

Abstract
The pathogenesis of type 2 diabetes (T2D) might change with increasing age. Here, we used a stratification based on age of diagnosis to gain insight into the genetics and causal risk factors of T2D across different age-groups. We performed genome-wide association studies (GWAS) on T2D and T2D subgroups based on age of diagnosis (<50, 50–60, 60–70, and >70 years) (total of 24,986 cases). As control subjects, participants were at least 70 years of age at the end of follow-up without developing T2D (N =187,130). GWAS identified 208 independent lead single nucleotide polymorphism (SNPs) mapping to 69 loci associated with T2D (P < 1.0e−8). Among others, SNPs mapped to CDKN2B-AS1 and multiple independent SNPs mapped to TCF7L2 were more strongly associated with cases diagnosed after age 70 years than with cases diagnosed before age 50 years. Based on the different case groups, we performed two-sample Mendelian randomization. Most notably, we observed that of the investigated risk factors, the association between BMI and T2D attenuated with increasing age of diagnosis. Collectively, our results indicate that stratification of T2D based on age of diag-nosis reveals subgroup-specific genetics and causal determinants, supporting the hypothesis that the pathogenesis of T2D changes with increasing age.
Introduction
Type 2 diabetes (T2D) is characterized by the failure of target tissues (e.g., muscle and adipose tissue) to respond appropriately to insulin, leading to increased plasma levels of glucose and ensuing vascular damage. T2D is strongly associated with obesity and a Western lifestyle (1). Importantly, data-driven statistical clustering of T2D patient characteristics (2) or genetic risk factors (3) have both identified five different subgroups of T2D, reinforcing the notion that T2D is not a single disease entity but, rather, is highly heterogeneous (4,5).
The pathophysiology of T2D comprises a vicious cycle, in which increasing insulin resistance drives increasing insulin secretion. When the insulin-secreting capacity of the pancreas is no longer sufficient to overcome the insulin resistance (e.g., due to progressing obesity and/or aging), glucose levels start to increase (6). However, the relative contribution of insulin resistance and insulin secretion capacity to the onset of T2D varies among individuals. In addition, previous research suggests that aging affects the pathophysiological mechanisms leading to impaired glucose homeostasis (7). More specifically, aging is thought to result in impaired β-cell function, which consequently results in impaired insulin secretion and development of T2D (8–11). These observations suggest that the pathophysiology and the genetics of T2D change with increasing age. This hypothesis is supported by recent observations that the impact of obesity is lesser in older-aged individuals with respect to the development of T2D (12) and that the increased risk of cardiovascular disease and mortality in T2D case subjects was diminished in those diagnosed after the age of 80 years (13). The genetic variants previously identified in genome-wide association studies (GWAS) on T2D (14) might thus cause different T2D subphenotypes characterized by differences based on age at diagnosis, as has been previously proposed (3).
Data from the UK Biobank, an open-access resource comprising a large prospective cohort, have become available for the study of a wide range of known and novel (genetic) risk factors for a variety of diseases, including T2D (15). Exploiting the large sample size of this cohort, we set out to test the hypothesis that stratification of T2D on the basis of the age of diagnosis will yield specific genetic risk profiles and genetic correlates with different traits. Mendelian randomization (MR), which uses genetic variants for exposures as instrumental variables (16), on these T2D subphenotypes was performed to provide insight into the causal risk profile of these T2D subphenotypes. In addition, we explored whether the association between the genetic liability for T2D and risk of coronary artery disease (CAD) was dependent on the age of T2D diagnosis.
Research Design and Methods
Information on the UK Biobank study populations is presented in Supplementary Material.
Outcome Definitions
For the current study, we made use of the objectively collected patient data available in the full UK Biobank cohort, which is predominantly based on hospital admission data and primary care data and to a lesser extent on self-reported data (data field 130709). Here, we used data on the date of the first occurrence of “non-insulin-dependent diabetes mellitus” (data field 130708). For the current study, we used the data available until 1 January 2020. On the basis of the date of first occurrence and the year of birth, we calculated the age of diagnosis of T2D. Based on this data, we classified the following case groups: participants diagnosed with T2D irrespective of the age of diagnosis, T2D diagnosed before the age of 50 years, T2D diagnosed between the age of 50 and 60 years, T2D diagnosed between the age of 60 and 70 years, and T2D diagnosed after the age of 70 years. We selected as control participants all participants without T2D occurrence (or occurrence of any other diabetes type) during the time when data were collected and who were older than 70 years at the moment data collection stopped. For all genetic association analyses, we used the same control population; cases of any T2D age-group not contributing to a certain analysis were set as missing.
Genome-Wide Association Analyses
Genome-wide association analyses were performed with use of linear mixed models implemented in the program BOLT_LMM (version 2.3.2) (17). We adjusted the analyses for age, sex, and the first 10 principal components and corrected for the genetic correlation matrix (to correct for familial relationships in the UK Biobank population). Analyses were done on the autosomal chromosomes only. Single nucleotide polymorphisms (SNPs) with a minor allele frequency <0.01 were excluded as well as SNPs with an imputation quality <0.3. SNPs reaching a P value threshold of <1e-8, which takes into account a total of five GWAS, were first processed using the online tool FUMA (18) to identify independent lead SNPs and to perform gene mapping. We determined lead SNPs that were independent of each other at R2 < 0.1 using the UK Biobank Release 2b UK10K as a reference panel population. On the basis of the results, which were composed of additive β estimates and SEs, we calculated the average difference between the consecutive age-groups and considered, for interpretation and replication purposes, an average difference in log odds of 0.04 as possibly relevant. Furthermore, to explore a possible changing genetic contribution to T2D dependent on the age of diagnosis, we performed linear regression on age (of diagnosis or the maximum age without T2D diagnosis) with a multiplicative interaction term between the SNP and T2D, with adjustment for sex and the first 10 principal components. Results visualization was performed with the R-based packages ggplot2 (19), EasyStrata (www.genepi-regensburg.de/easystrata) (20), and VennDiagram.
Genetic Correlations
Genetic correlations were calculated on the basis of the summary statistics from the GWAS analyses using linkage disequilibrium score regression (21,22). For this, we assessed the genetic correlation between the different case groups of T2D and the different phenotypes available in LD Hub (excluding the phenotypes retrieved from UK Biobank data) (https://ldsc.broadinstitute.org/ldhub/). To prevent false positive correlations in the presence of correlated traits, we corrected for multiple testing on the basis of the 193 different external phenotypes (α: 0.05/ 193 = 2.6e-4). For the current study, we were particularly interested in comparing the genetic correlates with T2D diagnosed before age 50 years and T2D diagnosed after age 70 years.
Replication
We performed replication analyses of the independent lead SNPs that had an average difference in β estimates between the consecutive age-groups in the population-based Estonian Biobank and BioMe Biobank. The analyses were performed with the same analysis protocol and study design as we used in the UK Biobank population, with case subjects stratified into the four T2D subgroups (age <50, 50–60, 60–70, and >70 years) and the control subjects age 70 years and older. A detailed description of the study design is presented in Supplementary Material. For the present efforts, we only used data of the (self reported) European sample of the BioMe Biobank. In addition, we calculated the P values for heterogeneity for the results as identified in the UK Biobank and the meta analyzed results (derived with R-based rmeta fixed-effects meta-analyses) of the replication cohorts using fixedeffects meta-analysis in the rmeta package in R.
MR Analyses
We performed two-sample MR with summary-based statistics of GWAS using the R-based statistical package TwoSampleMR (available from https://github.com/MRCIEU/TwoSampleMR) (23). This statistical package also contains a large library of exposures from published GWAS for use as instrumental variables, which is aligned with the online GWAS catalog. For the current study, we used the risk factors that have been described in relation to T2D in MR studies (24), restricted to the exposures that were derived from GWAS that identified at least three independent (R2 < 0.001) genetic instruments (SNPs) that reached genome-wide significance in the original genome-wide association analysis (P value <5e−8). Methods for MR analyses of summary-level data based on two study samples have previously been described in detail (25,26). Using inverse variance–weighted (IVW) analyses, we combined the effects of the individual genetic instruments to obtain a genetically determined association between exposure and outcome under the assumption of absence of horizontal pleiotropy. We performed the sensitivity analyses MR-Egger regression (27) and weighted median estimator analyses (28) to be able to correct for potential bias caused by horizontal pleiotropy. Because MR analyses in BioMe would likely be underpowered, we only performed these analyses in the UK Biobank and the Estonian Biobank.
Furthermore, we assessed the relations of the significant independent lead SNPs (P value <1e-8) from the different T2D GWAS analyses based on the age of diagnosis in relation to CAD. For this analysis, we used publicly available summary-statistics data of a GWAS conducted in participants of the CARDIoGRAMplusC4D (Coronary ARtery DIsease Genome wide Replication and Meta-analysis (CARDIoGRAM) plus The Coronary Artery Disease (C4D) Genetics) consortium, which included 60,801 CAD case and 123,504 control subjects (29), mainly of European ancestry. Cases of CAD were defined as the presence of a history of myocardial infarction, acute coronary syndrome, chronic stable angina, or coronary stenosis of >50%.
Data and Resource Availability
With the exception of the data from the UK Biobank (www.ukbiobank.ac.uk), the data sets analyzed in the current study are not publicly available due to legal restrictions but are available from the corresponding author upon reasonable request.
Results
Characteristics of the Study Population
Discovery analyses were conducted in 24,986 T2D case and 187,130 control subjects (Table 1). Of the T2D case subjects, 2,331 were diagnosed before age 50 years, 7,140 between age 50 and 60 years, 10,966 between age 60 and 70 years, and 4,549 after age 70 years. Of all case subjects, ∼38% were women, which was similar across the different age-groups. The characteristics of the replication cohorts Estonian Biobank and BioMe Biobank are presented in Supplementary Tables 1 and 2.
Table 1.
Characteristics of the Caucasian UK Biobank study population in the current study
| Control subjects | All case subjects | Stratification of case subject population by age of diagnosis (years) | ||||
|---|---|---|---|---|---|---|
| <50 | 50–60 | 60–70 | >70 | |||
| N | 187,130 | 24,986 | 2,331 | 7,140 | 10,966 | 4,549 |
| Age at study visit, years | 75.3 (3.2)* | 60.9 (6.8) | 50.6 (6.7) | 57.1 (6.2) | 63.0 (4.1) | 67.0 (2.2) |
| Age at diagnosis, years, median (IQR) | NA | 62.8 (56.4, 68.3) | 46.2 (42.8, 48.4) | 56.0 (53.3, 58.1) | 65.0 (62.5, 67.5) | 72.6 (71.2, 74.5) |
| % women | 54 | 38 | 40 | 37 | 38 | 40 |
Data are means (SD) unless stated otherwise. IQR, interquartile range; NA, not applicable.
Most recent date without a known diagnosis of diabetes.
Genome-Wide Association Analyses
The GWAS on T2D irrespective of age of diagnosis yielded 208 independent lead SNPs mapping to 69 different loci (P value <1e-8) (Supplementary Table 3). The GWAS on T2D diagnosed before the age of 50 years identified 3 independent SNPs (all mapped to TCF7L2), the GWAS on T2D diagnosed between age 50 and 60 years identified 21 independent SNPs (mapped to 8 loci), the GWAS on T2D diagnosed between age 60 and 70 years identified 57 independent SNPs (mapped to 21 loci), and the GWAS on T2D diagnosed after age 70 years identified 14 independent SNPs (mapped to 4 loci).
When we performed a lookup of SNPs identified in the overall T2D GWAS (irrespective of the age of diagnosis) in the other four GWAS using a conservative threshold of P < 4.81e-5 (calculated as follows: 0.05/[208 × 5]), we observed large overlap (Fig. 1), especially in those diagnosed between age 50 and 60 years and between 60 and 70 years.
Figure 1.
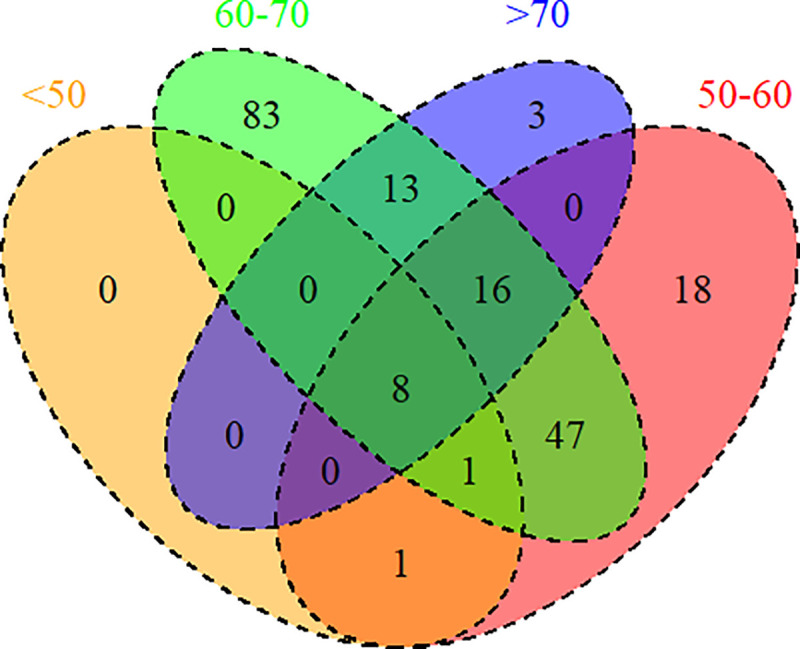
Venn diagram showing overlap of loci associated with diabetes diagnosed at different ages (years). The figure shows the overlap of the independent genetic associations identified in the GWAS on diabetes irrespective of age of diagnosis with those identified in the age-stratified genetic association analyses. We considered P < 4.81e−5 in this figure to be counted in a particular cell in the Venn diagram.
When we compared the effect sizes of alleles from SNPs identified in the overall T2D GWAS (irrespective of the age of diagnosis) with those of the T2D GWAS based on the age of diagnosis (Fig. 2), we observed larger variation in effect sizes of the SNP alleles between the cases diagnosed at younger and older age, whereas the age-groups 50–60 years and 60–70 years (which represented the largest case groups) showed the largest consistency. When we compared the youngest with the oldest T2D case group, we observed the largest variation in observed β (in log odds) estimates (Fig. 2E). For all of the investigated SNPs there was large consistency in the direction of effect.
Figure 2.

Between-subgroup comparisons of effect sizes between different T2D subgroups based on the age of diagnosis. Each dot represents an independent SNP as identified in the overall T2D GWAS (irrespective of the age of diagnosis). A: Presentation of the concordance in effect sizes as observed between the overall T2D GWAS (x-axis) and the GWAS for T2D case subjects diagnosed before age 50 years (y-axis). B: Presentation of the concordance in effect sizes as observed between the overall T2D GWAS (x-axis) and the GWAS for T2D case subjects diagnosed between ages 50 and 60 years (y-axis). C: Presentation of the concordance in effect sizes as observed between the overall T2D GWAS (x-axis) and the GWAS for T2D case subjects diagnosed between ages 60 and 70 years (y-axis). D: Presentation of the concordance in effect sizes as observed between the overall T2D GWAS (x-axis) and the GWAS for T2D case subjects diagnosed after age 70 years (y-axis). E: Presentation of the concordance in effect sizes as observed between the GWAS for T2D case subjects diagnosed before age 70 years (x-axis) and after age 50 years (y-axis). The plots were prepared with the R-based package ggplot2 (19).
There were 17 independent SNPs (mapped to eight loci) identified in the overall T2D GWAS that had different effect sizes dependent on the age of diagnosis (Supplementary Table 3). The loci, including loci mapped to SLC4C1, SLC6A1, RP11-58B2.1, PAM, C5orf30, CDKN2B-AS1, TCF7L2, and CCND2-AS1, had an average difference in the observed log odds between consecutive age groups of at least 0.04. For 12 (mapped to eight loci) of the 17 independent SNPs displaying differences in effect size, we observed also multiplicative interaction (Pinteraction < 0.05) between the SNP and T2D for age (of diagnosis or maximum age without T2D). Of these, SNPs mapped to SLCO4C1, SLC6A1, RP11-58B2.1, PAM, and CCND2-AS1 were more strongly associated with cases diagnosed before age 50 years than with cases diagnosed after age 70 years. SNPs mapped to CDKN2B-AS1 and multiple independent SNPs mapped to TCF7L2 were more strongly associated with cases diagnosed after age 70 years than with cases diagnosed before age 50 years.
In general (Supplementary Tables 4–7), the GWAS on the T2D subgroups based on age of diagnosis did not reveal additional loci that were not otherwise identified in the overall T2D GWAS with the exception of the rs11390198 SNP mapped to PROX1-AS1, which was only identified in relation to T2D diagnosed after age 70 years (Supplementary Table 7).
Results of the replication in European-ancestry participants are presented in Supplementary Table 8. Although none of the SNPs reached statistical significance in this relatively small sample (especially in those extreme groups based on age of diagnosis), directionality was consistent with that observed in UK Biobank for most of the identified associations (Pheterogeneity > 0.05 for most analyses).
Genetic Correlations
On the basis of the GWAS summary-level data, we calculated the genetic correlations with other phenotypes (Fig. 3 and Supplementary Table 9). T2D irrespective of age of diagnosis (Fig. 3A) was genetically strongest correlated with obesity-associated traits and glycemic traits. Similar results, but with lower significance, were observed in stratification of T2D based on age of diagnosis (Fig. 3B and C). When comparing the genetic correlations of T2D diagnosed before age 50 or after age 70 years (Fig. 3D), we generally observed much consistency in correlation with other traits. However, we observed some notable differences. Most notably, we observed that the genetic correlation was weaker (difference in correlation > 0.2) with obesity-related traits (e.g., BMI: genetic correlation = 0.56 and 0.34, respectively, for cases diagnosed before age 50 and cases diagnosed after age 70 years) and a stronger genetic correlation with longevity-associated traits (e.g., mother age of death: genetic correlation = −0.35 and −0.63 for cases diagnosed before age 50 and cases diagnosed after age 70 years) and with triglyceride levels (genetic correlation = 0.30 and 0.52 for cases diagnosed before age 50 and cases diagnosed after age 70 years).
Figure 3.

Genetic correlations with the external phenotypes. Plot was constructed with the R-based package corrplot. A–C: Presentation of the genetic correlation at the x-axis and the −log(P value) of the genetic correlation at the y-axis for T2D irrespective of age of diagnosis (A), T2D diagnosed before age 50 years (B), and T2D diagnosed after age 70 years (C). Genetic correlations with a P value <2.6e-4 are visualized in black (otherwise in gray). For visualization purposes, we only labeled the phenotypes that showed a genetic correlation of at least 0.5. D: Presentation of the concordance in the genetic correlations with the external phenotypes between T2D diagnosed before age 50 years and T2D diagnosed after age 70 years. Genetic correlations that were significant (P value <2.6e-4) in both case subgroups are visualized in black (otherwise in gray), and significant genetic correlations that showed a difference in genetic correlations of at least 0.2 in either direction are labeled. The plots were prepared with the R-based package ggplot2 (19). HOMA-IR, HOMA of insulin resistance; Leptin_not_adjBMI, leptin not adjusted for BMI.
MR Analyses
For the MR analyses, we examined 10 exposures in relation to T2D and the subphenotypes based on the age of diagnosis. Here (Supplementary Table 10), we specifically observed that, irrespective of age of diagnosis, years of schooling was inversely associated with the risk of developing T2D (log odds = −0.742 per additional year; P value = 8.8e−33) and that BMI (log odds = 0.964 per SD increase; P value = 1.16e−59), childhood BMI (log odds = 0.528 per SD increase; P value = 7.47e−5), systolic blood pressure (log odds = 0.018 per mmHg; P value = 5.34e−8), and insomnia (log odds = 0.164 regular vs. absent; P value = 2.45e−12) were associated with a higher risk of developing T2D. We did not observe major difference in effect estimates when we performed the sensitivity analyses MR-Egger and weighted median estimator.
Whereas particularly BMI and childhood BMI were strong risk factors for developing T2D before age 50 years (log odds = 1.310 per SD increase, P value = 2.62e−42, and log odds = 0.989 per SD increase, P value = 4.44e−12, respectively), these were less strongly associated with developing T2D after the age of 70 years (log odds = 0.681 per SD increase, P value 1.41e−15, and log odds = 0.227 per SD increase, P value = 0.13). This was also evidenced by observed interaction by the two age-groups (Pinteraction = 2.50e−6 and 4.77e−4, respectively). The observed inverse relationship with years of schooling (Pinteraction = 0.22) and the risk factor insomnia (Pinteraction = 0.37) did not differ between the age-groups. The results of the sensitivity analyses MR-Egger and weighted median estimator did not materially differ with the results from the IVW analyses. Furthermore, results were directionally consistent in the Estonian Biobank (Supplementary Table 11)
With use of summary-level data on CAD from the external CARDIoGRAMplusC4D consortium as outcome, genetically determined T2D (irrespective of age of diagnosis) was associated with an increased risk for CAD (IWV estimate 0.12 [OR 1.12; P value = 7.1e−14]). We did not observe a difference in association between genetic propensity to develop T2D and to develop CAD dependent on the age of diagnosis of the T2D (e.g., IVW estimate 0.10 [OR 1.11; P value = 0.021] with diagnosis before 50 years of age and IVW estimate 0.11 [OR 1.11; P value = 3.73e−8] with diagnosis after 70 years of age). Results did not materially differ in the sensitivity analyses MR-Egger and weighted median estimator (results not shown).
Discussion
For the current study, we investigated the hypothesis that the genetic basis of T2D may change with increasing age. Using data from European-ancestry participants from the large UK Biobank, and replication in the Estonian Biobank and BioMe Biobank (which largely showed directionally consistent results), we were able to identify specific genetic risk factors for T2D diagnosed at either younger or older age. More specifically, we observed a number of loci that were more strongly associated either with T2D diagnosed at older age (CDKN2B-AS1 and TCF7L2) or with T2D diagnosed at younger age (SLCO4C1, SLC6A1, RP11-58B2.1/LINC00492, PAM, and CCND2-AS1). On the basis of the summary-level data from these studies, we conducted two-sample MR analyses and particularly found that adiposity traits, which were previously associated with T2D in MR studies (24), were less strongly associated with T2D diagnosed at older age. Collectively, the stratification in our study based on age of diagnosis yielded novel insights that could contribute to precision medicine and the identification of specific pathophysiological mechanisms associated with the age of T2D onset.
In performance of genetic association analyses and MR analyses in older people, investigators will need to consider the issue of possible survival/selection or collider stratification bias (30). However, although no real-life examples are available, relatively simple simulations showed that the effect of collider stratification bias particularly started to influence the results after the age of 80 years and therefore minimally affected the current study (30). As aging might be considered as some kind of population stratification, we further explored the possible association between the genetic principal components (as usually used to correct for population stratification and which we also included in the statistical models) and T2D and age of diagnosis. However, we did not find evidence that the main genetic principal components (for which we also adjusted) were not associated with either T2D or age of diagnosis (Supplementary Table 12). Furthermore, if collider stratification was playing a significant role in the current study, we expected all results would have been biased in a similar direction (e.g., all effect sizes attenuated with increasing age of diagnosis). Therefore, we considered our results to likely reflect true differences dependent on the age of diagnosis.
With the most recent efforts from the DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) consortium, in a current sample of ∼900,000 individuals of European descent, >240 genetic risk loci have been identified to increase the risk of T2D, explaining 18% of the genetic heritability and enhancing the potential for clinical translation (14). Based on our stratification of T2D by age of diagnosis, we identified a partly distinct genetic background suggesting a different pathophysiology of T2D according to age. Specifically, we identified different genetic determinants for cases diagnosed before age 50 years and after age 70 years. Genetic variation in CDKN2a, which is close to our mapped CDKN2B-AS1 locus, which encodes for p16INK4a, a protein involved in cellular senescence, is consistently identified among the top genetic variants associated with T2D (14). In mouse models, increased expression of p16INK4a limits the regenerative capacity of β-cells (31). Together with our observation that this gene was more strongly associated with T2D in older age, this finding indicates a stronger role of cellular senescence in the older-aged individuals. In addition, although the well-known SNP mapped to TCF7L2 rs7903146 (14) did not differ in effect size across different age-groups, we observed that multiple (lower-frequency) variants were more strongly associated with T2D diagnosed at older age. The exact mechanisms of TCF7L2 leading to T2D is still under discussion but are likely to involve insulin secretion as observed in pleiotropy analyses (32). In addition, we observed PROX1-AS1 to be identified only in T2D case subjects age ≥70 years. Although previous GWAS identified PROX1-AS1 already (14), there is also suggestive evidence that this gene mainly involves insulin secretion (33). Taken together, these findings support the existing hypothesis that the pathogenesis of T2D in older patients is more crucially dependent on insulin secretion (7). Of the SNPs mapped to genes associated less strongly with T2D diagnosed at older age, all have been previously associated with T2D (14,34) with the exception of the RP11-58B2.1/LINC00492 locus. SLCO4C1 contributes to T2D progression and increases phenyl sulfate levels, a gut microbiota–derived metabolite (35), and SLC6A1 was specifically identified in efforts exploring the shared pathogenesis of T2D with Alzheimer disease (36). CCND2-AS1 has only recently been associated with T2D in the Million Veteran Program (34), and no mechanistic insights into the role of this gene in T2D pathogenesis have been described at this point. Despite our attempts for independent replication of our main findings, additional research is required to confirm these findings as well as to investigate their role in T2D pathogenesis at younger and older ages.
Using MR analyses (16), previous studies have studied multiple risk factors that were causally associated with T2D irrespective of the age of diagnosis (e.g., 24,26,37–39). In the current study, using previously described causal risk factors for T2D (24), we particularly observed that obesity-related traits were less strongly associated with T2D diagnosed at older age. This observation is in line with earlier observational findings in which the risk conferred by BMI decreased with increasing age of diagnosis (12,40). These findings suggest that tailoring a personalized genetic risk profile should involve taking into account different characteristics of T2D. However, our findings do not indicate that CAD risk associated with T2D changes with increasing age, which is in contrast with a previously proposed hypothesis (13). Although survival bias has previously been indicated to be a factor to consider in MR studies, such as ours (30), we did not find indications (e.g., consistent decrease in effect sizes) that this played a (major) role in the current study. Future studies, with use of additional characteristics (e.g., complications, treatment effectiveness, geriatric vulnerability), might further tailor toward a more individualized causal risk profile that might assist in the prevention of T2D from an aging perspective.
The current study made use of a large sample size that allowed stratification of the overall diagnosis of T2D based on age of diagnosis. However, the number of individuals within the age-groups <50 and >70 years was still limited. Nevertheless, as opposed to division of the study groups into equally sized groups, this strategy permitted us to keep sufficient contrast between the different case subgroups. Although our strategy resulted in an unequal distribution of the statistical power across the T2D subgroups, we obtained novel insights based on the effect sizes of the loci and genetic correlations dependent on the age of diagnosis. Furthermore, the replication sample included a limited number of cases, which likely resulted in limited replication of the results from UK Biobank. Importantly, most of the results were directionally consistent with the results observed in UK Biobank. Furthermore, our outcome variable could be affected by some degree of measurement error. However, as it is unlikely that the measurement error is associated with the genetic profile, it will only affect the effect estimates in the direction of the null and result in a reduced statistical power. In addition, our study was predominantly conducted in participants of European ancestry, as samples of other ancestries in the available Biobanks were too limited in size for drawing of any firm conclusions. As this was predominantly a proof-of-principle study based on age of diagnosis of T2D, findings should be followed up in future work in independent cohort settings.
In summary, in the current study we identified different genetic associations and causal risk factors for T2D after stratification for age of diagnosis. Our findings specifically contribute to our understanding of the pathophysiological mechanisms of T2D in different age-groups on a population level and to direct novel leads in precision medicine of T2D.
Article Information
Acknowledgments. The authors are grateful for all the studies and databases that made GWAS summary data available: ADIPOGen (Adiponectin genetics consortium), C4D, CARDIoGRAM, CKDGen (Chronic Kidney Disease Genetics) Consortium, dbGaP (database of Genotypes and Phenotypes), DIAGRAM, ENIGMA (Enhancing Neuro Imaging Genetics by Meta-Analysis), EAGLE (EArly Genetics and Lifecourse Epidemiology Eczema Consortium, excluding 23andMe), EGG (Early Growth Genetics) Consortium, GABRIEL (A Multidisciplinary Study to Identify the Genetic and Environmental Causes of Asthma in the European Community), GCAN (Genetic Consortium for Anorexia Nervosa), GEFOS (GEnetic Factors for OSteoporosis) Consortium, GIANT (Genetic Investigation of ANthropometric Traits), GIS (Genetics of Iron Status) consortium, GLGC (Global Lipids Genetics Consortium), GPC (Genetics of Personality Consortium), GUGC (Global Urate Genetics Consortium), HaemGen (haemotological and platelet traits genetics consortium), HRgene (Heart Rate consortium), IIBDGC (International Inflammatory Bowel Disease Genetics Consortium), ILCCO (International Lung Cancer Consortium), IMSGC (International Multiple Sclerosis Genetics Consortium), MAGIC (Meta-Analyses of Glucose and Insulin-related traits Consortium), MESA (Multi-Ethnic Study of Atherosclerosis), PGC (Psychiatric Genomics Consortium), Project MinE consortium, ReproGen (Reproductive Genetics Consortium), SSGAC (Social Science Genetic Association Consortium), TAG (Tobacco and Genetics Consortium), TRICL (Transdisciplinary Research in Cancer of the Lung) consortium, and UK Biobank. The authors are grateful for the contributions of Alkes Price (the systemic lupus erythematosus GWAS and primary biliary cirrhosis GWAS; University of Harvard, Boston, MA) and Johannes Kettunen (lipids metabolites GWAS; University of Oulu, Finland). The authors thank all of the participants contributing to the UK Biobank resources. Estonian Biobank analysis were performed in the High-Performance Computing Center of the University of Tartu. The authors thank all participants of Estonian Biobank.
Funding. The present UK Biobank project was conducted under project number 22474. Statistical analyses were performed using the facilities of the Dutch supercomputer (Surfsara, Inc. [access granted by the Dutch Research Council to R.N.]). D.v.H. was supported by the European Commission–funded (FP7 Health) project HUMAN (Health-2013-INNOVATION-1-602757). R.N. was supported by an innovation grant from the Dutch Heart Foundation (2019T103). R.M., K.L., A.M., and T.L. were supported by the Estonian Research Council grant PUT (PRG687). L.M. was supported by European Union’s Horizon 2020 Research and Innovation Programme under grant agreements 777107 and 847776, Green ICT programme under Norway Grants 2014–2021 (grant no. EU53928), and by the Estonian Research Council grant PUT (PRG184). T.E. was supported by the European Union through the European Regional Development Fund project no. MOBERA5 (Norface Network project no 462.16.107), the Estonian Research Council grant PUT (PUT1660), and the European Union through Horizon 2020 grant no. 810645. A.M. was supported by the European Union through the European Regional Development Fund (project no. 2014-2020.4.01.16-0125). The Mount Sinai Institute for Personalized Medicine BioMe Biobank Program is supported by The Andrea and Charles Bronfman Philanthropies. R.J.F.L. is funded by the National Institutes of Health (R01DK110113, R01DK107786, R01HL142302, R01DK124097).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. R.N. initialized the project, performed the analyses in UK Biobank, and wrote the initial draft of the manuscript. K.L. retrieved the data from Estonian Biobank and performed the analyses. R.A.J.S. retrieved the data from BioMe Biobank and performed the analyses and contributed to the revised draft manuscript. T.L. performed the replication analyses in Estonian Biobank. R.J.F.L. supervised the analyses in the BioMe Biobank and collected the data. R.M. supervised the analyses in the Estonian Biobank and was responsible for the data collection in Estonian Biobank. K.W.v.D. and D.v.H. initialized and supervised the project and drafted and commented on the manuscript. All authors approved submission of the submitted work. R.N. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Appendix
Estonian Biobank Research Team. Members of the Estonian Biobank Research Team are as follows: Andres Metspalu, Tõν Esko, and Lili Milani. All members are affiliated with Estonian Genome Centre, Institute of Genomics, University of Tartu.
Footnotes
A complete list of Estonian Biobank Research Team members appears in the appendix.
This article contains supplementary material online at https://doi.org/10.2337/figshare.14542332.
Contributor Information
Collaborators: Estonian Biobank Research Team:, Andres Metspalu, Tõnu Esko, and Lili Milani
References
- 1. Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus--present and future perspectives. Nat Rev Endocrinol 2011;8:228–236 [DOI] [PubMed] [Google Scholar]
- 2. Ahlqvist E, Storm P, Käräjämäki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 2018;6:361–369 [DOI] [PubMed] [Google Scholar]
- 3. Udler MS, Kim J, von Grotthuss M, et al.; Christopher D. Anderson on behalf of METASTROKE and the ISGC . Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med 2018;15:e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trischitta V, Prudente S, Doria A. Disentangling the heterogeneity of adulthood-onset non-autoimmune diabetes: a little closer but lot more to do. Curr Opin Pharmacol 2020;55:157–164 [DOI] [PubMed] [Google Scholar]
- 5. Wagner R, Heni M, Tabák AG, et al. Pathophysiology-based subphenotyping of individuals at elevated risk for type 2 diabetes. Nat Med 2021;27:49–57 [DOI] [PubMed] [Google Scholar]
- 6. Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005;365:1333–1346 [DOI] [PubMed] [Google Scholar]
- 7. Lee PG, Halter JB. The pathophysiology of Hyperglycemia in older adults: clinical considerations. Diabetes Care 2017;40:444–452 [DOI] [PubMed] [Google Scholar]
- 8. Chang AM, Smith MJ, Galecki AT, Bloem CJ, Halter JB. Impaired beta-cell function in human aging: response to nicotinic acid-induced insulin resistance. J Clin Endocrinol Metab 2006;91:3303–3309 [DOI] [PubMed] [Google Scholar]
- 9. Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 1999;104:787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab 2003;284:E7–E12 [DOI] [PubMed] [Google Scholar]
- 11. Basu R, Breda E, Oberg AL, et al. Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes 2003;52:1738–1748 [DOI] [PubMed] [Google Scholar]
- 12. Wright AK, Welsh P, Gill JMR, et al. Age-, sex- and ethnicity-related differences in body weight, blood pressure, HbA1c and lipid levels at the diagnosis of type 2 diabetes relative to people without diabetes. Diabetologia 2020;63:1542–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sattar N, Rawshani A, Franzén S, et al. Age at diagnosis of type 2 diabetes mellitus and associations with cardiovascular and mortality risks. Circulation 2019;139:2228–2237 [DOI] [PubMed] [Google Scholar]
- 14. Mahajan A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018;50:1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sudlow C, Gallacher J, Allen N, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 2008;27:1133–1163 [DOI] [PubMed] [Google Scholar]
- 17. Loh PR, Tucker G, Bulik-Sullivan BK, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet 2015;47:284–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag, New York, 2016 [Google Scholar]
- 20. Winkler TW, Kutalik Z, Gorski M, Lottaz C, Kronenberg F, Heid IM. EasyStrata: evaluation and visualization of stratified genome-wide association meta-analysis data. Bioinformatics 2015;31:259–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bulik-Sullivan B, Finucane HK, Anttila V, et al.; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3 . An atlas of genetic correlations across human diseases and traits. Nat Genet 2015;47:1236–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng J, Erzurumluoglu AM, Elsworth BL, et al.; Early Genetics and Lifecourse Epidemiology (EAGLE) Eczema Consortium . LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 2017;33:272–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuan S, Larsson SC. An atlas on risk factors for type 2 diabetes: a wide-angled Mendelian randomisation study. Diabetologia 2020;63:2359–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burgess S, Scott RA, Timpson NJ, Davey Smith G; EPIC- InterAct Consortium . Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 2015;30:543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noordam R, Smit RA, Postmus I, Trompet S, van Heemst D. Assessment of causality between serum gamma-glutamyltransferase and type 2 diabetes mellitus using publicly available data: a Mendelian randomization study. Int J Epidemiol 2016;45:1953–1960 [DOI] [PubMed] [Google Scholar]
- 27. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smit RAJ, Trompet S, Dekkers OM, Jukema JW, le Cessie S. Survival bias in Mendelian randomization studies: a threat to causal inference. Epidemiology 2019;30:813–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006;443:453–457 [DOI] [PubMed] [Google Scholar]
- 32. Masotti M, Guo B, Wu B. Pleiotropy informed adaptive association test of multiple traits using genome-wide association study summary data. Biometrics 2019;75:1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dupuis J, Langenberg C, Prokopenko I, et al.; DIAGRAM Consortium; GIANT Consortium; Global BPgen Consortium; Anders Hamsten on behalf of Procardis Consortium; MAGIC investigators . New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010;42:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vujkovic M, Keaton JM, Lynch JA, et al.; HPAP Consortium; Regeneron Genetics Center; VA Million Veteran Program . Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet 2020;52:680–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kikuchi K, Saigusa D, Kanemitsu Y, et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat Commun 2019;10:1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu Y, Ding X, She Z, et al. Exploring shared pathogenesis of Alzheimer’s disease and type 2 diabetes mellitus via co-expression networks analysis. Curr Alzheimer Res 2020;17:566–575 [DOI] [PubMed] [Google Scholar]
- 37. Dale CE, Fatemifar G, Palmer TM, et al.; UCLEB Consortium; METASTROKE Consortium . Causal associations of adiposity and nody fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: a Mendelian randomization analysis. Circulation 2017;135: 2373–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu J, van Klinken JB, Semiz S, et al. A Mendelian randomization study of metabolite profiles, fasting glucose, and type 2 diabetes. Diabetes 2017;66:2915–2926 [DOI] [PubMed] [Google Scholar]
- 39. Bos MM, Smit RAJ, Trompet S, van Heemst D, Noordam R. Thyroid signaling, insulin resistance, and 2 diabetes mellitus: a Mendelian randomization study. J Clin Endocrinol Metab 2017;102:1960–1970 [DOI] [PubMed] [Google Scholar]
- 40. Peters SA, Huxley RR, Sattar N, Woodward M. Sex differences in the excess risk of cardiovascular diseases associated with type 2 diabetes: potential explanations and clinical implications. Curr Cardiovasc Risk Rep 2015;9:36. [DOI] [PMC free article] [PubMed] [Google Scholar]