

Abstract
Rearrangements involving the neurotrophic receptor tyrosine kinase (NTRK) gene family have been reported in diverse tumor types, and NTRK‐targeted therapies have recently been approved. In this article, we report a case of a 26‐year‐old man with an NTRK2‐rearranged isocitrate dehydrogenase‐wild‐type glioblastoma who showed a robust but temporary response to the NTRK inhibitor larotrectinib. Rebiopsy after disease progression showed elimination of the NTRK2‐rearranged tumor cell clones, with secondary emergence of a PDGFRA‐amplified subclone. Retrospective examination of the initial biopsy material confirmed rare cells harboring PDGFRA amplification. Although mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma has been previously described, mosaicism involving a fusion gene driver event has not. This case highlights the potential efficacy of NTRK‐targeted treatment in glioblastoma and the implications of molecular heterogeneity in the setting of targeted therapy.
Key Points
This case highlights the efficacy of the NTRK inhibitor larotrectinib in treating NTRK‐rearranged glioblastoma.
This is the first case to demonstrate mosaicism in glioblastoma involving both a fusion gene and amplification for receptor tyrosine kinases.
Intratumoral heterogeneity in glioblastoma has significant implications for tumor resistance to targeted therapies.
Short abstract
This article reports a case of a 26‐year‐old man with an IDH‐wild type glioblastoma mosaic for both NTRK2 fusion and receptor tyrosine kinase amplification. He showed a robust but temporary response to the recently‐approved NTRK inhibitor larotrectinib with subsequent outgrowth of a PDGFRA‐amplified subclone.
Patient Story
A 26‐year‐old right‐handed man with no significant medical history presented with 2 weeks of progressive right upper and lower extremity weakness, loss of coordination, fatigue, and difficulty with expressive language. Physical examination revealed a flat affect, right upper extremity drift, and right lower extremity weakness. Magnetic resonance imaging (MRI) showed a heterogeneously enhancing 3.5‐cm mass lesion in the left frontal lobe, with T2 signal abnormality involving the left frontal white matter and extending across the corpus callosum into the right frontal white matter, as well as inferiorly along the left corticospinal tract and into the left cerebral peduncle (Fig. 1A).
Figure 1.

Radiologic and histopathologic findings on initial presentation and biopsy. (A): Postcontrast T1‐weighted magnetic resonance images show a contrast‐enhancing tumor centered in the left frontal lobe and involving the corpus callosum (white arrowheads). Top: axial; middle: coronal; bottom: sagittal. (B): H&E‐stained slides from the initial biopsy show a hypercellular, pleomorphic neoplasm with microvascular proliferation (top right; black arrowheads). By immunohistochemistry, the neoplastic cells are positive for OLIG2 (middle left) and negative for R132H‐mutant IDH (middle right; wild‐type pattern). Fluorescence in situ hybridization using green and red NTRK2‐specific break‐apart probes flanking the breakpoint (bottom row) shows split red and green signals (white brackets: split signals, white arrowheads: nonsplit signals), confirming NTRK translocation. Abbreviations: IDH, isocitrate dehydrogenase; NTRK, neurotrophic receptor tyrosine kinase.
A core biopsy of the lesion was performed. On histology, the tumor was densely cellular and composed of epithelioid cells with large, pleomorphic nuclei, with diffuse immunoreactivity for OLIG2 and focal immunoreactivity for GFAP (Fig. 1B). Microvascular proliferation was prominent. On the basis of these features, a diagnosis of glioblastoma was rendered. Subsequent testing showed that the tumor was isocitrate dehydrogenase (IDH) wild type, ATRX‐retained, and MGMT promoter unmethylated. No EGFR or MET amplifications were identified by fluorescence in situ hybridization. Molecular testing, performed on total nucleic acid extracted from formalin‐fixed paraffin‐embedded biopsy material in a Clinical Laboratory Improvement Amendments–certified laboratory at Massachusetts General Hospital, included a DNA‐based genotyping panel (SNaPshot next‐generation sequencing [NGS] v2.0) that targets 98 known oncogenes and tumor suppressors and detects single nucleotide variants, insertion/deletions, and copy numbers, as well as a separate RNA‐based NGS assay that detects rearrangements in 59 genes (Solid Fusion Assay v2.0) [1], revealed two variants in TP53 (ENST00000269305.4: c.375G>A, splice variant and c.524G>A, p.Arg175His), copy number loss of CDKN2A, and a gene fusion between KANK1 (exon 4) and NTRK2 (exon 16). NTRK2 rearrangement was confirmed by fluorescence in situ hybridization (FISH) using an NTRK2 break‐apart probe, defined as red‐green pair splits of more than two probe lengths in >15% of cells (Fig. 1B).
The patient received corticosteroids and levetiracetam for seizure prophylaxis in the postoperative period. He underwent subsequent bulk tumor resection with motor mapping, although part of the tumor was unresectable due to eloquent region involvement. On postoperative day 3, the patient was discharged to inpatient rehabilitation.
Following discussion at a multidisciplinary tumor board, the decision was made to treat with six weeks of temozolomide and concurrent radiation therapy (200 cGy per fraction, 30 total fractions), followed by treatment with the tropomyosin kinase (Trk) receptor inhibitor larotrectinib, given the presence of an NTRK2 rearrangement.
An MRI obtained after the end of chemotherapy/radiation and before starting larotrectinib showed residual enhancement of the remaining tumor without evidence of significant disease progression. Shortly thereafter, the patient began larotrectinib (100 mg b.i.d; Fig. 2). On day 49 of larotrectinib treatment, repeat MRI showed evidence of a significant treatment response, with the residual enhancing mass lesion, previously 25 mm in greatest extent, measuring less than 10 mm. These findings correlated with slight overall improvement in the patient's motor weakness on physical exam.
Figure 2.
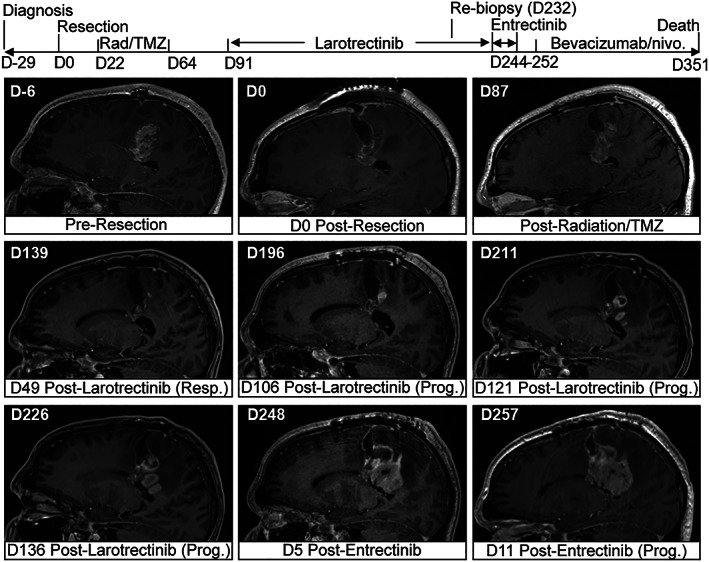
Clinical course demonstrating initial clinical response to larotrectinib. Top: clinical timeline and treatment course. Bottom: postcontrast T1‐weighted sagittal magnetic resonance imaging views at indicated days pre‐ or postsurgical resection. D indicates day number with respect to date of initial resection. Abbreviations: nivo., nivolumab; rad, radiation therapy; resp., response; prog., progression; TMZ, temozolomide.
Unfortunately, the patient's next MRI (performed on day 106 of larotrectinib treatment) showed interval enlargement of an enhancing focus along the resection cavity suggestive of disease progression. He also experienced additional seizures during this time. Rebiopsy of the enhancing lesion was performed, confirming recurrent/residual glioblastoma in the resection cavity. While the results of molecular testing on the new sample were pending, the patient was started on a different Trk inhibitor, entrectinib, approved for solid tumors with neurotrophic receptor tyrosine kinase (NTRK) fusions only a few weeks prior, due to its superior central nervous system penetration. Unexpectedly, the previously identified KANK1‐NTRK2 fusion was no longer identified by the Solid Fusion Assay; however, new amplifications in platelet‐derived growth factor receptor alpha (PDGFRA) and MYCN were seen on SNaPshot NGS, in addition to persistence of the previously identified CDKN2A copy number loss, and two TP53 variants. PDGFRA amplification was confirmed by FISH (Fig. 3). Entrectinib treatment was stopped, and the patient subsequently began treatment with combination bevacizumab and nivolumab.
Figure 3.

Disease progression with emergence of a PDGFRA‐amplified subclone. Top left: postcontrast T1‐weighted MRI showing increase in tumor size after initial response to larotrectinib. Top right: H&E stain on rebiopsied tissue shows a pleomorphic infiltrative neoplasm consistent with recurrent/residual glioblastoma. Bottom row: NTRK fluorescence in situ hybridization shows a non‐rearranged pattern (left), whereas numerous tumor cells show evidence for PDGFRA amplification (right) (red: PDGFRA‐specific probe; green: centromere control probe). Abbreviations: NTRK, neurotrophic receptor tyrosine kinase; PDGFRA, platelet‐derived growth factor receptor alpha.
In light of these findings, PDGFRA FISH was performed on the patient's original biopsy specimen. Rare nuclei (approximately 1 out of 100 cells) were identified within the 0.6 cm2 of tissue evaluated that harbored PDGFRA amplification and were mutually exclusive with abundant intermixed NTRK2‐rearranged tumor cells (Fig. 4).
Figure 4.

Presence of rare PDGFRA‐amplified tumor cells in initial biopsy material. Analysis of the initial biopsy confirms mutually exclusive rare tumor cells with PDGFRA amplification (bottom, middle) among more numerous NTRK2‐rearranged tumor cells (bottom left, right). White signal: PDGFRA‐specific probe; green and red signals: NTRK2 break‐apart probes. Abbreviations: NTRK, neurotrophic receptor tyrosine kinase; PDGFRA, platelet‐derived growth factor receptor alpha.
Molecular Tumor Board
Functional and Clinical Significance of NTRK Fusions and PDGFRa Amplification in Glioblastoma
The neurotrophic receptor tyrosine kinase gene family consists of NTRK1, NTRK2, and NTRK3, which encode the tropomyosin receptor kinase proteins TrkA, TrkB, and TrkC, respectively [2]. These serve as receptors for nerve growth factor, brain‐derived neurotrophic factor, and neurotrophins 3 and 4, and their signaling is involved in the development and homeostasis of the embryonic and adult nervous system [3, 4]. NTRK fusions, in which the carboxy‐terminal kinase domain of NTRK1, ‐2, or ‐3 is joined to the amino‐terminal of a partner gene, result in constitutive kinase activity, leading to continuous autophosphorylation of intracellular tyrosine residues and activation of downstream MAP kinase, PI3 kinase, and protein kinase C pathways. As such, NTRK fusions have been identified as driver mutations in diverse cancer types, including gliomas [5, 6, 7], and two NTRK‐targeted therapies, larotrectinib and entrectinib, have recently gained tumor type‐agnostic U.S. Food and Drug Administration (FDA) approvals.
The prevalence of recurrent gene fusion events in glioblastoma is estimated at 10%–15% [8] and may be more common in IDH‐wild‐type tumors [9]. NTRK fusions account for only a small minority of the total, with a prevalence in the adult population estimated at approximately 1%–2% [8]. Although some clinicopathologic features of NTRK‐rearranged gliomas have been described [10], the prognostic significance of NTRK fusions in glioblastoma is unclear, in part because of the scarcity of these cases to date.
In contrast to NTRK fusion events, amplification of receptor tyrosine kinases (RTKs), including EGFR, KIT, VEGFR2, PDGFRA, and MET, are seen in up to 50% of glioblastomas [11, 12, 13]. The most commonly amplified RTK gene is EGFR followed by PDGFRA [11, 12, 14]. Glioblastoma is characterized by significant heterogeneity, both within and across tumors. Several studies using single‐cell genomic and clonal analyses have documented intratumoral heterogeneity at the cellular and molecular level, with many patients displaying different glioblastoma subtypes within the same tumor [15, 16, 17, 18]. Mosaic amplification of multiple RTK genes within the same tumor in glioblastoma has been previously described in approximately 5% of cases [14]. This mosaicism was seen in adjacent intermingled cells with up to three different RTKs (EGFR, MET, PDGFRA) amplified in different cells in a mutually exclusive manner. The ratios of different amplified subclones varied between those having a dominant population and others showing evenly distributed subclones. To our knowledge, however, mosaicism involving gene fusions has not been previously described in glioblastoma. Our case underscores the diversity of molecular alterations that contribute to intratumoral heterogeneity, with significant implications for disease course and response to therapy.
Interpretation of the Molecular Results
The robust initial response to larotrectinib over several months, followed by disease progression and absence of an identifiable NTRK2 fusion on subsequent biopsy, suggests efficacy of targeting the tumor subclones with NTRK2 fusions. The predominance of PDGFRA‐amplified tumor cells in the follow‐up biopsy suggests that elimination of the NTRK2‐fusion‐harboring cells allowed the proliferation of the untargeted subclone. This case underscores the clinical importance of identifying tumor heterogeneity at initial diagnosis in predicting the response to targeted therapies and subsequent resistance.
Potential Strategies to Target the Pathway and Implications for Clinical Practice
Two small molecule Trk kinase domain inhibitors, larotrectinib and entrectinib, have recently gained FDA approval for solid tumors with NTRK fusions [19, 20, 21]. Larotrectinib is a specific inhibitor of TrkA, TrkB, and TrkC, whereas entrectinib shows additional activity against ALK and ROS1. Entrectinib in particular has been reported to show marked antitumor activity in metastatic and primary intracranial malignancies [22, 23]. Both drugs act through competitive inhibition of ATP binding within the Trk kinase domains. These approvals stemmed from the marked and durable response rates seen in two major basket trials that enrolled patients with NTRK fusions regardless of tumor histology or fusion partner, showing an overall response of 75% [20, 21]. In all nine patients who progressed after objective response or stable disease and underwent repeat testing, mutation of the Trk kinase domain was identified as the causative mechanism underlying acquired resistance to larotrectinib [21].
Few cases of NTRK inhibitor treatment for NTRK‐rearranged glioblastoma have been reported. Schram and colleagues noted a partial response to larotrectinib in a patient with multifocal glioblastoma harboring an EML4‐NTRK3 fusion in one disease focus, which had progressed from a previously treated IDH‐mutant anaplastic astrocytoma (World Health Organization grade III) [24]. In this case, disease progression of the non‐NTRK‐rearranged tumor subclones led to treatment discontinuation after several weeks. Retrospective whole exome sequencing of both the original tumor and three spatially distinct areas of the recurrent tumor revealed truncal mutations in all four specimens evaluated (IDH1, ATRX, and TP53) with the following alterations showing distinct regional differences: CDKN2A/B loss, temozolomide‐induced hypermutation, and PDGFRA amplification, although these alterations were not assessed on a single‐cell level, and rare cell populations may not be discernible by bulk sequencing methodologies. Per their report, two other patients with NTRK‐rearranged glioblastomas (BCR‐NTRK2, AFAP1‐NTRK1) who were treated with larotrectinib showed evidence of treatment response and continued on treatment for at least 4 months [24]. Buerki et al. reported a case of a 2‐month‐old girl with infantile glioblastoma harboring an in‐frame TPM3‐NTRK1 fusion who had been managed with larotrectinib for 20 months at the time of report, with evidence of tumor shrinkage [25]. Alharbi et al. also reported a case of 18‐month‐old with infantile glioblastoma that was found to harbor an ETV6‐NTRK3 fusion and showed significant tumor regression 8 weeks following larotrectinib treatment [26]. One patient with a glioneuronal tumor harboring a BCAN‐NTRK1 fusion showed a temporary clinical and radiologic response to entrectinib lasting 11 months before disease progression and treatment discontinuation [23].
PDGFRA‐specific inhibitors are in use for the treatment of a variety of tumors, including prostatic adenocarcinoma, neuroblastoma, meningioma, and certain sarcomas and hematopoietic neoplasms [27]; however, little is known about their efficacy in glioblastoma. One clinical trial investigating PDGFRA inhibition in glioblastoma has been completed, with results not yet reported (NCT00895180), and one is currently underway (NCT02626364). Imatinib mesylate, a nonspecific tyrosine kinase inhibitor with activity toward PDGFR, has shown overall limited success in phase II and III glioblastoma trials [28, 29, 30, 31, 32]. Nonetheless, although our patient did not ultimately receive PDGFR‐targeted treatment, identifying multiple driver mutations on initial tissue diagnosis would be useful in designing multitargeted drug combinations to prevent clonal selection.
Patient Update
The patient's recurrent disease was unfortunately aggressive and refractory to further treatment. After several weeks, he elected to pursue comfort measures and died 380 days after initial diagnosis.
Disclosures
Tyler E. Miller: Telomere Diagnostics, Inc. (C/A); Valentina Nardi: Loxo Oncology (SAB); John Iafrate: Invitae (OI, IP), Kinnate Therapeutics, Invitae, Oncoclinicas (SAB); Lauren L. Ritterhouse: Loxo Oncology (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Zheng Z, Liebers M, Zhelyazkova B et al. Anchored multiplex PCR for targeted next‐generation sequencing. Nat Med 2014;20:1479–1484. [DOI] [PubMed] [Google Scholar]
- 2. Cocco E, Scaltriti M, Drilon A. NTRK fusion‐positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018;15:731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mitre M, Mariga A, Chao MV. Neurotrophin signalling: Novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 2016;131:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bothwell M. Recent advances in understanding neurotrophin signaling. F1000Res 2016;5:F1000. [DOI] [PMC free article] [PubMed]
- 5. Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov 2015;5:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frattini V, Trifonov V, Chan JM et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 2013;45:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Solomon JP, Linkov I, Rosado A et al. NTRK fusion detection across multiple assays and 33,997 cases: Diagnostic implications and pitfalls. Modern Pathol 2020;33:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu T, Wang H, Huang X et al. Gene fusion in malignant glioma: An emerging target for next‐generation personalized treatment. Transl Oncol 2018;11:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferguson SD, Zhou S, Huse JT et al. Targetable gene fusions associate with the IDH wild‐type astrocytic lineage in adult gliomas. J Neuropathol Exp Neurol 2018;77:437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Torre M, Vasudevaraja V, Serrano J et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathologica Commun 2020;8:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Joensuu H, Puputti M, Sihto H et al. Amplification of genes encoding KIT, PDGFRα and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J Pathol 2005;207:224–231. [DOI] [PubMed] [Google Scholar]
- 12. Puputti M, Tynninen O, Sihto H et al. Amplification of KIT, PDGFRA, VEGFR2, and EGFR in gliomas. Mol Cancer Res 2006;4:927–934. [DOI] [PubMed] [Google Scholar]
- 13. Wullich B, Müller HW, Fischer U et al. Amplified met gene linked to double minutes in human glioblastoma. Eur J Cancer 1993;29:1991–1995. [DOI] [PubMed] [Google Scholar]
- 14. Snuderl M, Fazlollahi L, Le LP et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011;20:810–817. [DOI] [PubMed] [Google Scholar]
- 15. Patel AP, Tirosh I, Trombetta JJ et al. Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344:1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer M, Reimand J, Lan X et al. Single cell‐derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc National Acad Sci USA 2015;112:851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sottoriva A, Spiteri I, Piccirillo SGM et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc National Acad Sci USA 2013;110:4009–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Szerlip NJ, Pedraza A, Chakravarty D et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc National Acad Sci USA 2012;109:3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lange AM, Lo HW. Inhibiting TRK proteins in clinical cancer therapy. Cancers (Basel) 2018;10:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drilon A, Siena S, Ou SHI et al. Safety and antitumor activity of the multi‐targeted pan‐TRK, ROS1, and ALK inhibitor entrectinib (RXDX‐101): Combined results from two phase 1 trials (ALKA‐372‐001 and STARTRK‐1). Cancer Discov 2017;7:400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Drilon A, Laetsch TW, Kummar S et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. New Engl J Med 2018;378:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Farago AF, Le LP, Zheng Z et al. Durable clinical response to entrectinib in NTRK1‐rearranged non‐small cell lung cancer. J Thorac Oncol 2015;10:1670–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alvarez‐Breckenridge C, Miller JJ, Nayyar N et al. Clinical and radiographic response following targeting of BCAN‐NTRK1 fusion in glioneuronal tumor. NPJ Precis Oncol 2017;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schram AM, Taylor BS, Hechtman JF et al. Potential role of larotrectinib (LOXO‐101), a selective pan‐TRK inhibitor, in NTRK fusion‐positive recurrent glioblastoma. 2017;77(suppl):LB‐302a. [Google Scholar]
- 25. Buerki R, Banerjee A, Zamorski A et al. HGG‐15. Successful treatment of an NTRK‐fusion positive infantile glioblastoma with larotrectinib, a targeted TRK inhibitor. Neuro Oncol 2019;21(suppl 2):ii89–ii90. [Google Scholar]
- 26. Alharbi M, Mobark NA, Balbaid AAO et al. Regression of ETV6‐NTRK3 infantile glioblastoma after first‐line treatment with larotrectinib. JCO Precis Oncol 2020;4:796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal 2013;11:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wen PY, Yung WKA, Lamborn KR et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99‐08. Clin Cancer Res 2006;12:4899–4907. [DOI] [PubMed] [Google Scholar]
- 29. Raymond E, Brandes AA, Dittrich C et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: A European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J Clin Oncol 2008;26:4659–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reardon DA, Dresemann G, Taillibert S et al. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br J Cancer 2009;101:1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dresemann G, Weller M, Rosenthal MA et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol 2010;96:393–402. [DOI] [PubMed] [Google Scholar]
- 32. Dresemann G. Imatinib and hydroxyurea in pretreated progressive glioblastoma multiforme: A patient series. Ann Oncol 2005;16:1702–1708. [DOI] [PubMed] [Google Scholar]