

Abstract
Tumor antigens (Ags) are weakly immunogenic and elicit inadequate immune responses, thus induction of antigen-specific immune activation via the maturation of dendritic cells (DCs) is a strategy used for cancer immunotherapy. In this study, we examined the effect of Rv3628 from Mycobacterium tuberculosis (Mtb) on activation of DCs and anti-tumor immunity in vivo. Intravenous injection of mice with Rv3628 promoted DC activation of spleen and lymph nodes. More importantly, Rv3628 also induced activation of DCs and enhanced Ag presentation in tumor-bearing mice. In mice bearing ovalbumin (OVA)-expressing tumors, combination treatment with Rv3628 and OVA peptide promoted OVA-specific T cell activation and accumulation of interferon (IFN)-γ and tumor necrosis factor (TNF)-α-producing OT-I and OT-II cells in tumor-draining lymph nodes. Moreover, three different tumor Ags in three different tumor models showed enhanced anti-tumor activity with Rv3628 as adjuvant, including inhibition of growth of OVA-expressing B16 melanoma, CT26 carcinoma, and B16 melanoma tumors, and a synergistic effect with anti-programmed cell death protein 1 (PD-1) antibody treatment. Additionally, potential application against human tumors was indicated by similar activation of human peripheral blood DCs by Rv3628. Taken together, these data demonstrate that Rv3628 could be an effective adjuvant in tumor immunotherapy via enhanced capacity of DC activation and Ag presentation.
Keywords: Rv3628, Mycobacterium tuberculosis, adjuvant, dendritic cells, antigen presentation, immunotherapy
Graphical abstract
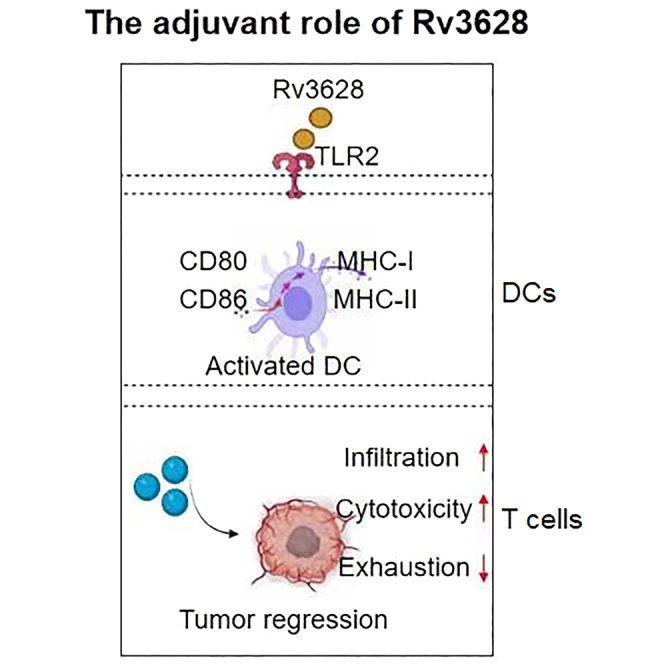
This study investigated the effect of Rv3628 from Mycobacterium tuberculosis (Mtb) on activation of dendritic cells (DCs) and anti-tumor immunity in vivo. The results demonstrate that Rv3628 activates DCs in TLR2-dependent manner, and could be an effective adjuvant in tumor immunotherapy via enhanced capacity of DC activation and antigen presentation.
Introduction
Cancer immunotherapy is currently at the cusp of becoming an important aspect of comprehensive cancer treatment in the clinic. A combined treatment method using adjuvants and tumor antigens (Ags) is desirable as a form of traditional immunotherapy. Since tumor Ags are poorly immunogenic, adjuvants are often added to treatments to enhance the activation of immune responses.1 Activation of cytotoxic T lymphocytes (CTLs) and T-helper (Th) cells is controlled by dendritic cells (DCs), the most powerful of the Ag-presenting cells (APCs).2, 3, 4 DCs express pattern recognition receptors, which contribute to phagocytosis of pathogens and activation of the DCs. Activation results in an expression of immunostimulatory and co-stimulatory molecules that in turn upregulate the production of pro-inflammatory cytokines and the migration of DCs into lymphoid organs. Within lymphoid organs, the DCs presentation of Ag results in the differentiation of naive T cells into effector T cells. Although pathogens typically trigger such immune activation, tumor Ags are weakly immunogenic and elicit inadequate immune responses. Moreover, the tumor microenvironment suppresses immune responses by several mechanisms, such as by promoting immune tolerance to tumor Ags.5 Therefore, induction of tumor Ag-specific immune responses demands additional stimulatory molecules or adjuvants.6
DCs can be split into two subtypes: plasmacytoid DCs (pDCs) and myeloid DCs (mDCs).7 The mDCs are further classified into two sub-populations that differ in functions for T cell activation. CD8α+ DCs in mice and BDCA3+ DCs in humans efficiently promote cross-presentation of Ags that associates with the major histocompatibility complex (MHC) class I on their surface to CD8 T cells.8, 9, 10, 11 In contrast, CD8α− DCs in mice and BDCA1+ DCs in humans present Ag that associates with MHC class II directly to CD4 T cells.12, 13, 14 The presentation of the Ag by these two complementary functions of DCs is essential for inducing Ag-specific immune activation for cancer immunotherapy and for vaccine efficacy, but with weak tumor Ags, enhancement by adjuvants is needed.
A previous study showed that Mycobacterium tuberculosis (Mtb) Rv3628 induced a Th1-biased immune response via DC activation and displayed vaccine potential against Mtb infection.15 Rv3628 is a unique soluble class I pyrophosphatase (PPase) that has been shown to be a strong immune activator that acts as a selective agonist of TLR2 in DCs and induces DCs maturation.15, 16, 17 The direct effects of Rv3628 on DCs in vivo have not been well characterized yet. In this study, in vivo administration of Rv3628 is shown to stimulate both CD8α+ and CD8α− DCs for enhanced CD4 and CD8 T cell responses to Ag and to act as a potent adjuvant for tumor Ags in the treatment of cancer in several mouse models. These results may provide important information for the development of a potential new therapeutic strategy to combat cancer.
Results
Endotoxin-free Rv3628
The purified Rv3628 protein was passed through a polymyxin B agarose column to remove any contaminating endotoxins prior to all experiments. The molecular mass of purified Rv3628 was confirmed as approximately 20 kDa by SDS-PAGE (Figure 1A). The endotoxin content, as measured by a limulus amoebocyte lysate (LAL) assay, was below 0.3 UE/μg in all Rv3628 preparations (Figure 1B). The treatment of bone marrow-derived DCs (BMDCs) with Rv3628, with or without ATP (an enhancer of interleukin [IL]-1β production), did not induce IL-1β production, which would otherwise have indicated activation of inflammasomes (Figure 1C). Furthermore, 80% of mice that were injected intravenously (i.v.) with Rv3628 at a dose of 250 mg/kg survived for longer than 72 h; however, all mice treated with 20 mg/kg lipopolysaccharide (LPS) died within 36 h (Figure 1D). The treatment with 250 mg/kg Rv3628 did not promote inflammation in the lungs (Figure 1E). Thus, these findings suggest that Rv3628 has much lower toxicity in mice than does LPS.
Figure 1.

Purification and cytotoxicity assay of soluble protein Rv3628
(A) The purified soluble protein Rv3628 was subjected to SDS-PAGE. (B) Endotoxin content was measured by a LAL assay and was below 0.3 UE/μg in Rv3628 preparations (n = 3, one-way ANOVA, mean ± SEM).(C) BMDCs were treated with the indicated doses of Rv3628 and LPS in the presence and absence of ATP. IL-1β production levels in the culture medium were measured 24 h after treatment (n = 3, one-way ANOVA, mean ± SEM).(D) Survival rates of mice after intravenous treatment with Rv3628 and LPS (n = 5).(E) H&E histology of lungs 24 h after treatment (n = 3, one-way ANOVA, mean ± SEM).
Rv3628-induced maturation of DCs in vitro and in vivo
We first examined the dose-dependent effect of Rv3628 in the activation of C57BL/6 mouse BMDC in vitro and spleen DCs in vivo. Single-cell suspensions from bone marrows were cultured with granulocyte-macrophage colony-stimulating factor (GM-CSF) for 7 days. The CD11c+ immature BMDCs were further treated with Rv3628 or with LPS as a positive control. The dendritic morphology of the BMDCs was dramatically changed after 24 h of Rv3628 treatment (Figure S1A). Identification of BMDC subsets among the live CD11c+ cells (Figure S1B) showed the expression of co-stimulatory molecules in BMDCs was significantly increased by treatment with 1–10 μg/mL Rv3628 (Figure 2A). Furthermore, at 10 μg/mL, Rv3628 upregulated the co-stimulatory molecules and MHC class I and II significantly more than treatment with negative-control proteins Rv1860 or Rv0222 (Figure S2A). To assess whether Rv3628 was recognized by Toll-like receptors (TLRs) in DCs, we compared the responses of cells isolated from wild-type (WT), TLR2-knockout (KO), and TLR4-KO mice and found that Rv3628 induced the expression of various surface molecules (Figure S2B) and augmented the secretion of pro-inflammatory cytokines (Figure S2C) in WT- and TLR4 KO-derived DCs compared with TLR2 KO-derived DCs. Thus, these data indicate that Rv3628 promotes DC activation in a TLR2-dependent manner. To assess splenic DC activation, C57BL/6 mice were injected i.v. with 2.5 mg/kg Rv3628 or with 1 mg/kg LPS as a control. The pDC subsets were defined in the lineage-negative (Lin−) CD11c+ cells among live splenocytes (Figure S3A) and inguinal lymph node (iLN) DCs (Figure S3B). 24 h after injection of Rv3628, expression levels of co-stimulatory molecules and MHC class I and II were significantly upregulated in splenic CD8α+ and CD8α− pDCs (Figure 2B). The activation of DCs was dose dependent: 1.0 mg/kg and 2.5 mg/kg Rv3628 promoted expression levels of MHC class I and II that were similar to those induced by LPS (Figures S4B and S5B). Furthermore, the higher levels of MHC class II and co-stimulatory molecules induced by Rv3628 were maintained for much longer than those that resulted from LPS treatment (Figures S4A and S5A). Matured DCs secrete pro-inflammatory cytokines.18 We found, for example, that the treatment of BMDCs with 10 μg/mL Rv3628 and the injection of mice with 2.5 mg/kg Rv3628 promoted marked increases in tumor necrosis factor (TNF)-α, IL-6, and IL-12p70 in the culture medium and in serum, respectively (Figures 2C and 2D). Activated DCs promote T cell stimulation,19 and we found that treatment of mice with Rv3628 induced a significant upregulation of splenocyte mRNA levels for interferon (IFN)-γ and T-bet, critical transcription factors for Th1 and cytotoxic T1 (Tc1) cell functions (Figure 2E). However, Th2- and Th17-related mRNA levels were not upregulated by Rv3628. These data confirmed that the activation of DCs by Rv3628 was of a type consistent with promotion of protective immune responses.
Figure 2.

Rv3628 promotes the activation of BMDCs and splenic DCs
(A) BMDCs were treated with 1, 5, and 10 μg/mL Rv3628 and with 100 ng/mL LPS as a positive control analyzed after 24 h. Mean fluorescence intensity levels of CD80, CD86, MHC class I, and MHC class II in BMDCs are shown (n = 6, one-way ANOVA, mean ± SEM). (B) C57BL/6 mice were injected subcutaneously with 2.5 mg/kg Rv3628 and with 1 mg/kg LPS as a positive control and splenocytes harvested after 24 h. Lineage markers included CD3, Thy1.1, B220, Gr-1, CD49b, and TER-119. Mean fluorescence intensity (MFI) of co-stimulatory molecules and MHC classes I and II in CD8α+ Lin− CD11c+ cells (upper) and CD8α− Lin− CD11c+ cells (lower) are shown (n = 6 mice, one-way ANOVA, mean ± SEM). (C) Concentrations of TNF-α, IL-6, and IL-12p70 in BMDC-cultured medium from (A) (n = 6, two-way ANOVA, mean ± SEM). (D) The serum concentration of TNF-α, IL-6, and IL-12p70 from (B) (n = 6 mice, two-way ANOVA, mean ± SEM). (E) Mean values of the mRNA expression levels in the spleen from (B) (n = 4 mice, two-way ANOVA, mean ± SEM).
Rv3628 enhanced Ag-specific immune activation
The activation of DCs by Rv3628 indicated a potential to induce strong Ag-specific immune responses. We injected C57BL/6 mice i.v. with 2.5 mg/kg ovalbumin (OVA), 2.5 mg/kg Rv3628, a combination of Rv3628 and OVA, or a combination of OVA and 1 mg/kg LPS as a positive control and 24 h later analyzed the presentation of OVA peptide on the surface of the spleen DCs subsets. The splenic DCs were divided into CD8a+ and CD8a− Lin− cells. As shown in Figure 3A, treatment with the combination of Rv3628 and OVA led to increases in the percentages of CD8a+ and CD8a− DCs presenting OVA257–264 peptide on the surface, whereas injection of Rv3628 alone did not. To assess the associated Ag-specific immune responses, we used an OVA-specific T cell proliferation assay after the transfer of carboxyfluorescein succinimidyl ester (CFSE)-labeled OT-I and OT-II T cells into CD45.1 congenic mice. Beginning 24 h after the cell transfer, the mice were treated for 3 days with 2.5 mg/kg OVA, 2.5 mg/kg Rv3628, phosphate-buffered saline (PBS) or the combination of Rv3628 and OVA. The combination of OVA and LPS was used as a positive control. The combined treatment with Rv3628 and OVA promoted substantial increases in the proliferation of OT-I and OT-II T cells, whereas Rv3628 alone did not promote the proliferation, as indicated by the degree of reduction of CFSE levels (Figures 3B and S6A). We also tested an OVA-specific T cell proliferation assay in which CFSE-labeled OT-I and OT-II T cells were transferred into CD45.2 congenic mice (Figure S6B). The levels of OVA peptide presentation and OT cell proliferation induced by the combination of Rv3628 and OVA were again similar to those induced by the combined treatment with LPS and OVA (Figures 3 and S6). These data suggested that Rv3628 enhanced the Ag presentation by DCs and promoted Ag-specific T cell proliferation in vivo.
Figure 3.

Rv3628-enhanced OVA presentation in DCs and OVA-specific T cell proliferation (A and B) CD45-1 and CD45-2 congenic C57BL/6 mice were injected i.v. with PBS, 2.5 mg/kg OVA, 2.5 mg/kg Rv3628, and the combination of Rv3628 and OVA. The combination of LPS and OVA was also injected into mice as a positive control. (A) The surface presentation of OVA Ag on the splenic CD8a+and CD8a− DCs was measured by labeling with anti-OVA (257–264) Abs 24 h after Rv3628 treatment (left). Mean numbers of cells positive for the presence of OVA peptide on the surface of the splenic CD8a+ and CD8a−DCs are shown (right) (n = 6 mice, two-way ANOVA, mean ± SEM). (B) CD45.1 C57BL/6 mice were injected with fluorescent-labeled OT-I and OT-II T cells and after 24 h injected with Ag. The proliferation of OT-I and OT-II T cells was assessed 72 h later in iLN (left) and the mean percentages of the proliferating cells are shown (right). Division index (DI) was calculated by using the proliferation assay tool in FlowJo software (n = 6 mice, two-way ANOVA, mean ± SEM).
Rv3628 induced DCs maturation in tumor-bearing mice
Since the tumor microenvironment promotes immune suppression20,21 and different subsets of DCs show different specialized functions for Ag presentation,11 we further evaluated the DC maturation effect of Rv3628 in tumor-bearing mice. C57BL/6 mice were injected subcutaneously (s.c.) on the right side with 1 × 106 B16 melanoma cells. When the tumors were well established, the mice were injected s.c. on the left side with 2.5 mg/kg of Rv3628 and the activation of DCs in the spleen, mediastinal LN (mLN), and inguinal iLN was analyzed 24 h later. The DC subsets were defined in live cells of the CD11c+ Lin− set (Figure S7B). Treatment with Rv3628 induced significant increases in the expression levels of CD80, CD86, and MHC class I and II molecules on the DCs of tumor-draining mLN and spleen (Figures 4A and S7A). Thus, Rv3628 can activate DCs in tumor-bearing mice. We next assessed whether Ag-specific immune activation of T cells was also promoted in tumor-bearing mice. CFSE-labeled OT-I and OT-II cells were transferred into B16 tumor-bearing CD45.2 congenic mice and after 24 h the mice received 2.5 mg/kg of OVA, 2.5 mg/kg of Rv3628, or a combination of 2.5 mg/kg OVA and 2.5 mg/kg Rv3628 in PBS. Control mice were treated with 2.5 mg/kg of OVA and 1 mg/kg of LPS. Three days after the transfer of cells, the tumor, spleen, mLN, and tumor-draining lymph nodes (drLNs) were harvested and OT-I and OT-II cell proliferation was analyzed (Figure S8A). The combined treatment with OVA and Rv3628 had induced a marked increase in the proliferation of OT-I and OT-II cells in the tumor mLN, which was significantly higher than that of the cells induced with OVA only (Figures 4B–4E). The combined treatment with Rv3628 and OVA also markedly increased the proportions of OT-I and OT-II cells producing IFN-γ and TNF-α (Figure 4F). We also transferred CFSE-labeled OT-I and OT-II cells into B16-bearing CD45.1 congenic mice and again observed that the combined treatment with OVA and Rv3628 induced an increase in the proliferation of OT-I and OT-II cells in the tumor drLN (Figures S8B). Thus, the injection of Rv3628 also promoted Ag-specific T cell activation, proliferation, and production of IFN-γ and TNF-α in tumor-bearing mice.
Figure 4.

Rv3628-promoted DCs maturation in the tumor microenvironment
(A) C57BL/6 CD45-2 mice were injected s.c. with 1 × 106 B16-OVA melanoma cells. Fifteen days after tumor injection, the mice were injected s.c. with PBS, 2.5 mg/kg Rv3628, and 1 mg/kg LPS for 24 h, and the mLN were harvested. The expression levels of co-stimulatory molecules and MHC classes I and II in CD8α+ DCs (upper) and CD8α− DCs (lower) are shown (n = 6 mice, one-way ANOVA, mean ± SEM). (B) CD45-1 mice bearing B16-OVA tumors were injected with fluorescent-labeled OVA-specific CD8+ OT-I and CD4+ OT-II cells and 24 h later injected with Ag. The proliferation of the cells in tumor-draining mLNs was measured 72 h later. (C–E) The division index (C), the mean percentages of the proliferating cells (D), and the absolute numbers of OT-I and OT-II cells (E) (n = 6 mice, two-way ANOVA, mean ± SEM). (F) The mean percentages of IFN-γ- and e TNF-α-producing OT-I and OT-II cells.
The combination of OVA and Rv3628 reduced B16-OVA tumor growth
Since the combination of Rv3628 and OVA could induce OVA-specific immune activation in tumor-bearing mice, we next examined the anti-tumor effect. C57BL/6 mice were injected s.c. on the right side with 5 × 105 B16-OVA cells. On day 7, when the tumors were well established, the mice were injected s.c. on the left side with PBS, 2.5 mg/kg OVA, 2.5 mg/kg Rv3628, or the combination of OVA and Rv3628. On days 14 and 21, the mice received the same treatment again. On day 28 (Figure 5A), the tumor volume, weight, and size were significantly smaller in mice treated with the combination than in the mice treated with PBS, OVA, or Rv3628 alone (Figures 5B–5D). In addition, Enzyme-linked Immunospot Assay (ELISPOT) analysis showed that a significantly higher proportion of splenocytes were producing IFN-γ in specific response to OVA peptides after the combined treatment with OVA and Rv3628 than after the separate treatments (Figure 5E). Mice treated with the combination of Rv3628 and OVA also survived much longer than those treated with PBS, OVA, or Rv3628 alone (Figure 5F) and there was substantially greater OVA-specific cytotoxicity against OVA peptide-pulsed target cells in mouse spleens (Figure 5G). These data showed that the combined treatment induced anti-tumor effects in mice by activating Ag-specific immune responses.
Figure 5.

The combination of OVA and Rv3628 treatment prevented B16-OVA tumor growth
(A–F) C57BL/6 mice were injected s.c. on the right side with 5 × 105 B16-OVA cells. Once tumors were well established, on days 7, 14, and 21 the mice received s.c. injections, on the left side, of PBS, 2.5 mg/kg OVA, 2.5 mg/kg Rv3628, and the combination of OVA and Rv3628. (A) Treatment schedule. (B) The size of the tumor masses on day 28 after B16-OVA tumor cell challenge (n = 3 mice). (C) The weight of the tumor masses on day 28 after tumor cell challenge (n = 3 mice, one-way ANOVA, mean ± SEM). (D) The curves of B16-OVA tumor growth in mice (n = 3 mice). (E) OVA257-264 and OVA323-339 peptide-specific IFN-γ production in splenocytes was analyzed by ELISPOT (upper) and the mean number of spots is shown (lower) (n = 3 mice, two-way ANOVA, mean ± SEM). (F) The survival rates of the mice (n = 6 mice). (G) Ag-specific cytotoxic activity was measured in vivo. The dot plots in the upper panel show the percentages of OVA peptide SIINFEK-loaded cells (CFSE+ cells) and control peptide-loaded cells (CMTMR+ cells) 24 h after injection into immunized mice. The mean percentages of Ag-specific lysis are shown (lower) (n = 6 mice, one-way ANOVA, mean ± SEM).
Rv3628-induced anti-tumor self-Ag immunity
We next evaluated the anti-tumor effect of Rv3628 with two model self-Ags, lysate of carcinoma CT26 cells in BALB/c mice and melanoma TRP2 in C57BL/6 mice, respectively22, 23, 24, 25. The BALB/c mice were injected s.c. with 1 × 106 CT26 cells and the C57BL/6 mice with 5 × 105 B16 cells. On days 7, 14, and 21 after the tumor cell injection, the BALB/c mice were treated contra-laterally with PBS, 100 μL of lysate of CT26 cells (1 × 107 cell/100μL), 2.5 mg/kg Rv3628, or a combination of Rv3628 and CT26 cell lysate. The C57BL/6 mice received a combination of Rv3628 and tyrosinase-related protein 2 (TRP2, a melanoma self-Ag) and relevant controls. In mice injected with the mixture of Rv3628 with self-Ag, the tumor growth was much slower (Figure 6B and S9B), and on day 25 or 28 the tumor mass was much smaller (Figures 6C, 6D, S9C, and S9D) compared with those injected with PBS, self-Ag, or Rv3628 alone. Ag-specific IFN-γ production in the drLN was substantially increased by the combined injection of Rv3628 and self-Ag (Figures 6E and 6F). These data showed that Rv3628 can induce tumor self-Ag-specific immune activation and inhibit tumor growth through the resultant self-Ag-specific immune responses.
Figure 6.

Rv3628 promotes cancer Ag-specific immune activation and immunity against CT26 carcinoma
(A–F) BALB/c mice were injected s.c. with 1 × 106 CT26 carcinoma cells. After 7, 14, and 21 days the mice were treated with PBS and 100 μL of lysate of CT26 cells (1 × 107 cell/100 μL), 2.5 mg/kg Rv3628. (A) The treatment schedule. (B) The curves of CT26 tumor growth in mice (n = 5 mice, two-tailed p value, unpaired t test, means ± SEM). (C) The weights of the tumor masses on day 25 after CT26 tumor cell injection (n = 5 mice, one-way ANOVA, mean ± SEM). (D) The size of the tumor masses on day 25 after CT26 tumor cell injection (n = 6 mice). (E) The concentration of IFN-γ in cultured medium from AH1 peptide-treated tumor-drLN cells after 72 h (n = 5 mice, one-way ANOVA, mean ± SEM). (F) AH1-specific IFN-γ production by tumor-drLN cells was analyzed by ELISPOT (left) and the mean number of spots is shown (right) (n = 5 mice, one-way ANOVA, mean ± SEM).
Enhancement of anti-PD-L1 antibody effect by Rv3628
Next, we examined the effect of Rv3628 on the enhancement of the anti-tumor effects that could be induced by immune checkpoint blockade antibodies (Abs). B16 tumor-bearing C57BL/6 mice were treated intraperitoneally (i.p.) with 7.5 mg/kg anti-PD-L1 Abs, 2.5 mg/kg Rv3628 and 2.5 mg/kg TRP2, or a combination of anti-PD-L1 Abs, TRP2, and Rv3628. The treatment was repeated every 5 days until 25 days after tumor injection. A combination of 1 mg/kg LPS, TRP2, and anti-PD-L1 Abs was administered as a positive control. The treatment with anti-PD-L1 Abs efficiently inhibited B16 tumor growth, and the tumor was completely abrogated by the combination of anti-PD-L1 Abs and Rv3628 (Figures 7B and 7C). Moreover, the TNF-α- and IFN-γ-producing tumorinfiltrating lymphocytes (TILs) in the tumor (Figure S10) were markedly increased by the combined treatment of anti-PD-L1 Abs, TRP2, and Rv3628 (Figure 7D). Thus, these data showed that the Rv3628 adjuvant could enhance the effects of anti-PDL1 Abs in tumor treatment.
Figure 7.

Enhancement of the anti-cancer effect of anti-PD-L1 Abs by additional treatment with Rv3628
(A–D) C57BL/6 mice were injected s.c. with 5 × 105 B16 cells. Five days after tumor injection, mice were treated i.p. with 7.5 mg/kg anti-PD-L1 Abs, TRP2 melanoma Ag plus 2.5 mg/kg Rv3628, or the combination of anti-PD-L1 Abs, TRP2 Ag, and Rv3628. The treatment was repeated every 5 days. (A) Treatment schedule. (B) The curves of B16 tumor growth in mice (n = 6 mice, two-tailed p value, unpaired t test, mean ± SEM). (C) The sizes of the tumor masses on day 25 after B16 tumor cell injection (n = 6 mice). (D) FACS profiles of intracellular IFN-γ and TNF-α in the CD8+ T cells obtained from tumors on day 25.
Rv3628 induces human peripheral blood DCs activation
Rv3628 effectively induced DC activation in mice; therefore, we examined its effect on healthy human peripheral blood DCs (PBDCs) to determine whether it could be used as an adjuvant for humans. BDCA1+ and BDCA3+ cells from live Lin− CD11c+ cells in human peripheral blood mononuclear cells (PBMCs) were classified as human PBDCs and the activation of these cells by Rv3628 treatment was analyzed (Figure 8A). The levels of co-stimulatory and MHC class I and II molecules in both BDCA1+ and BDCA3+ DCs were upregulated by Rv3628 treatment (Figure 8B). Thus, Rv3628 could also induce activation of human PBDCs.
Figure 8.

Rv3628 induces human PBDC activation
(A–C) PBMCs were treated with 10 μg/mL of Rv3628 or 100 ng/mL of LPS for 24 h. (A) The FACS gating of CD11c+ PBDCs after treatment. Additional lineage markers included CD3, CD14, CD16, CD19, CD20, and CD56. (B and C) The expression levels of co-stimulatory molecules and MHC classes I and II in BDCA1+ DCs (upper) and BDCA3+ DCs (lower) (n = 6 donors, one-way ANOVA, mean ± SEM or two-tailed p value, unpaired t test, mean ± SEM).
Discussion
Here we showed that protein Rv3628, purified from recombinant Escherichia coli, has strong adjuvant activity against tumors in mice. As such, it was important to control for potential endotoxin (LPS) contamination. LPS is a powerful adjuvant that cannot be used for humans due to its high toxicity.26 The possibility that endotoxin contamination contributed to the potent effect of Rv3628 was assessed and excluded (Figure 1). It was striking that, at similar low doses (about 1 mg/kg), Rv3628 was as potent an adjuvant as LPS but without toxicity, and this was observed with diverse tumor treatment models and three different tumor Ags, suggesting broad applicability. The observed effects were consistent with action of this protein as an agonist of TLR2 receptors on dendritic cells (Figure S2), as previously reported15 and as confirmed here by the effects on BMDC in vitro and pDCs in spleens in vivo (Figure 2), although the possibility of additional modes of action, for example involving its properties as a unique soluble metal-dependent class I pyrophosphatase (PPase) that converts pyrophosphate into orthophosphate, was not investigated. This is different to LPS, which can be recognized by both TLR2 and TLR4 (mainly by TLR4),27, 28, 29 and thus provides a possible explanation why Rv3628 is less toxic than LPS, further substantiating its value as a novel adjuvant for cancer immunotherapy.
Stimulation of a broad immune response was presaged by the observation that both CD8α+ and CD8α− DCs were stimulated by Rv3628 into an increased expression of both MHC class I and MHC class II and of co-stimulatory molecules CD80 and CD86. The stimulation of CD8α+ DCs is notable for the ability of these cells to promote Ag cross-presentation and Th-1 cytotoxic T cell generation. These responses were similar to those from low doses of the potent, but toxic, LPS adjuvant and, in contrast to LPS, the toxicity and IL-1-driven inflammatory responses were negligible (Figure 1). Increased production of T-bet and Th-1 cytokines indicated an associated differentiation toward supporting cellular immune responses (Figures 2C–2E). As expected, the enhanced in vivo presentation of OVA-specific peptides on both CD8α+ and CD8α− splenic DCs that was observed when Rv3628 was injected together with the OVA Ag (Figure 3A) was associated with stimulation of proliferation of both CD8+ and CD4+ Ag-specific T cells (Figure 3B).
Importantly, this immune stimulatory activity was also manifested in the tumors of B16-OVA-bearing mice. DCs in drLNs were presumed representative of those in the tumor itself and had increased Ag-presenting capacity, and the proliferation of Ag-specific CD4 and CD8 T cells producing Th-1 cytokines was markedly increased (Figure 4). This suggested that Rv3628 had a capacity to overcome immunosuppressive activity within tumors. Consistent with this, a significantly stronger anti-tumor activity was observed. IFN-γ production and lysis of Ag-loaded target splenocytes were increased, tumor growth was slowed, and mouse survival was prolonged (Figure 5).
Although the B16 OVA-expressing tumor is a standard model for studies of tumor therapy, tumors differ in the degree to which they are refractory to immune responses. Therefore, it is important that the potent adjuvant activity of Rv3628 was not an aberration consequent upon the selection of C57BL/6 B16-OVA as a model tumor. This was confirmed by observation of action against CT26 tumors in BALB/c mice. Injections of tumor lysate alone had a modest anti-tumor effect and this was markedly enhanced when injected together with Rv3628; drLNs acquired Ag-specific IFN-γ responses and tumor growth was markedly impaired (Figure 6).
Specific anti-tumor Abs are now sometimes injected into patients as part of tumor treatment.30, 31, 32 Accordingly, it was important to establish that the adjuvant action of Rv3628 could function without interference between adjuvant and Ab treatments. We demonstrated that this was achievable in the C57BL/6 mice carrying B16 tumors (Figure 7). Instead of OVA model Ag, we used a natural tumor peptide, TRP2.33 Injection of this peptide together with Rv3628 generated anti-tumor immunity, manifested in reduced tumor growth rates and in the appearance of Ag-specific CD8 T cells that produced IFN-γ and TNF-α in tumor-infiltrating lymphocytes. When injected in combination with an immune checkpoint inhibitory Ab, anti-PD-L1, the treatments were synergistic, giving an increase in the T cell response and a further decrease in tumor growth (Figure 7). Thus, Rv3628 not only can be an adjuvant for diverse tumor Ags but can also be compatible with the administration of checkpoint inhibitory Abs.
That the Rv3628 adjuvant has the potential for human use was shown by in vitro stimulation of human PBDCs. The stimulated cells had elevated expression levels of MHC class I, MHC class II, and co-stimulatory molecules. The observation that the increases in BDCA3+ DCs were similar to those in BDCA1+ DCs suggests that the superior capacity of BDCA3+ cells (analogous to the mouse CD8α+ DC subset) to cross-present Ag and induce Th1 cell responses may be enhanced by exposure to this adjuvant (Figure 8). Other mycobacterial proteins have been reported to have adjuvanticity against tumors in mice,34, 35, 36, 37 and direct comparisons remain to be undertaken.
In summary, our data show that the Rv3628 protein can play a critical role in DC maturation in mice, driving Th1 immune responses to a tumor Ag that inhibits tumor growth. It can also promote human PBDC activation. Thus, Rv3628 has potential as an adjuvant for use in developing tumor vaccines for human uses.
Materials and methods
Experimental animals, cell lines, and ethics statement
Mice and cell lines C57BL/6, TLR2-KO,TLR4-KO, OT-I, and OT-II TCR transgenic mice, C57BL/6-Ly5.1 (CD45.1) congenic mice, C57BL/6-Ly5.1 (CD45.2) congenic mice, and BALB/c mice were obtained from Shanghai Public Health Clinical Center (SPHCC) and kept under pathogen-free conditions. The mice were housed in a room at 20°C –22°C and 50%–60% humidity, and fed standard rodent chow and water. The study was carried out using the guidelines of the Institutional Animal Care and Use Committee at SPHCC. The mouse protocol was approved by the Ethics of Animal Experiments Committee of SPHCC. CO2 inhalation euthanasia was used to euthanize the mice. The murine carcinoma cell line CT26 (ATCC, CRL-2638), murine melanoma cell line B16F10 (ATCC, CRL-6475), and OVA-expressing B16F10 (B16-OVA) were cultured in RPMI 1640 (including 2 mM glutamine, 100 μg/mL streptomycin, 10% fetal bovine serum [FBS], 100 U/mL penicillin, and 1 M HEPES). All cell lines were cultured at 37°C in a humidified atmosphere of 5% CO2 and air.
Purification of soluble protein Rv3628 from E. coli
To produce recombinant Rv3628 protein, the corresponding gene was amplified by PCR using Mtb H37Rv (ATCC 27294) genomic DNA as a template and the following primers: forward, 5′-CATATGGTGCAATTCGACGTGACC-3′; reverse, 5′-GAATTCTCAGTGTGTACCGGCCTTGAAG-3′. The PCR product of Rv3628 was digested and inserted into a pET28a (+) plasmid (Novagen, Madison, WI). After cell disruption via sonication of the recombinant E. coli strain, soluble protein Rv3628 was purified using Ni-nitrilotriacetic acid (NTA) resin as previously described with slight modifications.38 To remove endotoxins, the dialyzed recombinant protein was incubated with Endotoxin Removal Agarose Resin (Yeasen, 20518ES10, China). Finally, the purified endotoxin-free recombinant protein was filter sterilized and frozen at −80°C. The protein concentration was estimated using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL) with BSA as a standard. Toxicity in mice was assessed in comparison with LPS by histopathology of lungs 24 h after the i.v. injection of graded doses.
Chemicals and Ags
Chicken OVA was obtained from Sigma-Aldrich. OVA solution was incubated with endotoxin removal agarose resin before use. OVA257–264 peptide (SIINFEKL), OVA323–339 (ISQAVHAAHAEINEAGR), TRP2180–188 peptide (SVYDFFVWL), AH1 peptide (SPSYVYHQF) were purchased from Phtdpeptides.
Abs
Anti-mouse CD3 (eFlour450,17A2), anti-mouse CD4 (APC-eFlour780, RM4-5), anti-mouse CD8a (AF700, 53-6.7), anti-mouse CD11c (percp-cy5.5,N418), CD80 (PE-cy7, 16-10A1), CD86 (APC-cy7, GL1), anti-MHC class I (PE, SF1-1.1.1), anti-MHC class II (APC, AMS-32.1), anti-IFN-γ (APC, XMG1.2), anti-mouse TNF-α (PE-cy7, MP6-XT22), OVA257-264 (SIINFEKL) peptide bound to H-2Kb monoclonal Ab (mAb) (PE-cy7, 25-D1.16), anti-mouse CD45.1 (PE, A20), and anti-mouse CD45.2 (AF700, 104) were from eBioscience. CD11c (APC, 3.9, #301613); CD80 (PE, 2D10, #207831); CD83 (BV421, HB15e, #147674); CD86 (PE-cy7, BU63, #202906); anti-HLA-A, B, C (BV605, W6/32, #212641); anti-HLA-DR, DP, DQ (Percpcy5.5, Tü39, #211013); anti-CD1c (APC/Fire750, L161, #331510); and anti-CD141 (BV785, M80, #344112) were obtained from BioLegend (San Diego, CA).
Flow cytometry analysis
Cells were washed with PBS containing 0.5% BSA, and then labeled with fluorescence-conjugated Abs by incubation on ice for 30 min followed by washing with PBS. Cells were analyzed with fluorescence-activated cell sorting (FACS) Aria II (Becton Dickinson) and FlowJo 10 software (Tree Star). Cellular debris was excluded from the analysis by forward- and side-scatter gating.
In vitro BMDC generation
The initial cultures were prepared as described previously.39,40 Bone-marrow nucleated cells (2 × 105 cells/mL) were cultured in 3 mL of modified RPMI 1640 medium containing 10% FBS in six-well plates; 50 ng/mL mGM-CSF was added to the medium to support the generation of BMDCs. Unless otherwise stated, the cells were cultured for 6 days at 37°C under 5% CO2. The cultured cells were washed twice in fresh medium before the subsequent experiments were conducted.
Analysis of splenic DCs
DCs were analyzed as described previously.39,40 Briefly, the tissues were cut into small fragments and digested with 2% FBS with 1 mg/mL collagenase for 20 min at room temperature. The cells from the digestion were centrifuged to pellets, which were resuspended in 2 mL of a 1.077 Histopaque (Sigma-Aldrich). An additional 2 mL of Histopaque and the culture medium were layered below and above the cell suspension, respectively, which was then centrifuged at 1,700 × g for 10 min. The low-density fraction (<1.077 g/cm3) was collected and incubated for 30 min with the following fluorescein isothiocyanate (FITC)-conjugated mAbs: anti-CD3 (17A2), anti-Thy1.1 (OX-7), anti-B220 (RA3–6B2), anti-Gr1 (RB68C5), anti-CD49b (DX5), anti-TER-119 (TER-119). The lineage-negative (Lin−) CD11c+ cells were defined as cDCs, which were further divided into CD8a+ and CD8a− cDCs. The analysis was performed using FACS Aria II (Becton Dickinson).
Analysis of lymphoid DCs
Mouse lymphoid DCs were collected and incubated for 30 min with the following FITC-conjugated mAbs: anti-CD3 (17A2), anti-Thy1.1 (OX-7), anti-B220 (RA3–6B2), anti-Gr1 (RB68C5), anti-CD49b (DX5), and anti-TER-119 (TER-119). The Lin− CD11c+ cells were defined as cDCs, which were further divided into CD8a+ and CD8a− cDCs. The analysis was performed using FACS Aria II (Becton Dickinson).
Intracellular cytokine staining
This was done as described in detail previously.41 In the intracellular cytokine staining, the cells were stained for surface molecules first, fixed and permeabilized with Cytofix/Cytoperm buffer (eBioscience), and subsequently incubated with Abs in Perm/Wash buffer (eBioscience) for 30 min.
Cytokine assays
IFN-γ, IL-6, TNF-α, IL-1β, and IL-12p70 concentrations in the sera and culture supernatants were measured using standard ELISA kits (BioLegend, San Diego, CA).
Tumor treatments
(1) C57BL/6 mice were injected s.c. with 5 × 105/200 μL B16 or B16-OVA melanoma cells. After 7 days, when tumors were well established, the animals were treated s.c. with 100 μL PBS alone, 2.5 mg/kg Ag in PBS, 2.5 mg/kg Rv3628, or a mixture of the same amounts of Ag and Rv3628 in 100 μL of PBS. Treatments were given on days 7, 14, and 21 after the tumor challenge. On day 28, mice were killed, and splenocytes were harvested for further analysis. (2) BALB/c mice were injected s.c. with 1 × 106 CT26 carcinoma cells. After 7, 14, and 21 days, the mice were treated with PBS and 100 μL of lysate of CT26 cells (1 × 107 cell/100 μL), and 2.5 mg/kg Rv3628. (3) C57BL/6 mice were injected s.c. with 5 × 105 B16 cells. Five days after tumor injection, mice were treated i.p. with 7.5 mg/kg anti-PD-L1 Abs or 2.5 mg/kg TRP2 (a melanoma-specific Ag) with 2.5 mg/kg Rv3628, or a combination of anti-PD-L1 Abs, TRP2, and Rv3628. The treatment was repeated every 5 days.
ELISPOT assay
Mouse IFN-γ ELISPOTs were performed according to the manufacturer's protocol (BD Biosciences). In short, spleens were harvested from treated mice and mononuclear cells isolated by density cut. The cells were seeded at 5 × 105 cells/well or 1 × 106 cells/well in a pre-coated plate. The cells were then stimulated with 2 mg/mL of the OVA257–264 peptide (SIINFEKL), OVA323–339 (ISQAVHAAHAEIN), TRP2180–188 peptide (SVYDFFVWL), or a negative-control peptide at 37°C for 24 h. ELISPOT plates were counted automatically using a CTL ELISPOT reader (CTL Europe), and the number of spots observed with a control peptide was subtracted from the number of spots with specific peptides. Each mouse was tested individually.
Real-time PCR
Total RNA was extracted from cells and reverse transcribed into cDNA by Oligo (dT) and M-MLV reverse transcriptase (Promega, Madison, WI). The cDNA was subjected to real-time PCR (Qiagen, Hilden, Germany) for 40 cycles with an annealing and extension temperature of 60°C, on a Light Cycler 480 Real-Time PCR System (Roche, Basel, Switzerland). Primer sequences were: mouse β-actin forward, 5ʹ-TGGATGACGATATCGCTGCG-3ʹ; β-actin reverse, 5ʹ-AGGGTCAGGATACCTCTCTT-3ʹ; T-bet forward, 5ʹ-CAACAAC CCCTTTGCCAAAG-3ʹ; T-bet reverse, 5ʹ-TCCCCCAAGCATTGACAGT-3ʹ; IFN-γ forward, 5ʹ-GGATGCATTCATGAGTATTGC-3ʹ; IFN-γ reverse, 5ʹ-CTTTTCCG CTTCCTGAGG-3ʹ; IL-4 forward, 5ʹ-ACAGGAGAAGGGACGCCAT-3ʹ; IL-4 reverse, 5ʹ-GAAGCCCTACAGACGAGCTCA-3ʹ; IL-17A forward, 5ʹ-GCGCAA AAGTGAGCTCCAGA-3ʹ; IL-17A reverse, 5ʹ-ACAGAGGGATATCTATCAGGG-3ʹ.
OT-I and OT-II T cell proliferation
Cell-isolation kits (Miltenyi Biotec, Bergisch Gladbach, Germany) were used to purify OVA-specific CD4 T cells and CD8 T cells from OT-II and OT-I mice, respectively. The cells were suspended in PBS/0.1% BSA containing 5 μM CFSE (Invitrogen, San Diego, CA) and incubated for 10 min. The CFSE-labeled cells (1 × 106) were injected i.v. into CD45.1 or CD45.2 (WT) congenic mice, or into B16 tumor-bearing mice, and 24 h later the mice were injected with either 100 μL of PBS alone or 2.5 mg/kg of OVA in PBS, or 2.5 mg/kg Rv3628, or a mixture of the same amounts of OVA and Rv3628 in 100 μL of PBS. At 72 h after treatment, tumor, splenic tumor drLN, and other tumor drLN were harvested and the proliferation of OT-I or OT-II T cells was determined by analyzing the CFSE fluorescence intensity using flow cytometry (FACS Fortessa, Becton Dickinson) and FlowJo 8.6 software (Tree Star).
In vivo cytotoxicity assay
C57BL/6 mice were immunized with PBS, 2.5 mg/kg OVA, 50 mg/kg Rv3628, or a combination of OVA and Rv3628 on days 7 and 14. Five days after the last treatment, target cells were transferred into the immunized mice. Target cells were splenocytes from naive mice. Half of the splenocytes were labeled with 10 mM Cell Tracker Orange CMTMR (Life Technologies, Carlsbad, CA; #M7510) and loaded with control peptide. The other half were labeled with 1 μM CFSE (Invitrogen, #C34554) and 1 μM SIINFEKL peptide was loaded. An equal mixture of the two target cell populations (a total of 10 × 106 cells) was intravenously injected into immunized mice. Splenocytes were collected from spleens 24 h after injection of the target cells. Percentage killing was calculated as described by FACS Fortessa (Becton Dickinson).
Hematoxylin and eosin staining
Lungs were infused with 1 mL of 4% paraformaldehyde solution and harvested. Samples were fixed in 4% paraformaldehyde solution overnight at 4°C. The fixed lung samples were embedded in paraffin and sectioned to 5-μm thickness from different areas across the lung. The sections were placed on glass slides, de-paraffinized and hydrated, then stained with hematoxylin and eosin (H&E) and examined microscopically for tissue damage.
Human PBDC analysis
The PBMCs were isolated from whole blood by a density-gradient centrifugation method using Ficoll Histopaque. The cells were stained with FITC-conjugated Lin− Abs, anti-CD3 (HIT3a, #300306), anti-CD14 (63D3, #367115), anti-CD16 (3G8, #302006), anti-CD19 (HIB19, #302206), anti-CD20 (2H7, #302304), and anti-CD56 (5.1H11, #362545). The Lin− CD11c+ cells were gated as PBDCs using flow cytometry with a FACS Fortessa (Becton Dickinson, Franklin Lakes, NJ). The PBDCs were further fractionated into BDCA1+ (L161, #331510) and BDCA3+ (M80, #344112) DCs cells.
Statistical analysis
Results were expressed as the mean ± standard error of the mean (SEM). Datasets were analyzed using two-tailed p values and unpaired t tests or one-way ANOVA or two-way ANOVA using GraphPad Prism 8. p values <0.05 were considered to be statistically significant.
Acknowledgments
This work was supported by grants from National Natural and Science Foundation of China (31771004, 82171815, 81770011), Chinese National Mega Science and Technology Program on Infectious Diseases (2018ZX10731301, 2018ZX10302301), and Shanghai Science and Technology Commission (19XD1403100, 20Y11903400).
Author contributions
X.Y.F., and J.W. conceived the project. H.Y., J.C.X., W.F.G., Z.Y.C., J.X.X., and J.W. carried out the experiments. J.W., Z.D.H., D.B.L., and X.Y.F. conducted the analyses. J.W., D.B.L., and X.Y.F. wrote the paper. X.Y.F., D.B.L., and S.H.L. provided overall project supervision and administration.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.10.003.
Supplemental information
References
- 1.Dubensky T.W., Jr., Reed S.G. Adjuvants for cancer vaccines. Semin. Immunol. 2010;22:155–161. doi: 10.1016/j.smim.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Palucka K., Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39:38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palucka K., Banchereau J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 6.Dang Y., Wagner W.M., Gad E., Rastetter L., Berger C.M., Holt G.E., Disis M.L. Dendritic cell-activating vaccine adjuvants differ in the ability to elicit antitumor immunity due to an adjuvant-specific induction of immunosuppressive cells. Clin. Cancer Res. 2012;18:3122–3131. doi: 10.1158/1078-0432.Ccr-12-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vremec D., Pooley J., Hochrein H., Wu L., Shortman K. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 2000;164:2978–2986. doi: 10.4049/jimmunol.164.6.2978. [DOI] [PubMed] [Google Scholar]
- 8.Ma Y., Shurin G.V., Gutkin D.W., Shurin M.R. Tumor associated regulatory dendritic cells. Semin. Cancer Biol. 2012;22:298–306. doi: 10.1016/j.semcancer.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jäger D., Jäger E., Knuth A. Immune responses to tumour antigens: implications for antigen specific immunotherapy of cancer. J. Clin. Pathol. 2001;54:669–674. doi: 10.1136/jcp.54.9.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naik S.H., Proietto A.I., Wilson N.S., Dakic A., Schnorrer P., Fuchsberger M., Lahoud M.H., O'Keeffe M., Shao Q.X., Chen W.F. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 2005;174:6592–6597. doi: 10.4049/jimmunol.174.11.6592. [DOI] [PubMed] [Google Scholar]
- 11.Schnorrer P., Behrens G.M., Wilson N.S., Pooley J.L., Smith C.M., El-Sukkari D., Davey G., Kupresanin F., Li M., Maraskovsky E. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc. Natl. Acad. Sci. U S A. 2006;103:10729–10734. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pooley J.L., Heath W.R., Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 2001;166:5327–5330. doi: 10.4049/jimmunol.166.9.5327. [DOI] [PubMed] [Google Scholar]
- 13.Jongbloed S.L., Kassianos A.J., McDonald K.J., Clark G.J., Ju X., Angel C.E., Chen C.J., Dunbar P.R., Wadley R.B., Jeet V. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010;207:1247–1260. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu L., Kwak M., Zhang W., Zeng L., Lee P.C., Jin J.O. Rehmannia glutinosa polysaccharide induces toll-like receptor 4 dependent spleen dendritic cell maturation and anti-cancer immunity. Oncoimmunology. 2017;6:e1325981. doi: 10.1080/2162402x.2017.1325981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim W.S., Kim J.S., Cha S.B., Kim H., Kwon K.W., Kim S.J., Han S.J., Choi S.Y., Cho S.N., Park J.H. Mycobacterium tuberculosis Rv3628 drives Th1-type T cell immunity via TLR2-mediated activation of dendritic cells and displays vaccine potential against the hyper-virulent Beijing K strain. Oncotarget. 2016;7:24962–24982. doi: 10.18632/oncotarget.8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benini S., Wilson K. Structure of the Mycobacterium tuberculosis soluble inorganic pyrophosphatase Rv3628 at pH 7.0. Acta Crystallogr. Sect F Struct. Biol. Cryst. Commun. 2011;67:866–870. doi: 10.1107/s1744309111023323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tammenkoski M., Benini S., Magretova N.N., Baykov A.A., Lahti R. An unusual, His-dependent family I pyrophosphatase from Mycobacterium tuberculosis. J. Biol. Chem. 2005;280:41819–41826. doi: 10.1074/jbc.M509489200. [DOI] [PubMed] [Google Scholar]
- 18.Banchereau J., Palucka A.K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 19.Moser M., Murphy K.M. Dendritic cell regulation of TH1-TH2 development. Nat. Immunol. 2000;1:199–205. doi: 10.1038/79734. [DOI] [PubMed] [Google Scholar]
- 20.Finn O.J. Cancer immunology. N. Engl. J. Med. 2008;358:2704–2715. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 21.Gajewski T.F., Schreiber H., Fu Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parkhurst M.R., Fitzgerald E.B., Southwood S., Sette A., Rosenberg S.A., Kawakami Y. Identification of a shared HLA-A∗0201-restricted T-cell epitope from the melanoma antigen tyrosinase-related protein 2 (TRP2) Cancer Res. 1998;58:4895–4901. [PubMed] [Google Scholar]
- 23.Toda M., Martuza R.L., Kojima H., Rabkin S.D. In situ cancer vaccination: an IL-12 defective vector/replication-competent herpes simplex virus combination induces local and systemic antitumor activity. J. Immunol. 1998;160:4457–4464. [PubMed] [Google Scholar]
- 24.Park H.B., Lim S.M., Hwang J., Zhang W., You S., Jin J.O. Cancer immunotherapy using a polysaccharide from Codium fragile in a murine model. Oncoimmunology. 2020;9:1772663. doi: 10.1080/2162402x.2020.1772663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W., Xu L., Park H.B., Hwang J., Kwak M., Lee P.C.W., Liang G., Zhang X., Xu J., Jin J.O. Escherichia coli adhesion portion FimH functions as an adjuvant for cancer immunotherapy. Nat. Commun. 2020;11:1187. doi: 10.1038/s41467-020-15030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raetz C.R., Whitfield C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heine H., Kirschning C.J., Lien E., Monks B.G., Rothe M., Golenbock D.T. Cutting edge: cells that carry A null allele for toll-like receptor 2 are capable of responding to endotoxin. J. Immunol. 1999;162:6971–6975. [PubMed] [Google Scholar]
- 28.Takeuchi O., Hoshino K., Kawai T., Sanjo H., Takada H., Ogawa T., Takeda K., Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 29.Mazgaeen L., Gurung P. Recent advances in lipopolysaccharide recognition systems. Int. J. Mol. Sci. 2020;21:379. doi: 10.3390/ijms21020379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.Cd-18-0367. [DOI] [PubMed] [Google Scholar]
- 31.Sato-Kaneko F., Yao S., Ahmadi A., Zhang S.S., Hosoya T., Kaneda M.M., Varner J.A., Pu M., Messer K.S., Guiducci C. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight. 2017;2:e93397. doi: 10.1172/jci.insight.93397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capasso A., Lang J., Pitts T.M., Jordan K.R., Lieu C.H., Davis S.L., Diamond J.R., Kopetz S., Barbee J., Peterson J. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J. Immunother. Cancer. 2019;7:37. doi: 10.1186/s40425-019-0518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bloom M.B., Perry-Lalley D., Robbins P.F., Li Y., el-Gamil M., Rosenberg S.A., Yang J.C. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J. Exp. Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mani R., Gupta M., Malik A., Tandon R., Prasad R., Bhatnagar R., Banerjee N. Adjuvant potential of poly-α-l-glutamine from the cell wall of Mycobacterium tuberculosis. Infect. Immun. 2018;86 doi: 10.1128/iai.00537-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung I.D., Shin S.J., Lee M.G., Kang T.H., Han H.D., Lee S.J., Kim W.S., Kim H.M., Park W.S., Kim H.W. Enhancement of tumor-specific T cell-mediated immunity in dendritic cell-based vaccines by Mycobacterium tuberculosis heat shock protein X. J. Immunol. 2014;193:1233–1245. doi: 10.4049/jimmunol.1400656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liso A., Benedetti R., Fagioli M., Mariano A., Falini B. Modulatory effects of mycobacterial heat-shock protein 70 in DNA vaccination against lymphoma. Haematologica. 2005;90:60–65. [PubMed] [Google Scholar]
- 37.Lukacs K.V., Lowrie D.B., Stokes R.W., Colston M.J. Tumor cells transfected with a bacterial heat-shock gene lose tumorigenicity and induce protection against tumors. J. Exp. Med. 1993;178:343–348. doi: 10.1084/jem.178.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byun E.H., Kim W.S., Shin A.R., Kim J.S., Whang J., Won C.J., Choi Y., Kim S.Y., Koh W.J., Kim H.J. Rv0315, a novel immunostimulatory antigen of Mycobacterium tuberculosis, activates dendritic cells and drives Th1 immune responses. J. Mol. Med. (Berl) 2012;90:285–298. doi: 10.1007/s00109-011-0819-2. [DOI] [PubMed] [Google Scholar]
- 39.Jin J.O., Zhang W., Du J.Y., Wong K.W., Oda T., Yu Q. Fucoidan can function as an adjuvant in vivo to enhance dendritic cell maturation and function and promote antigen-specific T cell immune responses. PLoS One. 2014;9:e99396. doi: 10.1371/journal.pone.0099396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W., Okimura T., Xu L., Zhang L., Oda T., Kwak M., Yu Q., Jin J.O. Ascophyllan functions as an adjuvant to promote anti-cancer effect by dendritic cell activation. Oncotarget. 2016;7:19284–19298. doi: 10.18632/oncotarget.8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin J.O., Han X., Yu Q. Interleukin-6 induces the generation of IL-10-producing Tr1 cells and suppresses autoimmune tissue inflammation. J. Autoimmun. 2013;40:28–44. doi: 10.1016/j.jaut.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.