

Abstract
Patients with familial arrhythmogenic cardiomyopathy typically present with ventricular arrhythmias or progressive heart failure. This paper characterizes a rare presentation of an underlying genetic cardiomyopathy with clinical manifestations mimicking an acute myocardial infarction in 2 siblings, each with the same mutation in the desmoplakin (DSP) gene. (Level of Difficulty: Advanced.)
Key Words: ACM, genetic cardiomyopathy, heart failure, myocardial injury, sudden cardiac death, troponin I
Abbreviations and Acronyms: ACM, arrhythmogenic cardiomyopathy; CMR, cardiac magnetic resonance; CP, chest pain; DSP, desmoplakin; ECG, electrocardiogram; LGE, late gadolinium enhancement; LV, left ventricle; RV, right ventricle; PVC, premature ventricular contraction; TnI, troponin I
Central Illustration
Chest pain (CP), ischemic electrocardiographic (ECG) changes, and troponin concentration elevation are hallmarks of an acute myocardial infarction. This case report describes 2 siblings with this typical presentation and angiographically normal coronary arteries. In both patients, a biventricular cardiomyopathy was identified in association with a heterozygous missense mutation in the desmoplakin (DSP) gene, implicated in arrhythmogenic cardiomyopathy (ACM) (1, 2).
Learning Objectives
-
•
To understand that ACM patients with a DSP mutation may exhibit stepwise disease progression marked by episodes of acute myocardial injury with chest pain, troponin I elevation, and ECG changes.
-
•
To recognize that familial ACM should be considered in the differential diagnosis of myocardial infarction with nonobstructive coronary arteries, myocarditis, and unspecified biventricular cardiomyopathy with troponin I elevation.
-
•
To understand that cardiac magnetic resonance imaging can help further characterize myocardial injury and findings that may be related to a progressive familial cardiomyopathy, including ACM.
Patients with ACM typically present with ventricular arrhythmias, sudden cardiac death, and/or heart failure (1, 2). Diagnostic evaluation is guided by the 2010 ARVC/D Task Force Criteria and includes genetic testing, depolarization and repolarization electrocardiographic abnormalities, arrhythmias, histological findings, and cardiac imaging (3).
The patients discussed here shared features consistent with the recognized expansion of arrhythmogenic right ventricular cardiomyopathy in the medical literature to include left-dominant, biventricular, and cardiocutaneous variants (2,4). It has been described that disease progression could occur stepwise during episodes of acute myocardial injury detected by cardiac magnetic resonance (CMR) imaging. Thus, ACM should be included in the differential diagnosis of patients with suspected acute myocarditis or myocardial infarction with nonobstructive coronary arteries.
Cases
Patient 1
Patient 1 was an asymptomatic white female who presented at age 20 after a psychosocial stressor with acute CP, anterolateral ST-segment elevation (Supplemental Figure 1), and elevated troponin I (TnI) (276 ng/ml: normal: <0.034 ng/ml) (Figure 1A). Cardiac catheterization and echocardiography demonstrated normal coronary arteries, anterolateral left ventricular (LV) hypokinesis, and decreased LV ejection fraction (EF) 35%. Coronary vasospasm was initially suspected, and the patient was prescribed beta-blocker therapy for cardiomyopathy. LVEF improved to 45% after 3 months. Twenty-four-hour Holter monitoring recorded 8,663 premature ventricular contractions (PVC) (Table 1).
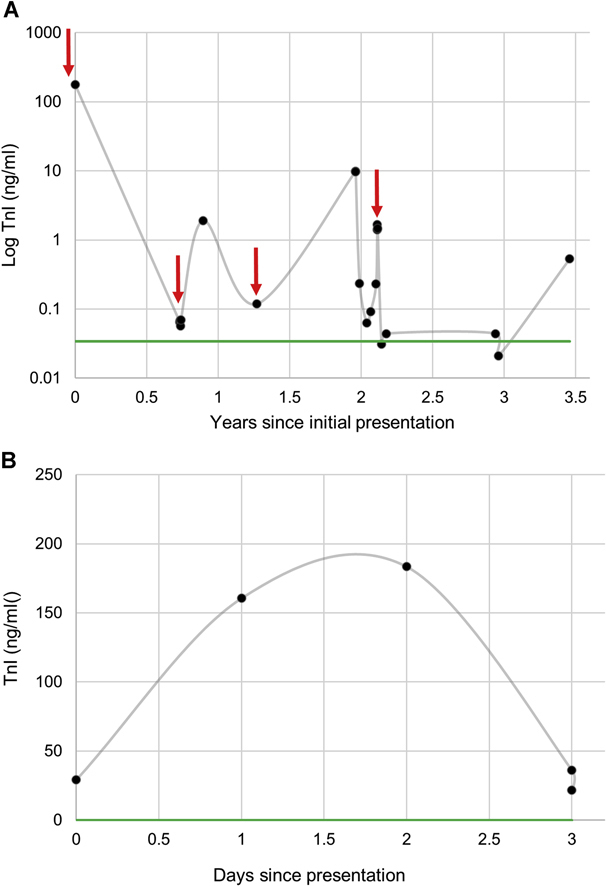
Figure 1.

Serial TnI Measurements in Patients 1 and 2
(A) Patient 1 had serial troponin I (TnI) levels drawn during follow-up visits and during acute episodes of chest pain (red arrows). Her baseline TnI level was elevated above normal (green line). Patient 2 had normal TnI baseline measurements. His serial TnI results from his hospitalization for chest pain (B) are shown to peak and decline spontaneously.
Table 1.
Demographic Information, Genotype, and Phenotype of Patients 1 and 2
| Patient 1 | Patient 2 | |
|---|---|---|
| Sex | Female | Male |
| Ancestry | European | European |
| Age (y) at initial presentation | 20 | 19 |
| Presenting symptom | Acute chest pain | Acute chest pain |
| Genotype | DSP (p.Thr564Ile) | DSP (p.Thr564Ile) |
| Cardiac phenotype | Biventricular cardiomyopathy (left-dominant) | Biventricular cardiomyopathy (left-dominant) |
| Cutaneous phenotype | Wooly hair, mild palmoplantar keratoderma | Wooly hair; hyperkeratotic palmar lesions |
| Peak TnI | 276 ng/ml | 184 ng/ml |
| Initial CMR | ||
| LV EF%/RV%, EF | 36/normal | 43/39 |
| LV EDV/ESV, ml/m2 | 113/61 | 116/67 |
| RV EDV/ESV, ml/m2 | Normal | 122/74 |
| Wall motion abnormalities | Regional LV hypokinesia/akinesia | Global RV hypokinesis |
| Follow-up ECG | Incomplete RBBB; nonspecific T-wave abnormality | Nonspecific T-wave abnormality |
| Ventricular arrhythmias | PVC (8,663/24 h) | NSVT PVC (1,149/24 h) |
Similarities between the clinical presentations of the 2 siblings, as well as key differences, are listed. CMR imaging volumes are presented as normalized to BSA, and all values were above the normal range except for RV EDV in Patient 2. Ventricular arrhythmias reported were identified on Holter monitoring.
BSA = body surface area; CMR = cardiac magnetic resonance; DSP = desmoplakin gene; EDV = end diastolic volume; ECG = electrocardiography; ESV = end systolic volume; LV = left ventricle; NSVT = non-sustained ventricular tachycardia; RBBB = right bundle branch block; RV = right ventricle; PVC = pre-mature ventricular contraction; TnI = troponin I.
Eight months later, CMR demonstrated anterolateral to inferolateral LV epicardial and mid-myocardial late gadolinium enhancement (LGE), anterolateral hypokinesis, and right ventricle (RV) wall thinning (Figure 2). Her RV endomyocardial biopsy was unremarkable.
Figure 2.

Cardiac Magnetic Resonance With Late Gadolinium Enhancement of Patients 1 and 2
Cardiac magnetic resonance 4-chamber (A and C) and left ventricular (LV) outflow track (B and D) views of Patient 1 (A and B) and Patient 2 (C and D) demonstrate extensive gadolinium uptake in the epicardium to mid-myocardium with sparing of the endocardium. This is consistent with extensive fibrotic scarring indicative of a biventricular arrhythmogenic cardiomyopathy. RV = right ventricle.
One year after her index event, Patient 1 had elevated C-reactive protein (CRP) (37.84 mg/dL) and TnI with LVEF of 56%. Follow-up CMR demonstrated progressive LV LGE in a nonischemic pattern and RV enlargement, basilar free wall hypokinesis and akinesis, small aneurysm formation, and dysfunction (RVEF 39%).
The patient’s deceased mother had a history of “viral” cardiomyopathy requiring heart transplantation at age 22 years, heightening suspicion for a familial cardiomyopathy. A comprehensive cardiomyopathy genetic screening of 76 genes (GeneDx) revealed a likely pathogenic heterozygous missense DSP mutation (p.Thr564Ile, c.1691 C>T), a variant previously identified in a patient with Carvajal syndrome, a cardiocutaneous genetic condition (5). Notably, Patient 1 had wooly hair and mild palmoplantar keratoderma. Genetic testing was not performed on the patient’s mother, although her hair was described as similar in texture, and her father was negative for the mutation. She had no other family history of sudden cardiac death or cardiomyopathy.
The patient met diagnostic criteria for ACM, and a primary prevention implantable cardioverter defibrillator was placed but later explanted due to infection. In 3 years, she experienced 3 additional episodes of CP with elevated TnI (Figure 1A) and CRP concentrations.
Patient 2
Patient 2, the asymptomatic 18-year-old brother of Patient 1, was evaluated for the familial cardiomyopathy. His physical examination was notable for wooly hair and palmar hyperkeratosis. CMR demonstrated extensive subepicardial biventricular LGE and decreased biventricular function (LVEF: 43%; RVEF: 39%). Holter monitoring revealed nonsustained ventricular tachycardia and a PVC burden of 1,149/24 h (Table 1). TnI concentration was normal (<0.01 ng/mL). Targeted genetic testing identified the heterozygous DSP mutation carried by his sister. He was initiated on beta-blocker therapy and an angiotensin-converting enzyme inhibitor.
Four months later, at age 19, Patient 2 presented with acute CP after physical exertion with anterolateral ST-segment elevation and TnI elevation (peak: 184 ng/mL) (Figures 1B and 3). Emergent coronary angiography demonstrated normal coronary arteries without vasospasm, and CMR showed progressive biventricular apical and anterolateral LGE. New regional myocardial wall motion abnormalities corresponded with ECG changes (Figures 2 and 3). Viral serology analysis results were negative, except for Epstein-Barr antibodies (immunoglobulin M [IgM] and IgG). His CP and ST-segment elevation resolved spontaneously, and TnI decreased within 2 days to 21.95 ng/mL (Figures 1B and 3). A primary prevention implantable cardioverter-defibrillator was implanted for ACM with extensive LV involvement, and prophylactic anticoagulation therapy was initiated, given the potential for LV thrombus formation with a new apical akinesis.
Figure 3.

Electrocardiograms of Patient 2 Before, During, and After Initial Presentation
Patient 2’s admission electrocardiogram (B) showed ST-segment elevation in the anterolateral leads (I and aVL) and across the precordium (V2 to V6) with reciprocal inferior lead changes in the setting of chest pain and elevated troponin I. (A) A prior electrocardiogram and one from 24 hours after his admission (C) are included for comparison.
Six months after the index event and weeks after anticoagulation was discontinued, echocardiography revealed an LV thrombus and LVEF of 42%. CMR demonstrated subepicardial and mid-myocardial LV LGE and decreased biventricular function (LVEF: 44%, RVEF: 42%). He experienced no additional episodes of CP, and TnI remained normal.
Discussion
Two siblings with biventricular cardiomyopathy and an associated DSP mutation presented with signs and symptoms mimicking acute myocardial infarction. Prior to their initial presentations, both siblings were asymptomatic. Emergent coronary angiography showed no identifiable thromboembolic or vasospastic epicardial coronary occlusion to explain ongoing anterolateral ST-segment elevation and marked TnI elevation. Clinical evaluation was also not consistent with Takotsubo cardiomyopathy or viral myocarditis. CMR in Patient 2 noted new regional anteroapical akinesis but no LV thrombus. The striking degree of ST-segment elevation and TnI release and spontaneous resolution were, thus, likely indicative of primary myocardial injury.
Serial CMR demonstrated a progressive familial biventricular cardiomyopathy in both siblings. The pathophysiology of myocyte death and fibrofatty infiltration in ACM is incompletely understood. It has been proposed that the disease progresses in “hot phases” representative of acute necrosis rather than gradual apoptosis (1,4,6, 7, 8). This is consistent with the injury sequence observed in these 2 patients. Further research is needed to better understand triggers for myocardial injury in ACM.
Viral myocarditis has been proposed as a stimulus for acute myocyte death in ACM (4,8,9). In these patients, no recent infections or clear viral prodrome were identified. Given the comparable presentations in 2 related individuals with the same DSP mutation at a similar age, viral myocarditis as the trigger for myocardial injury was thought to be unlikely. This does not exclude myocarditis related to a noninfectious inflammatory process, which has been previously observed in ACM (8,9). Overall, these cases are most consistent with other rare reports of DSP mutation-associated cardiomyopathies presenting with acute myocardial infarction-like symptoms without coronary artery disease (4,7, 8, 9, 10).
Conclusions
Paroxysmal TnI elevation and CP in some ACM patients with a DSP mutation suggest the disease may progress stepwise through episodes of acute myocardial injury. ACM should be considered in the differential diagnosis of myocardial infarction or myocarditis in the presence of significant TnI elevation. Additionally, patients who do not initially meet diagnostic criteria for ACM may still benefit from clinical surveillance, particularly with CMR, which is uniquely capable of characterizing myocardial injury.
Funding Support and Author Disclosures
Supported by the Minneapolis Heart Institute Foundation and Medtronic, Inc. The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
Appendix
For a supplemental figure, please see the online version of this paper.
Appendix
References
- 1.Corrado D., Link M.S., Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376:61–72. doi: 10.1056/NEJMra1509267. [DOI] [PubMed] [Google Scholar]
- 2.Sen-Chowdhry S., McKenna W.J. When rare illuminates common: how cardiocutaneous syndromes transformed our perspective on arrhythmogenic cardiomyopathy. Cell Adhes Commun. 2014;21:3–11. doi: 10.3109/15419061.2013.876415. [DOI] [PubMed] [Google Scholar]
- 3.Marcus F.I., McKenna W.J., Sherrill D. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sen-Chowdhry S., Syrris P., Prasad S.K. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 5.Boulé S., Fressart V., Laux D. Expanding the phenotype associated with a desmoplakin dominant mutation: Carvajal/Naxos syndrome associated with leukonychia and oligodontia. Int J Cardiol. 2012;161:50–52. doi: 10.1016/j.ijcard.2012.06.068. [DOI] [PubMed] [Google Scholar]
- 6.Patrianakos A.P., Protonotarios N., Nyktari E. Arrhythmogenic right ventricular cardiomyopathy/dysplasia and troponin release. Myocarditis or the “hot phase” of the disease? Int J Cardiol. 2012;157:e26–e28. doi: 10.1016/j.ijcard.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Smith E., Lakdawala N.K., Papoutsidakis N. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation. 2020;141:1872–1884. doi: 10.1161/CIRCULATIONAHA.119.044934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bariani R., Cipriani A., Rizzo S. “Hot phase” clinical presentation in arrhythmogenic cardiomyopathy. EP Europace. 2020:euaa343. doi: 10.1093/europace/euaa343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopez-Ayala J.M., Pastor-Quirante F., Gonzalez-Carrillo J. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2015;12:766–773. doi: 10.1016/j.hrthm.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Bauce B., Basso C., Rampazzo A. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005;26:1666–1675. doi: 10.1093/eurheartj/ehi341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.