

Abstract
The list of long non-coding RNAs (lncRNAs) that participate in the function of ovarian granulosa cells (GCs) is rapidly expanding, but the mechanisms through which lncRNAs regulate GC function are not yet fully understood. Here, we recognized a minimally expressed lncRNA RP4-545C24.1 (which we named DDGC) in GCs from patients with biochemical premature ovarian insufficiency (bPOI). We further explored the role of lncRNA DDGC in GC function and its contribution to the development of bPOI. Mechanistically, silencing DDGC downregulated RAD51 by competitively binding with miR-589-5p, and this resulted in significant inhibition of DNA damage repair capacity. In addition, decreased expression of DDGC promoted ubiquitin-mediated degradation of Wilms tumor 1 (WT1) protein through interactions with heat shock protein 90 (HSP90), which led to aberrant differentiation of GCs. Moreover, DDGC was able to ameliorate the etoposide-induced DNA damage and apoptosis in vivo. Taken together, these findings provide new insights into the contribution of lncRNAs in POI pathogenesis.
Keywords: premature ovarian insufficiency, granulosa cells, lncRNA, WT1, RAD51
Graphical abstract
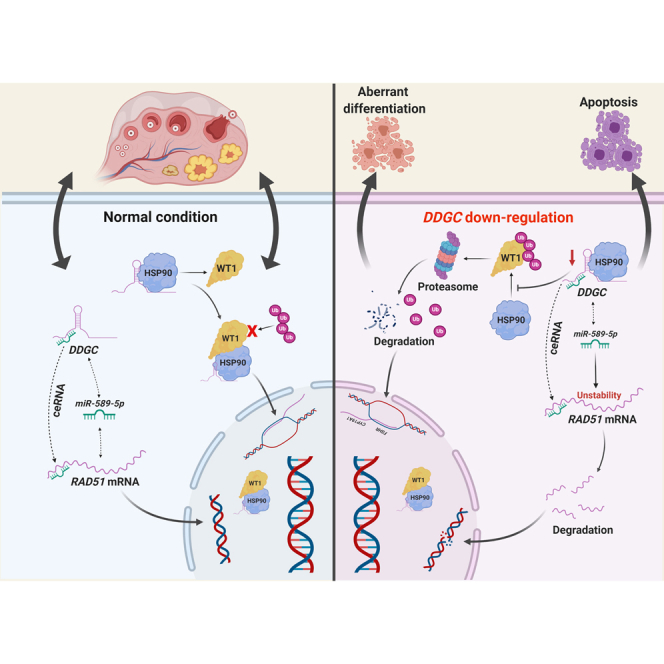
The mechanisms through which lncRNAs participate in the pathogenesis of premature ovarian insufficiency (POI) are rarely reported. We have identified an lncRNA, DDGC, that plays dual roles in the DNA repair and differentiation of ovarian somatic cells, providing new insights into the contribution of lncRNAs in POI pathogenesis.
Introduction
Substantial advances in high-throughput sequencing technologies have shown that the majority of the human genome is transcribed into long non-coding RNAs (lncRNAs) rather than into proteins.1,2 LncRNAs are highly heterogeneous RNA molecules with lengths exceeding 200 nt, and they play multiple roles in diverse cellular processes.3,4 Our growing understanding of the tissue and cell specificity of lncRNAs suggests that lncRNAs might also be involved in the development of pathological disorders such as cancers, immune dysfunction, and cardiovascular disease.5, 6, 7 However, clarifying the regulatory role of lncRNAs in ovarian function has not been studied as intensely.
Premature ovarian insufficiency (POI) is one of the most common reproductive disorders, and is defined as the decline or loss of ovarian function before the age of 40 years, affecting ∼1% of women of childbearing age, and it is accompanied by long-term health complications.8,9 A broad range of etiological factors have been studied, such as genetic, autoimmune, and infectious factors,10 but these well-known causes are behind fewer than half of all cases, implying that other unknown mechanisms or regulators might contribute to the pathogenesis of POI.
The pathology of POI is a multilayered process that is characterized by unbalanced follicle formation, activation, and exhaustion. Granulosa cells (GCs), as a prominent component of ovarian somatic cells, play an essential role in determining the growth or atresia of follicles,11, 12, 13 and loss-of-function mutations in genes regulating the proliferation or differentiation of GCs, such as BMP15,14 FSHR,15 and WT1,16 have been shown to be causative for POI. Also, recent studies in human GC lines have revealed the association between lncRNAs and polycystic ovary syndrome (PCOS).17,18 However, the mechanisms through which dysfunctional lncRNAs participate in POI development remain poorly understood.
In the present work, RNA sequencing (RNA-seq) identified the differentially expressed lncRNA DDGC in GCs from biochemical (b) POI patients and further revealed its regulatory roles in DNA damage repair processes and differentiation in vitro and in vivo. Mechanistically, DDGC sponged miR-589-5p to stabilize RAD51 mRNA, and DDGC also regulated the ubiquitin-mediated degradation of the Wilms tumor 1 (WT1) protein through direct interaction with the heat shock 90 (HSP90) protein. Our study suggests that lncRNA DDGC is involved in DNA damage repair and in the timely differentiation of GCs and thus provides a new epigenetic perspective on POI pathogenesis.
Results
LncRNA DDGC is significantly minimally expressed in GCs from bPOI patients
A total of 78 lncRNAs were differentially expressed in GCs between the eight patients and the nine controls (Table 1, Figures 1A and 1B) according to RNA-seq analyses (GSE158526).19 To validate the RNA-seq results, six lncRNAs with high conservative properties as shown by the UCSC resources20 were chosen for further detection by qRT-PCR. As shown in Figures 1C and S1, three lncRNAs—RP4-545C24.1, AC112721.1, and ZNF674-AS1—were minimally expressed, and three lncRNAs—RP11-3D4.3, SNAI3-AS1, and GS1-358P8.4—were upregulated in GCs from bPOI patients (Table 2). Among them, lncRNA RP4-545C24.1, located at 7q35 and with limited known functions, showed the greatest fold change (Table S1, fold change = 2.12). We assessed the coding potential of RP4-545C24.1 using the Coding-Potential Calculator21 and Coding-Potential Assessment Tool,22 which confirmed the non-coding characteristic of RP4-545C24.1 (Figure S2A). Furthermore, RP4-545C24.1 had a relatively high expression level in the ovary according to the Genotype-Tissue Expression database23 (Figure S2B), which was similar to its expression profile in 21-week human fetal tissues (Figure 1D), suggesting a potential regulatory role in ovarian function. In addition, the single-cell RNA-seq analysis24 showed that the expression of DDGC in human GCs was increased with follicle development (Figure S2C). Therefore, we focused on RP4-545C24.1 for subsequent experiments. For simplicity, we refer to RP4-545C24.1 as DDGC (DNA damage repair and differentiation of GC regulatory RNA), given its roles in DNA damage repair and differentiation in GCs.
Table 1.
Baseline characteristics of study participants for RNA-seq
| Characteristic | Control (n = 9) | bPOI (n = 8) | p |
|---|---|---|---|
| Age (years) | 32.00 ± 4.15 | 34.00 ± 4.41 | 0.351 |
| BMI (kg/m2) | 21.36 ± 1.89 | 22.33 ± 2.08 | 0.329 |
| bFSH (IU/L) | 6.37 ± 0.85 | 15.90 ± 3.73 | <0.0001 |
| bLH (IU/L) | 4.67 ± 2.02 | 7.55 ± 2.95 | 0.032 |
| bE2 (pg/mL) | 33.89 ± 17.77 | 28.93 ± 11.93 | 0.516 |
| AMH (ng/mL) | 4.09 ± 2.08 | 0.63 ± 0.17 | <0.001 |
bFSH, basal FSH; bLH, basal luteinizing hormone; bE2, basal estradiol.
Data are presented as mean ± SD, Students' t test.
Figure 1.

LncRNA DDGC is significantly minimally expressed in GCs from bPOI patients
(A) RNA-seq heatmap of lncRNAs with significantly altered expression in GCs from controls (n = 9) or bPOI patients (n = 8). LncRNAs selected for validation are marked by a red box. (B) Volcano plot showing the lncRNAs with significantly altered expression. (C) The qRT-PCR analysis of the expression levels of DDGC in GCs from controls (n = 27) or bPOI patients (n = 17). Error bars show the mean ± SD. ∗∗p < 0.01. (D) The qRT-PCR analysis of the expression levels of DDGC in 21-week fetal tissues. The expression level of DDGC in ovary was set to 1. GAPDH was regarded as an endogenous control for (C) and (D).
Table 2.
Baseline characteristics of study participants for qRT-PCR
| Characteristic | Control (n = 27) | bPOI (n = 17) | p |
|---|---|---|---|
| Age (years) | 31.48 ± 3.96 | 32.65 ± 3.97 | 0.347a |
| BMI (kg/m2) | 21.85 ± 2.14 | 22.44 ± 3.76 | 0.510a |
| bFSH (IU/L) | 6.31 ± 1.34 | 11.95 (11.33, 12.64) | <0.0001b |
| bLH (IU/L) | 5.83 ± 1.51 | 5.76 ± 1.85 | 0.893a |
| bE2 (pg/mL) | 36.11 ± 16.02 | 36.76 ± 17.04 | 0.898a |
| AMH (ng/mL) | 3.21 ± 1.34 | 0.513 ± 0.30 | <0.0001a |
bFSH, basal FSH; bLH, basal luteinizing hormone; bE2, basal estradiol.
Data are presented as mean ± SD.
Students' t test. Data are presented as median (interquartile range).
Mann-Whitney U test.
Silencing DDGC impairs DNA damage repair by downregulating RAD51
Because DDGC is a transcript with limited known functions, we analyzed transcriptome data by RNA-seq (GSE158555) in KGN cells upon downregulation of DDGC with small interfering RNA (siRNA) (Figure S3A). As shown in Figures 2A and S3B, a total of 728 genes were differentially expressed as a consequence of DDGC silencing. Analysis of Kyoto Encyclopedia of Genes and Genomes pathways found that the most significantly enriched pathways included cell-cycle-related pathways, the Fanconi anemia pathway, and the homologous recombination (HR) pathway (Figure 2B), which were confirmed by qRT-PCR analysis (Figures S3C and S3D). Loss-of-function experiments were performed to determine the effect of DDGC knockdown in GCs.
Figure 2.

Silencing of DDGC impairs DNA damage repair through downregulation of RAD51
(A) RNA-sequencing heatmap of differentially expressed mRNAs between DDGC-silenced (n = 2) and negative control KGN cells (n = 2). (B) KEGG analysis of RNA-sequencing data for KGN cells after si-DDGC and si-NC transfection. (C) The etoposide (ETO)-induced γH2AX foci formation in KGN and SVOG cells after si-DDGC and si-NC transfection. Scale bars, 15 μm. (D) After exposure to ETO, the expression levels of γH2AX were measured by western blot in KGN and SVOG cells after si-DDGC and si-NC transfection. (E) The levels of cleaved PARP induced by ETO were measured by western blot in KGN and SVOG cells after si-DDGC and si-NC transfection. Protein levels were quantified by ImageJ software and normalized to the loading controls. Error bars show the mean ± SD from three repeated experiments. n.s., not significant; ∗p < 0.05, ∗∗∗p < 0.001. (F) TUNEL assays of KGN and SVOG cells with si-DDGC and si-NC transfection, followed by the addition of ETO for 12 h. Error bars show the mean ± SD from three repeated experiments. ∗∗∗p < 0.001. Scale bars, 50 μm (KGN) or 20 μm (SVOG). (G) Western blots of protein levels of several essential genes related to DNA damage repair in KGN and SVOG cells after si-DDGC and si-NC transfection. (H) Western blots of protein levels of γH2AX in KGN and SVOG cells with si-RAD51 and si-NC transfection after ETO exposure. (I) After exposure to camptothecin (CPT), the γH2AX levels were measured by western blot in si-DDGC-, si-RAD51-, and si-NC-transfected KGN cells.
As shown in Figures S3E–S3I, CCK8, 5-ethynyl-2′-deoxyuridine (EdU) staining, and flow cytometry analysis showed no significant impact of DDGC silencing on cell-cycle progression or proliferation of GCs. To measure DNA damage repair capacity and to evaluate the cellular response to DNA damage induced by etoposide (ETO), γH2AX levels were measured by western blot and immunofluorescence as an indicator of double-strand breaks (DSBs). After exposure to ETO, γH2AX foci were observed and disappeared as the repair process progressed (Figure 2C). DDGC silencing significantly prolonged the time to repair ETO-induced DSBs in both KGN and SVOG cells (Figure 2D), which suggested that DDGC silencing inhibits the progress of DSB repair in GCs. As a consequence, silencing of DDGC significantly increased GC apoptosis as evidenced by increased levels of cleaved poly ADP-ribose polymerase protein (PARP) (Figure 2E) and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) signal (Figure 2F). Collectively, these findings suggest that downregulation of DDGC inhibits DNA damage repair in GCs.
To clarify the molecular mechanism underlying this phenomenon, we measured the DNA repair proteins that were differentially expressed after DDGC knockdown. As shown in Figure 2G, silencing DDGC led to a significantly decreased RAD51 protein level, but there was no significant effect on BRCA2, BARD1, or BRCA1 protein levels. Therefore, we next explored whether downregulated RAD51 was responsible for the impaired DNA damage repair seen in DDGC-silenced cells. Downregulation of RAD51 by siRNA (Figure S3J) impaired DNA damage repair of KGN and SVOG cells (Figure 2H), which mimicked the phenotype upon DDGC silencing. Moreover, overexpression of RAD51 attenuated the levels of γH2AX induced by ETO after DDGC silencing (Figures S3K and S3L). Considering the vital role of RAD51 in the HR repair pathway, we validated the impact of DDGC silencing on the repair of DNA damage induced by camptothecin, which can be repaired only through the HR pathway, and not the non-homologous end-joining pathway. Consistent with the results of ETO treatment, γH2AX was observed after camptothecin-induced damage and then decreased as the damage was repaired (Figure 2I). Silencing DDGC or RAD51 extended the time for repairing camptothecin-induced DSB in KGN cells as expected (Figure 2I), which further demonstrated that the HR repair process was impaired in DDGC-silenced or RAD51-silenced cells. Collectively, our findings suggest that RAD51 is the critical target gene through which DDGC participates in DNA damage repair processes in GCs.
DDGC is necessary for the stabilization of RAD51 mRNA by serving as a sponge for miR-589-5p
DDGC was predominantly localized in the cytoplasm of KGN and SVOG cells (Figures 3A and 3B), implying that it might serve as a competitive endogenous RNA (ceRNA) by binding to microRNAs (miRNAs) and mediating mRNA degradation.25 Cellular transcription and translation were inhibited by actinomycin D and cycloheximide, respectively. Then we conducted qRT-PCR and western blot assays to observe the degradation rate of RAD51 mRNA and protein after DDGC knockdown. Results showed that silencing DDGC promoted the degradation of RAD51 mRNA (Figure 3C), while the protein degradation rate remained unchanged compared with that of negative control cells (Figure 3D). Together, these results suggest that DDGC is necessary for RAD51 mRNA stabilization rather than for degradation of RAD51 protein.
Figure 3.

DDGC is necessary for the stabilization of RAD51 mRNA by serving as a sponge for miR-589-5p
(A) The qRT-PCR analysis of the expression levels of DDGC in the cytoplasm and nucleus of KGN cells. MALAT1 served as the cytoplasmic reference for lncRNA, and lamin B1 and β-actin were used as the nuclear and cytoplasmic reference, respectively. (B) RNA fluorescence in situ hybridization (FISH) assay showing the subcellular localization of DDGC in KGN and SVOG cells. Scale bars, 10 μm. (C) RAD51 mRNA expression in si-DDGC- and si-NC-transfected KGN and SVOG cells was measured by qRT-PCR at different time points after the addition of actinomycin D (CHD). (D) Western blots of the RAD51 protein levels in si-DDGC- and si-NC-transfected KGN cells at different time points after the addition of cycloheximide (CHX). Protein levels of RAD51 were quantified by ImageJ software and normalized to the loading controls. Error bars show the mean ± SD from three repeated experiments. (E) RIP assay of the enrichment of DDGC after anti-Ago2 immunoprecipitation. (F) The relative expression of DDGC in si-RAD51- and si-NC-transfected KGN and SVOG cells was measured by qRT-PCR. (G) qRT-PCR was applied to analyze the relative expression levels of DDGC and RAD51 mRNA in miRNA mimics and negative-control-transfected KGN cells. (H) Western blots of the RAD51 protein levels in miRNA-mimics and negative-control-transfected KGN and SVOG cells. (I, J) Luciferase activities were analyzed at 48 h after co-transfection of reporter vectors and miR-589-5p or negative control in (I) HEK293 or (J) HeLa cells. Data are presented as the mean ± SD from three repeated experiments. (K) Enrichment of DDGC and RAD51 after biotin-miR-589-5p or biotin-negative control pull-down. (L) Enrichment of miR-589-5p pulled down by biotin-labeled probes directed to DDGC and biotin-labeled negative control probes. Error bars show the mean ± SD. The qRT-PCR values in (A), (C), (E–G), (K), and (L) were obtained from three repeated experiments. n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Because argonaute RISC catalytic component 2 protein (Ago2) is a central element of the RNA-induced silencing complex involved in miRNA-regulated mRNA instability, we performed RNA immunoprecipitation (RIP) assays utilizing anti-Ago2 antibody and found that endogenous DDGC was preferentially co-precipitated by Ago2 compared with IgG (Figure 3E). Moreover, silencing of RAD51 mRNA also reduced DDGC expression (Figure 3F). These results suggest that DDGC serves as a ceRNA to regulate RAD51 mRNA stability and DNA damage repair. Prediction using the publicly available starBase v.2.0 tool26 showed that three miRNAs (miR-384, miR-589-5p, miR-653-5p) contained seed regions complementary to both DDGC and RAD51 mRNA. Thus, we transfected KGN cells with mimics of the three candidate miRNAs, and the expression of lncRNAs DDGC and RAD51 was detected via qRT-PCR and western blot. As shown in Figures 3G and 3H, both miR-384 and miR-589-5p could downregulate the expression of DDGC and RAD51 simultaneously. To further determine whether these two miRNAs bind to DDGC and RAD51 mRNA, we established luciferase reporters comprising either wild-type (WT) or mutant (MT) DDGC or the 3′ UTR of RAD51 mRNA (Figures S4A and S4B). As shown in Figures 3I and 3J, miR-589-5p overexpression significantly suppressed the luciferase activity of WT reporter vectors but not MT vectors in human embryonic kidney (HEK) 293 and HeLa cells. In contrast, miR-384 mimics had no effect on either candidate target sequence (Figure S4C). Thus, we focused on miR-589-5p in seeking further evidence for this interaction. A biotin-labeled RNA pull-down assay showed that miR-589-5p precipitated endogenous DDGC and RAD51 mRNA in KGN cells (Figure 3K). Moreover, miR-589-5p was pulled down by biotin-labeled antisense probes directed to DDGC (Figure 3L). These results indicate that DDGC directly “absorbs” miR-589-5p in order to regulate the stability of RAD51 mRNA.
Silencing of DDGC leads to the aberrant differentiation of GCs by downregulating WT1
Steroid hormone synthesis is indispensable for folliculogenesis, especially the conversion of androgen to estradiol, which is a characteristic of differentiated GCs.27 Therefore, we further explored whether DDGC participated in estradiol synthesis. Interestingly, we found that the level of estradiol was significantly increased after DDGC silencing (Figure 4A), as were the markers of GC differentiation, i.e., FSHR and CYP19A1 (Figures 4B and 4C). In addition, genes that are involved in steroid hormone synthesis and GC differentiation were also found to be differentially expressed in the KGN RNA-seq analysis (GSE158555). These findings suggest that the aberrant differentiation of GCs can be attributed to DDGC silencing.
Figure 4.

Silencing of DDGC leads to the aberrant differentiation of GCs through downregulation of WT1
(A) The estradiol (E2) level in the supernatant of KGN cells was measured after si-DDGC and si-NC transfection. Error bars show the mean ± SD from three repeated experiments. ∗∗∗p < 0.001. (B) The qRT-PCR analysis of relative mRNA levels of FSHR and CYP19A1 in KGN cells with si-DDGC and si-NC transfection. The qRT-PCR values were obtained from three repeated experiments. Error bars show the mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001. (C) The protein levels of FSHR and CYP19A1 in si-DDGC- and si-NC-transfected KGN cells were measured by western blot. (D) The protein levels of FOXO1 and WT1 in si-DDGC- and si-NC-transfected KGN cells were measured by western blot. (E) The estradiol levels were measured in negative control, DDGC-silenced, WT1-silenced, and co-transfected KGN cells. Error bars show the mean ± SD from three repeated experiments. n.s., not significant; ∗∗p < 0.01. (F) The protein levels of FSHR and CYP19A1 in negative control, DDGC-silenced, WT1-silenced, and co-transfected KGN cells were measured by western blot.
Because forkhead box O1 (FOXO1) and WT1 are two critical transcription factors governing GC differentiation, we explored whether FOXO1 and WT1 were involved in the DDGC silencing-mediated aberrant differentiation of GCs. Intriguingly, WT1, but not FOXO1, was significantly downregulated after DDGC silencing (Figure 4D). Moreover, silencing WT1 resulted in an apparent increased level of estradiol and expression of FSHR and CYP19A1 (Figures S4D, 4E, and 4F), which mimicked the results of DDGC silencing. In contrast, overexpression of WT1 led to a significant decline in the levels of estradiol and FSHR and CYP19A1 expression, which partially rescued the impact of DDGC silencing on GC differentiation (Figures S4D, 4E, and 4F). In general, these data suggest that the aberrant differentiation of GCs after DDGC silencing can be attributed to insufficient WT1 expression.
DDGC combines with HSP90 and mediates the degradation of the WT1 protein
To elucidate the mechanism through which DDGC silencing leads to decreased WT1 expression, we observed the changes in WT1 protein and mRNA after DDGC silencing. The WT1 protein was notably diminished, whereas the level of WT1 mRNA was not significantly changed (Figure 5A), which indicated that DDGC might regulate WT1 mRNA translation or protein degradation. As shown in Figure 5B, treatment of cells with cycloheximide after DDGC silencing resulted in a notably shorter half-life of WT1 protein compared with controls, which suggested that DDGC mediates the degradation of WT1 protein. In addition to this, chloroquine and MG132 were used to block the autophagy and proteasome pathways in GCs, respectively. As shown in Figure 5C, treatment of cells with MG132 resulted in increased levels of endogenous WT1 protein compared with control cells, whereas chloroquine treatment had no impact on the decreasing level of WT1 protein upon DDGC knockdown (Figure S4E), thus indicating that DDGC modulates WT1 protein via the ubiquitin-proteasome pathway but not through autophagy. Indeed, the ubiquitination of the WT1 protein was dramatically increased in DDGC-silenced cells (Figure 5D). In summary, silencing of DDGC led to aberrant differentiation of GCs by promoting the ubiquitination and subsequent degradation of WT1.
Figure 5.

DDGC combines with HSP90 and mediates the degradation of the WT1 protein
(A) The qRT-PCR analysis of the relative WT1 mRNA levels in si-DDGC- and si-NC-transfected KGN cells. The qRT-PCR values were obtained from three repeated experiments. Error bars show the mean ± SD. n.s., not significant. (B) Western blots of the WT1 protein levels in si-DDGC- and si-NC-transfected KGN cells at different time points after the addition of CHX. Protein levels of WT1 were quantified by ImageJ software and normalized to the loading controls. Error bars show the mean ± SD from three repeated experiments. ∗p < 0.05. (C) Western blots of protein levels of WT1 in KGN cells with si-DDGC or si-NC transfection, followed by the addition of MG132 for 24 h. (D) The polyubiquitination level of WT1 in si-DDGC- and si-NC-transfected KGN cells at 24 h after the addition of MG132 was measured by western blot. The bottom blot depicts the input of the cell lysates. (E) Western blots of protein levels of WT1 in KGN cells at 12 h after the addition of 17-AAG and MG132. (F) Western blots of protein levels of WT1 in si-DDGC- and si-NC-transfected KGN cells after anti-HSP90 immunoprecipitation. (G) Western blots of the proteins pulled down by biotin-labeled DDGC and DDGC-AS. (H) RIP assays were conducted using the anti-HSP90 antibodies in KGN cells, and RT-PCR was used to measure DDGC.
Because the chaperone activity of HSP90 is essential for WT1 protein stability,28 we focused on HSP90 to elucidate the possible mechanism underlying the DDGC-mediated ubiquitination of WT1. Consistent with the results presented above, diminished WT1 protein was observed in KGN cells treated with the HSP90 inhibitor 17-AAG, which could be rescued by MG132 (Figure 5E), indicating that inhibition of HSP90 chaperone activity promotes the ubiquitin-proteasome degradation of WT1 in GCs. As a molecular chaperone, HSP90 interacts with hundreds of proteins to help maintain their activity.29 Therefore, we hypothesized that DDGC might act as an intermediate in the interaction between HSP90 and WT1. Intriguingly, immunofluorescence showed that HSP90 and WT1 were co-distributed in the nuclei of KGN cells (Figure S4F). DDGC silencing had no impact on the expression of HSP90 (Figure S4G); however, it impaired the interaction between HSP90 and WT1 in both nucleus and cytoplasm (Figure 5F). Together, these results suggest that DDGC mediates the interaction between HSP90 and WT1.
We further explored whether DDGC could directly bind to HSP90. The biotin-labeled RNA pull-down assay and subsequent western blot showed that HSP90 bound directly to DDGC, but not to DDGC-AS (Figure 5G). Furthermore, the RIP assay indicated an endogenous interaction between DDGC and HSP90 in KGN cells (Figure 5H). Collectively, these results suggest that DDGC is essential for the HSP90-WT1 interaction, and when DDGC is downregulated, HSP90 can no longer efficiently bind to WT1, leading to subsequent WT1 degradation.
DDGC regulates the expression of Rad51 and DNA damage repair in vivo
To further verify the regulatory role of DDGC on the expression of RAD51 and WT1, DDGC was introduced into in vitro-cultured mouse ovaries (Figure S4H) by infection or mouse ovaries (in vivo) by ovarian bursa injection with adenovirus expressing DDGC (Figure 6A). Results showed that overexpressing DDGC significantly decreased the protein levels of ETO-induced γH2AX and cleaved PARP in in vitro-cultured ovaries (Figure 6B); however, it had no impact on the ubiquitination of WT1 (Figure S4I). In vivo experiments found that the mRNA and protein levels of Rad51 were significantly increased in ovaries overexpressing DDGC (Figures 6C and 6D), indicating the regulation of DDGC on Rad51 expression in vivo. Meanwhile, no significant change was observed in the protein levels of WT1, FSHR, and CYP19A1 after DDGC overexpression (Figure S4J). The above-mentioned results suggested the protective role of DDGC in mouse ovaries upon DNA damage. Then, we explored the therapeutic effect of DDGC within mouse ovaries. After intraperitoneal injection of ETO, lower γH2AX and cleaved PARP protein levels, as well as TUNEL signals, were detected in ovaries overexpressing DDGC (Figures 6E and 6F). These data indicate the therapeutic effect of DDGC in mouse ovaries bearing DNA damage.
Figure 6.

DDGC regulates the expression of Rad51 and DNA damage repair in vivo
(A) The RT-PCR analysis of the expression of DDGC in mouse ovaries infected with adenovirus. (B) The protein levels of γH2AX and cleaved PARP in DDGC-overexpressing and negative control in vitro-cultured ovaries were measured by western blot at 48 h after the addition of ETO. (C) The qRT-PCR analysis of the relative mRNA levels of Rad51 in DDGC-overexpressing and negative control mouse ovaries. The qRT-PCR values were obtained from three repeated experiments. Error bars show the mean ± SD. (D) Western blots of the protein levels of RAD51 in DDGC-overexpressing and negative control mouse ovaries. (E) Western blots of the protein levels of γH2AX and cleaved PARP in DDGC-overexpressing and negative control mouse ovaries at 48 h after the addition of ETO. (F) The apoptosis of DDGC-overexpressing and negative control mouse ovaries at 48 h after the addition of ETO was measured by TUNEL assay. Scale bar, 100 μm. Error bars show the mean ± SD. Protein levels of (B), (D), and (E) were quantified by ImageJ software and normalized to the loading controls. n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
In the ovary, the roles and mechanisms of lncRNAs have not been extensively explored until recently. The latest reports have shown a relationship between certain lncRNAs and PCOS; for instance, LINC-01572:28 and Lnc-OC1 participate in PCOS development by mediating GC proliferation.17,30 However, few lncRNAs have been explored in POI pathogenesis. Here, we identified the lncRNA DDGC, which plays dual roles in both DNA damage repair progression and the differentiation of GCs. Our mechanistic study showed that DDGC modulated the stability of RAD51 mRNA and WT1 protein simultaneously (Figure 7). Interestingly, a similar mode of action is seen for the lncRNAs GUARDIN and OVAAL, which have recently been identified to play dual roles in the senescence and survival of cancer cells.31,32 The dual functions of these lncRNAs are carried out at the same time in a non-interfering manner, which provides deeper insights into their RNA regulatory effects. However, a lack of such molecules in GCs has impeded our understanding of the pathogenic mechanism behind the development of POI. Our findings are the first to show that RAD51 and WT1, two pathogenic genes for POI, are post-transcriptionally modulated by lncRNA DDGC in GCs, providing new insights into the contribution of lncRNAs to POI pathogenesis.
Figure 7.

The schematic model for the post-transcriptional regulation and function of lncRNA DDGC
In mammals, abnormal GC function contributes to follicle atresia and POI,13 and if DSBs in GCs are not repaired correctly or promptly this can also lead to cell death and then atretic follicles and premature ovarian aging.33,34 Variants within a series of DNA repair genes, such as MCM8,35 MSH5,36 and BRCA2,37,38 have been proved as causative factors for POI. In addition, lncRNA HCP5 was previously shown to participate in the development of POI by mediating the process of DNA damage repair in GCs.39 In this study, our results show that downregulation of DDGC inhibited the DNA damage repair activity of GCs by degrading RAD51 mRNA and impairing the HR pathway, thereby leading to GC apoptosis. Thus, our results shed new light on how lncRNAs regulate the process of DNA damage repair in GCs.
In the present study, we identified a ceRNA network through which lncRNA DDGC regulates the expression of the DNA repair gene RAD51. RAD51 is the eukaryotic ortholog of the bacterial recombinase RecA, which plays a central role in the error-free HR pathway of DNA repair.40,41 The latest studies have shown that Rad51-mediated HR is the predominant pathway in oocytes for repairing DSBs,42 especially in primordial follicles.43 Furthermore, through whole-exome sequencing, a pathogenic variant of RAD51 has been identified in POI patients with primary amenorrhea.44 Consistent with this, our results show the impact of dysregulated RAD51 on GC function and survival, not only strengthening the role of DNA repair deficiency in the pathogenesis of POI, but also highlighting DDGC as an important modulator of RAD51. To our knowledge, this is the first report of a ceRNA mechanism involved in POI pathogenesis.
The second mechanism through which silencing of DDGC interferes with GC function is the accelerated degradation of the WT1 protein by binding to HSP90, and this leads to aberrant GC differentiation. WT1 is a known causative gene for POI16,45 due to its role in regulating genes responsible for GC differentiation, including SF1, FSHR, and CYP19A1.45,46 However, the post-transcriptional regulation of WT1 in the ovary has not been previously described. We show here that the chaperone activity of HSP90 governs the stability of the WT1 protein in GCs. In addition, we show the essential role of DDGC in the HSP90-WT1 interaction, thereby protecting the WT1 protein from ubiquitin-mediated protein degradation. Similarly, lncRNA GLCC1 and GALNT5 UTR-associated RNA have been reported to interact with HSP90 and to mediate the ubiquitination of substrate proteins such as c-Myc and IKK in colorectal cancer and gastric cancer.47,48 We have identified DDGC as a novel regulator of WT1 ubiquitination by binding to HSP90 in GCs, suggesting the protein binding domain within DDGC. Based on the current findings, it is reasonable to hypothesize that lncRNA DDGC might maintain the efficient combination of HSP90 and WT1 through the secondary structure formed by itself. Spatial structure is an essential feature of non-coding RNAs and is closely related to cellular conditions and processes, such as capping, splicing, polyadenylation, and folding.49,50 For example, the lncRNA HOTAIR tethers multiple histone-modifying complexes through its intricate secondary structure.51,52 However, inappropriate modification could be made by endonucleases and spliceosomes in mouse GCs due to the low conservative property of the DDGC sequence, resulting in incorrect folding and inaccurate structure formation. In the present study, protein levels of WT1 remain unchanged upon DDGC overexpression in vivo, which might be because DDGC of adenovirus origin could not form the homologous spatial structure in mouse ovaries. Further studies are warranted to elucidate the physiological role of DDGC in vivo and whether this lncRNA-protein regulatory mechanism exists in mouse GCs.
In summary, we have demonstrated here that lncRNA DDGC participates in POI pathogenesis by post-transcriptionally regulating the stability of RAD51 mRNA and WT1 protein. In particular, our study has expanded on the two regulatory mechanisms of lncRNAs in POI, and we propose a new epigenetic mechanism in the etiology of POI in which the two causative genes, RAD51 and WT1, are regulated simultaneously. Therefore, DDGC might be a potential therapeutic approach for POI.
Materials and methods
Clinical samples
The participants in this study were recruited from the Reproductive Hospital affiliated with Shandong University after written informed consent was obtained. The groups contained 25 bPOI patients and 36 age and BMI-matched healthy women (control group). All of the participants were undergoing in vitro fertilization or intracytoplasmic sperm injection and embryo transfer (IVF/ICSI-ET) treatment because of tubal or male factors. The bPOI patients were recruited according to the following criteria: (1) <40 years of age; (2) menstrual cycle 23–35 days; (3) basal follicle-stimulating hormone (bFSH) 10 < serum bFSH < 25 (IU/L); (4) serum anti-Müllerian hormone (AMH) <1.1 ng/mL. Women with chromosomal abnormalities or carrying POI-causative mutations, or with an experience of ovarian radiotherapy, chemotherapy, or surgery, were precluded. The study was under approval from the Institutional Review Board (IRB) of Reproductive Medicine of Shandong University. The clinical indicators of all participants are summarized in Tables 1 and 2. GCs were collected from participants and prepared as previously described.39
RNA-seq analysis
The total RNA was purified from the GCs of eight bPOI patients and nine controls utilizing the TRIzol reagent (TaKaRa, China). The degradation, contamination, purity, concentration, and integrity of total RNA were monitored by agarose gels, spectrophotometer instruments (IMPLEN, USA), a Qubit RNA detection kit (Life Technologies, USA), and an RNA Nano 6000 Assay Kit (Agilent Technologies, USA). The input RNA for sample preparations was set as 3 μg RNA per sample. An rRNA Removal Kit (Epicentre, USA) and subsequent ethanol precipitation were utilized to remove rRNA. Subsequently, the rRNA-free RNA was used to generate the libraries for sequencing using a NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB, USA) according to the manufacturer's instructions. An AMPure XP system (Beckman Coulter, Beverly, MA, USA) was utilized to purify the library fragments and subsequently collect cDNA fragments with length ranging from 150 to 200 bp. The generation of the cluster was conducted via the TruSeq PE Cluster Kit v.3-cBot-HS (Illumina) following the manufacturer's guidelines. Then, the sequencing was performed on the Illumina HiSeq 4000 platform with 150-bp paired-end reads generated. Low-quality reads, as well as reads containing adapter and poly-N, were removed, thereby generating clean reads. Coding potential analysis was performed utilizing CNCI, CPC, Pfam-sca, and phyloCSF software. The Cuffdiff (v.2.1.1) was applied to calculate the fragments per kilobase of transcript per million mapped reads (FPKMs) of both lncRNAs and mRNAs in each sample. Differentially expressed transcripts were assigned as p-adjust <0.05. Significantly enriched gene ontology (GO) terms were assigned as p < 0.05. Significantly enriched KEGG pathways were determined by KOBAS software. The raw data have been uploaded as GEO: GSE158526.
For KGN RNA-seq analysis, the total RNA was extracted from KGN cells at 48 h after transfection with DDGC siRNA (si-DDGC) or with negative control siRNA (si-NC). RNA-seq and data analyses were carried out by Annoroad (China). A Kaiao K5500 spectrophotometer (Kaiao, China) was used to check the purity of total RNA. The input RNA for sample preparations was set as 2 μg RNA per sample. The library RNA concentration was diluted to 1 ng/μL after quantification using a Qubit RNA Assay Kit in Qubit 3.0. The Agilent Bioanalyzer 2100 system (Agilent Technologies) and StepOnePlus Real-Time PCR System were utilized to quantify the insert size. The cluster was generated via HiSeq PE Cluster Kit v.4-cBot-HS (Illumina) following the manufacturer's guidelines. Subsequently, the sequencing was conducted on an Illumina platform. The remaining steps were the same as in the above paragraph. The raw data have been uploaded as GEO: GSE158555.
Isolation of RNA and qRT-PCR assays
The expression of DDGC in GCs was validated from an independent cohort consisting of 17 bPOI patients and 27 controls. The extraction of RNA from GCs or cell lines was conducted according to protocols as described above. The PrimeScript RT Reagent Kit (TaKaRa) was utilized to reverse transcribe all mRNAs and lncRNAs, whereas miRNA-specific reverse transcription primers (GenePharma, China) were used to reverse transcribe miRNAs individually. The SYBR Green Master Mix (TaKaRa) was applied for quantitative RT-PCR, and GAPDH was regarded as an endogenous control for qRT-PCR of mRNAs and lncRNAs. U6 was regarded as an endogenous control for qRT-PCR of miRNAs. The qRT-PCR assay per sample was run in triplicate, and the 2−ΔΔCt method was applied to analyze all obtained values. Table S2 shows the primers used for qRT-PCR analysis.
Cell lines, cell culture, and transfection of vectors
The KGN GC tumor cell line was purchased from RIKEN BRC (Japan).53 SVOG (luteinized GC line) was generously provided by Professor Peter C.K. Leung. The culture medium was prepared, to which 10% fetal bovine serum (FBS) (Gibco, USA) and 1% penicillin/streptomycin (P/S) (Invitrogen, USA) were added. KGN and SVOG cells were grown in DMEM/F12 culture medium (HyClone, USA), and the HEK293 and HeLa (human cervix carcinoma) cells were cultured in DMEM high-glucose culture medium (HyClone). All cells were incubated under humid conditions at 37°C and 5% CO2.
The siRNA duplexes and miRNA mimics applied in this study were generated at GenePharma. The cDNA sequence encoding human RAD51 was subcloned into the pEGFP-N1 vector (pEGFP-N1-RAD51), and an empty vector served as the control (pEGFP-N1). Adenoviruses for overexpressing WT1 (Ad-WT1) were designed by Cyagen (China). The virus that expresses only GFP was regarded as a negative control (Ad-null) and cells were infected with 1 × 106 PFU/mL for the in vitro experiments. Transfection of siRNA duplexes, pEGFP-N1 vectors, and miRNA mimics was conducted using the Lipofectamine 3000 reagent (Invitrogen) following the manufacturer's protocols. Downstream experiments were conducted 48 h after transfection or infection. Table S3 shows the sequences of all siRNAs involved in this study.
CCK8 assay
KGN cells were seeded onto 96-well dishes at a density of 4,000 cells/well. Cell Counting Kit-8 (CCK8, Beyotime, China) was used to measure the viability of KGN cells at different times after transfection.
EdU assay
KGN cells were seeded onto 96-well plates and fixed at 48 h after transfection. Then, a cell proliferation assay (EdU, RiboBio, China) was performed in triplicate following the manufacturer's protocols.
Cell-cycle analysis by flow cytometry
KGN cells were cultured in 10-cm2 plates and collected at 48 h after transfection. Then, 70% prechilled ethanol was utilized to fix cells at 4°C for 2 h. Subsequently, cells were stained with 7-AAD (Multi Sciences, China) followed by flow cytometry cell-cycle analysis.
Western blot
Cell pellets were lysed by SDS or RIPA solution incorporating an inhibitor of proteinase (Cell Signaling Technology, USA), and total protein was extracted immediately following the manufacturer's instructions. Approximately 20 μg of protein was loaded and separated on NuPAGE bis-tris mini gels (Invitrogen) or SDS polyacrylamide gels and subsequently transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). Then, the PVDF membranes were blocked after treatment with 5% non-fat milk at room temperature for 1 h. Blocked membranes were mixed with a solution of specific primary antibodies at 4°C overnight. Subsequently, secondary antibodies (Proteintech, China) were added and reacted with the primary antibodies at room temperature for 1 h. After staining via ECL chemiluminescence kit (Millipore), protein bands were detected by a ChemiDoc MP imaging system (Bio-Rad, USA). Table S4 shows the antibodies used for western blot.
DNA damage assay
KGN and SVOG cells were cultured in 12-well plates. Cells were harvested immediately at 48 h after transfection, or were exposed to 5 μg/mL ETO or 10 μM camptothecin for 6 h, followed by recovery for 2 or 4 h. The expression of the DNA damage marker γH2AX was measured by western blot.
Immunofluorescence
KGN and SVOG cells were fixed by treatment with paraformaldehyde at a concentration of 4% at 37°C for 20 min. Permeabilization and blocking were accomplished after treatment with 0.3% Triton X-100 and 10% BSA at room temperature for 30 min. The samples were incubated at 4°C overnight with specific primary antibodies. Subsequently, the antibodies were labeled at room temperature through treatment with Alexa Fluor secondary antibody for 1 h. Nuclei were stained with DAPI. A laser confocal microscope (ANDOR, UK) was utilized to capture images. Table S4 shows the antibodies used for immunofluorescence.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling assays
At 48 h after transfection, ETO was added to the culture medium of KGN and SVOG cells at a concentration of 5 μg/mL for 12 h to induce DNA damage, followed by TUNEL assays using a TUNEL kit (KeyGen, China) following the manufacturer's protocols.
Nuclear and cytoplasmic component separation
The nuclear and cytoplasmic components were isolated via the PARIS™ Kit (Invitrogen) following the manufacturer's protocols. After purification, proteins or RNAs were subjected to western blot or qRT-PCR analysis. For qRT-PCR analysis, the input RNA was set at 100% to normalize the expression levels of DDGC along with the reference genes in the cytoplasmic and nuclear fractions.
Fluorescence in situ hybridization
DDGC probes were synthesized by RiboBio and labeled with Cy3. Then, assays were conducted using a fluorescence in situ hybridization (FISH) kit (RiboBio) following the manufacturer's protocols. The samples were observed using a laser confocal microscope (ANDOR, UK).
Actinomycin D exposure
KGN and SVOG cells were cultured in 12-well plates. At 24 h after transfection, cellular transcription was blocked for 0, 2, 4, 8, or 12 h through the addition of actinomycin D to the culture medium at the final concentration of 10 μM. The qRT-PCR assay was performed to measure RAD51 mRNA expression levels.
Cycloheximide exposure
KGN cells were cultured in 12-well plates. At 24 h after transfection, cellular translation was blocked for 0, 2, or 4 h through the addition of cycloheximide to the culture medium at the final concentration of 100 μg/mL. Cells were harvested and the proteins were analyzed by western blot.
RNA immunoprecipitation
RIP assays were carried out using the EZ-Magna RIP kit (Millipore) following the manufacturer's protocols. The cells (107 per sample) were lysed by RIP lysis solution containing inhibitors of RNase and protease. After one period of freezing and melting, specific primary antibodies or homologous IgG (negative control) was added to cell lysates and incubated overnight at 4°C. TRIzol reagent and subsequent qRT-PCR were used to extract and quantify the immunoprecipitated RNA as described above. The antibodies used for RIP assays are listed in Table S4.
Dual luciferase reporter assay
The pmirGLO plasmids containing approximately 150 bp of RAD51 mRNA 3′ UTR or DDGC sequences with WT or deleted miRNA binding sites were generated and purchased from Promega (Madison, WI, USA). Plasmids and miRNA mimics or negative controls were co-introduced into HEK293 or HeLa cells. At 48 h after transfection, the cells were lysed and the luciferase activity of HEK293 or HeLa cells was assessed based on the protocols of the Dual-Luciferase Reporter Assay System (Promega).
Biotin-labeled RNA pull-down assay
For the biotin-labeled miRNA pull-down assays, bio-589-5p (biotin-labeled miR-589-5p) or bio-NC (empty vector) was designed by GenePharma and subsequently introduced into KGN cells. Cells were lysed followed by incubation with Pierce IP lysis buffer (Invitrogen) supplemented with inhibitors of protease and RNase. For the DDGC pull-down assay, KGN cells were lysed followed by incubation with the biotinylated DDGC probe (Bio-DDGC-probe) or negative control probe (Bio-NC-probe) or with the in vitro-transcribed DDGC sense strand and anti-sense strand (DDGC-AS). In vitro transcription was performed to generate biotin-labeled RNA via the MEGAscript T7 transcription kit (Invitrogen). Then magnetic streptavidin beads (Invitrogen) were added to the cell lysates, followed by incubation with RNA capture buffer at room temperature for 2 h. TRIzol reagent and qRT-PCR were used to isolate and quantify the RNA bound to the beads. Co-precipitated proteins were analyzed by western blot.
Detection of estradiol
KGN cells were cultured in 12-well plates. The KGN cell culture medium was replaced with DMEM without phenol red at 48 h after transfection, followed by treatment with 10 nM androstenedione for 24 h. The Roche Cobas e 801 system was used to determine the concentration of estradiol from the supernatant.
Chloroquine and MG132 exposure
KGN cells were cultured and transfected in 12-well plates. Subsequently, the cellular pathway of autophagy or proteasome was blocked through treatment with 50 μM chloroquine or 10 μM MG132 for 24 h, respectively. Then, the cells were harvested and the proteins were analyzed by western blot.
Co-immunoprecipitation and ubiquitination assay
KGN cells were harvested with Pierce IP lysis buffer containing proteinase protease. Proteins were extracted and subsequently treated with the specific primary antibodies at 4°C overnight. PureProteome Protein A/G magnetic beads (Millipore) were applied to precipitate the proteins at room temperature for 1 h. Loading buffer was used to resuspend the reaction mixtures after immunoprecipitation. Immunoprecipitated proteins were denatured and further analyzed by western blot. Table S4 shows the antibodies for co-immunoprecipitation.
17-AAG exposure
KGN cells were cultured in 12-well plates, followed by exposure to 10 μM 17-AAG alone or combined with 10 μM MG132 for 12 h, and the cells were harvested and the proteins were analyzed by western blot.
Ovary culture in vitro
All experiments involving mice were approved by the IRB of Reproductive Medicine of Shandong University and conformed to NIH regulatory standards. The in vitro culture system was established as previously described.54 Briefly, 21-day-old female mice with C57BL/6 background were purchased from Charles River Laboratories (China) and sacrificed by cervical dislocation. After isolation by microdissection, mouse ovaries were cultured in six-well culture dishes (NEST, China) with a plate (Sigma, USA). The culture medium contained 1,200 μL DMEM/F12 (Gibco) plus 1 μg/μL AlbuMAX II (Gibco), 0.18% L-ascorbic acid (Sigma), 1% ITS (Sigma, USA), and penicillin/streptomycin. Ovaries were incubated under humidified conditions at 37°C and 5% CO2. Adenoviruses for overexpressing DDGC (Ad-DDGC) and empty virus (Ad-NC) were synthesized by HanBio (China). A total of 1 μL Ad-DDGC or Ad-NC at 1 × 1011 PFU/mL was introduced into the medium. In vitro-cultured ovaries were harvested or treated with 5 μg/mL ETO or 10 μM MG132 at 48 h after infection, followed by H&E staining, co-immunoprecipitation, or western blot.
In vivo assays
A total of 10 μL Ad-DDGC or Ad-NC at 1 × 1011 PFU/mL was injected into 21-day-old female mouse ovarian bursa. Seven days after injection, the expression levels of RNA and protein were analyzed by RT-PCR and western blot. DNA damage was induced by intraperitoneal injection of ETO at 10 μg/g. The ovaries were harvested for western blot and TUNEL assay at 48 h after ETO injection. The TUNEL assays were performed using a TUNEL detection kit (KeyGen, China) following the manufacturer's protocols.
Statistics
All the statistical analyses were performed by SPSS software and GraphPad Prism 7 software. Except where indicated otherwise, the statistical significance was determined by the two-tailed Student t test. A p < 0.05 was considered statistically significant.
Data availability
The data reported in this study have been deposited in GEO: GSE158526 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158526) and GSE158555 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158555).
Acknowledgments
This study was supported by the National Key Research & Developmental Program of China (2016YFC1000604, 2017YFC1001100). The authors thank all participants.
Author contributions
Y.Q. and S.Z. contributed to the study design and direction. X.W. and L.X. contributed to analysis and interpretation of the results. Y.D. participated in designing the experiment. P.C.K.L., G.L., and W.-Y.C. participated in providing the SVOG cell line. D.L. and W.X. contributed to the experiment completion and manuscript draft. All of the authors read, revised, and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.10.015.
Supplemental information
References
- 1.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deveson I.W., Hardwick S.A., Mercer T.R., Mattick J.S. The dimensions, dynamics, and relevance of the mammalian noncoding transcriptome. Trends Genet. 2017;33:464–478. doi: 10.1016/j.tig.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Kopp F., Mendell J.T. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fatica A., Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 5.Slack F.J., Chinnaiyan A.M. The role of non-coding RNAs in oncology. Cell. 2019;179:1033–1055. doi: 10.1016/j.cell.2019.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atianand M.K., Caffrey D.R., Fitzgerald K.A. Immunobiology of long noncoding RNAs. Annu. Rev. Immunol. 2017;35:177–198. doi: 10.1146/annurev-immunol-041015-055459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poller W., Dimmeler S., Heymans S., Zeller T., Haas J., Karakas M., Leistner D.M., Jakob P., Nakagawa S., Blankenberg S. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur. Heart J. 2018;39:2704–2716. doi: 10.1093/eurheartj/ehx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coulam C.B., Adamson S.C., Annegers J.F. Incidence of premature ovarian failure. Obstet. Gynecol. 1986;67:604–606. [PubMed] [Google Scholar]
- 9.European Society for Human, R. Embryology Guideline Group on, POI. Webber L., Davies M., Anderson R., Bartlett J., Braat D., Cartwright B., Cifkova R., de Muinck Keizer-Schrama S. ESHRE Guideline: management of women with premature ovarian insufficiency. Hum. Reprod. 2016;31:926–937. doi: 10.1093/humrep/dew027. [DOI] [PubMed] [Google Scholar]
- 10.Qin Y., Jiao X., Simpson J.L., Chen Z.J. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum. Reprod. Update. 2015;21:787–808. doi: 10.1093/humupd/dmv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robker R.L., Richards J.S. Hormone-induced proliferation and differentiation of granulosa cells: a coordinated balance of the cell cycle regulators cyclin D2 and p27Kip1. Mol. Endocrinol. 1998;12:924–940. doi: 10.1210/mend.12.7.0138. [DOI] [PubMed] [Google Scholar]
- 12.Zhou J., Kumar T.R., Matzuk M.M., Bondy C. Insulin-like growth factor I regulates gonadotropin responsiveness in the murine ovary. Mol. Endocrinol. 1997;11:1924–1933. doi: 10.1210/mend.11.13.0032. [DOI] [PubMed] [Google Scholar]
- 13.Matsuda F., Inoue N., Manabe N., Ohkura S. Follicular growth and atresia in mammalian ovaries: regulation by survival and death of granulosa cells. J. Reprod. Dev. 2012;58:44–50. doi: 10.1262/jrd.2011-012. [DOI] [PubMed] [Google Scholar]
- 14.Di Pasquale E., Beck-Peccoz P., Persani L. Hypergonadotropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP15) gene. Am. J. Hum. Genet. 2004;75:106–111. doi: 10.1086/422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H., Xu X., Han T., Yan L., Cheng L., Qin Y., Liu W., Zhao S., Chen Z.J. A novel homozygous mutation in the FSHR gene is causative for primary ovarian insufficiency. Fertil. Steril. 2017;108:1050–1055.e1052. doi: 10.1016/j.fertnstert.2017.09.010. [DOI] [PubMed] [Google Scholar]
- 16.Wang H., Li G., Zhang J., Gao F., Li W., Qin Y., Chen Z.J. Novel WT1 missense mutations in han Chinese women with premature ovarian failure. Sci. Rep. 2015;5:13983. doi: 10.1038/srep13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao J., Xu J., Wang W., Zhao H., Liu H., Liu X., Liu J., Sun Y., Dunaif A., Du Y. Long non-coding RNA LINC-01572:28 inhibits granulosa cell growth via a decrease in p27 (Kip1) degradation in patients with polycystic ovary syndrome. EBioMedicine. 2018;36:526–538. doi: 10.1016/j.ebiom.2018.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Che Q., Liu M., Zhang D., Lu Y., Xu J., Lu X., Cao X., Liu Y., Dong X., Liu S. Long noncoding RNA HUPCOS promotes follicular fluid androgen excess in PCOS patients via aromatase inhibition. J. Clin. Endocrinol. Metab. 2020;105:dgaa060. doi: 10.1210/clinem/dgaa060. [DOI] [PubMed] [Google Scholar]
- 19.Li D., Wang X., Li G., Dang Y., Zhao S., Qin Y. LncRNA ZNF674-AS1 regulates granulosa cell glycolysis and proliferation by interacting with ALDOA. Cell Death Discov. 2021;7:107. doi: 10.1038/s41420-021-00493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang Y.J., Yang D.C., Kong L., Hou M., Meng Y.Q., Wei L., Gao G. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017;45:W12–W16. doi: 10.1093/nar/gkx428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L., Park H.J., Dasari S., Wang S., Kocher J.P., Li W. CPAT: coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Consortium G.T. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Yan Z., Qin Q., Nisenblat V., Chang H.M., Yu Y., Wang T., Lu C., Yang M., Yang S. Transcriptome landscape of human folliculogenesis reveals oocyte and granulosa cell interactions. Mol. Cell. 2018;72:1021–1034 e1024. doi: 10.1016/j.molcel.2018.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 26.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Hayek S., Clarke H.J. Control of oocyte growth and development by intercellular communication within the follicular niche. Results Probl. Cell Differ. 2016;58:191–224. doi: 10.1007/978-3-319-31973-5_8. [DOI] [PubMed] [Google Scholar]
- 28.Bansal H., Bansal S., Rao M., Foley K.P., Sang J., Proia D.A., Blackman R.K., Ying W., Barsoum J., Baer M.R. Heat shock protein 90 regulates the expression of Wilms tumor 1 protein in myeloid leukemias. Blood. 2010;116:4591–4599. doi: 10.1182/blood-2009-10-247239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schopf F.H., Biebl M.M., Buchner J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017;18:345–360. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 30.Wu G., Yang Z., Chen Y., Li X., Yang J., Yin T. Downregulation of Lnc-OC1 attenuates the pathogenesis of polycystic ovary syndrome. Mol. Cell. Endocrinol. 2020;506:110760. doi: 10.1016/j.mce.2020.110760. [DOI] [PubMed] [Google Scholar]
- 31.Hu W.L., Jin L., Xu A., Wang Y.F., Thorne R.F., Zhang X.D., Wu M. GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability. Nat. Cell Biol. 2018;20:492–502. doi: 10.1038/s41556-018-0066-7. [DOI] [PubMed] [Google Scholar]
- 32.Sang B., Zhang Y.Y., Guo S.T., Kong L.F., Cheng Q., Liu G.Z., Thorne R.F., Zhang X.D., Jin L., Wu M. Dual functions for OVAAL in initiation of RAF/MEK/ERK prosurvival signals and evasion of p27-mediated cellular senescence. Proc. Natl. Acad. Sci. U S A. 2018;115:E11661–E11670. doi: 10.1073/pnas.1805950115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang D., Zhang X., Zeng M., Yuan J., Liu M., Yin Y., Wu X., Keefe D.L., Liu L. Increased DNA damage and repair deficiency in granulosa cells are associated with ovarian aging in rhesus monkey. J. Assist. Reprod. Genet. 2015;32:1069–1078. doi: 10.1007/s10815-015-0483-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Titus S., Li F., Stobezki R., Akula K., Unsal E., Jeong K., Dickler M., Robson M., Moy F., Goswami S. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci. Transl. Med. 2013;5:172ra121. doi: 10.1126/scitranslmed.3004925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dou X., Guo T., Li G., Zhou L., Qin Y., Chen Z.J. Minichromosome maintenance complex component 8 mutations cause primary ovarian insufficiency. Fertil. Steril. 2016;106:1485–1489 e1482. doi: 10.1016/j.fertnstert.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 36.Guo T., Zhao S., Zhao S., Chen M., Li G., Jiao X., Wang Z., Zhao Y., Qin Y., Gao F. Mutations in MSH5 in primary ovarian insufficiency. Hum. Mol. Genet. 2017;26:1452–1457. doi: 10.1093/hmg/ddx044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg-Shukron A., Rachmiel M., Renbaum P., Gulsuner S., Walsh T., Lobel O., Dreifuss A., Ben-Moshe A., Zeligson S., Segel R. Essential role of BRCA2 in ovarian development and function. N. Engl. J. Med. 2018;379:1042–1049. doi: 10.1056/NEJMoa1800024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin Y., Zhang F., Chen Z.J. BRCA2 in ovarian development and function. N. Engl. J. Med. 2019;380:1086. doi: 10.1056/NEJMc1813800. [DOI] [PubMed] [Google Scholar]
- 39.Wang X., Zhang X., Dang Y., Li D., Lu G., Chan W.Y., Leung P.C.K., Zhao S., Qin Y., Chen Z.J. Long noncoding RNA HCP5 participates in premature ovarian insufficiency by transcriptionally regulating MSH5 and DNA damage repair via YB1. Nucleic Acids Res. 2020;48:4480–4491. doi: 10.1093/nar/gkaa127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X., Heyer W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.West S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 42.Ma J.Y., Feng X., Tian X.Y., Chen L.N., Fan X.Y., Guo L., Li S., Yin S., Luo S.M., Ou X.H. The repair of endo/exogenous DNA double-strand breaks and its effects on meiotic chromosome segregation in oocytes. Hum. Mol. Genet. 2019;28:3422–3430. doi: 10.1093/hmg/ddz156. [DOI] [PubMed] [Google Scholar]
- 43.Stringer J.M., Winship A., Zerafa N., Wakefield M., Hutt K. Oocytes can efficiently repair DNA double-strand breaks to restore genetic integrity and protect offspring health. Proc. Natl. Acad. Sci. U S A. 2020;117:11513–11522. doi: 10.1073/pnas.2001124117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo W., Guo T., Li G., Liu R., Zhao S., Song M., Zhang L., Wang S., Chen Z.J., Qin Y. Variants in homologous recombination genes EXO1 and RAD51 related with premature ovarian insufficiency. J. Clin. Endocrinol. Metab. 2020;105:dgaa505. doi: 10.1210/clinem/dgaa505. [DOI] [PubMed] [Google Scholar]
- 45.Gao F., Zhang J., Wang X., Yang J., Chen D., Huff V., Liu Y.X. Wt1 functions in ovarian follicle development by regulating granulosa cell differentiation. Hum. Mol. Genet. 2014;23:333–341. doi: 10.1093/hmg/ddt423. [DOI] [PubMed] [Google Scholar]
- 46.Chen M., Zhang L., Cui X., Lin X., Li Y., Wang Y., Wang Y., Qin Y., Chen D., Han C. Wt1 directs the lineage specification of sertoli and granulosa cells by repressing Sf1 expression. Development. 2017;144:44–53. doi: 10.1242/dev.144105. [DOI] [PubMed] [Google Scholar]
- 47.Tang J., Yan T., Bao Y., Shen C., Yu C., Zhu X., Tian X., Guo F., Liang Q., Liu Q. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-Myc. Nat. Commun. 2019;10:3499. doi: 10.1038/s41467-019-11447-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo H., Zhao L., Shi B., Bao J., Zheng D., Zhou B., Shi J. GALNT5 uaRNA promotes gastric cancer progression through its interaction with HSP90. Oncogene. 2018;37:4505–4517. doi: 10.1038/s41388-018-0266-4. [DOI] [PubMed] [Google Scholar]
- 49.Mortimer S.A., Kidwell M.A., Doudna J.A. Insights into RNA structure and function from genome-wide studies. Nat. Rev. Genet. 2014;15:469–479. doi: 10.1038/nrg3681. [DOI] [PubMed] [Google Scholar]
- 50.Bevilacqua P.C., Ritchey L.E., Su Z., Assmann S.M. Genome-Wide analysis of RNA secondary structure. Annu. Rev. Genet. 2016;50:235–266. doi: 10.1146/annurev-genet-120215-035034. [DOI] [PubMed] [Google Scholar]
- 51.Somarowthu S., Legiewicz M., Chillon I., Marcia M., Liu F., Pyle A.M. HOTAIR forms an intricate and modular secondary structure. Mol. Cell. 2015;58:353–361. doi: 10.1016/j.molcel.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai M.C., Manor O., Wan Y., Mosammaparast N., Wang J.K., Lan F., Shi Y., Segal E., Chang H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishi Y., Yanase T., Mu Y., Oba K., Ichino I., Saito M., Nomura M., Mukasa C., Okabe T., Goto K. Establishment and characterization of a steroidogenic human granulosa-like tumor cell line, KGN, that expresses functional follicle-stimulating hormone receptor. Endocrinology. 2001;142:437–445. doi: 10.1210/endo.142.1.7862. [DOI] [PubMed] [Google Scholar]
- 54.Yan H., Zhang J., Wen J., Wang Y., Niu W., Teng Z., Zhao T., Dai Y., Zhang Y., Wang C. CDC42 controls the activation of primordial follicles by regulating PI3K signaling in mouse oocytes. BMC Biol. 2018;16:73. doi: 10.1186/s12915-018-0541-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data reported in this study have been deposited in GEO: GSE158526 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158526) and GSE158555 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158555).