” expression of the endogenous antibody, since expression from sequential IgH promoters has previously been shown to favor the promoter most proximal to the Eμ enhancer.78 However, this was not formally demonstrated in this study.73 Finally, it is not known to what extent expression of the endogenous LC could interfere with proper assembly of the inserted linked LC-HC cassette.
A similar universal editing approach was also reported by Hartweger et al.,74 who inserted an LC + VH cassette at the same location in both human and mouse B cells, using CRISPR-Cas9 and long single-stranded DNA (ssDNA) homology donors (Figure 4A; Table 1). Instead of a peptide linker, their design included the ribosome-skipping P2A motif to promote bicistronic expression of LC and HC. However, since this approach does not physically link LC and HC, erroneous HC-LC cross-pairings remained a possibility. To reduce this possibility, the authors targeted the endogenous LC by including an additional gRNA targeting the Igκ constant region (Figure 4B). Such disruption of the endogenous LC may also help to select for productively edited cells, since a functional BCR is necessary for B cell survival.79 Igκ was targeted due to its simpler constant region structure (Figure 3C) and because it is expressed in ∼95% of murine B cells. However, the more balanced 2:1 ratio of Igκ and Igλ in humans means that pre-sorting for Igκ+ cells may be necessary to apply this approach in human B cells.80 Finally, to further prevent transcripts being generated that included the endogenous rearranged VDJ sequences and that could lead to HC-HC heterodimers, the authors included a poly(A) signal upstream of their cassette.74
Using this editing approach, the authors found that the vast majority of engineered B cells only expressed a BCR from one IgH allele, with a small number of dual-expressing cells that could result from either biallelic editing or monoallelic editing of the nonproductively rearranged allele.74 Moreover, mice that received B cells engineered with an anti-HIV bnAb were able to produce HIV-specific IgG that was capable of neutralizing HIV in vitro, suggesting that the engineered B cells could produce functional antibodies with a predefined specificity after vaccination.
In sum, both of the studies by Moffett et al.73 and Hartweger et al.74 showed evidence for antigen-specific immune responses in vivo from engineered murine B cells. However, other functions of engineered B cells were not demonstrated in these studies, such as the long-term persistence of LLPCs or anamnestic responses driven by memory B cells.
Ex vivo culture of B cells and genome editing
The selection of ex vivo culture conditions for successful B cell editing includes several considerations. The activation of B cells ex vivo is expected to be necessary for genome editing approaches relying on homology-directed repair, since the required DNA repair proteins are only present in the G2/S phases of the cell cycle.81, 82, 83 However, the nature and strength of B cell activation can also affect downstream programming. For example, strong CD40L signaling has been shown to bias memory B cells to bypass the GC and differentiate directly into antibody-secreting cells upon antigen re-exposure.84 Thus, the potential impact of ex vivo stimulation on B cell programming may be an important consideration for achieving optimal functionality of engineered B cells.
These or similar mechanisms may have been involved in the lack of antigen responsiveness observed by Moffett et al.73 While protection from RSV infection was observed in their murine models, viral challenge did not seem to boost anti-RSV antibody titers relative to uninfected mice. Rather, spontaneous antibody secretion and development of engineered plasma cells and resting class-switched B cells (suggested to be memory B cells) occurred at similar rates regardless of whether mice were challenged with RSV. This lack of response to the viral antigen could be a consequence of the antigen load, the route of administration (intranasal) used in the viral challenges, or because a strong ex vivo B cell activation protocol was used, based on CD40L feeder cells, that resulted in differentiated B cells that would not be antigen responsive after adoptive transfer.
Hartweger et al. also evaluated an alternate B cell activation stimulus, anti-RP105 (a Toll-like receptor 4 [TLR4] homolog that can trigger B cell activation).85,86 This resulted in higher antibody titers after adoptive transfer compared to B cells activated with CD40L feeder cells.74 By day 21, the antibody titers had started to decline but were still detectable, whereas longer time points or recall responses were not assessed. Additionally, it is unclear whether the antibody secretion they observed was driven by vaccination or merely spontaneous secretion as seen by Moffett et al.,73 since transfer of engineered cells without vaccination was not investigated.74
Taken together, these results suggest that the method of ex vivo activation prior to editing may affect the ability of engineered B cells to respond in vivo, but further studies would be necessary to clearly demonstrate antigen-specific B cell responses, including B cell memory and antibody evolution.
Toward complete functionality in engineered B cells
More recently, Huang et al.75 and Nahmad et al.76 have further characterized the properties of the B cells resulting from the universal editing approach described above (Figure 4A; Table 1). Their studies have now demonstrated that HIV-specific engineered B cells are capable of prime-boost responses to vaccination, and that the inserted antibody sequences can undergo SHM.
Huang et al.75 explored both dual editing and single insertion strategies in murine B cells, finding that the dual insertion of an engineered HC at the IgH locus and an engineered LC at the Igκ locus was about 10-fold less efficient than inserting a single multicistronic cassette containing a 2A peptide at IgH. For homology donor delivery, the authors found plasmid DNA to be similarly effective to AAV6 in murine B cells activated with LPS (a natural TLR4 ligand), albeit with significantly increased toxicity. After adoptive transfer to wild-type mice, cells engineered in this way to express the anti-HIV bnAb VRC01 adopted a memory phenotype reminiscent of GC-independent memory B cells.
Upon vaccination, both total HIV-specific antibody titers and VRC01 concentrations (measured with an anti-idiotypic antibody) exhibited escalating responsiveness to multiple rounds of vaccination.75 This suggests that engineered B cells were capable of producing functional memory cells. After the third boost, peak serum VRC01 levels of ∼2,400 μg/mL were observed in one animal, surpassing previously reported peak levels of ∼100–200 μg/mL with AAV vectors in mice and nonhuman primates,87, 88, 89 and far exceeding the ∼0.01–2 μg/mL reported in mice with lentiviral vector approaches.46, 47, 48 Engineered cells persisted for at least 41 weeks after adoptive transfer,75 similar to the longevity observed with lentivirus-modified HSPCs.48 While these results are promising, large animal studies are required to further demonstrate persistence of engineered B cells, comparable to the >6 years reported for approaches based on AAV vectors expressing antibodies in nonhuman primates.88
Moreover, despite the GC-independent phenotype of adoptively transferred B cells observed by Huang et al.,75 vaccination triggered the engineered cells to differentiate into GC B cells, GC-dependent memory B cells, and plasma cells. Class switching of VRC01-expressing B cells into IgG1, IgG2, and IgA was also observed, but only after vaccination. In addition, SHM was observed across the length of the inserted cassette, resulting in up to 4% sequence divergence in some IgG1 clones.
Interestingly, in contrast with the findings of Hartweger et al.,74 comparable antibody responses were observed when the murine B cells were pre-stimulated with LPS or CD40L,75 suggesting that further studies will be required to understand the impact of ex vivo activation on downstream B cell functionality. However, in sum, the observations from these studies indicate that ex vivo-engineered B cells can support a broad range of B cell functions and mediate durable serum antibody secretion in vivo after vaccination.
Despite these impressive observations, Huang et al.75 also reported that a significant percentage of VCR01-engineered murine cells expressed both endogenous and engineered LCs on their cell surface, which could generate potentially autoreactive antigen-binding domains (Figure 3D). As reported in other studies (Figure 4B; Table 1), this could potentially be mitigated by adopting approaches that physically link the LCs and HCs, or by disrupting the endogenous Igκ. However, adoptive transfer of naive or LPS-stimulated B cells into a mouse model of autoreactivity resulted in specific deletion of autoreactive B cells,75 suggesting that peripheral clonal deletion mechanisms could also help to protect from this undesirable outcome.
At the same time, Nahmad et al.76 similarly reported evidence of a full range of B cell functions for this universal editing strategy (Figure 4A; Table 1). They engineered the IgH locus with a multicistronic cassette that expressed the anti-HIV bnAb 3BNC117 and included an upstream poly(A) signal. To edit human B cells, anti-RP105 activation and AAV6 vectors were used, while murine B cells received LPS activation and AAV-DJ vectors. After adoptive transfer and vaccination, 3BNC117-expressing murine B cells were specifically recruited to GCs where they made up over 90% of the HIV-reactive B cells. This suggests that B cells engineered with a highly effective anti-HIV antibody were not restricted by competition from endogenous naive B cells that shared antigen reactivity. The authors also found that circulating 3BNC117 concentrations and engineered B cell numbers could be further boosted by a second round of vaccination. After boost immunization, class switching of 3BNC117 to IgA and the antiviral murine subclass IgG2c was observed.
Deep sequencing of the inserted 3BNC117 VH region revealed evidence of both SHM and clonal selection, particularly after the boost immunization, which could be further enhanced by the introduction of silent mutations in the 3BNC117 sequence to encode additional AID hotspot motifs.76 Interestingly, a comparison of two HIV Env proteins with differing affinity for 3BNC117 showed that immunization with a higher affinity HIV antigen increased the magnitude of the B cell response and the rate of SHM; however, this effect did not impact the observed frequency of secreted 3BNC117 or the affinity of circulating anti-HIV antibodies in the mice.
Finally, to minimize the risks of antibody cross-pairing, Nahmad et al.76 evaluated the feasibility of both Igκ knockout and using a linker between the LC and VH domains (Figure 4B). Their studies found that both engineering approaches were possible, although they did not evaluate the impact of these on the rates of HC-LC cross-pairing. However, these measures were not necessary to generate functionally reprogrammed B cells after adoptive transfer.
In summary, for the first time, these studies75,76 support that ex vivo genome editing of the IgH locus of murine B cells can be used to express highly functional preformed antibodies. Engineered B cells respond to multiple rounds of vaccination with typical prime-boost kinetics and enter GCs to undergo SHM and clonal selection that are suggestive of affinity maturation. In conjunction with the results from the earlier pioneering studies, these findings suggest that despite the many challenges presented by the complexity of the Ig locus, site-specific genome editing of this locus is a viable approach to reprogramming B cells that maintain the myriad functions of the naturally generated cells.
Looking forward
With a platform for B cell engineering seemingly established, where does the field go from here? As a start, further refinement of the approach is likely necessary, in particular to better understand the risks of mismatched antibody cross-pairings and how to prevent them from happening. One additional approach to avoid this could be by engineering HSPCs upstream of B cell lineage commitment. Studies in transgenic mice have shown that expression of a preformed antibody can prevent de novo VDJ recombination during B cell development,90,91 leading to a naive B cell that would only express the engineered antibody. However, this approach would likely not be suitable for the many therapeutic monoclonal antibodies that target self-antigens (such as in cancer and autoimmunity) or that may have characteristics of self-reactivity, as in the case of anti-HIV bnAbs.92 Here, such an approach is expected to lead to clonal deletion, anergy, or receptor editing during B cell development in the bone marrow.91,93, 94, 95, 96, 97
As another possibility to prevent HC-LC cross-pairings, we are developing a B cell genome editing approach based on HC antibodies (Figure 4). These mimic the structure of camelid single-domain antibodies, which are dimers comprising only HCs, and which additionally lack the CH1 exon that is necessary for cross-pairing of HC and LC.77,98 We have observed that after genome editing, such CH1 null HC antibodies can seemingly be expressed alongside natural human antibodies with no impact on their functionality (unpublished data). In this way we abrogate the risk of forming novel antigen-binding domains due to erroneous HC-LC cross-pairings, although additional engineering would be required to prevent HC-HC cross-pairings.
While much has been accomplished in mouse studies to date, the next steps for B cell engineering will require large animal models that more closely mimic human immunology. These will include long-term studies to evaluate the durability of engineered B cell memory and antibody secretion. A major goal of such studies will be to optimize the ex vivo activation methods used to prime B cells for genome editing, based on the current requirement for cells to be in the G2/S phases of the cell cycle to support HDR-mediated gene insertion.81, 82, 83 In mouse studies, Huang et al.75 were able to generate antigen-responsive murine B cells using both LPS and CD40L-based activation methods, while Moffett et al.73 and Hartweger et al.74 were less successful using CD40L. However, these findings in murine B cells may not perfectly translate to human or nonhuman primate B cells, and may need to be empirically determined.
Alternatively, although current genome editing approaches based on HDR require B cell activation, it may be possible to develop site-specific insertion approaches using non-homologous end joining (NHEJ) or homology-mediated end joining (HMEJ) pathways, which are more ubiquitously available across the cell cycle.99, 100, 101 Finally, the optimal editing and activation methods used may vary depending on the desired functionality of the resulting engineered B cell. Altogether, further study and optimization of ex vivo activation and editing in primate and human B cells will likely be required.
Of note, most of the studies published so far on B cell editing have focused on generating HIV-specific B cells. HIV represents a particularly exciting target for these therapies, not only because of the need for prolonged antibody expression to suppress this chronic viral infection, but also because HIV is a highly mutagenic virus that constantly changes to escape antigen-specific antibody responses.102,103 Thus, it is envisioned that engineered B cells, with the capacity for evolution through SHM and affinity maturation, may be able to keep pace with virus mutations in a way that is not available for recombinant or gene therapy vector-delivered antibodies of fixed specificities. Of course, it remains to be seen whether engineered B cells programmed with a highly active bnAb (or combination of bnAbs)104 will fare better than natural B cells in being able to keep up with HIV’s rate of mutagenesis over time.105,106
Beyond HIV, a wide variety of disorders are currently treated with monoclonal antibodies, and there is increasing interest in exploring whether engineered B cells could be a vehicle for prolonged expression of such therapeutics. This includes cancers where long-term surveillance would be desirable to prevent relapse and certain autoimmune disorders. In addition to antibody expression, the immunogenic29,107 or tolerogenic40,108 functions of the B cell could also be harnessed to further improve the therapeutic potential of the engineered cells.
In considering potential applications of engineered B cells, one can clearly see parallels to CAR T cells: in both cases the adaptive immune response is reprogrammed by the introduction of target-specific antibodies. CAR T cells replace the function of the T cell receptor (TCR) with a specific antibody, allowing the engineered cells to now recognize a specific target cell. In addition, this strategy also allows them to bypass the normal requirement to interact with MHC molecules on the target cell, which can limit the deployment of TCR-based therapies. In current CAR T cell products, both CD4 and CD8 CAR T cells are engineered.109 However, the loss of TCR-MHC interactions means that engineered CD4 CAR T cells may not retain the ability to communicate in an antigen-specific manner with antigen-presenting cells or B cells, which is necessary to coordinate the overall immune response. Nevertheless, CD4 CAR T cells do enhance the efficacy of CAR T cell therapies,110,111 perhaps though more general pro-inflammatory pathways.112
In contrast, B cells that are engineered to express both secreted antibodies and the matched transmembrane BCR retain the native signaling complex responsible for directing clonal responses.113,114 In this way, engineered B cells can potentially harness the functionality of the lymphocyte in a more authentic manner than CAR T cells, and thus have the potential to access a broader range of the natural functions of the cell.
Going forward, if engineered B cells are to be realized as a therapy that can be deployed at scale, rather than a boutique autologous cell therapy, it will be necessary to innovate ways to deliver them to patients. One possibility is an off-the-shelf allogeneic engineered B cell product, manufactured from induced pluripotent stem cell-derived B cells,115 as is being explored for CAR T cells.12 Alternatively, the generation of large numbers of engineered allogeneic B cells may be feasible even without that approach, since some studies have suggested that CD40-stimulated B cells have a nearly limitless capacity for proliferation ex vivo.116 However, this may not be effective in an allogeneic setting if the reprogrammed B cells remain dependent on T cell help after adoptive transfer. Interestingly, Pesch et al.117 have begun to develop chimeric BCRs that incorporate both antigen recognition and signaling moieties into a single molecule, which could potentially be used to produce T cell-independent engineered B cells.
A parallel approach would be to remove the need for ex vivo manipulation of B cells altogether and directly engineer the cells in vivo. Nahmad et al.118 reported a proof of principle of this possibility, achieved by injecting mice with dual AAV6 vectors, expressing Staphylococcus aureus Cas9 under the control of a B cell-specific promoter in one vector and the gRNA plus homology template in the second vector. Cells doubly transduced in vivo were capable of generating 3BNC117-expressing B cells that exhibited memory responses. As this field continues to develop, it is likely that even more innovative approaches will be developed to reduce costs and improve the deployability of engineered B cells.
Conclusions
It is an exciting time for the nascent field of B cell engineering. Published results to date have demonstrated that several strategies to genome edit the Ig locus can result in expression of functional preformed antibodies that reprogram the B cell. With certain of these approaches, the engineered B cells also express the surface BCR and respond to antigens, secrete antibodies, clonally expand even in the presence of endogenous B cells, and enter GCs to undergo CSR to produce diverse antibody classes and SHM to further evolve antibody specificity. As a result, genome-edited B cells have potential as an engineered cell therapy against chronic infectious diseases such as HIV, and they may also find utility treating certain cancers and autoimmune disorders. The necessary further experiments to move this into human therapies are already underway, so that engineered B cells may soon accompany CAR T cells as a powerful new reprogrammed immune cell therapy.
Acknowledgments
This work was supported by National Institutes of Health grants HL129902 and HL156247 to P.M.C.
Author contributions
G.L.R. and P.M.C. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Hirakawa M.P., Krishnakumar R., Timlin J.A., Carney J.P., Butler K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020;40 doi: 10.1042/BSR20200127. BSR20200127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.June C.H., Sadelain M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dana H., Chalbatani G.M., Jalali S.A., Mirzaei H.R., Grupp S.A., Suarez E.R., Rapôso C., Webster T.J. CAR-T cells: Early successes in blood cancer and challenges in solid tumors. Acta Pharm. Sin. B. 2021;11:1129–1147. doi: 10.1016/j.apsb.2020.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhlmann A.S., Peterson C.W., Kiem H.P. Chimeric antigen receptor T-cell approaches to HIV cure. Curr. Opin. HIV AIDS. 2018;13:446–453. doi: 10.1097/COH.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maldini C.R., Claiborne D.T., Okawa K., Chen T., Dopkin D.L., Shan X., Power K.A., Trifonova R.T., Krupp K., Phelps M. Dual CD4-based CAR T cells with distinct costimulatory domains mitigate HIV pathogenesis in vivo. Nat. Med. 2020;26:1776–1787. doi: 10.1038/s41591-020-1039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis G.I., Sheppard N.C., Riley J.L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 2021;22:427–447. doi: 10.1038/s41576-021-00329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyquem J., Mansilla-Soto J., Giavridis T., van der Stegen S.J., Hamieh M., Cunanan K.M., Odak A., Gönen M., Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qasim W., Zhan H., Samarasinghe S., Adams S., Amrolia P., Stafford S., Butler K., Rivat C., Wright G., Somana K. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017;9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 10.Benjamin R., Graham C., Yallop D., Jozwik A., Mirci-Danicar O.C., Lucchini G., Pinner D., Jain N., Kantarjian H., Boissel N., UCART19 Group Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet. 2020;396:1885–1894. doi: 10.1016/S0140-6736(20)32334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Y., Zhou Y., Zhang M., Ge W., Li Y., Yang L., Wei G., Han L., Wang H., Yu S. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2021;27:2764–2772. doi: 10.1158/1078-0432.CCR-20-3863. [DOI] [PubMed] [Google Scholar]
- 12.Sadeqi Nezhad M., Abdollahpour-Alitappeh M., Rezaei B., Yazdanifar M., Seifalian A.M. Induced pluripotent stem cells (iPSCs) provide a potentially unlimited T cell source for CAR-T cell development and off-the-shelf products. Pharm. Res. 2021;38:931–945. doi: 10.1007/s11095-021-03067-z. [DOI] [PubMed] [Google Scholar]
- 13.Lu R.M., Hwang Y.C., Liu I.J., Lee C.C., Tsai H.Z., Li H.J., Wu H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020;27:1. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zahavi D., Weiner L. Monoclonal antibodies in cancer therapy. Antibodies (Basel) 2020;9:34. doi: 10.3390/antib9030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yasunaga M. Antibody therapeutics and immunoregulation in cancer and autoimmune disease. Semin. Cancer Biol. 2020;64:1–12. doi: 10.1016/j.semcancer.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Burton D.R., Mascola J.R. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat. Immunol. 2015;16:571–576. doi: 10.1038/ni.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Julg B., Barouch D. Broadly neutralizing antibodies for HIV-1 prevention and therapy. Semin. Immunol. 2021;51:101475. doi: 10.1016/j.smim.2021.101475. [DOI] [PubMed] [Google Scholar]
- 18.Hernandez I., Bott S.W., Patel A.S., Wolf C.G., Hospodar A.R., Sampathkumar S., Shrank W.H. Pricing of monoclonal antibody therapies: Higher if used for cancer? Am. J. Manag. Care. 2018;24:109–112. [PubMed] [Google Scholar]
- 19.Zhan W., Muhuri M., Tai P.W.L., Gao G. Vectored immunotherapeutics for infectious diseases: Can rAAVs be the game changers for fighting transmissible pathogens? Front. Immunol. 2021;12:673699. doi: 10.3389/fimmu.2021.673699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuchs S.P., Martinez-Navio J.M., Rakasz E.G., Gao G., Desrosiers R.C. Liver-directed but not muscle-directed AAV-antibody gene transfer limits humoral immune responses in rhesus monkeys. Mol. Ther. Methods Clin. Dev. 2019;16:94–102. doi: 10.1016/j.omtm.2019.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Navio J.M., Fuchs S.P., Pantry S.N., Lauer W.A., Duggan N.N., Keele B.F., Rakasz E.G., Gao G., Lifson J.D., Desrosiers R.C. Adeno-associated virus delivery of anti-HIV monoclonal antibodies can drive long-term virologic suppression. Immunity. 2019;50:567–575.e5. doi: 10.1016/j.immuni.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Priddy F.H., Lewis D.J.M., Gelderblom H.C., Hassanin H., Streatfield C., LaBranche C., Hare J., Cox J.H., Dally L., Bendel D. Adeno-associated virus vectored immunoprophylaxis to prevent HIV in healthy adults: A phase 1 randomised controlled trial. Lancet HIV. 2019;6:e230–e239. doi: 10.1016/S2352-3018(19)30003-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu L.L., Suscovich T.J., Fortune S.M., Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018;18:46–61. doi: 10.1038/nri.2017.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saunders K.O. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front. Immunol. 2019;10:1296. doi: 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hua Z., Hou B. The role of B cell antigen presentation in the initiation of CD4+ T cell response. Immunol. Rev. 2020;296:24–35. doi: 10.1111/imr.12859. [DOI] [PubMed] [Google Scholar]
- 26.Junker F., Gordon J., Qureshi O. Fc gamma receptors and their role in antigen uptake, presentation, and T cell activation. Front. Immunol. 2020;11:1393. doi: 10.3389/fimmu.2020.01393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petitprez F., de Reyniès A., Keung E.Z., Chen T.W., Sun C.M., Calderaro J., Jeng Y.M., Hsiao L.P., Lacroix L., Bougoüin A. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 28.Helmink B.A., Reddy S.M., Gao J., Zhang S., Basar R., Thakur R., Yizhak K., Sade-Feldman M., Blando J., Han G. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griss J., Bauer W., Wagner C., Simon M., Chen M., Grabmeier-Pfistershammer K., Maurer-Granofszky M., Roka F., Penz T., Bock C. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019;10:4186. doi: 10.1038/s41467-019-12160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabrita R., Lauss M., Sanna A., Donia M., Skaarup Larsen M., Mitra S., Johansson I., Phung B., Harbst K., Vallon-Christersson J. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 31.Brase J.C., Walter R.F.H., Savchenko A., Gusenleitner D., Garrett J., Schimming T., Varaljai R., Castelletti D., Kim J., Dakappagari N. Role of tumor-infiltrating B cells in clinical outcome of patients with melanoma treated with dabrafenib plus trametinib. Clin. Cancer Res. 2021;27:4500–4510. doi: 10.1158/1078-0432.CCR-20-3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crotty S., Felgner P., Davies H., Glidewell J., Villarreal L., Ahmed R. Cutting edge: Long-term B cell memory in humans after smallpox vaccination. J. Immunol. 2003;171:4969–4973. doi: 10.4049/jimmunol.171.10.4969. [DOI] [PubMed] [Google Scholar]
- 33.Yu X., Tsibane T., McGraw P.A., House F.S., Keefer C.J., Hicar M.D., Tumpey T.M., Pappas C., Perrone L.A., Martinez O. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature. 2008;455:532–536. doi: 10.1038/nature07231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roco J.A., Mesin L., Binder S.C., Nefzger C., Gonzalez-Figueroa P., Canete P.F., Ellyard J., Shen Q., Robert P.A., Cappello J. Class-switch recombination occurs infrequently in germinal centers. Immunity. 2019;51:337–350.e7. doi: 10.1016/j.immuni.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammarlund E., Thomas A., Amanna I.J., Holden L.A., Slayden O.D., Park B., Gao L., Slifka M.K. Plasma cell survival in the absence of B cell memory. Nat. Commun. 2017;8:1781. doi: 10.1038/s41467-017-01901-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhenni R., Phan T.G. The geography of memory B cell reactivation in vaccine-induced immunity and in autoimmune disease relapses. Immunol. Rev. 2020;296:62–86. doi: 10.1111/imr.12862. [DOI] [PubMed] [Google Scholar]
- 37.Laidlaw B.J., Cyster J.G. Transcriptional regulation of memory B cell differentiation. Nat. Rev. Immunol. 2021;21:209–220. doi: 10.1038/s41577-020-00446-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akkaya M., Kwak K., Pierce S.K. B cell memory: Building two walls of protection against pathogens. Nat. Rev. Immunol. 2020;20:229–238. doi: 10.1038/s41577-019-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turner J.S., Benet Z.L., Grigorova I.L. Signals 1, 2 and B cell fate or: Where, when and for how long? Immunol. Rev. 2020;296:9–23. doi: 10.1111/imr.12865. [DOI] [PubMed] [Google Scholar]
- 40.Wang X., Herzog R.W., Byrne B.J., Kumar S.R.P., Zhou Q., Buchholz C.J., Biswas M. Immune modulatory cell therapy for hemophilia B based on CD20-targeted lentiviral gene transfer to primary B cells. Mol. Ther. Methods Clin. Dev. 2017;5:76–82. doi: 10.1016/j.omtm.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X., Moghimi B., Zolotukhin I., Morel L.M., Cao O., Herzog R.W. Immune tolerance induction to factor IX through B cell gene transfer: TLR9 signaling delineates between tolerogenic and immunogenic B cells. Mol. Ther. 2014;22:1139–1150. doi: 10.1038/mt.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su Y., Zhang A.H., Noben-Trauth N., Scott D.W. B-cell gene therapy for tolerance induction: Host but not donor B-cell derived IL-10 is necessary for tolerance. Front. Microbiol. 2011;2:154. doi: 10.3389/fmicb.2011.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott D.W. Gene therapy for immunologic tolerance: Using bone marrow-derived cells to treat autoimmunity and hemophilia. Curr. Stem Cell Res. Ther. 2011;6:38–43. doi: 10.2174/157488811794480753. [DOI] [PubMed] [Google Scholar]
- 44.Levy C., Fusil F., Amirache F., Costa C., Girard-Gagnepain A., Negre D., Bernadin O., Garaulet G., Rodriguez A., Nair N. Baboon envelope pseudotyped lentiviral vectors efficiently transduce human B cells and allow active factor IX B cell secretion in vivo in NOD/SCIDγc−/− mice. J. Thromb. Haemost. 2016;14:2478–2492. doi: 10.1111/jth.13520. [DOI] [PubMed] [Google Scholar]
- 45.Luo X.M., Maarschalk E., O’Connell R.M., Wang P., Yang L., Baltimore D. Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood. 2009;113:1422–1431. doi: 10.1182/blood-2008-09-177139. [DOI] [PubMed] [Google Scholar]
- 46.Hur E.M., Patel S.N., Shimizu S., Rao D.S., Gnanapragasam P.N., An D.S., Yang L., Baltimore D. Inhibitory effect of HIV-specific neutralizing IgA on mucosal transmission of HIV in humanized mice. Blood. 2012;120:4571–4582. doi: 10.1182/blood-2012-04-422303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fusil F., Calattini S., Amirache F., Mancip J., Costa C., Robbins J.B., Douam F., Lavillette D., Law M., Defrance T. A lentiviral vector allowing physiologically regulated membrane-anchored and secreted antibody expression depending on B-cell maturation status. Mol. Ther. 2015;23:1734–1747. doi: 10.1038/mt.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuhlmann A.S., Haworth K.G., Barber-Axthelm I.M., Ironside C., Giese M.A., Peterson C.W., Kiem H.P. Long-term persistence of anti-HIV broadly neutralizing antibody-secreting hematopoietic cells in humanized mice. Mol. Ther. 2019;27:164–177. doi: 10.1016/j.ymthe.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hung K.L., Meitlis I., Hale M., Chen C.Y., Singh S., Jackson S.W., Miao C.H., Khan I.F., Rawlings D.J., James R.G. Engineering protein-secreting plasma cells by homology-directed repair in primary human B cells. Mol. Ther. 2018;26:456–467. doi: 10.1016/j.ymthe.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo B., Zhan Y., Luo M., Dong H., Liu J., Lin Y., Zhang J., Wang G., Verhoeyen E., Zhang Y., Zhang H. Engineering of α-PD-1 antibody-expressing long-lived plasma cells by CRISPR/Cas9-mediated targeted gene integration. Cell Death Dis. 2020;11:973. doi: 10.1038/s41419-020-03187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peterson M.L. Mechanisms controlling production of membrane and secreted immunoglobulin during B cell development. Immunol. Res. 2007;37:33–46. doi: 10.1007/BF02686094. [DOI] [PubMed] [Google Scholar]
- 52.Bayles I., Milcarek C. Plasma cell formation, secretion, and persistence: The short and the long of it. Crit. Rev. Immunol. 2014;34:481–499. doi: 10.1615/critrevimmunol.2014012168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martincic K., Alkan S.A., Cheatle A., Borghesi L., Milcarek C. Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat. Immunol. 2009;10:1102–1109. doi: 10.1038/ni.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson A.M., Carew N.T., Smith S.M., Milcarek C. RNA splicing in the transition from B cells to antibody-secreting cells: The influences of ELL2, small nuclear RNA, and endoplasmic reticulum stress. J. Immunol. 2018;201:3073–3083. doi: 10.4049/jimmunol.1800557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu K.K., Aguilar K., Tsai J., Galimidi R., Gnanapragasam P., Yang L., Baltimore D. Use of mutated self-cleaving 2A peptides as a molecular rheostat to direct simultaneous formation of membrane and secreted anti-HIV immunoglobulins. PLoS ONE. 2012;7:e50438. doi: 10.1371/journal.pone.0050438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng Y., Seija N., Di Noia J.M., Martin A. AID in antibody diversification: There and back again. Trends Immunol. 2020;41:586–600. doi: 10.1016/j.it.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wuerffel R., Wang L., Grigera F., Manis J., Selsing E., Perlot T., Alt F.W., Cogne M., Pinaud E., Kenter A.L. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27:711–722. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X., Zhang Y., Ba Z., Kyritsis N., Casellas R., Alt F.W. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature. 2019;575:385–389. doi: 10.1038/s41586-019-1723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Senigl F., Maman Y., Dinesh R.K., Alinikula J., Seth R.B., Pecnova L., Omer A.D., Rao S.S.P., Weisz D., Buerstedde J.M. Topologically associated domains delineate susceptibility to somatic hypermutation. Cell Rep. 2019;29:3902–3915.e8. doi: 10.1016/j.celrep.2019.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doudna J.A. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–236. doi: 10.1038/s41586-020-1978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maeder M.L., Gersbach C.A. Genome-editing technologies for gene and cell therapy. Mol. Ther. 2016;24:430–446. doi: 10.1038/mt.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jasin M., Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013;5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sather B.D., Romano Ibarra G.S., Sommer K., Curinga G., Hale M., Khan I.F., Singh S., Song Y., Gwiazda K., Sahni J. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci. Transl. Med. 2015;7:307ra156. doi: 10.1126/scitranslmed.aac5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang J., Exline C.M., DeClercq J.J., Llewellyn G.N., Hayward S.B., Li P.W., Shivak D.A., Surosky R.T., Gregory P.D., Holmes M.C., Cannon P.M. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015;33:1256–1263. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu C.M., Roth T.L., Baglaenko Y., Ferri D.M., Brauer P., Zuniga-Pflucker J.C., Rosbe K.W., Wither J.E., Marson A., Allen C.D.C. Genetic engineering in primary human B cells with CRISPR-Cas9 ribonucleoproteins. J. Immunol. Methods. 2018;457:33–40. doi: 10.1016/j.jim.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson M.J., Laoharawee K., Lahr W.S., Webber B.R., Moriarity B.S. Engineering of primary human B cells with CRISPR/Cas9 targeted nuclease. Sci. Rep. 2018;8:12144. doi: 10.1038/s41598-018-30358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laoharawee K., Johnson M.J., Moriarity B.S. CRISPR/Cas9-mediated genome engineering of primary human B cells. Methods Mol. Biol. 2020;2115:435–444. doi: 10.1007/978-1-0716-0290-4_24. [DOI] [PubMed] [Google Scholar]
- 69.Laoharawee K., Johnson M.J., Lahr W.S., Peterson J.J., Webber B.R., Moriarity B.S. Genome engineering of primary human B cells using CRISPR/Cas9. J. Vis. Exp. 2020 doi: 10.3791/61855. Published online November 3, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rogers G.L., Huang C., Clark R.D.E., Seclén E., Chen H.-Y., Cannon P.M. Optimization of AAV6 transduction enhances site-specific genome editing of primary human lymphocytes. Mol. Ther. Methods Clin. Dev. 2021 doi: 10.1016/j.omtm.2021.09.003. Published online September 9, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Voss J.E., Gonzalez-Martin A., Andrabi R., Fuller R.P., Murrell B., McCoy L.E., Porter K., Huang D., Li W., Sok D. Reprogramming the antigen specificity of B cells using genome-editing technologies. eLife. 2019;8:e42995. doi: 10.7554/eLife.42995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Greiner V., Bou Puerto R., Liu S., Herbel C., Carmona E.M., Goldberg M.S. CRISPR-mediated editing of the B cell receptor in primary human B cells. iScience. 2019;12:369–378. doi: 10.1016/j.isci.2019.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moffett H.F., Harms C.K., Fitzpatrick K.S., Tooley M.R., Boonyaratanakornkit J., Taylor J.J. B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 2019;4:eaax0644. doi: 10.1126/sciimmunol.aax0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hartweger H., McGuire A.T., Horning M., Taylor J.J., Dosenovic P., Yost D., Gazumyan A., Seaman M.S., Stamatatos L., Jankovic M., Nussenzweig M.C. HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. J. Exp. Med. 2019;216:1301–1310. doi: 10.1084/jem.20190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang D., Tran J.T., Olson A., Vollbrecht T., Tenuta M., Guryleva M.V., Fuller R.P., Schiffner T., Abadejos J.R., Couvrette L. Vaccine elicitation of HIV broadly neutralizing antibodies from engineered B cells. Nat. Commun. 2020;11:5850. doi: 10.1038/s41467-020-19650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nahmad A.D., Raviv Y., Horovitz-Fried M., Sofer I., Akriv T., Nataf D., Dotan I., Carmi Y., Burstein D., Wine Y. Engineered B cells expressing an anti-HIV antibody enable memory retention, isotype switching and clonal expansion. Nat. Commun. 2020;11:5851. doi: 10.1038/s41467-020-19649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muyldermans S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 78.Delpy L., Decourt C., Le Bert M., Cogné M. B cell development arrest upon insertion of a neo gene between JH and Eμ: Promoter competition results in transcriptional silencing of germline JH and complete V(D)J rearrangements. J. Immunol. 2002;169:6875–6882. doi: 10.4049/jimmunol.169.12.6875. [DOI] [PubMed] [Google Scholar]
- 79.Lam K.P., Kühn R., Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 80.Murphy K., Travers P., Walport M., Janeway C. Garland Science; 2008. Janeway’s Immunobiology, Seventh Edition; p. 268. [Google Scholar]
- 81.Branzei D., Foiani M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 82.Heyer W.D., Ehmsen K.T., Liu J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lomova A., Clark D.N., Campo-Fernandez B., Flores-Bjurström C., Kaufman M.L., Fitz-Gibbon S., Wang X., Miyahira E.Y., Brown D., DeWitt M.A. Improving gene editing outcomes in human hematopoietic stem and progenitor cells by temporal control of DNA repair. Stem Cells. 2019;37:284–294. doi: 10.1002/stem.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koike T., Harada K., Horiuchi S., Kitamura D. The quantity of CD40 signaling determines the differentiation of B cells into functionally distinct memory cell subsets. eLife. 2019;8:e44245. doi: 10.7554/eLife.44245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Divanovic S., Trompette A., Atabani S.F., Madan R., Golenbock D.T., Visintin A., Finberg R.W., Tarakhovsky A., Vogel S.N., Belkaid Y. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat. Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miura Y., Shimazu R., Miyake K., Akashi S., Ogata H., Yamashita Y., Narisawa Y., Kimoto M. RP105 is associated with MD-1 and transmits an activation signal in human B cells. Blood. 1998;92:2815–2822. [PubMed] [Google Scholar]
- 87.Gardner M.R. Promise and progress of an HIV-1 cure by adeno-associated virus vector delivery of anti-HIV-1 biologics. Front. Cell. Infect. Microbiol. 2020;10:176. doi: 10.3389/fcimb.2020.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martinez-Navio J.M., Fuchs S.P., Mendes D.E., Rakasz E.G., Gao G., Lifson J.D., Desrosiers R.C. Long-term delivery of an anti-SIV monoclonal antibody with AAV. Front. Immunol. 2020;11:449. doi: 10.3389/fimmu.2020.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Balazs A.B., Chen J., Hong C.M., Rao D.S., Yang L., Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. 2011;481:81–84. doi: 10.1038/nature10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nussenzweig M.C., Shaw A.C., Sinn E., Danner D.B., Holmes K.L., Morse H.C., 3rd, Leder P. Allelic exclusion in transgenic mice that express the membrane form of immunoglobulin mu. Science. 1987;236:816–819. doi: 10.1126/science.3107126. [DOI] [PubMed] [Google Scholar]
- 91.Goodnow C.C., Crosbie J., Adelstein S., Lavoie T.B., Smith-Gill S.J., Brink R.A., Pritchard-Briscoe H., Wotherspoon J.S., Loblay R.H., Raphael K. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 92.Haynes B.F., Fleming J., St Clair E.W., Katinger H., Stiegler G., Kunert R., Robinson J., Scearce R.M., Plonk K., Staats H.F. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. 2005;308:1906–1908. doi: 10.1126/science.1111781. [DOI] [PubMed] [Google Scholar]
- 93.Verkoczy L., Diaz M., Holl T.M., Ouyang Y.B., Bouton-Verville H., Alam S.M., Liao H.X., Kelsoe G., Haynes B.F. Autoreactivity in an HIV-1 broadly reactive neutralizing antibody variable region heavy chain induces immunologic tolerance. Proc. Natl. Acad. Sci. USA. 2010;107:181–186. doi: 10.1073/pnas.0912914107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nemazee D.A., Bürki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 95.Kumar K.R., Mohan C. Understanding B-cell tolerance through the use of immunoglobulin transgenic models. Immunol. Res. 2008;40:208–223. doi: 10.1007/s12026-007-8008-7. [DOI] [PubMed] [Google Scholar]
- 96.Hippen K.L., Schram B.R., Tze L.E., Pape K.A., Jenkins M.K., Behrens T.W. In vivo assessment of the relative contributions of deletion, anergy, and editing to B cell self-tolerance. J. Immunol. 2005;175:909–916. doi: 10.4049/jimmunol.175.2.909. [DOI] [PubMed] [Google Scholar]
- 97.Hartley S.B., Crosbie J., Brink R., Kantor A.B., Basten A., Goodnow C.C. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–769. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- 98.Achour I., Cavelier P., Tichit M., Bouchier C., Lafaye P., Rougeon F. Tetrameric and homodimeric camelid IgGs originate from the same IgH locus. J. Immunol. 2008;181:2001–2009. doi: 10.4049/jimmunol.181.3.2001. [DOI] [PubMed] [Google Scholar]
- 99.Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yao X., Wang X., Hu X., Liu Z., Liu J., Zhou H., Shen X., Wei Y., Huang Z., Ying W. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017;27:801–814. doi: 10.1038/cr.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nami F., Basiri M., Satarian L., Curtiss C., Baharvand H., Verfaillie C. Strategies for in vivo genome editing in nondividing cells. Trends Biotechnol. 2018;36:770–786. doi: 10.1016/j.tibtech.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 102.Albert J., Abrahamsson B., Nagy K., Aurelius E., Gaines H., Nyström G., Fenyö E.M. Rapid development of isolate-specific neutralizing antibodies after primary HIV-1 infection and consequent emergence of virus variants which resist neutralization by autologous sera. AIDS. 1990;4:107–112. doi: 10.1097/00002030-199002000-00002. [DOI] [PubMed] [Google Scholar]
- 103.Ouyang Y., Yin Q., Li W., Li Z., Kong D., Wu Y., Hong K., Xing H., Shao Y., Jiang S. Escape from humoral immunity is associated with treatment failure in HIV-1-infected patients receiving long-term antiretroviral therapy. Sci. Rep. 2017;7:6222. doi: 10.1038/s41598-017-05594-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mendoza P., Gruell H., Nogueira L., Pai J.A., Butler A.L., Millard K., Lehmann C., Suárez I., Oliveira T.Y., Lorenzi J.C.C. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. 2018;561:479–484. doi: 10.1038/s41586-018-0531-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sheng Z., Schramm C.A., Connors M., Morris L., Mascola J.R., Kwong P.D., Shapiro L. Effects of Darwinian selection and mutability on rate of broadly neutralizing antibody evolution during HIV-1 infection. PLoS Comput. Biol. 2016;12:e1004940. doi: 10.1371/journal.pcbi.1004940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moir S., Fauci A.S. B-cell responses to HIV infection. Immunol. Rev. 2017;275:33–48. doi: 10.1111/imr.12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Largeot A., Pagano G., Gonder S., Moussay E., Paggetti J. The B-side of cancer immunity: The underrated tune. Cells. 2019;8:449. doi: 10.3390/cells8050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang L., Fu Y., Chu Y. Regulatory B cells. Adv. Exp. Med. Biol. 2020;1254:87–103. doi: 10.1007/978-981-15-3532-1_8. [DOI] [PubMed] [Google Scholar]
- 109.Salmikangas P., Kinsella N., Chamberlain P. Chimeric antigen receptor T-cells (CAR T-cells) for cancer immunotherapy—Moving target for industry? Pharm. Res. 2018;35:152. doi: 10.1007/s11095-018-2436-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boulch M., Cazaux M., Loe-Mie Y., Thibaut R., Corre B., Lemaître F., Grandjean C.L., Garcia Z., Bousso P. A cross-talk between CAR T cell subsets and the tumor microenvironment is essential for sustained cytotoxic activity. Sci. Immunol. 2021;6:eabd4344. doi: 10.1126/sciimmunol.abd4344. [DOI] [PubMed] [Google Scholar]
- 113.Kwak K., Akkaya M., Pierce S.K. B cell signaling in context. Nat. Immunol. 2019;20:963–969. doi: 10.1038/s41590-019-0427-9. [DOI] [PubMed] [Google Scholar]
- 114.Tanaka S., Baba Y. In: B Cells in Immunity and Tolerance. Wang J.-Y., editor. Springer Singapore; 2020. B cell receptor signaling; pp. 23–36. [Google Scholar]
- 115.French A., Yang C.T., Taylor S., Watt S.M., Carpenter L. Human induced pluripotent stem cell-derived B lymphocytes express sIgM and can be generated via a hemogenic endothelium intermediate. Stem Cells Dev. 2015;24:1082–1095. doi: 10.1089/scd.2014.0318. [DOI] [PubMed] [Google Scholar]
- 116.Wiesner M., Zentz C., Mayr C., Wimmer R., Hammerschmidt W., Zeidler R., Moosmann A. Conditional immortalization of human B cells by CD40 ligation. PLoS ONE. 2008;3:e1464. doi: 10.1371/journal.pone.0001464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pesch T., Bonati L., Kelton W., Parola C., Ehling R.A., Csepregi L., Kitamura D., Reddy S.T. Molecular design, optimization, and genomic integration of chimeric B cell receptors in murine B cells. Front. Immunol. 2019;10:2630. doi: 10.3389/fimmu.2019.02630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nahmad A.D., Lazzarotto C.R., Zelikson N., Kustin T., Tenuta M., Huang D., Reuveni I., Horovitz-Fried M., Dotan I., Rosin-Arbesfeld R. In vivo engineered B cells retain memory and secrete high titers of anti-HIV antibodies in mice. bioRxiv. 2021 doi: 10.1101/2021.04.08.438900. [DOI] [PMC free article] [PubMed] [Google Scholar]