

SUMMARY
Immunoglobulin heavy chain (IgH) locus-associated G-rich long noncoding RNA (SμGLT) is important for physiological and pathological B cell DNA recombination. We demonstrate that the METTL3 enzyme-catalyzed N6-methyladenosine (m6A) RNA modification drives recognition and 3’-end processing of SμGLT by the RNA exosome, promoting class switch recombination (CSR) and suppressing chromosomal translocations. The recognition is driven by interaction of the MPP6 adaptor protein with nuclear m6A reader YTHDC1. MPP6 and YTHDC1 promote CSR by recruiting AID and the RNA exosome to actively transcribing SμGLT. Direct suppression of m6A modification of SμGLT or of m6A reader YTHDC1 reduces CSR. Moreover, METTL3, an essential gene for B cell development in the bone marrow and germinal center, suppresses IgHassociated aberrant DNA breaks and prevents genomic instability. Taken together, we propose coordinated and central roles for MPP6, m6A modification, and m6A reader proteins in controlling long noncoding RNA processing, DNA recombination and development in B cells.
eTOC BLURB
B-lymphocytes generate vast number of antibodies to neutralize pathogenic insults. Somatic mutations of antibody genes are important for antibody diversity but also causes lymphomagenesis. Here, the authors demonstrate that N6-methyladenosine modification of the SμGLT long-noncoding RNA expressed from antibody gene locus drives somatic mutations but suppresses lymphomagenic genomic alterations.
Graphical Abstract
INTRODUCTION
Generation of diversity in the immune system requires ncRNA transcription and metabolism, particularly in B cells (Ansel, 2013; Nair et al., 2020). Two mechanisms employed to generate diversity are V(D)J recombination and class switch recombination (CSR). V(D)J recombination is the selection of gene segments of the V(D)J exons that together express the antigen-binding pocket of the antibody molecule, while CSR determines the C-terminal constant region of the antibody molecule for optimal antibody function (Keim et al., 2013; Schatz and Ji, 2011; Shang and Meng, 2021). Both these noncoding RNA transcription dependent DNA recombination processes provide opportunities to examine RNA processing mechanism(s) of ncRNAs and associated regulatory events. Our laboratory has demonstrated previously that ncRNAs expressed in the immunoglobulin switch regions, known as IgH switch region transcripts (IgS transcripts), are robustly targeted for degradation by the cellular 3’−5’ RNA exonuclease complex known as the RNA exosome. As the RNA exosome processes nascently transcribed IgS transcripts, it binds and recruits the DNA cytidine deamination enzyme Activation-Induced cytidine Deaminase (AID) to catalyze DNA single-strand nicks on both strands of the DNA (Basu et al., 2011; Feng et al., 2020). These closely placed DNA nicks cause DNA double-strand breaks that ultimately are necessary for CSR. On its own, the RNA exosome complex lacks the ability to unwind RNA/RNA hybrids or RNA/DNA hybrids that accumulate during the transcription of IgS regions. Previous studies have identified the RNA helicases MTR4 and SETX as the factors responsible for unwinding these RNA/RNA and RNA/DNA hybrids (Kazadi et al., 2020; Lim et al., 2017), and it is likely other RNA helicases contribute as well. Once unwound, the IgS transcripts are immediate targets for RNA exosome-mediated degradation. Cryo-EM microscopy studies biophysically demonstrated that the complexation of the MTR4 protein with the RNA exosome promotes the decay of the RNA present in a DNA/RNA hybrid configuration through the exosome complex’s 3’ end processing activity. These observations have vast implications outside of IgH recombination biology (Lim et al., 2017; Weick et al., 2018). Studies have pointed out that RNA/DNA hybrids accumulating at various promoters, enhancers, and gene bodies can cause somatic DNA mutagenesis that could be oncogenic (Lim et al., 2017; Pefanis et al., 2014). However, these mutations are avoided by rapid RNA decay mediated by MTR4 and the RNA exosome complex (Lim et al., 2017; Pefanis et al., 2014).
How specific ncRNAs are identified by the RNA exosome complex remains unanswered. Association of nascently expressed RNA with RNA binding proteins (like splicing factors) and/or the formation of secondary RNA structures may tag the ncRNAs either for preservation or degradation. Here we interrogate the role of the N6-methyladenosine (m6A) RNA modification in the recruitment of the RNA exosome complex. We provide evidence that the RNA exosome cofactor MPP6 and the nuclear m6A-modified RNA reader YTHDC1 function together in B cells to recruit the RNA exosome complex to the IgH switch region germline transcript (SμGLT long noncoding RNA) to promote processing and programmed DNA recombination. We also emphasize the biological impact of the collaboration of m6A modification with RNA exosome-mediated RNA processing in (a) the catalysis of antibody gene diversity and (b) the protection of genomic integrity. Although the presence of m6A RNA modifying enzymes in biology is firmly established (Meyer and Jaffrey, 2017; Schwartz, 2016; Yue et al., 2015), evidence that the m6A modification actually plays a direct role in biology is debated. Here, we provide comprehensive evidence of the involvement of m6A modification and its reader YTHDC1 in catalyzing important biochemical and biological processes in the nucleus of B cells.
RESULTS
MPP6 interacts with the RNA exosome complex and facilitates IgH class switch recombination
We aimed to determine what parameters drive the co-localization of the RNA exosome complex, its helicase cofactor MTR4 (Lim et al., 2017; Lubas et al., 2011; Puno and Lima, 2018; Weick et al., 2018), and AID at IgH germline transcripts. Based on RNA exosome complex purification experiments from primary B cells, we previously reported MPP6 as an interacting protein of the RNA exosome/AID complex in B cells using mass spectrometry (Lim et al., 2017), a finding initially reported in Sacchromyces cerevisiae and mammalian cells (Falk et al., 2017; Wasmuth et al., 2017). MPP6 recruits the NEXT (Nuclear EXosome Targeting) complex to the RNA exosome, allowing the MTR4 helicase component of the NEXT complex to unwind ncRNAs from RNA/DNA or RNA/RNA hybrid configurations for exosome-mediated decay (Falk et al., 2017; Lim et al., 2017; Schilders et al., 2005; Wasmuth et al., 2017; Weick et al., 2018). To determine whether MPP6 is responsible for nucleation of the RNA exosome complex with AID during CSR, we generated Mpp6 mutant (MPP6mut, Fig. S1A) CH12F3 B cells and evaluated CSR to IgA with and without cytokine stimulation. MPP6mut cells had a clear defect in CSR (Fig. 1A, B) with detectable but not major changes in proliferation (Fig. 1C) and did show a loss of expression of complete MPP6 protein (Fig. 1D). Using 3D-STORM imaging technology, we found that the nuclear distance of MPP6 from the RNA exosome was 49 nm via nearest neighbor analysis under normal conditions (Fig. 1E, Fig. S1D). Given that two interacting proteins (direct or indirect) are placed between 39 nm and 205 nm in our assays (Lim et al., 2017), it is likely that MPP6 and the RNA exosome form a complex in B cells. Consistent with previous studies regarding RNA exosome and MPP6 interaction (Falk et al., 2017; Wasmuth et al., 2017), recombinant MPP6 was found to migrate in the excluded volume of an S200 gel filtration column (from 293T cells, Fig. S1B) when incubated with purified macromolecular RNA exosome under in vitro conditions, providing further evidence that MPP6 is an interacting protein of the RNA exosome complex. MTR4 is a bona fide RNA helicase component of the RNA exosome complex (Makino et al., 2015) and also functions in unwinding RNA/RNA and RNA/DNA hybrids to control AID-mediated somatic mutations in the B cell genome (Lim et al., 2017). We evaluated whether MPP6 function is important for physical proximity of MTR4 (representing the RNA exosome complex) and AID by using 3D-STORM to identify whether association between the RNA exosome and AID was affected by the loss of MPP6. Using MTR4 as a proxy for RNA exosome location, we found that MPP6mut cells (Fig. 1G) had an increase in nuclear distance between AID and MTR4 compared to the parental CH12F3 control (Fig. 1F), as quantified in Fig 1H, I. These results suggest a role for MPP6 as a bona fide component of the RNA exosome/MTR4/AID complex with a role in driving programmed DNA rearrangements during class switch recombination.
Figure 1. MPP6 promotes programmed DNA recombination at the IgH locus and associates with m6A RNA reader proteins.

(A) Fluorescence-activated cell sorting (FACS) assessment of class switch recombination (CSR) to IgA in Mpp6 mutant (MPP6mut) and parental control CH12F3 cells stimulated with (stimulated) and without (unstimulated) LPS, TGFβ, and IL-4. (B) Percent IgA CSR of MPP6mut and parental control CH12F3 cells stimulated for class switch recombination with LPS, TGFβ, and IL-4. Data from four different MPP6mut clones are provided with each clone being assayed three times for CSR efficiency. Student’s t-test; two-tailed. N=3. (**P<0.01, ***P<0.001). (C) Proliferation test of parental control cells and MPP6mut cells using VPD450 dye. (D) Western blot analysis showing diminished protein in MPP6mut clones compared with parental (“CH12F3WT”) cells. RPA32 expression shown as a housekeeping control. (E) Histogram of the distribution of interactions of MPP6 and RNA exosome calculated in the (right) nuclear sub-compartment and (left) cytoplasm of activated B cells. (Nearest neighbor analysis, Matlab Software). Please see methods section for details of statistical evaluation. (F) Reconstructed STORM image of stimulated parental control CH12F3 cells with AID (green), MTR4 (red), and DAPI (blue)-labeled nucleus. Scale bar, 1 μm. (G) Reconstructed STORM image of stimulated MPP6mut CH12F3 cells with AID (green), MTR4 (red), and DAPI (blue)-labeled nucleus. Scale bar, 1 μm. (H) Histogram of the distribution of interactions between AID and MTR4 in the nuclear sub-compartment from (upper) parental control CH12F3 cells and (lower) MPP6mut CH12F3 cells. Data quantitated in (I). Student’s t-test; two-tailed. N=4. (**P<0.01) (Nearest neighbor analysis, Matlab Software). Please see methods section for details of statistical evaluation. Data evaluated from three independent experiments, with 4 independent set of data acquisition (from three independent experiments) analyzed in each experiment. (J) Flag immunoprecipitation reactions (IPs) were performed on HEK293T cells to demonstrate interaction of MPP6 with m6A reader protein YTHDC1 and RNA helicase MTR4. MPP6-FLAG was immunoprecipitated using FLAG tag and blotted with anti-FLAG or anti-YTHDC1 or anti-MTR4 antibodies. YTHDC1 and MTR4 interactions with FLAG-tagged MPP6 were observed. The negative controls for the immunoprecipitation reaction were transfected with empty vector (EV) that only express FLAG but not MPP6.
MPP6 associates with m6A reader protein YTHDC1 and lncRNA SμGLT.
Next, we wanted to interrogate what allows MPP6, the RNA exosome, and AID to form a complex in B cells, leading us to question whether RNA base modifications, another form of RNA surveillance, play a role. The N6 position of adenosine in RNAs can be methylated by multiple methyltransferases, yielding the common m6A modification. CAPAM (also known as PCIF1) targets 2’-O-methyladenosine that is found adjacent to the 5’ cap of RNA to produce m6Am, and the METTL3/METTL14 heterodimer is responsible for internal m6A modifications (Heck and Wilusz, 2019; Roundtree et al., 2017a). Recent studies have indicated that, in the context of exosome-substrate intronic RNAs, N6-methyladenosine modification of RNA (m6A RNA) by the METTL3/METTL14 complex plays an important role in decay (Fish et al., 2019). Motivated by these observations, we interrogated whether MPP6 plays a role in recognizing or recruiting components of the m6A RNA modification pathway to ncRNAs to catalyze processing or decay. We immuno-precipitated MPP6 in 293T cells and found that the nuclear m6A reader protein YTHDC1 co-immunoprecipitated with MPP6 (Roundtree et al., 2017b) (Fig. 1J). As an additional control to this experiment, we tested and demonstrated that MPP6 immunoprecipitates also contained MTR4, the bona fide RNA exosome helicase cofactor that can be considered as a marker representing the RNA exosome complex. To further confirm this interaction, we also show that immunoprecipitation of HA-tagged tagged YTHDC1 brings down MPP6 and MTR4, when co-expressed in 293T cells (Fig. S1C). Thus, we conclude that MPP6 can co-complex with the m6A reader protein YTHDC1 and the RNA exosome complex (represented by MTR4).
We then examined the exosome-sensitive lncRNA SμGLT, whose expression and processing are necessary for CSR (Lorenz et al., 1995), to address whether the abovementioned complex interacts with CSR-associated lncRNAs. We demonstrated that although in RNA-immunoprecipitation assays both MPP6 and YTHDC1 interact with SμGLT in CH12F3 cells (Fig. S1E (left panel), YTHDC1 interaction with SμGLT is not MPP6-dependent (Fig. S1E, center panel) but MPP6 interaction with SμGLT is YTHDC1-dependent (Fig. S1E, right panel) These observations indicate that RNA methylation identifies nuclear ncRNAs via intermediate interaction between the RNA exosome cofactor MPP6 and the m6A reader YTHDC1, eventually promoting RNA processing.
Loss of m6A methyltransferases results in the enriched expression of ncRNA subsets
Accumulation of ncRNAs in cells can cause various problems such as activation of cytoplasmic RNA sensors for expression of proinflammatory cytokines (Rehwinkel and Gack, 2020) and induction of RNA-mediated genomic instability (Nair et al., 2020; Pefanis et al., 2014), highlighting the importance of the regulation of RNA-processing mechanisms. Since m6A reader proteins bind to MPP6, we postulated that RNA methylation of various ncRNAs may be important for RNA exosome complex recruitment to, and eventual decay of, these ncRNAs. To interrogate whether METTL3 is indeed important for decay of other lncRNAs expressed in the B cell genome, we evaluated enrichment of long intergenic ncRNAs (lincRNAs) (Rinn and Chang, 2012), enhancer RNAs (eRNAs) (Kim et al., 2010; Li et al., 2016; Pefanis and Basu, 2015; Pefanis et al., 2014) and transcription start site-associated RNAs (TSS-RNAs) (Preker et al., 2008; Seila et al., 2008) in RNA exosome mutant (Exosc3C/C) primary, METTL3-deficient (Mettl3f/f +OHT) primary, and METTL14-mutant (Mettl14mut) CH12F3 B cells. We observed stabilization of TSS-RNAs, eRNA, and lincRNAs following loss of the Mettl3 gene (represented as Mettl3f/f +OHT) (Fig. 2A), though significantly less pronounced than transcript stabilization seen upon loss of the RNA exosome subunit gene Exosc3 (represented as Exosc3C/C). This is likely due to a partial redundancy with other mechanisms of RNA exosome targeting to substrates, and also due to 5’end RNA decay by the XRN1/2 complex pathway We evaluated individual classes of ncRNAs: eRNAs in the lncCSRIgA locus or in the 3’ regulatory region (3’RR, a well-known superenhancer that drives IgH recombination) (Fig. 2B), well-established lincRNAs Neat1 and Malat1 (Fig. 2C), and TSS-RNAs in the Cd83 and Cd79b loci (important genes for B cell development) (Fig. 2D) were prevented from decay in Exosc3C/C and Mettl3f/f +OHT B cells with overlapping signals of m6A modification. Additional examples of lncRNAs and TSS-RNAs for which decay is prevented in Exosc3C/C and Mettl3f/f +OHT B cells or Mettl14mut CH12F3 B cells are shown in Fig. S2A and Fig. S3, respectively (details of all METTL3-sensitive ncRNAs are in Table S1). Taken together, these results establish that various families of RNA exosome-sensitive ncRNAs are also targets of m6A modification, although all lincRNA, TSS-RNA, or eRNA levels are not altered as robustly as seen in exosome-deficient B cells.
Figure 2. METTL3 m6A methyltransferase and RNA exosome target overlapping sets of noncoding RNAs.

(A) Differential expression analysis for various RNA classes. Vertical bars indicate standard errors of log2 fold change between controls and EXOSC3-deficient (Exosc3C/C), METTL3-deficient (Mettl3f/f +OHT). The increase in lncRNA expression in METTL3-deficient B cells can be observed but the increase is substantially lower than that seen when RNA exosome is depleted from B cells (Exosc3C/C). (B) RNA-seq profile and m6A-IP-seq profile at enhancerassociated regions (lncCSRIgA and IgH 3’RR) in ex vivo-stimulated splenic B cells. (C) RNA-seq profile and m6A-IP-seq profile at lncRNA regions (Neat1 and Malat1) in activated B cells. (D) RNA-seq profile and m6A-IP-seq profile at IgH translocation hotspot genes (Cd83 and Cd79b) in ex vivo-stimulated splenic B cells. Black boxes and arrows indicate xTSS-RNA.
METTL3 activity is important for processing of G-rich SμGLT and CSR
We wanted to assess further the role of METTL3 during B cell development, a process requiring a series of DNA recombination events. We generated a Mettl3f/f Mb1cre/+ mouse model where the Mettl3 gene is deleted in the bone marrow early in B cell development. METTL3 depletion in bone marrow developing B cells leads to a clear block at the pro-B cell to pre-B cell stage with a dramatic decrease in pre-B cells (Fig. S4A). Moreover, B cell numbers in the periphery were significantly decreased, as observed via decreases in follicular and marginal zone B cells in the spleen (Fig. S4C) as well as B cells in germinal centers present in Peyer’s patches (Fig. S4E), a reproducible result in three independent experiments (Fig. S4B, D, F). Taken together, we report METTL3 functions in B cell development and survival both in the bone marrow and in the germinal center reaction. To assess the role of METTL3 and m6A RNA modification in DNA recombination in primary B cells, we used the CSR model system where splenic B cells could be isolated, cultured, and stimulated ex vivo, an approach that overrides the aforementioned early B cell developmental defects. We isolated Mettl3f/f Rosacre splenic B cells for ex vivo stimulation with or without 4-hydroxytamoxifen (OHT) to compare CSR in METTL3-deficient (Mettl3f/f +OHT) and METTL3-proficient (Mettl3f/f −OHT) primary B cells. As expected, RNA-seq results showed that exon 4 of the Mettl3 RNA is deleted in the Mettl3f/f +OHT samples, but not in the Mettl3f/f −OHT or Exosc3C/C samples (Fig. 3A), accompanied by a decrease in total METTL3 transcript. Transcription through the IgH switch sequence, also known as IgH-Sμ germline transcription that produces SμGLT, is essential for catalysis of CSR (Shinkura et al., 2003; Yu et al., 2003). RNA exosome-sensitive SμGLT have been shown to have additional roles in AID recruitment to the IgH region, and thus their biology is of paramount importance in CSR (Ribeiro de Almeida et al., 2018; Zheng et al., 2015). We evaluated Mettl3f/f +OHT cells for levels of SμGLT and found a significant increase in transcript levels mirroring that seen in the absence of RNA exosome activity (Fig. 3B). Moreover, the increase in germline transcripts was observed on both strands of the IgH-Sμ, as is often seen for bidirectionally transcribed noncoding regions of the genome (those originating from two divergently transcribed promoters/enhancers placed in close proximity) in RNA exosome-deficient mammalian cells (Fig. 3B and (Lim et al., 2017; Pefanis and Basu, 2015). We evaluated the levels of m6A RNA in CSR-stimulated B cells and found that IgH-Sμ transcripts are m6A-modified (Fig. 3B, bottom panel). Thus, the SμGLT transcripts accumulate in the absence of m6A methylation, as they are protected from 3’ end processing and exosome-mediated decay. One should note that transcriptome reconstitution of METTL3-deficient cells was performed at early time points (48 hrs) following treatment of Mettl3f/f cells with OHT to avoid secondary effects on RNA metabolism that are caused by the wide range of functions METTL3 performs in B cells.
Figure 3. METTL3-deficient primary B cells have a defect in CSR and accumulate Sμ germline transcripts and demonstrate reduced AID proximity with RNA exosome.

(A) RNA-seq analyses of EXOSC3-proficient (Exosc3+/+), EXOSC3-deficient (Exosc3C/C), METTL3-proficient (Mettl3f/f −OHT) and METTL3-deficient (Mettl3f/f +OHT) B cells. RNA-seq tracks demonstrate that the METTL3 mRNA is expressed in Exosc3+/+, Exosc3C/C, Mettl3f/f OHTcells but is markedly reduced in Mettl3f/f +OHT B cells, demonstrating robust deletion of METTL3 activity. The Mettl3 gene is in the negative strand and exon 4 is completely lost due to deletion. (B) RNA seq analyses from transcriptomes described in (A) demonstrate accumulation of SμGLT in both exosome-deficient (Exosc3C/C) and METTL3-deficient (Mettl3f/f +OHT) B cells. (C) In METTL3-deficient B cells, class switch recombination to IgG3 is decreased following LPS stimulation and to IgG1 following LPS+IL4 stimulation and anti-CD40 ab+IL4 stimulation. As a control, effects in AID-deficient B cells (AID−/−) are shown. AID-deficient B cells do not show any CSR. (D) 6 sets of IgG1 stimulation experiments of (C) presented with P values. Student’s t-test; two-tailed. N=7. Error bars = Standard error. (***P<0.001) (E) Proliferation of Mettl3f/f −OHT and Mettl3 KO cells is shown in ex vivo culture conditions. (F) DNA/RNA hybrid immunoprecipitation seq (DRIP-seq) was performed for Mettl3f/f −OHT, Mettl3f/f +OHT, DIS3+/+ and DIS3C/C (KO) active B cells. As shown in the integrated genome viewer tracks, there is a decrease in DNA/RNA hybrids in the IgH-Sμ region in Mettl3f/f +OHTB cells (compared to control Mettl3f/f −OHTB cells; top two panels), whereas in DIS3C/C B cells there is accumulation of DNA/RNA hybrids (bottom two panels). (G) Distribution of AID and MTR4 (representing the RNA exosome complex) is perturbed in B cells using 3D-STORM microscopyassessing single molecule imaging. (H) The relative distance of AID and MTR4 molecules is increased following Mettl3-deletion (in +4OHT cells) under conditions of stimulation to IgG3 (top), IgG1 (middle), and IgA (bottom). (Nearest neighbor analysis, Matlab Software). Please see methods section for details of statistical evaluation.
Next, we observed inefficient CSR to IgG1 (following stimulation by both LPS and IL4 or anti-CD40 antibody and IL4) and IgG3 (following LPS-mediated stimulation) in METTL3-deficient splenic B cells (Fig. 3C). The decreased IgG1 CSR was observable in multiple experiments (Fig. 3D). Dye dilution staining showed some detectable but mild changes in cell proliferation kinetics at these early time points of assay (Fig. 3E). To investigate whether R-loop structures are affected at IgH-Sμ, we performed DRIP-seq. In exosome-deficient B cells (DIS3C/C) we found increased levels of R-loops but in Mettl3f/f +OHT cells there was a decrease in R-loops (Fig. 3F). Consistent with previous studies showing that m6A reader proteins stabilize R-loops (Abakir et al., 2020), we postulate that lack of m6A leads to decreased recruitment of reader proteins at SμGLT leading to rapid loss of R-loop structures and diminished recruitment of AID. Finally, STORM imaging indicates that there is a loss of proximity of AID to MTR4 in METTL3-deficient cells (Fig. 3G, H), adding to the evidence that reduced m6A+ SμGLT leads to a lack of AID activity for CSR.
Depletion of methylation of SμGLT reduces CSR efficiency
M6A RNA methylation occurs at various coding and noncoding RNAs. Accordingly, a lack of METTL3 activity could cause CSR defects due to various pleiotropic effects as reflected in loss of developing B cells and GC B cells in Mettl3f/f Mb1Cre mice (Fig. S4). We performed m6A-IP seq and Oxford Nanopore-seq on RNA exosome-deficient (Exosc3C/C) B cells with both assays employed to identify m6A modification sites particularly on RNA exosome-sensitive SμGLT. While m6A-IP-seq identified many RNA-fragments in the B cell transcriptome as containing m6A sites, Nanopore-seq identified fewer sites but with greater precision. Overall, 86% of the sites identified by Nanopore-seq were also represented in RNA fragments present in m6A-IP seq data sets (Fig. S5A). G-rich exosome-sensitive RNAs were represented at higher significance in m6A-IP samples (Fig. S5B) and G-rich patches were enriched 50–500 bases surrounding m6A sites identified by Nanopore-seq (Fig. S5C).
We next focused directly on our goal to understand m6A mediated SμGLT lncRNA regulation, As shown in Fig. 4A, multiple peaks that overlap m6A modification in both m6A-IPseq and Nanopore-seq were defined, indicating that SμGLT are methylated at multiple sites. A closer view of m6A peaks on various fragments of SμGLT can be seen in Fig. S6A and Fig. S6B; multiple peaks representing m6A sites were observed. To investigate the importance of modifications in SμGLT, we generated and utilized a fusion protein of catalytically dead Cas13b (dCas13b) with m6A RNA demethylases FTO and ALKBH5, (illustrated in Fig 4B.) Through m6Abased immunoprecipitation followed by SμGLT PCR, we were able to show that one gRNA (gRNA-4; Fig. 4A) was able to target both ALKBH5 and FTO to SμGLT and drive demethylation (Fig. 4C). Strikingly, both demethylases, when co-transfected with gRNA to SμGLT, also were able to suppress CSR partly but significantly (FTO in Fig. 4D and ALKBH5 in Fig. 4E). The expression of either dCas13b-FTO or dCas13b-ALKBH5 did not interfere with the level of AID expressed in these CH12F3 cells (Fig. 4F) or suppress CH12F3 cell proliferation (Fig. 4G). It is likely that targeting RNA demethylase to the SμGLT leads to demethylation, reduced RNA exosome/AID recruitment, and suppression of CSR. Given that Mettl3f/f +OHT cells may have some proliferation or gene expression changes (Zheng et al., 2020) that could indirectly affect CSR, the observations shown in Fig. 4 provide direct molecular demonstration of the important role of m6A modification in catalysis of CSR.
Figure 4. Directed demethylation of m6A modification of SμGLT reduces IgH class switch recombination without affecting B cell proliferation.

(A) Oxford-Nanopore RNA sequencing of exosome-deficient B cells reveals enrichment of SμGLT and identification of m6A sites. Top two panels demonstrate increased SμGLT in EXOSC3-deficient (Exosc3C/C) B cells. The identification of m6A sites in the SμGLT is shown via Oxford-Nanopore seq of the Exosc3C/C B cell transcriptome (middle panel) and m6A-IP-seq (bottom panel). The overlap of the sites identified by Oxford-Nanopore-seq with m6A-IP-seq was used to design gRNAs for Cas13-fused ALKBH5/FTO targeting (red arrow). (B) Schematic representation of ALKBH5 demethylase and/or FTO demethylase targeting to SμGLT using dead Cas13b fusion protein. (C) Demonstration of a substantial demethylation of SμGLT can be accomplished with the system following m6A RNA-IP coupled with SμGLT qPCR using either of the two dCas13b fused demethylases. 4 independent experiments were performed. Student’s t-test; two-tailed. N=4. (**** = P ≤ 0.0001) (D) Relative decrease in CSR activity of dCas13b-FTOexpressing CH12F3 cells that are transfected with gRNA-4. Left panel demonstrates FACS plots of dCas13b-FTO-expressing GFP+ cells (unstimulated, stimulated, and gRNA-4 introduced prior to stimulation). Right panel shows average and mean of three experiments demonstrating a defect in CSR following introduction of gRNA-4 in dCas13b-FTO expressing cells. 3 independent experiments were performed. Student’s t-test; two-tailed. N=3. (***P<0.001). (E) Relative decrease in CSR activity of dCas13b-ALKBH5-expressing CH12F3 cells that are transfected with gRNA-4. Left panel demonstrates FACS plots of dCas13b-ALKBH5-expressing GFP+ cells (unstimulated, stimulated, and gRNA-4 introduced prior to stimulation). Right panel shows average and mean of three experiments demonstrating a defect in CSR following introduction of gRNA-4 in dCas13b-ALKBH5 expressing cells. 3 independent experiments were performed. Student’s t-test; two-tailed. N=3. (**P<0.01). (F) The level of SμGLT is not altered by expression of dCas13b-FTO or dCas13b-ALKBH5, as measured by qPCR, indicating CSR in (D and E) is not affected due to reduced transcription in the IgH locus. Student’s t-test; two-tailed. N=3. (G) dCas13b-FTO or dCas13b-ALKBH5 overexpression did not alter CH12F3 proliferation, as observed by the VPD450 dye dilution method.
YTHDC1 m6A reader is important for CSR and SμGLT processing.
RNA exosome cofactor MPP6 binds to the nuclear m6A reader YTHDC1 (Fig. 1J) for interaction with lncRNA SμGLT (Fig. S1E). Consistent with our RNA-IP (Fig. S1E, biophysical modeling of YTHDC1 and SμGLT demonstrates potential interaction (Fig. 5A; see modeling details in methods section). Accordingly, we interrogated whether YTHDC1 is important for CSR. For this purpose, we generated mutant CH12F3 clones of YTHDC1 that express a truncated form (YTHDC1mut genotype shown in Fig. S6C). The knockout clones proliferated almost comparably to control CH12F3 WT cells with some minor deficiencies (Fig. 5B), and western blots demonstrate the truncation (Fig. 5C). Following stimulation for CSR to the IgA isotype, the YTHDC1mut clones demonstrated decreased CSR, both at 48 hrs and 72 hrs post stimulation (Fig. 5D, and three independent experiments in Fig. 5E). As would be expected, the YTHDC1mut clones had increased levels of SμGLT expression (data from RNA-seq for two separate YTHDC1mut clones, Fig. 5F). The YTHDC1mut cells do not exhibit any significant expression defect of MPP6, MTR4, or RNA exosome (shown through expression of Exosc10 and Exosc3), indicating that the increased levels of SμGLT expression is not due to changes in other components of RNA exosome pathway (Fig. S6D). We performed 3D-STORM on YTHDC1mut CH12F3 clones and observed loss of proximity of AID and MTR4 (Fig. 5G and I represent the distribution change in one YTHDC1mut clone; Fig. 5H represents distribution from two separately generated YTHDC1mut clones.) Overall, these observations provide additional evidence that m6A modification of lncRNAs (SμGLT in the case of IgH) promotes recruitment of the RNA exosome complex and AID to sites of DNA deamination for catalysis of IgH CSR.
Figure 5. m6A reader YTHDC1 is important for CSR and proximity of AID/MTR4-containing recombinational complex.

(A) A structural reconstruction of the SμGLT interaction with YTHDC1 (structure previously published) identifying potential interaction sites. (B) YTHDC1mut CH12F3 clones proliferate similarly to control CH12F3 cells, as evaluated by the VPD450 dye dilution method. (C) Western blotting identifies CH12F3 cells expressing a truncated version of the YTHDC1 protein (YTHDC1mut) lacking the RNA binding site. (D) FACS plots demonstrating decreased CSR occurs in YTHDC1mut clones. (E) 2 separate YTHDC1mut clones evaluated for effect on CSR, 5 independent experiments were performed. Student’s t-test; two-tailed. N=6. Error bars = Standard error (**P value < 0.01). (F) Increased levels of SμGLT in YTHDC1mut CH12F3 clones (cl1 and cl2). (G) Nearest neighbor analyses of AID and MTR4 in YTHDC1mut clones over three different data sets. YTHDC1mut clones demonstrate that the distance of the two proteins increases following loss of YTHDC1 activity, from 150 nm to 494 nm. Data are quantitated in (H) with a compilation of average differences in distance between AID and MTR4 proteins across the nucleus in 4 set of data acquisition from three independent run experiments. Student’s t-test; two-tailed. N=4. (**P value <0.01). (Nearest neighbor analysis, Matlab Software). Please see methods section for details of statistical evaluation. Data plotted from the aggregated values of two independent clones evaluated, with four cells analyzed in each experiment. (I) Images of AID-MTR4 distribution in the nucleus of parental and YTHDC1mut CH12F3 clones with AID in green, MTR4 in red, and the nucleus stained with DAPI (blue). Closeup images shown in the right-hand panel, with scale bars indicated in the figure.
METTL3 loss correlates with decreased CSR junctions, increased off-target DNA translocations, and use of alternative DNA repair pathways
RNA exosome-mediated ncRNA degradation is important to efficiently catalyze DNA CSR through the formation of DNA double-strand breaks in IgH switch sequences. If m6A does indeed promote RNA decay by RNA exosome activity, we postulate that METTL3-deficient cells would show decreased levels of DNA breaks that are necessary for CSR. Furthermore, in the absence of proper recombination events, breaks in the IgH-Sμ region would be expected to find translocation partners elsewhere in the B cell genome. Various ncRNAs accumulate at previously reported sites of genomic instability (Chiarle et al., 2011; Klein et al., 2011), sites that harness DNA mutations and translocations via AID-dependent and AID-independent mechanisms (Rothschild and Basu, 2017), in METTL3-deficient and EXOSC3-deficient B cells (Figure S7). To evaluate directly IgH recombination and IgH-Sμ-associated chromosomal translocations, we performed IgH linear amplification-mediated high-throughput genome-wide translocation sequencing (LAM-HTGTS, (Frock et al., 2015)) assays in METTL3-proficient and METTL3-deficient B cells. In this assay, the IgH switch sequence Sμ undergoes AID-mediated DNA double-strand breaks that either recombine to downstream switch regions for CSR or, if unregulated, can translocate to other regions of the B cell genome, potentially instigating pathological events. As seen in Figs. 6A and 6C, METTL3-deficient B cells have a clear decrease in levels of switch region DNA breaks at IgH-Sμ, or at downstream switch sequences like Sγ1 or Sε. Multiple experiments were performed to demonstrate reliably that in the absence of METTL3 there is a clear decrease in IgH DNA recombination events (Fig. 6B, left panel). Additionally, we found increased translocations between IgH-Sμ and other regions of the B cell genome (Fig. 6B, right panel), indicating an increase in genomic instability in the absence of METTL3.
Figure 6. Decreased programmed DNA recombination and increased aberrant DNA recombination in m6A methyltransferase activity-deficient B cells along with alternative DNA end joining.

(A) LAM-HTGTS assessment of DNA breaks at switch sequences (using Sμ bait) in the IgH locus in splenic B cells from Mettl3f/f Rosacre mice stimulated ex vivo for 72hr with LPS and IL4 after treatment with (+) or without (−) 4-hydroxytamoxifen (OHT). (B) Percent translocations inside (CSR) and outside (non-CSR) of the IgH locus in +OHT and −OHT splenic B cells as measured by LAM-HTGTS. Welch’s t-test; two-tailed. N=3. (C) LAM-HTGTS assessment of DNA breaks in the IgH locus as a whole. (D) Programmed mutagenesis of the IgH locus is dependent on IgH 3D-architecture and interaction of 3’RR and E enhancers. Hi-C analyses of Mettl3f/f −OHT and Mettl3f/f +OHT cells demonstrate no discernable changes in 3D-architecture and 3’RR/Eμ interaction at early time points of METTL3-deletion. (E) Insulation score analyses and (F) aggregated peak analyses (APA, only of common loops) of Mettl3f/f −OHT and Mettl3f/f +OHT B cells do not demonstrate changes in TAD boundaries genome-wide. Welch’s t-test; two-tailed. (G) Increased levels of microhomologies in METTL3-deficient (+OHT) cells at junctions of Sμ when recombined with downstream switch sequences or translocated to other regions of the B cell genome. Percent of microhomology joining events in Sμ translocations. Welch’s t-test; N=3. (H) Some examples of the microhomologies as reported in (G).
We have shown previously (Laffleur et al., 2021) that decreased stability of the IgH topologically-associated domain (TADIgH) leads to increased chromosomal translocations but this possibility was ruled out for Mettl3f/f +OHT cells. As shown in Fig. 6D, the IgH TAD is similar in Mettl3f/f −OHT and Mettl3f/f +OHT cells. Moreover, parameters that evaluate genome architecture and TAD boundaries like insulation score (Fig. 6E) or aggregated peak score (Fig. 6F) are not altered in Mettl3f/f +OHT cells. These observations are consistent with the fact that ncRNAs whose decay is important for TADIgH integrity, CTCF-binding element ncRNAs (cbeRNAs), are not G-rich (Laffleur et al., 2021), and thus should not rely on METTL3 activity for exosome-mediated processing in this model. Given that we successfully eliminated all known studied parameters that can affect CSR including switch region transcription decrease and TADIgH architectural alterations, the direct role of m6A SμGLT modification in RNA exosomedependent IgH recombination is solidly established. Finally, if R-loops are not stabilized in METTL3-deficient cells (Fig. 3F) at IgH-Sμ, we would expect that AID-mediated deamination would not be sufficiently clustered as to promote robust staggered DNA breaks on both strands (the preferred mechanism of CSR), thus producing single strand overhangs that induce microhomology-mediated end joining. Consistent with this expectation, Mettl3f/f +OHT cells have increased microhomology-mediated DNA end joining at IgH-Sμ both during CSR and during translocations (Fig. 6G, H). Taken together, METTL3-mediated modification of ncRNAs is important for the decay of a subset of ncRNAs, and the absence of such can lead to a decrease in programmed DNA recombination, increased off-target chromosomal translocations, and use of the alternative end joining pathway (modeled in Fig. 7A), as we have demonstrated at the IgH locus. Supporting these conclusions, we also evaluated the levels of exosome-sensitive ncRNAs expressed at overlapping sites of B cell genomic instability (Pefanis et al., 2014) and found increased accumulation of ncRNAs (Fig. S7) in METTL3-deficient B cells. Examples of accumulated genomic instability (assayed by TC-seq) that overlap METTL3-sensitive and exosome-sensitive ncRNAs at many other B cell loci (Sμ, Cd79b, Cd83, Pim1, Adssl1. etc.) are shown in Fig. S7.
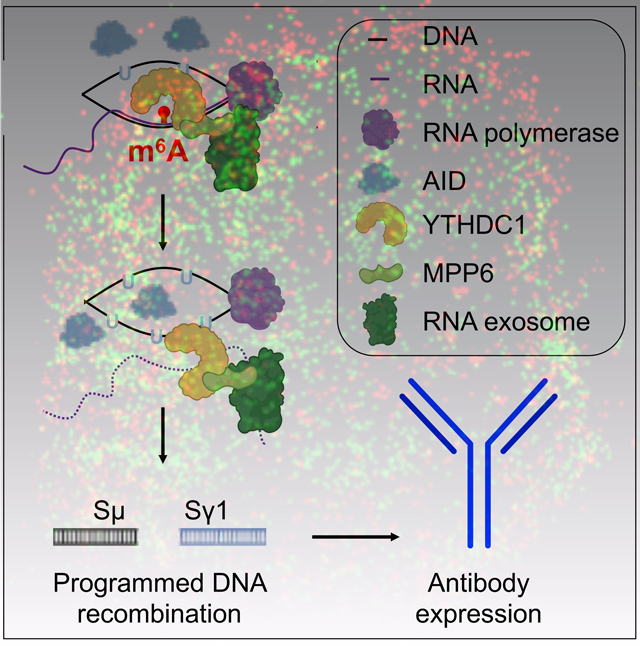
Figure 7. Schematic representation of m6A RNA modification in exosome sensitive RNA recognition expressed at the IgH locus.

m6A RNA is recognized on DNA/RNA hybrids by YTHDC1 and MPP6. YTHDC1 association to nascently synthesized SμGLT stabilizes larger R-loops and promotes non-template strand DNA deamination. Thereafter, YTHDC1/MPP6 recruits RNA exosome to promote RNA decay/processing, leading to AID-mediated strand-symmetric mutations. In the absence of METTL3 (or m6A modification), AID mutations occur less often on the non-template DNA strand (due to unstable R-loops) and template strand (due to weak RNA exosome recruitment). Reduced strand-specific deamination creates asymmetric mutations that ultimately lead to inefficient CSR, increased microhomologies, and chromosomal translocations.
DISCUSSION
Accumulation of exosome-sensitive ncRNAs eventually subverts gene expression control in mammalian cells through methods including induction of RNA/DNA hybrid-associated genomic instability (Lim et al., 2017; Wahba et al., 2011) often at promoters and superenhancers (Pefanis et al., 2014; Pefanis et al., 2015), activation of cytoplasmic RNA sensors (Eckard et al., 2014), lack of efficient DNA double-strand break repair (Marin-Vicente et al., 2015; Pefanis et al., 2015), and alterations of 3D-genomic architecture (Laffleur et al., 2021). Similarly, defects in RNA methylation have been associated with alterations in various biological processes (Choe et al., 2018; Dominissini et al., 2012; Geula et al., 2015; Lee et al., 2019; Li et al., 2017; Wang et al., 2014; Zaccara et al., 2019). The question arises whether the exosome and RNA methylation pathways converge for regulating expression and function of ncRNA subsets. In this study, we identify m6A RNA modification of exosome sensitive SμGLT lncRNA as an important mechanism for regulating programmed DNA recombination and suppression of aberrant DNA recombination. A central observation in this study is the role of N6-methyladenosine in the nucleation of the RNA exosome, MTR4, and AID on the G-rich SμGLT lncRNA. We postulate that RNA methylation alters the tertiary structure of the ncRNA subsets in a way that makes them more sensitive to MTR4-mediated presentation to the RNA exosome central channel for decay. Biophysical evaluation of m6A-modified ncRNA (compared to unmodified RNA) being presented by MTR4 to the central channel of the RNA exosome complex will be required to understand further how this could occur. We also postulate that the interaction of MPP6 with the RNA exosome complex and with the m6A nuclear reader (YTHDC1) provides specificity to the RNA exosome pathway for identifying m6A ncRNAs (outlined in Fig. 7).
M6A on RNA slows the kinetics of RNA/RNA hybridization (Shi et al., 2019), potentially slowing RNA secondary structure formation on the G-rich Sμ and thus facilitating DNA/RNA hybridization in switch regions. Switch sequences are well known to form many types of secondary DNA structures like G-quadruplexes or stem loops as parts of a larger R-loop (Yu et al., 2003). Perturbation of DNA/RNA hybrid dynamics at IgH-Sμ due to reduced stability of DNA/RNA hybrids in METTL3-deficient B cells (lack of YTHDC1 recruitment due to loss of m6A on nascent SμGLT) may reduce AID mutational frequency, increase asymmetric nicks, leading to increased microhomology-mediated DNA double-strand break repair (Fig. 6G, H) and increased representation of chromosomal translocations (Fig. 6B). These possibilities are outlined in Fig. 7.
Although RNA methylation enzymes have been demonstrated to play roles in various biological events, rarely are m6A RNA modification sites directly demonstrated to do the same. To this end, we successfully demethylated SμGLT m6A sites by targeting RNA demethylases FTO and ALKBH5 using a guide RNA-mediated dCas13b system and overlapped phenotypic outcomes with complete loss of METTL3 enzyme (Fig. 4). These experiments are particularly important since they directly show m6A RNA modification of SμGLT lncRNA is causative for CSR, and not due to cellular proliferation or gene expression alterations that are observable in METTL3-deficient, MPP6-deficient, or YTHDC1-deficient cells. Thus, our study provides a rare demonstration of lncRNA regulation by m6A modification in the context of an important biological event. In addition to promoting RNA decay, other roles of METTL3 and m6A modifications of mRNA and lncRNAs are possible. Depending upon the reader protein that interacts with a specific mRNA, the m6A modification can also promote stability and prevent decay. As an example, m6A-modified c-Myc mRNA is stabilized, not decayed, promoting proliferation of hematopoietic stem cells (Cheng et al., 2019; Lee et al., 2019). Thus, the context and regulatory activity of m6A on a specific RNA are extremely important to understand its role in a specific biological function; these mechanisms cannot be generalized. In this study, we provide evidence that the G-tracts of the SμGLT, which also generate secondary RNA structures, have a role in recruiting degradative/3’ end processing complexes. Future studies will focus on additional RNA sequence- and secondary structure-based cues that direct the role of m6A in stabilizing mRNAs versus promoting their decay. Finally, questions arise regarding the direct contribution of METTL3 activity on CSR and maintaining genomic integrity. METTL3-deficient cells in vivo suffer a proliferative disadvantage (Fig. S4), perhaps due to gene expression alterations and lncRNA function perturbations that lead to defects in cellular proliferation. In our studies, due to the fact that B cells do undergo CSR outside the germinal center (Roco et al., 2019) and that the lymphoma B cell line CH12F3 bypasses cell cycle checkpoints, we are able to somewhat override the proliferative disadvantage (although some defects persist) of METTL3-deficient B cells and study the role of m6A modification in lncRNA (particularly SμGLT) biology and CSR. We provide direct evidence of a role for m6A in CSR by perturbing m6A status of SμGLT using dCas13b-mediated RNA editing (Fig. 4). These results should be independent of any other METTL3-function in B cells. We note, based on our studies with the METTL3f/f Rosacre mouse model, that B cells are dependent upon METTL3 activity for development and proliferation. Deletion of METTL3 in vivo leads to a block in early B cell development and a strong loss of germinal center B cells (Fig. S4). METTL3 activity has been ascribed to DNA double-strand break repair (Zhang et al., 2020); it is likely that in the absence of METTL3, B cells will show genomic instability due to various AID-independent DNA damage mechanisms (Rothschild and Basu, 2017). To study these effects, one will need model systems that perturb m6A RNA modification sites on relevant mRNAs or lncRNAs (as done for SμGLT) to override a more pleiotropic effect of METTL3 deletion. We note that it is unlikely that SHM frequency in the germinal center will depend on the YTHDC1/MPP6 axis, since the VDJ GLTs are not G-rich and may not rely upon the described mechanism for catalyzing exosome-mediated VDJ GLT decay.
Our study has linked nuclear RNA processing by the RNA exosome with the m6A RNA modification status of RNAs. Specifically, SμGLT lncRNA activity is directly controlled by its m6A modification status. Our study provides direct mechanistic association of m6A RNA modification within two aspects of immune cell biology: (a) generating better antibodies, and (b) prevention of lymphoma-associated chromosomal alterations. It is likely that additional RNA modifications will play important roles in lncRNA biology, CSR, and the B cell immune response. Thus, future studies will lead to a unifying RNA epigenetics code that determines post-transcriptional RNA control, akin to the DNA epigenetics code that is well accepted to control transcription.
Limitation of the study.
Although m6A modification promotes processing/decay of SμGLT lncRNA, there should be additional contributing mechanisms. Deletion of METTL3 or YTHDC1 activities does not lead to stabilization of SμGLT lncRNA at similar levels as seen following the loss of RNA exosome activity. Similarly, depletion of the m6A pathway components partially affects CSR but does not diminish class switch to the levels seen resulting from loss of the AID protein. Accordingly, it is likely that recognition of SμGLT lncRNA by RNA exosome and other regulatory proteins may be dependent on additional RNA modifications awaiting discovery. In addition, there are m6A independent pathways that control CSR efficiency. Detecting RNA modifications is an evolving topic and with passing time technical skills will improve. Here we have used a combination of m6A-IP seq and Nanopore-seq to identify m6A modifications in the B cell transcriptome and specifically on SμGLT. We further enhanced m6A detection of less stable and lowly expressed lncRNAs by using RNA decay-ablated Exosc3-depleted cells. But the detection of m6A sites remains incomplete and additional studies with new techniques will be performed in the future.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact.
Request for resources and reagents should be directed to and will be fulfilled by the lead author, Uttiya Basu, PhD (ub2121@cumc.columbia.edu)
Material Availability.
This study did not generate new unique reagents. Derivative cell lines and plasmids will be provided following request.
Data and code availability.
All RNA-seq, DRIP-seq, HTGTS and HiC data has been provided in NIH repository with accession number PRJNA693506.
Original codes were not generated in this study.
Any additional information required to reanalyze the data reported in this work will be available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mettl3F/F mice were previously published (Weizmann Institute of Science) (Geula et al., 2015); Rosa26creERT2 (i.e. tamoxifen-dependent cre) mice which we refer to as Rosacre were supplied by Prof. Pierre Chambon (University of Strasbourg); Mb1cre mice were provided by Dr. Michael Reth (Max Planck Institute) (Hobeika et al., 2006); AID−/− mice were obtained from Prof. Tasuku Honjo (Kyoto University) (Muramatsu et al., 2000). 6- to 12-week-old mice were used for experiments. All mouse experiments were approved by the IACUC of Columbia University. The housing conditions were 22°C, 50% relative air humidity, and 12-h light cycles (from 7 am to 7 pm). Both male and female mice were used and sex of the animals did not affect experimental outcomes. Please refer to Supplementary Table S2 for mouse and cell line nomenclature.
METHOD DETAILS
Primary B cell isolation and stimulation.
After red blood cell lysis, CD43- cells were isolated from spleens using CD43 magnetic beads and LS column (Miltenyi Biotec). CD43 negative B lymphocytes from Mettl3f/f;RosaCre and AID knockout spleens were plated at day 0 at a concentration of 5*105 per ml of medium (RPMI supplemented with 15% FBS, 1x non-essential amino acids, 1x sodium pyruvate, a 1:100 dilution of 1M HEPES, 1x L-glutamine, 2x penicillin/streptomycin and 1x tissue culture grade beta-mercaptoethanol). In vitro B cell knockout and stimulations were carried out by adding 100 nM 4-OHT (Sigma) (or alcohol without 4-OHT) to the respective dish. Simultaneously, with the addition of the 4-OHT, the following cytokines were added: for assay of class switch recombination to IgG1, LPS (Sigma-Aldrich) (20 ug/ml) and Il4 (Peprotech) (20 ng/ml, for assay of class switch recombination to IgG3, LPS was added (Sigma-Aldrich) (20 ng/ml), and for assay of class switch recombination to IgG1, CD40 (BD 553721) (1 ug/ml) and IL4 were added. Cells were analyzed by flow cytometry using B220-PE (BD) and anti-IgG1-FITC or IgG3-FITC (BD). Cells were routinely analyzed via flow cytometry to monitor cell activation and viability.
Proliferation.
Cells were initially incubated for 24 h with LPS and tamoxifen to knock out Mettl3 expression (for primary B cells). Cells subsequently were stained with the cell proliferation dye VPD450 (BD) and treated with LPS and IL-4 for a further 72 h with analysis every 24 h.
CRISPR/Cas9 knockout.
Guide RNAs were designed using gene sequence information and DNA oligonucleotides of target guide RNA sequences (upstream and downstream pairs) were annealed, allowed to cool slowly, and then ligated into the BbsI site of Addgene vector pSpCas9(BB)-2A-GFP (Addgene). Each vector was transfected into CH12F3 cells by nucleofection (Lonza). 24h after the electroporation step, transfected cells were isolated into 96 well culture plates by cell sorter (BD FACSAria II) and incubated for 1 week. Genomic DNA of each clone was used as a template for PCR/sequencing screening to seek mutated clones by insertion or deletion. Where necessary, PCR products of mutated clones were inserted into pGEM-T Easy (Promega) vector and their mutated regions were confirmed by Sangersequence analysis.
Class switch assay.
CH12F3 cells were stimulated with LPS, IL-4, and TGFβ for class switch to IgA. Class switch data were collected at 48 and 72 hr.
RNAseq.
In vitro, primary or CH12F3 B cells were stimulated for 48 h with LPS + IL-4 or LPS +IL-4 + TGFβ, respectively, before RNA extraction. RNA quality was evaluated by Bioanalyzer (Agilent) and RNA samples were sequenced using the RNA Ribozero 90M 100PE sequencing kit (Illumina) at the Columbia University Genome Center.
RTA (Illumina) was used for base calling and bcl2fastq2 (version 2.17) for converting BCL to fastq format, coupled with adaptor trimming. The sequenced reads were mapped to the reference genome (mm9) by using hisat2 (version 2.1.0) (Kim et al., 2015) with default parameters. We used StringTie (version 1.3.3b) (Pertea et al., 2015) to assemble the alignments into potential transcripts. Finally, RNA-seq replicates were combined for visualization in IGV. The normalization method for IGV tracks is “reads per genomic content” (RPGC), i.e. the read count was normalized as if 1× depth sequencing were performed for all samples. The peak annotation module in Homer (Heinz et al., 2010) was employed to annotate RNA-seq transcripts. Mettl3 or Exosc3 substrate non-coding RNAs were defined as non-coding transcripts with 2-fold higher FPKM values in Exosc3C/C than in Exosc3C/+ or Mettl3Δ/Δ than in Mettl3WT cells. Since transcripts with extremely low-level expression may demonstrate huge changes in fold, transcripts with FPKM < 0.1 in both samples were filtered out.
Co-IP and Western blot.
4 million 293T cells (gift of Stephen Goff, HHMI and Columbia University) were plated on day 0 in 10 cm tissue culture dishes and incubated overnight in DMEM supplemented with 10% fetal bovine serum, 2mM L-Glutamine, 1 mM sodium pyruvate, 100 micromolar nonessential amino acids, 5 mM HEPES, 100 ug/ml penicillin/streptomycin and 5.5×10−5 M beta-mercaptoethanol. 16h after plating the medium on the cells was changed to fresh medium and 2 hrs subsequently cells were transfected with (or without) 20 ug of the construct (human)MPP6-myc-DDK (Origene) using Lipofectamine2000 (Life Technologies) following the manufacturer’s protocol. 6h after the end of the transfection the medium was completely changed. 48h subsequent to the medium change cells were washed with ice cold PBS and harvested by centrifugation with the supernatant subsequently aspirated, and cells resuspended in lysis buffer (20 mM Tris HCl, pH 7.5, 75 mM KCl, 5 mM MgCl2, 10% glycerol, 0.5% NP40, 1mM DTT with one “Complete Mini EDTA-free Protease Inhibitor Cocktail” tablet per 10 ml of lysis buffer. Cells were vortexed and then stored on ice for 10 minutes during which time they were dounced 100 times in a 1 ml dounce homogenizer. Subsequently the cells were centrifuged at maximum speed at 4°C for 30 minutes. The supernatant was transferred to a new tube and aliquots were taken, combined with Laemmli buffer, and snapfrozen on dry ice for use as input. The remainder of the supernatant was dialyzed overnight in dialysis buffer (20 mM Tris HCl pH 7.5, 150 mM NaCl, 2 mM beta-mercaptoethanol, 10% glycerol, and 0.5 mM EDTA). After dialysis, the material was pre-cleared with DEAD-dextran and Protein G Sepharose 4 Fastflow for 4 hrs. The material was centrifuged at maximum speed at 4°C for 10 minutes and the supernatant was transferred to a new tube to which anti-DDKaffinity resin (Pierce) was added. The tubes were allowed to rotate overnight at 4°C. The next day, the material was centrifuged (4°C, 500 x g) and the slurry was washed 3x in wash solution (20 mM Tris HCl, pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.01% NP40, and 1 mM DTT.) Subsequently, the material was eluted from the slurry by incubation for 6.5 hrs in 3M NaSCN. The material was combined with 4x Laemmli buffer, boiled for 5 minutes, and loaded on a 10% SDS-Page gel. The gel was transferred to activated PVDF membrane overnight at 55V at 4°C. Subsequently, the membrane was blocked in 5% non-fat dry milk in PBS-Tween (0.1%) and then probed in 5% NFDM with the various antibodies indicated, always per the manufacturer’s instructions. Antibodies used were anti-YTHDF3 (Santa Cruz Biotechnology), anti-YTHDC1 (Cell Signaling). Appropriate secondary antibodies were conjugated with horse radish peroxidase (HRP). Western blots were stripped between probings by incubating the membrane at 50°C for 15 minutes with gentle rocking in stripping solution (100 mM beta-mercaptoethanol, 2% sodium dodecyl sulfate, 62.5 mM Tris-HCl pH 6.7) prior to reblocking in 5% non-fat dry milk in PBS-Tween (0.1%) and then exposure to the next antibody.
DRIPseq.
We followed a published DRIP-seq protocol (Sanz and Chédin, 2019). Briefly, we cultured Rosacre Mettl3F/F cells with and without 4-hydroxytamoxifen and LPS for 24 hours, stimulated with LPS+IL-4 for 2 days, then performed DNA extraction and digested using a cocktail of restriction enzymes (BamHI, EcoRI, HindIII, NcoI, SpeI and XbaI) and Rnase III (3U) overnight at 37°C in buffer 2.1 (NEB); RNase H (NEB) was added to the negative controls. Then we performed S9.6 IP (using 10 μg of S9.6 antibody per IP) and eluted. The immunoprecipitated DNA fragments were validated by qPCR at the Sμ region. DNA fragments were sonicated using the Bioruptor plus sonication system (Diagenode, 3 pulses, low intensity, 30 sec) before library preparation. DNA concentration was evaluated using the Qubit™ 1X dsDNA HS Assay Kit (Life Technologies). DNA fragments were prepared for the final library following the protocol for the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina, manufactured by NEB. Adaptor-ligated and final DNA products were cleaned up using AMPure XP beads (Beckman Coulter). Barcoding was performed using NEBNext® Multiplex Oligos from Illumina® (NEB).
LAM-HTGTS.
Rosacre Mettl3F/F B splenocytes were stimulated with LPS and with and without tamoxifen for 24 hrs, followed by stimulation with LPS and IL-4 for 3 days prior to DNA extraction. The LAM-HTGTS method was adapted from the previously published protocol (Hu et al., 2016). Briefly, genomic DNA was sonicated with 3 pulses of 5 sec and 25% amplitude using the Sonic dismembrator model 500 (Fisher Scientific). LAM-PCR was performed using Phusion High-Fidelity DNA Polymerase (Thermo Scientific) and Sμ 5’ biotinylated bait. 8 independent PCRs were run on ~5 μg of sonicated DNA each, with 90 cycles of primer elongation. These biotinylated DNA fragments were purified using Dynabeads C1 streptavidin beads (Invitrogen) at room temperature, on a rotisserie, overnight. On bead ligation was performed with T4 DNA ligase (NEB) and annealed bridge adaptors for 1 h at 25°C, 2 h at 22°C, and overnight at 16°C. 15 cycles of nested PCR were performed on the DNA-bead complexes using an Sμ nested primer and a reverse universal primer I7. These pairs of primers also contain barcodes and adaptors for the next steps in the procedure. PCR products were cleaned with the QIAquick Gel Extraction Kit (Qiagen) and incubated with NspI (NEB) for 1 h at 37°C to digest and remove germline DNA. The final tagged PCR was performed using P5-I5 and P7-I7 primers for 15 cycles and purified using Select-a-Size DNA Clean & Concentrator Kit (Zymo Research) with a cutoff of 200 bp. Library quality was evaluated by Bioanalyzer (Agilent) and sequencing performed using the Illumina Miseq Reagent Kit v2 (500 cycles) following the manufacturer’s instructions.
We used FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to examine the sequencing quality and Cutadapt (Martin, 2011) to remove adapter sequences at the 3’ end of the raw reads. The Transloc pipeline (Hu et al., 2016) was used with default parameters to obtain all possible translocation events. The primer sequences for HTGTS are provided in Table S3.
Oxford Nanopore sequencing.
Rosacre Exosc3C/C and Rosacre Exosc3+/+ B splenocytes were stimulated with LPS and tamoxifen for 24 hrs, followed by stimulation with LPS and IL-4 for 48 hrs prior to RNA extraction. Total RNA was sent to De Novo Genomics for library preparation, direct RNA sequencing, and analysis.
Direct RNA sequencing:
Total RNA (30μg) was isolated from the samples labelled above, rRNA (NEBNext® rRNA Depletion Kit) and poly(A) (NEBNext® Poly(A) mRNA Magnetic Isolation Module) depleted using manufacturer’s protocol. rRNA/poly(A) depleted RNA was then poly(A) tailed using E. coli Poly(A) Polymerase (NEB, M0276) following the manufacturer’s protocol. Direct RNA sequence libraries were prepared using Direct RNA sequencing Kit (ONT) using the manufacturer’s protocol. Direct RNA sequencing libraries were sequenced using R.9.5.1 RevD Flow cells (ONT) for 48 hrs on a GridIONx5 (ONT) platform.
Transcript database generation:
Illumina short-read sequencing from samples were trimmed of known sequencing adaptors and low-quality scores using BBMap bbduk with the following settings; qtrim=lr trimq=20 minlen=75 ktrim=r k=23 mink=11 hdist=1 (Bushnell, 2014). Processed reads were aligned to mouse genome and transcriptome GRCm38 (Cunningham et al., 2019) using STAR (v2.7.5a) with the following settings; --twopassMode Basic –sjdbGTFfile (Dobin et al., 2013). The splice junction database output from STAR was then used with FLAIR (v1.5.1) to identify RNA transcripts in the direct RNA sequencing data using default settings (Tang et al., 2020). Transcripts FASTA sequences were extracted from the final GTF file outputted from FLAIR using GFFRead (Pertea and Pertea, 2020).
RNA modification detection:
Transcripts generated from FLAIR were used as the reference sequence for Tombo (v1.5.1) (Stoiber et al., 2017). Tombo resquiggle command was run using the transcript database generated from FLAIR. To detect modifications, the Rosacre Exosc3+/+ sample was compared to the Rosacre Exosc3C/C and Rosacre Mettl3F/F with the following Tombo commands; detect_modifications level_sample_compare. The positions of potential modified m6A sites were extracted using Tombo with the following command; text_output browser_files -motif-description “DRACH:3:m6A”. Potential modified DRACH transcript coordinates were converted back to genome coordinates using R Bioconductor packages GenomicFeatures and rtracklayer (Lawrence et al., 2009; Lawrence et al., 2013).
HiC.
The in situ Hi-C protocol from the 4DN Nucleosome Consortium was followed. Briefly, five million cells were crosslinked for each experiment and DpnII was used for DNA digestion followed ligation. To shear the DNA after ligation, ultra-sonication (Sonic dismembrator model 500 (Fisher Scientific)) was used with 10 timed on-pulses of 5 sec. each. The Hi-C library was prepared from Mettl3Δ/Δ and Mettl3WT B cells.
Raw fastq files were demultiplexed by Barcode Splitter (version 0.18.0) (Leach and Parsons, 2017) with parameter “--mismatches 2”. Read pairs with barcode detected in at least one end were retained and subject to adapter trimming by Cutadapt (Martin, 2011) with parameters “--trim-n -q 10,10 -u 6 -U 6 -m 5 -e 0.1”. Juicer pipeline (Durand et al., 2016b) was employed to align the chimeric reads to the reference genome (mm9), build the Hi-C map which can be visualized by Juicebox (Durand et al., 2016a), and detect the significant loops and contact domains genome-wide. Vanilla coverage (VC) (Lieberman-Aiden et al., 2009)-normalized observation over expectation (O/E) values were used to compare the contact intensity between genomic regions in different conditions and to quantify the intensity of chromatin contacts.
3D-STORM Super-Resolution Sample Preparation.
CH12F3 B cells and splenic B cells from various genotypes were prepared using CD43 microbead (Miltenyi Biotec) negative selection and cultured in RPMI 1640 containing 15% FBS (for primary cell culture) (10% for CH12F3 cell lines), 1x non-essential amino acids, 1x sodium pyruvate, 2 mM L-glutamine, 55μM β-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin and 25 mM HEPES. Isolated B cells were cultured with 20 μg ml−1 LPS (Sigma) and 20ng ml−1 IL4 (R&D system) for three days. B cells from days 1 and 3 were immobilized on MatTek 35mm high precision glass bottom culture dishes (P35G-0.170-14C) (previously washed and coated with poly-L-lysine (Sigma P4707) for 5 minutes before being washed and allowed to air dry) and subsequently fixed with 4% paraformaldehyde (Electron Microscopy Sciences) and 0.1% glutaraldehyde (Electron Microscopy Sciences) in PBS for 10 min. at room temperature, washed three times in 1x PBS (without calcium or magnesium), and then blocked and permeabilized in 0.1% (v/v) Triton X-100 supplemented with 2% normal goat serum (NGS) in 1x PBS for 1 hour and immunostained with primary antibodies overnight in 0.1% NGS and 0.05% Triton X-100 at 4°C in gentle rocking. Primary antibodies employed were anti-MTR4 (Abcam, rabbit polyclonal), and anti-AID (Basu et al., 2011) (Novus Biological, goat polyclonal). The cells were then washed in PBS three times and stained with photo-switchable secondary antibodies compatible for STORM imaging for 2 hr. Secondary antibodies varied according to the experiment: either donkey anti-goat Alexa fluor 647 and donkey anti-rabbit Alexa fluor 488 (Life Technologies A21447 and A21206, respectively) or donkey anti-rabbit Alexa fluor 647 and donkey anti-goat Alexa fluor 488 (Life Technologies A31573 and A11055, respectively.) Cells were then washed three times in PBS, counterstained with DAPI at a dilution of 1:500 for 10 minutes at room temperature, and then fixed again with 4% paraformaldehyde and 0.1% glutaraldehyde in PBS for 10 min. at room temperature. Cells were stored in PBS after the refixation until imaging. Immediately before STORM imaging, PBS was removed, and freshly prepared imaging buffer was added instead. The imaging buffer consisted of 50mM cysteamine (Sigma), 3% v/v oxyfluor (Oxyrase), 20% v/v sodium DL-Lactate (Sigma) in PBS with pH checked to insure it was between 8–8.5 prior to application to the cells. The two colors (Alexa fluor 488 and Alexa fluor 647) were imaged either simultaneously or sequentially, depending on the version of the NIS-Elements STORM imaging software from Nikon being used. Imaging buffer helped to keep the dye molecules in a transient dark state. Subsequently, individual dye molecules were excited stochastically with a high laser power at their excitation wavelength (488 nm for Alexa fluor 488 or 647 nm for Alexa fluor 647, respectively) to induce blinking on millisecond timescales. STORM images and the correlated high-power confocal stacks were acquired via a CFI Apo TIRF 100 × objective (1.49 NA) on a Nikon Ti-E inverted microscope equipped with a Nikon N-STORM system, an Agilent laser launch system, an Andor iXon Ultra 897 EMCCD (with a cylindrical lens for astigmatic 3D-STORM imaging) camera, and an NSTORM Quad cube (Chroma). This setup was controlled by Nikon NIS-Element AR software with N-STORM module. To obtain images, the field of view was selected based on the live EMCCD image under 488-nm illumination. 3D STORM datasets of 30,000 frames were collected. STORM images were processed to acquire coordinates of localization points using the N-STORM module in NIS-Elements AR software. Identical settings were used for every image. Each localization is depicted in the STORM image as a Gaussian peak, the width of which is determined by the number of photons detected. After the STORM images were acquired, images of the DAPI staining were taken.
3D-STORM Super-Resolution Data Statistical Analysis
Proximity of AID and Mtr4 is calculated by using algorithm “Nearest Neighbors Search” in the MATLAB (Math works). The 50nm range was used to maximize detection of co-localization. Distribution of the interaction between RNA exosome complex and its cofactors were represented in the form of frequency histogram plot by using MATLAB (Math works) software. We compared the distribution of interaction between AID and Mtr4 (nucleus or cytoplasm) by using a paired Student’s t test in MATLAB software. Comparison of the distribution of paired interaction of AID and Mtr4 between two different conditions were performed by one-way ANOVA (Tukey-Kramer test) method in MATLAB software. All of the 3D STORM imaging was performed in three different B cells (from independent experiments) and repeated three or more times. Data are presented as the means ± SEM or SD (indicated in the legend for each figure). Statistical precision measures (mean ± SEM/SD) and statistical significance are reported in the Figures and the Figure Legends when necessary. All error bars indicate SD (p values: ** < 0.01, *** < 0.001).
Cas13-FTO/ALKBH5 fusion protein expression.
Fusion protein constructs (dPspCas13bmFto and dPspCas13b-mAlkbh5) were designed and generated in the pcDNA3.1(+)-P2A-eGFP vector backbone (GenScript). Guide RNAs were designed using the sequence information for the Sμ transcript. For transient transfection experiments, CH12F3 cells were electroporated with both the fusion protein construct and the gRNA construct via Amaxa (Lonza). Cells were then stimulated with LPS, IL-4, and TGFβ for class switch to IgA.
For generation of the Cas13-FTO stable line, the fusion protein construct was linearized by digestion with PvuI and electroporated into CH12F3 cells for integration into the genome. Cells were then bulk sorted twice for GFP+ IgA− cells, then single cell sorted for selection of single clones with stable GFP expression. These cells were then electroporated with the gRNA construct and stimulated with LPS, IL-4, and TGFβ for class switching to IgA. The gRNA sequences for these experiments are provided in Table S3.
SμGLT and m6A reader protein YTHDC1 interaction prediction.
We first predicted the tertiary structure of SμGLT from its primary sequence via program HDOCK (Yan et al., 2020). The 3D structure of YTHDC1 was downloaded from Protein Data Bank (PDB) (Berman et al., 2000) using YTHDC1’s accession code 4R3I. The docking models between the two molecules were generated by HDOCK with intrinsic binding affinity scores. The model with the highest score was selected and visualized using program PyMol (https://pymol.org/).
RNA Immunoprecipitation (RIP).
RIP was performed as described previously (Peritz et al., 2006). Briefly, 5X106 cells were lysed in RIP buffer (100mM KCl, 5mM MgCl2, 10mM HEPES,pH 7.0, 0.5% Nonidet P-40, 1mM DTT, 100 Uml-1 RNase inhibitor, 2mM Vanadyl ribonucleoside complexes solution, 25 μl ml-1 protease inhibitor cocktail). Cell lysate was pelleted by centrifugation at 16,000g for 15 min at 4°C. Each antibody (5μg) was added to each lysate and incubated overnight in a rotator at 4°C. Then 50μl of Protein A/G agarose (Thermo Scientific) was added to each reaction and incubated for 4h on a rotator at 4°C. Beads were washed four times with RIP buffer and another wash four times with RIP buffer including 1M urea. Beads were resuspended in TRI reagent, and RNA was extracted for analysis. For the RIP analysis of the MPP6, anti-FLAG M2 magnetic beads and control beads were used, and 25μl of anti-FLAG M2 magnetic beads or control beads were added to each lysate.
QUANTIFICATION AND STATISTICAL ANALYSIS
N values for each experiment are specified in the figure legends. N represents the number of experiments done (the number of animals or cell clones used) and is described in the figure legends and results section. P values were calculated using Student’s t-test or Welch’s t-test, which is also specified in the figure legends, and the center of calculations is the mean of all values with dispersion values being standard error as specified in the figure legends.
Supplementary Material
Supplemental Table S1 (related with Figure 2): Coordinates of ncRNAs that are expressed at higher levels in Mettl3f/f +OHT B cells, in comparison to Mettl3f/f −OHT B cells.
Supplemental Table S2 (related with STAR Methods): Nomenclature of mouse and cell lines used in this study.
Supplemental Table S3 (related with STAR Methods): Oligonucleotides used in this study for generating mutant CH12F3 clones, dCas13-FTO mediated Sμ GLT knockdown and for HTGTS experiments.
KEY RESOURCES TABLE
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| anti-IgA | Invitrogen | 12-4204-82 |
| anti-MPP6 | Sigma | HPA026948-100UL |
| anti-RPA32 | Kind gift from Dr. Riccardo Dalla-Favera | N/A |
| anti-AID | Novus Biologicals | NB100-93454 |
| anti-MTR4 | Abcam | ab70551 |
| Donkey anti-goat Alexa fluor 647 | Life Technologies | A21447 |
| Donkey anti-rabbit Alexa fluor 488 | Life Technologies | A21206 |
| Donkey anti-rabbit Alexa fluor 647 | Life Technologies | A31573 |
| Donkey anti-goat Alexa fluor 488 | Life Technologies | A11055 |
| anti-YTHDC1 | Cell Signaling | 87489 |
| anti-YTHDF2 | Novus Biologicals | NBP2-31785 |
| anti-YTHDF3 | Santa Cruz | SC377119 |
| anti-hnRNPC1/C2 | Abcam | AB133607 |
| anti-IgG3 | Fisher Scientific | 553403 |
| anti-IgG1 | Fisher Scientific | 562026 |
| S9.6 | Kerafast | ENH001 |
| anti-m6A | Abcam | ab208577 |
| Normal Rabbit IgG | Cell Signalling | 2729S |
| Beta-Actin | Santra Cruz | sc-47778 |
| Strep-Tactin | IBA Lifesciences | 2-1502-001 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| VPD450 | BD Bioscience | 562158 |
| 4-OHT | Sigma-Aldrich | H6278 |
| LPS | Sigma-Aldrich | L4130 |
| IL-4 | R&D Systems | 404-ML-010 |
| CD40L | BD Bioscience | 553721 |
| TGFb | R&D Systems | 240-B-010 |
| Lipofectamine2000 | Invitrogen | 11668019 |
| anti-DDK-affinity resin | Pierce | A36801 |
| Poly-L-lysine | Sigma-Aldrich | P4707 |
| Cysteamine | Sigma-Aldrich | M9768 |
| Oxyfluor | Oxyrase | OF0005 |
| DL-Lactate | Sigma-Aldrich | L1375 |
| Phusion high-fidelity polymerase | Thermo Scientific | F530 |
| T4 DNA ligase | NEB | M0202S |
| E. coli Poly(A) Polymerase | NEB | M0276 |
| Protease inhibitor cocktail tablets | Roche | 11873580001 |
| HA Epitope Tag Ab | Thermo Scientific | 26181 |
| Protein A/G agarose | Thermo Scientific | 20421 |
| Micro particles based on polystyrene, Magnetic | Sigma-Aldrich | 49664-5ML |
| Vanadyl ribonucleoside complexes | Sigma-Aldrich | 94742-1ML |
| anti-FLAG M2 magnetic beads | Sigma-Aldrich | M8823-1ML |
| Q5® Site-Directed Mutagenesis Kit | NEB | E0554S |
| SuperScript IV | Thermo Scientific | 18091050 |
| TRI Reagent | Sigma-Aldrich | T9424-200ML |
| Ambion RNase III | Thermo Fisher | AM2290 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP | Addgene | 48138 |
| pGEM-T Easy | Promega | A1360 |
| (human)MPP6-myc-DDK | Origene | RC205667 |
| pC0043-PspCas13b crRNA backbone | Addgene | 103854 |
| dPspCas13b-mFto plasmid | This paper | N/A |
| dPspCas13b-mAlkbh5 plasmid | This paper | N/A |
| pcDNA3.1(+)-P2A-eGFP backbone | GenScript | N/A |
| NEB® Stable Competent E. coli | NEB | C3040I |
| hYTHDC1 cDNA Plasmid | R&D Systems | RDEH3421-3xHA |
| Oligonucleotides | ||
| See Supplementary table S3 | N/A | N/A |
| Critical Commercial Assays | ||
| CD43 magnetic beads | Miltenyi biotec | 130-049-801 |
| Mouse B Cell Nucleofector Kit | Lonza | VPA-1010 |
| Qubit 1X dsDNA HS Assay Kit | Invitrogen | Q33230 |
| NEBNext® Ultra II DNA Library Prep Kit for Illumina | NEB | E7645S |
| AMPure XP Beads | Beckman Coulter | A63880 |
| NEBNext® Multiplex Oligos from Illumina | NEB | E6609S |
| Magna MeRIP™ m6A Kit | EMD-Millipore | 17-10499 |
| Dynabeads C1 streptavidin beads | Invitrogen | 65001 |
| QIAquick Gel Extraction Kit | Qiagen | 28704 |
| Select-a-size DNA Clean & ConcentratorKit | Zymo Research | D4080 |
| NEBNext® rRNA Depletion Kit | NEB | E6310 |
| NEBNext® Poly(A) mRNA Magnetic Isolation Module | NEB | E7490S |
| Direct RNA sequencing Kit | ONT | SQK-RNA002 |
| Experimental Models: Cell lines | ||
| HEK293T | Kind gift from Dr. Stephen Goff | N/A |
| CH12F3 | Nakamura et al., 1996 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mettl3 f/f | Kind gift from Dr. Jacob Hanna | N/A |
| Exosc10COIN/COIN | Pefanis et al., 2014 | N/A |
| Mb1cre | Kind gift from Dr. Michael Reth | N/A |
| Rosa26creERT2 | Kind gift from Dr. Pierre Chambon | N/A |
| AID−/− | Kind gift from Dr. Tasuku Honjo | N/A |
| Software and Algorithms | ||
| MATLAB (R2020b) | N/A | https://www.mathworks.com/products/matlab.html |
| Hisat2 (v2.1.0) | Kim et al., 2015 | N/A |
| StringTie (v1.3.3b) | Pertea et al., 2015 | N/A |
| IGV | N/A | http://software.broadinstitute.org/software/igv/ |
| Homer | Heinz et al., 2010 | N/A |
| FastQC | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt | Martin, 2011 | N/A |
| Transloc pipeline | Hu et al., 2016 | N/A |
| BBMap | Bushnell, 2014 | https://sourceforge.net/projects/bbmap/ |
| STAR (v2.7.5a) | Dobin et al. 2013 | N/A |
| FLAIR (v1.5.1) | Tang et al., 2020 | N/A |
| GFFRead | Pertea and Pertea, 2020 | N/A |
| Tombo (v1.5.1) | Stoiber et al., 2017 | N/A |
| GenomicFeatures | Lawrence et al., 2013 | N/A |
| rtracklayer | Lawrence et al., 2009 | N/A |
| Barcode Splitter (v0.18.0) | Leach and Parsons, 2017 | https://bitbucket.org/princeton_genomics/barcode_splitter |
| Juicer | Durand et al., 2016b | N/A |
| Juicebox | Durand et al., 2016a | N/A |
| Vanilla Coverage | Lieberman-Aiden et al., 2009 | N/A |
| Prism 6 software (GraphPad) | N/A | https://www.graphpad.com/scientific-software/prism/ |
| Deposited Data | ||
| Next-Generation Sequencing data | This paper | PRJNA693506 |
| Mendeley data: uncropped western blot and microscopy images | This paper | https://data.mendeley.com/datasets/y4cpkdz77d/1 |
| Nanopore Seq data | This paper | http://52.20.87.23/public/m6A_project/Nanopore-seq/index.html |
HIGHLIGHTS.
N6 methyladenosine (m6A) guides IgH lncRNA SμGLT 3’ end processing by RNA exosome.
M6A RNA modification is important for IgH class switch recombination
M6A RNA modification is important of genomic stability of B cells.
YTHDC1 and MPP6 guide m6A recognition of G-rich SμGLT.
ACKNOWLEDGEMENTS
We thank Dr. Tasuku Honjo (Kyoto University) for providing CH12F3 cells and AID−/− mice, Dr. Michael Reth (University of Freiburg) for Mb1-Cre mice, Dr. Frederick Alt (Harvard University) for HTGTS protocol and guidance, Dr. Chuan He (University of Chicago) for discussions and the dCas13b construct, the Columbia Genome Center for high-throughput genome sequencing, the Department of Microbiology and Immunology core FACS facility and HICCC core microscopy facility. This work was supported by grants to U.B. (NIAID (1R01AI099195, 1RO1 AI143897-01A1 and R01AI134988)), Leukemia & Lymphoma Society, and the Pershing Square Sohn Cancer Research Alliance and L.N. (NIAID (5T32AI10671)).
Footnotes
DECLARATION OF INTEREST
Authors declare no competing interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abakir A, Giles TC, Cristini A, Foster JM, Dai N, Starczak M, Rubio-Roldan A, Li M, Eleftheriou M, Crutchley J, et al. (2020). N 6-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nature genetics 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansel KM (2013). RNA regulation of the immune system. Immunol Rev 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U, Meng F.-l., Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR., Wesemann DR., et al. (2011). The RNA Exosome Targets the AID Cytidine Deaminase to Both Strands of Transcribed Duplex DNA Substrates. Cell 144, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000). The Protein Data Bank. Nucleic Acids Res 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B. (2014). BBMap (sourceforge.net).
- Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, Schurer A, Gourkanti S, Brüning JC, Vu LP, et al. (2019). m 6 A RNA Methylation Maintains Hematopoietic Stem Cell Identity and Symmetric Commitment. Cell Reports 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho Y-J, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. (2011). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, Du P, Kim W, Tang S, Sliz P, et al. (2018). mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, et al. (2019). Ensembl 2019. Nucleic Acids Res 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D., Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016a). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell systems 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016b). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, Hartley JL, Crow YJ, and Stetson DB (2014). The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat Immunol 15, 839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk S, Bonneau F, Ebert J, Kögel A, and Conti E. (2017). Mpp6 Incorporation in the Nuclear Exosome Contributes to RNA Channeling through the Mtr4 Helicase. Cell Reports 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Seija N, Di Noia JM, and Martin A. (2020). AID in Antibody Diversification: There and Back Again. Trends Immunol 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish L, Navickas A, Culbertson B, Xu Y, Nguyen HCB, Zhang S, Hochman M, Okimoto R, Dill BD, Molina H, et al. (2019). Nuclear TARBP2 Drives Oncogenic Dysregulation of RNA Splicing and Decay. Mol Cell 75, 967–981 e969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, and Alt FW (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature biotechnology 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S., Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Heck AM, and Wilusz CJ (2019). Small changes, big implications: The impact of m(6)A RNA methylation on gene expression in pluripotency and development. Biochim Biophys Acta Gene Regul Mech 1862, 194402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, and Reth M. (2006). Testing gene function early in the B cell lineage in mb1-cre mice. Proceedings of the National Academy of Sciences 103, 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Meyers RM, Dong J, Panchakshari RA, Alt FW, and Frock RL (2016). Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nature Protocols 11, 853–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazadi D, Lim J, Rothschild G, Grinstein V, Laffleur B, Becherel O, Lavin MJ, and Basu U. (2020). Effects of senataxin and RNA exosome on B-cell chromosomal integrity. Heliyon 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim C, Kazadi D, Rothschild G, and Basu U. (2013). Regulation of AID, the B-cell genome mutator. Genes Dev 27, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-K., Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activityregulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. (2011). Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffleur B, Lim J, Zhang W, Chen Y, Pefanis E, Bizarro J, Batista CR, Wu L, Economides AN, Wang J, et al. (2021). Noncoding RNA processing by DIS3 regulates chromosomal architecture and somatic hypermutation in B cells. Nature genetics 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, and Carey V. (2009). rtracklayer: an R package for interfacing with genome browsers. Bioinformatics (Oxford, England) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach R, and Parsons L. (2017). Barcode Splitter, version 0.18.0 (Princeton, NJ: ). [Google Scholar]
- Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, Hanna JH, and Ding L. (2019). Stage-specific requirement for Mettl3 -dependent m 6 A mRNA methylation during haematopoietic stem cell differentiation. Nature Cell Biology 21, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H-B, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y, et al. (2017). m 6 A mRNA methylation controls T cell homeostasis by targeting the IL7/STAT5/SOCS pathways. Nature 548, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, and Rosenfeld MG (2016). Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet 17, 207–223. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E.,van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Giri PK, Kazadi D, Laffleur B, Zhang W, Grinstein V, Pefanis E, Brown LM, Ladewig E, Martin O, et al. (2017). Nuclear Proximity of Mtr4 to RNA Exosome Restricts DNA Mutational Asymmetry. Cell 169, 523–537.e515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SJ, Boyle PJ, Chinen M, Dale RK, and Lei EP (2013). Genome-wide localization of exosome components to active promoters and chromatin insulators in Drosophila. Nucleic Acids Res 41, 2963–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Jung S, and Radbruch A. (1995). Switch transcripts in immunoglobulin class switching. Science 267. [DOI] [PubMed] [Google Scholar]
- Lubas M, Christensen Marianne S., Kristiansen Maiken S., Domanski M, Falkenby Lasse G., Lykke-Andersen S, Andersen Jens S., Dziembowski A, and Jensen Torben H. (2011). Interaction Profiling Identifies the Human Nuclear Exosome Targeting Complex. Molecular Cell 43, 624–637. [DOI] [PubMed] [Google Scholar]
- Makino DL, Schuch B, Stegmann E, Baumgärtner M, Basquin C, and Conti E. (2015). RNA degradation paths in a 12-subunit nuclear exosome complex. Nature 524. [DOI] [PubMed] [Google Scholar]
- Marin-Vicente C, Domingo-Prim J, Eberle AB, and Visa N. (2015). RRP6/EXOSC10 is required for the repair of DNA double-strand breaks by homologous recombination. J Cell Sci 128, 1097–1107. [DOI] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17, 10–12. [Google Scholar]
- Meyer KD, and Jaffrey SR (2017). Rethinking m 6 A Readers, Writers, and Erasers. Annu Rev Cell Dev Biol 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo (2000). Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102. [DOI] [PubMed] [Google Scholar]
- Nair L, Chung H, and Basu U. (2020). Regulation of long non-coding RNAs and genome dynamics by the RNA surveillance machinery. Nature Reviews Molecular Cell Biology 21, 123136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefanis E, and Basu U. (2015). RNA Exosome Regulates AID DNA Mutator Activity in the B Cell Genome. Adv Immunol 127, 257–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, Economides AN, and Basu U. (2014). Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature 514, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefani E., Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu Z-P, et al. (2015). RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell 161, 774–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peritz T, Zeng F, Kannanayakal TJ, Kilk K, Eiríksdóttir E, Langel U, and Eberwine J. (2006). Immunoprecipitation of mRNA-protein complexes. Nature protocols 1. [DOI] [PubMed] [Google Scholar]
- Pertea G, and Pertea M. (2020). GFF Utilities: GffRead and GffCompare. F1000Research 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, and Jensen TH (2008). RNA exosome depletion reveals transcription upstream of active human promoters. Science 322, 1851–1854. [DOI] [PubMed] [Google Scholar]
- Puno MR, and Lima CD (2018). Structural basis for MTR4-ZCCHC8 interactions that stimulate the MTR4 helicase in the nuclear exosome-targeting complex. Proceedings of the National Academy of Sciences of the United States of America 115, E5506–E5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, and Gack MU (2020). RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro de Almeida C, Dhir S, Dhir A, Moghaddam AE, Sattentau Q, Meinhart A, and Proudfoot NJ (2018). RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol Cell 70, 650–662 e658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, and Chang HY (2012). Genome regulation by long noncoding RNAs. Annu Rev Biochem 81, 145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, Ellyard J, Shen Q, Robert PA, Cappello J, et al. (2019). Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild G, and Basu U. (2017). Lingering Questions about Enhancer RNA and Enhancer Transcription-Coupled Genomic Instability. Trends Genet 33, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, and He C. (2017a). Dynamic RNA Modifications in Gene Expression Regulation. Cell 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA.,Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P, et al. (2017b). YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, and Chédin F. (2019). High-resolution, strand-specific R-loop mapping via S9.6based DNA–RNA immunoprecipitation and high-throughput sequencing. Nature Protocols 14, 1734–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz DG, and Ji Y. (2011). Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol 11, 251–263. [DOI] [PubMed] [Google Scholar]
- Schilders G, Raijmakers R, Raats JM, and Pruijn GJ (2005). MPP6 is an exosomeassociated RNA-binding protein involved in 5.8S rRNA maturation. Nucleic Acids Res 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S. (2016). Cracking the epitranscriptome. RNA 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, and Sharp PA (2008). Divergent transcription from active promoters. Science 322, 1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, and Meng FL (2021). Repair of programmed DNA lesions in antibody class switch recombination: common and unique features. Genome instability & disease. [DOI] [PMC free article] [PubMed]
- Shi H, Liu B, Nussbaumer F, Rangadurai A, Kreutz C, and Al-Hashimi HM (2019). NMR Chemical Exchange Measurements Reveal That N6-Methyladenosine Slows RNA Annealing. J Am Chem Soc 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, and Alt FW (2003). The influence of transcriptional orientation on endogenous switch region function. Nature Immunology 4. [DOI] [PubMed] [Google Scholar]
- Stoiber M, Quick J, Egan R, Lee JE, Celniker S, Neely RK, Loman N, Pennacchio LA, and Brown J. (2017). De novo Identification of DNA Modifications Enabled by GenomeGuided Nanopore Signal Processing.
- Tang AD.,Soulette CM, van Baren MJ, Hart K, Hrabeta-Robinson E, Wu CJ, and Brooks AN (2020). Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, and Vuica-Ross M. (2011). RNase H and Multiple RNA Biogenesis Factors Cooperate to Prevent RNA:DNA Hybrids from Generating Genome Instability. Molecular Cell 44, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmuth EV, Zinder JC, Zattas D, Das M, and Lima CD (2017). Structure and reconstitution of yeast Mpp6-nuclear exosome complexes reveals that Mpp6 stimulates RNA decay and recruits the Mtr4 helicase. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick E-M, Puno MR, Januszyk K, Zinder JC, DiMattia MA, and Lima CD (2018). Helicase-Dependent RNA Decay Illuminated by a Cryo-EM Structure of a Human Nuclear RNA Exosome-MTR4 Complex. Cell 173, 1663–1677.e1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Tao H, He J, and Huang S-Y (2020). The HDOCK server for integrated protein–protein docking. Nature Protocols 15, 1829–1852. [DOI] [PubMed] [Google Scholar]
- Yu K, Chédin F, Hsieh C-L, Wilson TE, and Lieber MR (2003). R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology 4, 442–451. [DOI] [PubMed] [Google Scholar]
- Yue Y, Liu J, and He C. (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S, Ries RJ, and Jaffrey SR (2019). Reading, writing and erasing mRNA methylation. Nature Reviews Molecular Cell Biology 20, 608–624. [DOI] [PubMed] [Google Scholar]
- Zhang C, Chen L, Peng D, Jiang A, He Y, Zeng Y, Xie C, Zhou H, Luo X, Liu H, et al. (2020). METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Molecular Cell 79. [DOI] [PubMed] [Google Scholar]
- Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, and Chaudhuri J. (2015). Non-coding RNA Generated following Lariat Debranching Mediates Targeting of AID to DNA. Cell 161, 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Zhang L, Cui XL, Yu X, Hsu PJ, Lyu R, Tan H, Mandal M, Zhang M, Sun HL, et al. (2020). Control of Early B Cell Development by the RNA N 6-Methyladenosine Methylation. Cell Reports 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 (related with Figure 2): Coordinates of ncRNAs that are expressed at higher levels in Mettl3f/f +OHT B cells, in comparison to Mettl3f/f −OHT B cells.
Supplemental Table S2 (related with STAR Methods): Nomenclature of mouse and cell lines used in this study.
Supplemental Table S3 (related with STAR Methods): Oligonucleotides used in this study for generating mutant CH12F3 clones, dCas13-FTO mediated Sμ GLT knockdown and for HTGTS experiments.
Data Availability Statement
All RNA-seq, DRIP-seq, HTGTS and HiC data has been provided in NIH repository with accession number PRJNA693506.
Original codes were not generated in this study.
Any additional information required to reanalyze the data reported in this work will be available from the Lead Contact upon request.