

Summary
Here, we describe a detailed step-by-step protocol for the expression, purification, quantification, and activity determination of key enzymes for molecular detection of pathogens. Based on previous reports, we optimized the protocol for LbCas12a, Taq DNA polymerase, M-MLV reverse transcriptase, and TEV protease to make it compatible with minimal laboratory equipment, broadly available in low- and middle-income countries. The enzymes produced with this protocol have been successfully used for molecular detection applications.
For complete details on the use and execution of this protocol, please refer to Alcántara et al. (2021a, 2021b).
Subject areas: Health Sciences, Microbiology, Molecular Biology, CRISPR, Protein Biochemistry, Protein expression and purification, Biotechnology and bioengineering
Graphical abstract
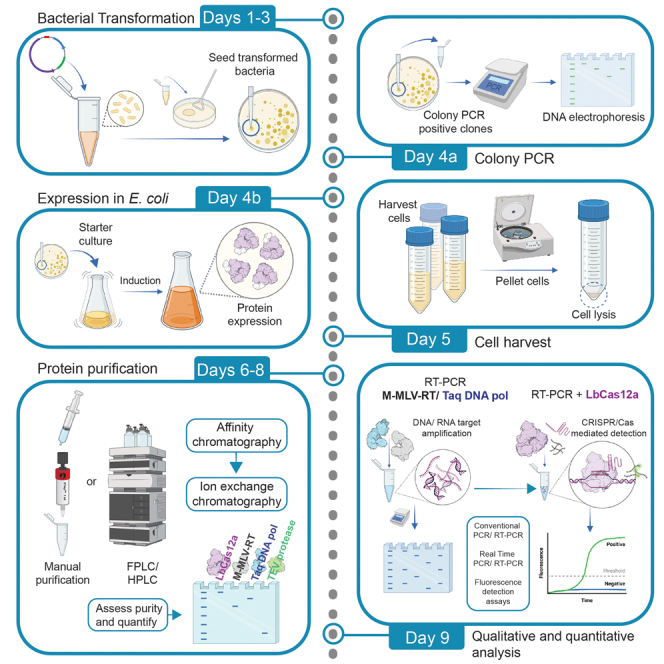
Highlights
-
•
Open-source and low-cost production schemes of Taq, M- MLV RT, and Cas12a enzymes
-
•
Compatible with basic equipment to produce enzymes for molecular detection
-
•
Enzyme preparations validated for SARS-CoV-2 detection
Here, we describe a detailed step-by-step protocol for the expression, purification, quantification, and activity determination of key enzymes for molecular detection of pathogens. Based on previous reports, we optimized the protocol for LbCas12a, Taq DNA polymerase, M-MLV reverse transcriptase, and TEV protease to make it compatible with minimal laboratory equipment, broadly available in low- and middle-income countries. The enzymes produced with this protocol have been successfully used for molecular detection applications.
Before you begin
This segment of the protocol describes the methodology to obtain enzyme-expressing E. coli for key recombinant proteins for use in molecular detection of pathogens. To obtain competent cells use your technique of choice. Here, the Mix & Go E.coli Transformation kit (Zymo Research, Cat. No.: T3002) was used. The E. coli BL21(DE3) strain was used for protein expression.
Note: For the composition and recipes of buffers and media, see Data S1.
Obtaining enzyme-expressing E. coli
Timing: three days
Note: pRK793 plasmid encoding TEV protease in E.coli BL21(DE3) pRIL cells is commercially available (Addgene, Cat. No.: 8827) (Kapust et al., 2001); thus, the transformation step can be omitted.
-
1.
Media preparation
The following media volumes are required to obtain enzyme-expressing E. coli, including the experimental assay (i.e., transformation with the plasmid of interest), positive and negative controls. Prepare Super Optimal Broth (SOB) and Super Optimal broth with Catabolite repression (SOC) media for bacterial transformation, and Luria-Bertani (LB) medium for bacterial growth under antibiotic selection (See Data S1 for media recipes).-
a.SOB medium: Prepare a volume of 50 mL. Dispense and sterilize 10 mL aliquots. Store at 4°C for no more than one month.
-
b.SOC medium: Prepare it fresh for the transformation process. Do not store. Add 400 μL of 10% glucose to 9.6 mL of SOB medium, to get a final concentration of 0.4% glucose.
-
c.LB medium:
-
i.Taq and RT: Prepare 250 mL LB agar with 30 μg/mL Kanamycin (Kan) + 17 μg/mL Chloramphenicol (Cam) for 10 plates.
-
ii.Cas12a: Prepare 150 mL LB agar with 100 μg/mL Ampicillin (Amp) for 6 plates.
-
iii.Tev: Prepare 150 mL LB agar with 100 μg/mL Amp + 30 μg/mL Cam for 6 plates.
-
i.
-
a.
-
2.
Bacterial transformation
Here we used the Mix & Go E.coli Transformation kit (Zymo Research, Cat. No. T3002) which allows the preparation of chemically competent cells and highly efficient DNA transformation with a brief heat shock at 37°C (i.e., at the SOC addition step, as detailed below). However, other heat shock transformation alternatives as well as electroporation can be performed instead.Note: Pre-warm SOC medium for 1 hour at 37°C before use.-
a.Transform 100 μL of competent E. coli BL21(DE3) cells. Use with 10 ng of the corresponding plasmid DNA (try to keep volume of DNA less than 5% of the total volume).
-
b.Mix gently and incubate on ice for 5–10 min.
-
c.Add four volumes of pre-warmed (at 37°C for about 1 h) SOC medium (400 μL of SOC for 100 μL of competent cells).
-
d.Incubate at 37°C for 1 h with gentle mixing (200–300 RPM) to let transformed cells recover from the treatment and begin to express the plasmid-encoded antibiotic resistance gene (Figure 1).
 Pause point: During the one-hour incubation in step (d), pre-warm the LB agar plates with the corresponding antibiotics for 1 hour at 37°C.
Pause point: During the one-hour incubation in step (d), pre-warm the LB agar plates with the corresponding antibiotics for 1 hour at 37°C. -
e.Plate 50–100 μL of each transformation tube in the pre-warmed LB agar plates (at 37°C for 1 h) with the corresponding antibiotics. Use an L-shaped loop to spread evenly (Figure 1).
-
f.Incubate at 37°C for 14–16 h. If just a few or none transformed colonies did grow on the LB plate, see troubleshooting section – problems 1 and 2.Note: Maintain sterility throughout the plating process. The working surface and the L-shaped loop can be both sterilized with 70% ethanol or using a Bunsen burner. Disposable sterile L-shaped loops can also be used.Note: Include the following negative controls: empty plate (plate control for contamination), untransformed cells (transformation control), and untransformed cells in plates without the selection antibiotic (cell viability control). If available, use a positive control for transformation with any available plasmid carrying the corresponding resistance marker gene.Alternatives: Many cells are provided with plasmids which could be used as positive controls for transformation.Note: Isolated colonies must be observed, otherwise see the troubleshooting section – problems 1 and 2.
-
g.Select three to five 2–4 mm well-isolated bacterial colonies to test and mark them on the plate using a permanent marker.
-
h.To confirm transformation, perform colony PCR with specific primers for the plasmid or gene of interest. Pick one-tenth of each selected colony with a sterile pipette tip and dip the tip into the PCR reaction mixture (Figure 1). Include a positive control using a plasmid dilution (5 ng plasmid DNA per PCR reaction).Note: Taking more cells can result in inhibition of the PCR reaction.
Set of primers used for colony PCR:Note: In this protocol, universal primers for the T7 promoter and terminator (polA gene, pol gene and cas12a gene), as well as 5TpET28 and M13F primer (p1 gene) are used for amplification.Primer Sequence (5’ – 3′) Annealing temperature (oC) T7 forward TAATACGACTCACTATAGG 55 T7 terminator GCTAGTTATTGCTCAGCGG 5TpET28 GGAATTGTGAGCGGATAAC 55 M13F GTAAAACGACGGCCAG -
i.Run the following PCR thermocycling program:
PCR cycling conditions
Step Temperature (°C) Time Cycles Initial denaturation 95 3 min 1 Denaturation 95 30 s 20 Annealing 55 30 s Extension 72 -- min∗ Final extension 72 5 min 1 Hold 10 ∞ ∗Extension time varies depending on the indicated gene:polA gene (Taq DNA polymerase): 3 minpol gene (M-MLV Reverse transcriptase): 3 mincas12a gene (LbCas12a): 5 min 30 sp1 gene (TEV protease): 2 min 30 s -
j.Analyze the PCR products by running 5 μL of each PCR sample on a 1% (w/v) agarose gel. Here we used 1 μL of a 6X stock of SafeGreen for visualization of DNA bands. The expected band sizes are polA 2775 bp, pol 2255 bp, cas12a 5200 bp, and p1 2079 bp. A GeneRuler™ 1 kb DNA ladder was used (ThermoFisher) (Figure 1).Alternatives: Any other low-mutagenic equivalent dyes, such as SYBR®Safe, EvaGreen® or non-hazardous non-cytotoxic like GelRed® or GelGreen® can also be used. Alternatively, stain with Ethidium Bromide (EtBr) (0.5 μg/ml final concentration on the agarose gel).
-
k.Spread one verified colony in the respective agar LB agar plate with antibiotics. Incubate at 37°C for 14–16 h. Store the plates at 4°C for back-up until the end of the protein expression workflow.Note: Pick an isolated colony from (k) and start an overnight culture in 50 mL LB with the corresponding antibiotic and incubate at 37°C for 14–16 hours at 150 RPM. From this overnight culture prepare 1500 μL 50% glycerol stocks in 2 mL cryovials (750 μL culture/ 750 μL sterile 100% glycerol). Store at −80°C (Figure 1).
-
a.
Figure 1.

Transformation and screening of bacterial colonies for protein expression in E. coli
The scheme summarizes all steps to obtain and verify by colony PCR the transformed BL21 E. coli cells that express the key molecular biology enzymes Taq DNA Polymerase, M-MLV Reverse Transcriptase, and LbCas12a. Additionally, BL21 cells obtained from Addgene and carrying the plasmid encoding the TEV protease were checked by colony PCR. The TEV protease is required to cleave LbCas12a from the expressing fusion protein MBP-LbCas12a. Ab: antibiotic.
General experimental considerations
-
3.
Consider using fresh or non-long term stored competent E. coli cells. The BL21(DE3)pLysS strain was used in the described protocol. However, other E. coli strains could be evaluated if they are compatible with the expression plasmids.
-
4.
It is important to maintain the cold chain over the course of recombinant enzyme production. If a cold room is available, the chromatography and dialysis steps should be performed in this facility. Alternatively, protein fractions must be kept as much as possible on ice.
-
5.
It is highly recommended to perform the cell lysis, chromatography, and dialysis steps, on the same day. Avoid freezing unpurified protein fractions.
-
6.
Protein expression could be performed with your lab standardized conditions (i.e., temperature, induction time, or IPTG inducer concentration). We recommend performing an analytical/pilot assay before a preparative one to evaluate protein expression performance.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E.coli BL21(DE3)pLysS | Promega | Cat # L1195 |
| Chemicals, peptides, and recombinant proteins | ||
| Ampicillin | Calbiochem | Cat # 171254-5GM |
| Chloramphenicol | Sigma-Aldrich | Cat # C0378-25G |
| Kanamycin | Sigma-Aldrich | Cat # K1876-25G |
| Isopropyl ß-D-1 thiogalactopyranoside | Thermo Fisher Scientific | Cat # R0392 |
| HEPES | Merck Millipore | Cat # 391338-1KG |
| Tryptone | Liofilchem | Cat # 611004 |
| Yeast extract | Liofilchem | Cat # 611005 |
| Glucose | Merck Millipore | Cat # 1083371000 |
| Ammonium acetate | Merck Millipore | Cat # 101116 |
| Potassium chloride | Merck Millipore | Cat # 104936 |
| Magnesium chloride hexahydrate | Merck Millipore | Cat # 105833 |
| Magnesium sulfate | Fisher Scientific | Cat # M63-500 |
| Bromophenol blue | Sigma-Aldrich | Cat # 2830-OP |
| Coomassie Brilliant blue R250 | Sigma-Aldrich | Cat # 1125530025 |
| Absolute ethanol | Sigma-Aldrich | Cat # 1009832500 |
| Glacial acetic acid | Sigma-Aldrich | Cat # 1000631000 |
| Tween 20 | Sigma-Aldrich | Cat # P9416-50ML |
| NP-40 | Merck Millipore | Cat # 492016-100ML |
| TEMED | Merck Millipore | Cat # 8930-100ML |
| Triton X-100 | Sigma-Aldrich | Cat # T8787-50ML |
| Maltose monohydrate | LOBA Chemie | Cat # 04495 |
| ß-mercaptoethanol | Merck Millipore | Cat # 444203 |
| Lysozyme | PanReac Applichem | Cat # A3711 |
| Trehalose | Sigma-Aldrich | Cat # T9449-25G |
| cOmplete® Protease Inhibitor Cocktail | Roche | Cat # 11697498001 |
| Sodium chloride | Merck Millipore | Cat # 106404 |
| Imidazole | Sigma-Aldrich | Cat # I0125 |
| Glycerol 85% | Sigma-Aldrich | Cat # 104094 |
| EDTA | Merck Millipore | Cat # 324503 |
| SureCast™ Ammonium Persulfate (APS) | Invitrogen | Cat # HC2005 |
| Dodecyl sulfate sodium salt | Merck Millipore | Cat # 1137601000 |
| Tris, Hydrochloride, Molecular Biology Grade | Merck Millipore | Cat # US1648317 |
| Tris base ULTROL grade | Calbiochem | Cat # 643811 |
| Dithiothreitol (DTT) | Thermo Fisher Scientific | Cat # R0682 |
| dNTP Mix (10 mM each) | Thermo Fisher Scientific | Cat # R0194 |
| SafeGreen | abm | Cat # G108-G |
| TriTrack DNA loading dye 6X | Thermo Fisher Scientific | Cat # R1161 |
| Agarose powder | Cleaver Scientific | Cat # CSL-AG500 |
| Polyacrylamide (40% acrylamide and bis-acrylamide) | Merck Millipore | Cat # 1006381000 |
| OmniPur Water DEPC treated, sterile, nuclease-free | Sigma-Aldrich | Cat # 9610-1L |
| DNaseI | PanReac Applichem | Cat # A3778 |
| Unstained Protein Standard, Broad Range (10–200 kDa) ladder | New England Biolabs | Cat # P7717S |
| AmpliSize Molecular Ruler | Biorad | Cat # 1708200 |
| GeneRuler 1 kb DNA Ladder | Thermo Scientific | Cat # SM0311 |
| GeneRuler 100 bp DNA Ladder | Thermo Scientific | Cat # SM0242 |
| Unstained Protein Molecular Weight Marker (14.4–116 kDa) ladder | Thermo Scientific | Cat # 26610 |
| Bovine Serum Albumin, lyophilized powder | Sigma-Aldrich | Cat # A9418-100G |
| Critical commercial assays | ||
| BSA protein standard | Sigma-Aldrich | Cat # P0834 |
| RevertAid First Strand cDNA Synthesis kit | Thermo Scientific | Cat # K1622 |
| Standard Taq Buffer | New England Biolabs | Cat # M0273S |
| NEBuffer™ r2.1 (10X) | New England Biolabs | Cat # B6002S |
| Mix&Go! E. coli transformation kit | Zymo Research | Cat # T3002 |
| Oligonucleotides | ||
| ssDNA oligo: T7 promoter forward primer TAATACGACTCACTATAGG | Commercial | N/A |
| ssDNA oligo: T7 terminator reverse primer GCTAGTTATTGCTCAGCGG | Commercial | N/A |
| ssDNA oligo: M13F forward primer GTAAAACGACGGCCAG | Commercial | N/A |
| ssDNA oligo: 5TpET28 reverse primer GGAATTGTGAGCGGATAAC | Commercial | N/A |
| Recombinant DNA | ||
| pRK793 plasmid | Kapust et al., 2001 | Addgene plasmid # 8827 |
| pMBP-LbCas12a Plasmid | Chen et al., 2018 | Addgene plasmid # 113431 |
| pET-28a_6H-MMLV_RT_D524N_-6H | Graham et al., 2021 | https://gitlab.com/tjian-darzacq-lab/bearmix |
| pET-28a_6H-TAQ_E602D | Graham et al., 2021 | https://gitlab.com/tjian-darzacq-lab/bearmix |
| Software and algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| GraphPad Prism version 9 | GraphPad Software, La Jolla, CA | www.graphpad.com |
| Other | ||
| Membrane filter 0.45 μM pore size | Merck | Cat # 10418024 |
| HisTrap FF Crude | Cytiva | Cat # 11000458 |
| HiTrap SP Sepharose FF | Cytiva | Cat # 17505401 |
| HiTrap Capto SP ImpRes | Cytiva | Cat # 17546851 |
| MBPTrap HP | Cytiva | Cat # 28918778 |
| D-TubeTM Dialyzer Maxi, MWCO 12–14 kDa | Merck | Cat # 71510 |
| 96-well flat clear bottom black polystyrene TC-treated microplates | Corning® | Cat # 3603 |
| NanoDrop One microvolume UV-Vis spectrophotometer | Thermo Fisher | Cat # 840-329700 |
| safeVIEW: LED/Blue Light Transilluminator | Cleaver Scientific | Cat # SAFEVIEW |
| Thermal Cycler T100 | BioRad | Cat # 1861096 |
| AkTATM Start | Cytiva | N/A |
| CytationTM 5 Cell Imaging Multi-Mode Reader | BioTek Instruments | N/A |
| SynergyTM H1 Hybrid Multi-Mode Reader | BioTek Instruments | N/A |
| Supplemental data | Mendeley Data | https://doi.org/10.17632/wynyf35xf3.1 |
Materials and equipment
Obtaining enzyme-expressing E. coli
In this protocol expendable materials including microcentrifuge tubes, culture plates, glassware for media preparation, and pipette tips are not included in the key resources table, as these materials must be available in the laboratory. Furthermore, basic equipment of a microbiology and biochemistry laboratory is needed, including an autoclave, bacteria culture incubator and water bath, conventional benchtop microcentrifuge and minicentrifuges, heating block, power source and agarose and SDS-PAGE electrophoresis equipment.
For colony PCR, DNA Taq polymerase, and M-MLV reverse transcriptase enzymatic activity assessment, we used a PCR thermocycler from Bio-Rad.. Alternatively, any PCR thermocycler can be used to corroborate enzymatic activity. For visualization of the PCR products we used SafeGreen dye and a blue light transilluminator (Cleaver Scientific). Other equivalent dyes, such as SYBR®Safe, EvaGreen®, GelRed®, GelGreen® or staining by Ethidium Bromide can also be used. Images from DNA gels, as from protein gels, were obtained using a gel documentation system (Cleaver Scientific), coupled to a digital camera or smartphone, and analyzed with ImageJ.
Expression in E. coli and protein purification
For collecting cells after protein expression, we used a preparative refrigerated centrifuge (Awel) using 50 mL conical tubes. Alternatively, tubes or bottles for larger volumes can be used depending on rotor availability (i.e., Sorvall or Beckman systems). Protein purification was performed using a Fast Protein Liquid Chromatography (FPLC) system (Amersham). Additionally, a manual procedure using disposable syringes of 10 mL is described.
For protein quantification, we used a Nanodrop One spectrophotometer (Thermofisher) and a SynergyTM H1 Hybrid Multi-Mode plate reader (Biotek) for Bradford analysis. The latter was also used for corroborating Cas12a enzymatic activity in the fluorescence detection assay optimized and reported on (Alcántara et al., 2021a, 2021b). Details on bacteria strains used are described within the adequate step and in the key resources table. All reagents and their corresponding identifiers used in this protocol, as the alternatives, have been included in the key resources table. All media preparation instructions for culture and the required buffer recipes for protein purification are described in the Data S1.
Step-by-step method details
The present work describes adapted protocols for the production of LbCas12a (henceforth Cas12a) (Chen et al., 2018; Zetsche et al., 2015), Taq DNA polymerase (Taq) (Chien et al., 1976; Graham et al., 2021; Lawyer et al., 1989), M-MLV reverse transcriptase (RT) (Graham et al., 2021; Kotewicz et al., 1985, 1988), and TEV protease (Tev)(Kapust et al., 2001; Reischl et al., 1997). These enzymes were intended to be used for molecular detection of SARS-CoV-2 viral RNA by an RT-PCR coupled to CRISPR/Cas detection (Alcántara et al., 2021a, 2021b). However, they can be used in a variety of molecular biology applications, such as nucleic acid amplification, DNA target detection, and in vitro DNA digestion. Protocols were adapted to use minimal laboratory equipment, broadly available in low- and middle-income countries.
Expression in E. coli
The following culture and reagent volumes are calculated for one liter (1 L) of protein expression (Figure 2). Scale proportionally if required. Consider additional 200 mL of LB medium to be used as controls (scaling is not required). Sterilize the media by autoclaving at 15 psi and 121°C for 15 min. Wait for the media to cool down to approximately 45°C (approximately 20 min) and proceed to add the corresponding antibiotic.
-
1.Media preparation
-
a.Total medium conditions for Taq and RT enzymes: Prepare 2.2 L of LB broth with 30 μg/mL Kan + 17 μg/mL Cam.
-
b.Total medium conditions for the Cas12a enzyme: Prepare 1.2L of LB broth with 100 μg/mL Amp.
-
c.Total medium condition for the Tev enzyme: Prepare 1.2 L LB broth with 100 μg/mL Amp + 30 μg/mL Cam.
-
a.
-
2.Starter culture
-
a.Transfer a freshly transformed colony using a pipette tip to a 250 mL flask containing 50 mL of LB with appropriate selection antibiotic(s) (see above section: obtaining enzyme-expressing E. coli) (Figure 2).
-
b.Incubate at 37°C for 14–16 h with shaking at 150 RPM.
-
a.
-
3.Induction of protein expression
-
a.Inoculate 1 L of fresh media (LB with the corresponding antibiotics) with the starter culture to reach a starting optical density at 600 nm (OD600) of 0.05.
-
b.Incubate at 37°C with shaking at 150 RPM until OD600 = 0.5 (1.5 h) for Tev and LbCas12a or OD600 = 0.8 for Taq and RT (2 h).
-
c.Take a 200 μL sample for pre-induction monitoring.
-
d.IPTG induction:
-
i.For Taq, Tev, and RT, add IPTG to a final concentration of 1 mM and incubate at 37°C for 3 h at 150 RPM.
-
ii.For Cas12a, incubate on ice or at 4°C for 10–15 min. Add IPTG to a final concentration of 0.2 mM and incubate at 22°C–25°C for 14–16 h at 150 RPM.
-
i.
-
e.For monitoring the induction of protein expression, measure the OD600 and take 200 μL of culture at 2–3-time intervals (each hour for Taq, Tev, and RT). For Cas12a, take a sample before cell harvest (store on ice) (Figure 3A). Keep culture samples on ice.
-
a.
-
4.Cell harvest
-
a.Keep flasks with induced cultures on ice. Use 50 mL conical tubes for collecting cells. Avoid loading more than 45 mL of cell culture in each tube. Centrifuge the culture at 3000 × g for 5 min at 4°C. Discard the supernatant.Alternatives: Tubes or bottles for larger volumes can be used depending on rotor availability (i.e. Sorvall or Beckman systems).
-
b.Weigh the cell pellet using an empty tube to tare the balance.
-
c.Wash cells with 10 mL per gram of cell pellet weight (1:10 volume buffer/ gram cells) using TAKM or HAKM buffer (Data S1). Gently resuspend until the pellet is homogenized.
-
d.Centrifuge at 3000 × g for 15 min at 4°C. Discard the supernatant and weigh the cell pellet.
-
e.Resuspend the cells in 5 mL per gram of cell pellet weight with the indicated buffers prepared with cOmplete™ protease inhibitor cocktail (Use 1 tablet for 50 mL of each buffer) (pre-cooled at 4°C, Data S1):
-
i.RT: MB1 buffer
-
ii.Taq: TaB1 buffer
-
iii.Cas12: HAKM
-
iv.Tev: TeB1 buffer
-
i.
-
f.Add lysozyme at 100 μg/mL final concentration to the resuspended cells and mix.
-
g.Add DNase I at 40 μg/mL final concentration to the resuspended cells and mix.
-
h.Add MgCl2 at 10 mM final concentration to Taq, RT, and Tev resuspended cells and mix.Pause point: Resuspended bacterial cells can be stored at −80°C.
-
a.
-
5.Induction confirmation
-
a.Centrifuge the 200 μL samples obtained before induction and during induction at 3000 × g for 5 min at 4°C. Discard the supernatant (Figure 3A).
-
b.Resuspend the cell pellets with SDS sample buffer (Data S1) as follows:
-
c.Heat the SDS-treated samples at 95°C for 10 min, spin briefly.
- d.
-
e.Run the gel for 5 min at 100 V followed by 55 min at 200 V.
-
f.Stain the gel using Coomassie blue dye or other alternatives available (Sasse and Gallagher, 2009).
-
g.Destain the gel by incubating it in 80 mL of destaining solution (Data S1) at 22°C–25°C with agitation for 3 h. Change the destaining solution several times until most of the Coomassie blue dye is removed from the gel matrix. Proteins will appear as blue bands. If protein bands are not visible, see the troubleshooting section – problem 3.
-
a.
Figure 2.

Protein expression workflow in E. coli
Figure 3.

Analysis of protein expression by SDS-PAGE
(A) The workflow shows the sample collection from pre-induction to up to 3 intervals of post-induction. Cells collected from 200 μL aliquots were lysed and the cell lysates were analyzed by SDS-PAGE gel electrophoresis. Proteins were visualized by Coomassie blue staining. Stained acrylamide gels show M-MLV RT (B), Taq DNA polymerase (C), MBP-LbCas12a fusion protein (D), and the TEV protease (E). The bands corresponding to each protein molecular weight are highlighted by a dotted-stroke box, showing increased band intensity from pre- to post-induction samples. Due to the presence of the auto-cleavage site, two different molecular weight bands are observed on (E), corresponding to the MBP tag protein and TEV protease. The SDS-PAGE gel percentage used is shown in the bottom right corner of the gel picture.
Protein purification
-
6.Cell lysis
-
a.Use two or three freeze-thaw cycles for cell lysis (Figure 4A). Thaw the frozen cell resuspension on ice and manually mix every 3 min. Once thawed, re-freeze it at −80°C for 15 min. Thaw it again at 4°C on ice, mixing every 3 min.Pause point: Take advantage of this time to cool down (4°C) the centrifuge with an appropriate rotor for cell debris removal.
-
b.Proceed as indicated for each protein (Figure 4A):M-MLV reverse transcriptase (RT)
-
i.Adjust NaCl concentration to 370 mM using a 4 M stock. Mix gently.
-
ii.Adjust to 20 mL per gram of cell pellet weight with MB1 buffer containing 370 mM NaCl.
-
iii.Centrifuge at 11000 × g for 45 min at 4°C.Alternatives: Other tubes can be used depending on rotor availability (i.e. Sorvall or Beckman systems). If using 50 mL conical tubes for collecting cells, avoid loading more than 45 mL of cell lysates in each tube. Make sure that the selected tubes resist the applied G forces.
-
iv.Transfer the supernatant to a new 50 mL tube. Maintain the clarified lysate at 4°C. Keep the cell pellet for SDS-PAGE analysis in case the protein is undetected in the supernatant.
Taq DNA polymeraseNote: Avoid freezing or storing crude lysates. -
v.Heat the lysis suspension at 80°C for one hour.
-
vi.Adjust to 20 mL per gram of cell pellet weight with TaB1 buffer.
-
vii.Adjust glycerol to 10% final concentration.
-
viii.Add imidazole to a 10 mM final concentration (use a 2 M imidazole stock solution, pH 8.0).
-
ix.Add β-mercaptoethanol to a 6 mM final concentration.
-
x.Centrifuge at 11000 × g for 45 min at 4°C.
-
xi.Transfer the supernatant to a new 50 mL tube. Maintain the clarified lysate at 4°C.
LbCas12a and TEV proteaseNote: Avoid freezing or storing crude lysates. -
xii.Centrifuge at 11000 × g for 45 min at 4°C.
-
xiii.Transfer the supernatant to a new 50 mL tube. Maintain the clarified lysate at 4°C.Note: Avoid freezing or storing crude lysates.
Break Point: Final purification steps differ from this point onwards as they depend on the desired protein, and whether the purification is manual or automated. Filter the supernatant with a 0.45 μm syringe filter before purification. This will avoid clogging of the column in both manual and automated systems.Pause point: During centrifugation, take advantage of this time to set up the first step of affinity chromatography (See Methods video S1). Optimize your steps and timing to reduce as much as possible the time between cell lysis and loading the lysate onto the first chromatographic column.
-
i.
-
a.
Figure 4.

Cell lysis and protein purification
(A) The workflow summarizes the most important steps of the cell lysis on the first part of day 1 (Day 1a). Each protein and the corresponding buffers have been allocated a specific color: M-MLV-RT (black), Taq DNA polymerase (blue), MBP-LbCas12a (purple) and TEV protease (green).
(B) The top-to-bottom scheme is divided into affinity (Day 1b) and ion exchange chromatography (Day 2b). Each protein and the corresponding buffers have been colored as in (A). A right-sided, purple-colored scheme has been included for MBP-LbCas12a representing the extra steps required for its purification. Extra steps for TEV protease purification have been highlighted in green. The days and the processes including day transitions are shown in light blue.
-
7.
Chromatography (Figure 4B)
All the steps described below can be performed using a Fast Protein Liquid Chromatography (FPLC) system (Madadlou et al., 2016). A manual procedure using disposable syringes of 10 mL (with a flow rate of 1 mL/min, 30–40 drops/min) can also be performed (See Methods video S1). All the required buffer recipes are described in Data S1.Note: For manual elution consider preparing gradient changes in buffer salt concentration at 50 mM increasing steps, according to the required concentration range for each protein.-
a.M-MLV reverse transcriptase (RT)Day 1: Affinity Chromatography (Figure 4B)
-
i.Remove the storage solution (20% ethanol) from the HisTrap FF Crude column (1 mL column volume (CV)) with 10 CV of ultra-pure water. For the automated process we used a flow rate of 1 mL/min, and for the manual process ∼1 mL/min or 1 drop every 3 s) (See Methods video S1)
-
ii.Equilibrate the column with 10 CV of MB1 buffer.
-
iii.Filter the clarified supernatant with a 0.45 μm filter to avoid column clogging and load the sample onto the column. Collect the flow-through (FT) in 15 mL conical tubes (See Tip 1 on Methods video S5).Note: Keep all purification intermediates until SDS-PAGE analysis. A 20 μL sample of each step can be prepared with SDS sample buffer during pauses.
-
iv.Wash with 10 CV of MB2 buffer followed by 10 CV of MB1 buffer. Collect washes in 15 mL conical tubes (Methods video S2).Note: Steps v. through vii, are shown in Methods video S3.
-
v.Elute the protein with 8 mL of MB3 buffer. Collect 0.5 mL fractions in 1.5 mL microcentrifuge tubes.
-
vi.Clean the column with 20 CV of MB1 buffer, 10 CV of ultra-pure water, and store it with 5 CV of 20% ethanol.
-
vii.Assess purification performance by 10% SDS-PAGE (100 V for 5 min followed by 200 V for 50 min). Coomassie blue staining can be used. Figure 5A shows the expected results. If protein band is not observed, see troubleshooting section – problem 4.Alternatives: Each fraction can be analyzed by absorbance at 280 nm or by the Bradford assay to identify the presence of protein.Note: M-MLV reverse transcriptase shows an expected size of 71 kDa. For this protocol the Unstained Protein Standard, Broad Range (10–200 kDa) (NEB) ladder was used (see key resources table).
-
viii.Select and pool fractions considering quantity and purity as seen in Figure 5A (eluted fractions 2, 3 and 4). Dialyze using 1 L of MB4 buffer on ice (or cold room set at 4°C if available) for 16 ± 2 h using a D-Tube™ dialyzer Maxi, molecular weight cutoff (MWCO) from 12 to 14 kDa (Merck).Note: Each dialyzer tube (D-Tube™ dialyzer Maxi) can contain up to 3 mL, equivalent to three 1 mL-fractions. Any dialysis technique or other dialyzer tubes with higher capacity can be used in this step.
-
ix.To measure the post-chromatography protein yield, calculate the ratio between recovered protein over total input protein by absorbance at 280 nm, Bradford assay or alternatively by pixel densitometry of the SDS-PAGE gel.Day 2: Ion Exchange Chromatography (Figure 4B; Methods video S4)
-
x.Collect the protein from the dialyzer tube in 1.5 mL tubes, centrifuge at 15000 × g for 15 min at 4°C and transfer the supernatant to new 1.5 mL tubes. If a large protein pellet is observed, see troubleshooting section - problem 5.
-
xi.Remove the storage solution (20% ethanol) from the HiTrap SP column (1 mL CV) with 10 CV of ultra-pure water.
-
xii.Equilibrate the column with 10 CV of MB4 buffer.
-
xiii.Load the column with the RT protein sample purified by affinity chromatography (step 7a., Day 1., viii.).Note: Keep all purification intermediates until SDS-PAGE analysis. A 20 μL sample of each step can be prepared with SDS sample buffer during pauses.
-
xiv.Wash the loaded column with 10 CV of MB4 buffer. Collect it in a 15 mL conical tube.
-
xv.Elution step using the FPLC system: Set a gradient from 0% to 100% (i.e., in 20 CV) of MB5 buffer. Collect 0.5 mL fractions in 1.5 mL microcentrifuge tubes.Alternatives: If using the manual mode, prepare 50 mL of buffer stocks from 100 mM to 1 M NaCl (MB5 buffer) by varying the NaCl concentration in 50 mM increments. Use 2 CV of each stock during elution.
-
xvi.Clean the column with 10 CV of MB5 buffer, 10 CV of MB4 buffer, 10 CV of ultra-pure water, and store it with 5 CV of 20% ethanol.
-
xvii.Assess purification performance by 10% SDS-PAGE (100 V for 5 min followed by 200 V for 50 min). Figure 5B shows the expected results. If protein band is not observed, see troubleshooting section – problem 4.Note: M-MLV reverse transcriptase shows an expected size of 71 kDa (Figure 5A). For this protocol the Unstained Protein Standard, Broad Range (10–200 kDa) (NEB) ladder was used (see key resources table).
-
xviii.Pool fractions considering quantity and purity for dialysis with 1 L of MB6 buffer on ice (or 4°C cold room if available) for 16 ± 2 h placing the protein in a D-Tube™ dialyzer Maxi.
-
xix.(Day 3) Collect the protein from the dialyzer tube into 1.5 mL tubes, centrifuge at 15000 × g for 15 min at 4°C and transfer the supernatant to new 1.5 mL tubes. If a large protein pellet is observed, see troubleshooting section – problem 5.
-
xx.Quantify the protein by densitometry analysis using BSA as a standard or by the Bradford assay (See enzyme quantification section). Store 20 μL aliquots at −80°C. Flash freeze in liquid nitrogen or dry ice ethanol/methanol/isopropanol bath if available.
-
i.
-
b.Taq DNA polymeraseDay 1: Affinity Chromatography (Figure 4B)
-
i.Remove the storage solution (20% ethanol) from the HisTrap FF Crude column (1 mL column volume) with 10 CV of ultra-pure water (See Methods video S1).
-
ii.Equilibrate the column with 10 CV of TaB1 buffer.
-
iii.Filter the clarified supernatant with a 0.45 μm filter and load the sample on the column (See Tip 1 on Methods video S5). Collect the flow-through (FT) in conical 15 mL tubes.Note: Keep all purification intermediates until SDS-PAGE analysis. A 20 μL sample of each step can be prepared with SDS sample buffer during pauses.
-
iv.Wash with 10 CV of TaB2 buffer followed by 10 CV of TaB3 buffer. Collect them in 15 mL conical tubes (Methods video S2).Note: Steps v. through vii., are shown in Methods video S3.
-
v.Elute the protein with 8 mL of TaB4 buffer. Collect 0.5 mL fractions in 1.5 mL microcentrifuge tubes.
-
vi.Clean the column with 5 CV of TaB1 buffer, 10 CV of ultra-pure water, and store it with 5 CV of 20% ethanol.
-
vii.Assess purification performance by 10% SDS-PAGE (at 100 V for 5 min followed by 200 V for 50 min). Coomassie blue staining can be used. Figure 6A shows the expected results. If protein band is not observed, see troubleshooting section – problem 4.Note: Taq DNA polymerase shows an expected size of 94 kDa. For this protocol the Unstained Protein Standard, Broad Range (10–200 kDa) (NEB) ladder was used.
-
viii.Pool fractions considering quantity and purity for dialysis against 1 L of TaB5 buffer on ice (or 4°C cold room if available) for 16 ± 2 h placing the protein in a D-Tube™ dialyzer Maxi, MWCO from 12 to 14 kDa. Any available dialysis technique or dialyzer tube can also be used.Day 2: Ion Exchange Chromatography (Figure 4B; Methods video S4)
-
ix.Collect the protein from the dialyzer tube into 1.5 mL tubes, centrifuge at 15000 × g for 15 min at 4°C and transfer the supernatant to new 1.5 mL tubes. If a large protein pellet is observed, see troubleshooting section – problem 5.
-
x.Remove the storage solution (20% ethanol) from the HiTrap Heparin HP column with 10 CV of ultra-pure water.
-
xi.Equilibrate the column with 10 CV of TaB5 buffer.
-
xii.Load the column with the Taq protein sample purified by affinity chromatography (step 7b., Day 1., viii.).Note: Keep all purification intermediates until SDS-PAGE analysis. A 20 μL sample of each step can be prepared with SDS sample buffer during pauses.
-
xiii.Wash twice with 10 CV of TaB5 buffer. Collect them in 15 mL conical tubes.
-
xiv.Elution step using the FPLC system: Set a gradient from 0% to 100% (i.e., 20 CV) of TaB6 buffer. Collect 0.5 mL fractions in 1.5 mL microcentrifuge tubes.Alternatives: If using the manual mode, prepare 50 mL buffer stocks from 100 mM to 1 M NaCl (TaB6 buffer) by varying the NaCl concentration in 50 mM increments. Use 2 CV of each stock during elution.
-
xv.Clean the column with 10 CV of TaB5 buffer, 10 CV of ultra-pure water, and store it with 5 CV of 20% ethanol.
-
xvi.Assess purification performance by 10% SDS-PAGE (100 V for 5 min followed by 200 V for 50 min). Coomassie blue staining can be used. Figure 6B shows the expected results. If protein band is not observed, see troubleshooting section – problem 4.Note: Taq DNA polymerase shows an expected size of 94 kDa (Figure 6). For this protocol the Unstained Protein Standard, Broad Range (10–200 kDa) (NEB) ladder was used.
-
xvii.Pool fractions considering quantity and purity for dialysis against 1 L of TaB7 buffer (at 4°C or cold room if available) for 16 ± 2 h placing the protein in a D-Tube™ dialyzer Maxi.
-
xviii.(Day 3) Collect the protein from the dialyzer tube into 1.5 mL tubes, centrifuge at 15000 × g for 15 min at 4°C and transfer the supernatant to new 1.5 mL tubes. If a large protein pellet is observed, see troubleshooting section – problem 5.
-
xix.Quantify the protein by densitometry analysis using BSA as a standard or by the Bradford assay (See enzyme quantification section). Store 20 μL aliquots at −80°C. Flash freeze in liquid nitrogen or dry ice ethanol/methanol/isopropanol bath if available.
-
i.
-
c.Cas12aDay 1: Affinity Chromatography (Figure 4B)
-
i.Remove the storage solution (20% ethanol) from the HisTrap FF crude column (1 mL column volume) with 10 CV of ultra-pure water (See Methods video S1).
-
ii.Equilibrate the column with 10 CV of CB1 buffer.
-
iii.Filter the clarified supernatant with a 0.45 μm filter and load the sample on the column (see Tip 1 on Methods video S5). Collect the flow-through (FT) in 15 mL tubes.Note: Keep all purification intermediates until SDS-PAGE analysis. A 20 μL sample of each step can be prepared with SDS sample buffer during pauses.
-
iv.Wash with 10 CV of CB2 buffer followed by 10 CV of CB3 buffer. Collect them in 15 mL conical tube (Methods video S2).Note: Steps v. through vii., are shown in Methods video S3.
-
v.Elute the protein with 8 mL of CB4 buffer. Collect 0.5 mL fractions in 1.5 mL microcentrifuge tubes.
-
vi.Clean the column with 5 CV of CB2 buffer, 10 CV of ultra-pure water, and store it with 5 CV of 20% ethanol.
-
vii.Assess purification performance by 7.5% SDS-PAGE (100 V for 5 min followed by 200 V for 50 min). Coomassie blue staining can be used. Figure 7A shows the expected results. If protein band is not observed, see troubleshooting section – problem 4.Note: MBP-LbCas12a shows an expected size of 172 kDa. For this protocol the Unstained Protein Standard, Broad Range (10–200 kDa) (NEB) ladder was used.
-
viii.Pool the protein-containing fractions considering quantity and purity, for TEV protease digestion and dialysis.
-
ix.Add TEV protease to a 2.21 μM final concentration.
-
x.Dialyze Tev-treated fractions of LbCas12a against 1 L of CB5 buffer on ice (or cold room set at 4°C if available) for 16 ± 2 h placing the protein in a D-TubeTM dialyzer Maxi.Day 2a: MBP Affinity Chromatography (Figure 4B)
-
xi.Collect the protein from the dialyzer tube into 1.5 mL tubes, centrifuge at 15000 × g for 15 min at 4°C and transfer the supernatant to new 1.5 mL tubes. If a large protein pellet is observed, see troubleshooting section – problem 5.
-
xii.Assess TEV protease cleavage performance and protein purity by 12.5% SDS-PAGE (100 V for 5 min followed by 200 V for 50 min). Use Coomassie blue for staining. Figure 7B shows the expected results.
-
xiii.Remove the storage solution (20% ethanol) from the MBPTrap HP column (1 mL column volume) with 10 CV of ultra-pure water.
-
xiv.Equilibrate the column with 10 CV of CB6 buffer.
-
xv.Load the column with the Tev-treated protein and collect the FT. Keep it on ice.
-
xvi.Assess purification performance by 12.5% SDS-PAGE (100 V for 5 min followed by 200 V for 50 min). Use Coomassie blue for staining.
-
xvii.Wash the column with 10 CV of CB7 buffer to remove the MBP trapped in the column.
-
xviii.Clean the column with 10 CV of CB6 buffer, 10 CV of ultra-pure water, and store it with 5 CV of 20% ethanol.Day 2b: Ion Exchange Chromatography (Figure 4B)
-
xix.Remove the storage solution (20% ethanol) from the HiTrap Capto SP ImpRes Sepharose column (1 mL column volume) with 10 CV of ultra-pure water.
-
xx.Equilibrate the column with 10 CV of CB6 buffer.
-
xxi.Load the FT collected from the MBPTrap HP to the column HiTrap Capto SP ImpRes Sepharose column (7c, Day2a, step xv.), and wash with 10 CV of CB6 buffer. Collect it in a 15 mL conical tube.Note: Keep all purification intermediates until SDS-PAGE analysis. A 20 μL sample of each step can be prepared with SDS sample buffer during pauses.
-
xxii.Elution step using the FPLC system: Set a gradient from 0% to 100% (i.e., 20 CV) of CB8 buffer. Collect 0.5 mL fractions in 1.5 mL microcentrifuge tubes.Alternatives: If using the manual mode, prepare 50 mL buffer stocks from 125 mM to 1 M KCl (CB8 buffer) by varying the salt concentration in 50 mM increments. Use 2 CV of each stock during elution.
-
xxiii.Clean the column with 10 CV of CB8 buffer, 10 CV of CB6 buffer, 10 CV of ultra-pure water, and store it with 5 CV of 20% ethanol.
- xxiv.
-
xxv.Pool fractions considering quantity and purity for dialysis against 1 L of CB9 buffer (or 4°C cold room if available) for 16 ± 2 h placing the protein in a D-Tube™ dialyzer Maxi.
-
xxvi.(Day 3) Collect the protein from the dialyzer tube into 1.5 mL tubes, centrifuge at 15000 × g for 15 min at 4°C and transfer the supernatant to new 1.5 mL tubes. If a large protein pellet is observed, see troubleshooting section – problem 5.
-
xxvii.Quantify the protein by densitometry analysis using BSA as a standard or by the Bradford assay (See enzyme quantification section). Store 20 μL aliquots at −80°C. Flash freeze in liquid nitrogen or dry ice ethanol/methanol/isopropanol bath if available.
-
i.
-
d.TEV proteaseDay 1: Affinity Chromatography (Figure 4B)
-
i.Remove the storage solution (20% ethanol) from the HisTrap FF Crude column (1 mL column volume) with 10 CV of ultra-pure water (See Methods video S1).
-
ii.Equilibrate the column with 10 CV of TeB2 buffer.
-
iii.Filter the clarified supernatant with a 0.45 μm filter and load the sample on the column (See Tip 1 on Methods video S5). Collect the FT in 15 mL tubes.Note: Keep all purification intermediates until SDS-PAGE analysis. A 20 μL sample of each step can be prepared with SDS sample buffer during pauses.
-
iv.Wash with 10 CV of TeB3 buffer. Collect it in 15 mL conical tubes.
-
v.Elute the protein with 20 CV of TeB4 buffer. Collect 0.5 mL fractions in 1.5 mL microcentrifuge tubes.
-
vi.Clean the column with 5 CV of TeB2 buffer, 10 CV of ultra-pure water, and store it with 5 CV of 20% ethanol.
-
vii.Assess purification performance by 12.5% SDS-PAGE (100 V for 5 min and 200 V for 50 min). Figure 8 shows the expected results. If protein band is not observed, see troubleshooting section – problem 4.Note: TEV protease has an expected size of 28 kDa (Figure 8). For this protocol, the Unstained Protein Molecular Weight Marker (14.4–116 kDa) (Thermo Scientific) ladder was used.
-
viii.Pool fractions considering quantity and purity for dialysis against 1 L of TeB5 buffer for 16 ± 2 h placing the protein in D-Tube™ dialyzer Maxi on ice (or 4°C cold room if available).
-
ix.(Day 2) Collect the protein from the dialyzer tube into 1.5 mL tubes, centrifuge at 15000 × g for 15 min at 4°C and transfer the supernatant to new 1.5 mL tubes. If a large protein pellet is observed, see troubleshooting section – problem 5.
-
x.Quantify the protein by densitometry analysis using BSA as a standard or by the Bradford assay (See enzyme quantification section). Store 20 μL aliquots at −80°C. Flash freeze in liquid nitrogen or dry ice ethanol/methanol/isopropanol bath if available.Note:Methods video S5 shows tips to improve manual chromatography performance.
-
i.
-
a.
Figure 5.

Purification of M-MLV reverse transcriptase (RT)
HisTrap FF and HiTrap SP columns were used for affinity and ion-exchange chromatography, respectively.
(A) The samples collected for cell lysis monitoring, the column post-loading flow-through (FT) and the washes, together with the main fractions obtained from affinity chromatography (AC) were analyzed by 10% SDS-PAGE gel electrophoresis and visualized by Coomassie blue staining.
(B) The post-dialysis samples from AC, the FT and the washes, together with the obtained fractions from the buffer salt concentration gradient obtained from ion-exchange chromatography (IEC) were analyzed as in (A). The salt concentrations at which the protein eluted are shown. The white dotted-stroke box shows the fractions selected and pooled for dialysis. The molecular weight of the protein is shown in the right and the SDS-PAGE gel percentage used is shown in the bottom left corners of the gel pictures.
Figure 6.

Purification of Taq DNA polymerase
HisTrap FF and HiTrap Heparin HP columns were used for affinity and ion-exchange chromatography, respectively.
(A) The samples collected for cell lysis monitoring, the column post-loading FT and the washes, together with the main fractions obtained from AC were analyzed by 10% SDS-PAGE gel electrophoresis and visualized by Coomassie blue staining.
(B) The post-dialysis samples from AC, the FT and the washes, together with the obtained fractions from the buffer salt concentration gradient obtained from IEC were analyzed as in (A). The salt concentrations at which the protein eluted are shown. The white dotted-stroke box shows the fractions selected and pooled for dialysis. Protein precipitation was observed on AC post-dialysis pooled fractions (P1 and P2). The molecular weight of the protein is shown in the right and the SDS-PAGE gel percentage used is shown in the bottom corners of the gel pictures.
Figure 7.

Affinity chromatography of MBP-LbCas12a, TEV digestion and purification of LbCas12a
HisTrap FF and MBPTrap HP columns (A and B) were used for affinity chromatography and a HiTrap Capto SP ImpRes Sepharose column (C) for ion-exchange chromatography.
(A) The samples collected for cell lysis monitoring, the column post-loading flow-through (FT) and the washes, together with the main fractions obtained from AC were analyzed by SDS-PAGE gel electrophoresis and visualized by Coomassie blue staining.
(B) The post-dialysis pooled samples were digested with TEV protease and loaded into the MBPTrap HP column. The white top dotted-stroke boxes show the bands resulting from the (MBP)LbCas12a before and after TEV protease treatment; and the bottom ones show the 28 kDa and 42 kDa bands corresponding to the TEV protease and the MBP tag proteins, respectively. The pre- and post-cleavage molecular weights are shown in the right.
(C) The post-dialysis samples from (B), the collected FT from (C) and the washes, together with the obtained fractions from the buffer salt concentration gradient obtained from IEC were analyzed as in (A). The salt concentrations at which the protein eluted are shown. The white dotted-stroke box shows the fractions selected and pooled for dialysis. The band corresponding to LbCas12a is marked with an arrow, while the MBP band is marked with a star symbol. The resulting molecular weights are on the right (A-B) and on the left corner (C) of the picture. The SDS-PAGE gel percentage used is shown in the bottom corners of the gel pictures.
Figure 8.

Purification of TEV protease
The HisTrap FF column was used for affinity chromatography. The samples collected for cell lysis monitoring, the column post-loading FT and the washes, together with the main fractions obtained from AC were analyzed by SDS-PAGE gel electrophoresis and visualized by Coomassie blue staining. The molecular weight of the TEV protease is shown in the left and the SDS-PAGE gel percentage used is shown in the bottom corner of the gel pictures.
Enzyme quantification
-
8.Quantify the purified aliquots according to the method of your preference.Note: We recommend quantifying all four enzymes using the same quantification method. Here we describe three methods for protein quantification that can be used depending on equipment availability.Alternatives: A quality control assay can be performed (Raynal et al., 2014). As some buffers might interfere with concentration measurement, consider the buffer used to choose a quantification assay.
-
a.Absorbance at 280 nmNote: Here we use the NanoDrop spectrophotometer. Alternatively, any spectrophotometer that reads in the UV range can be used with appropriate quartz cuvettes.
-
i.Clean the pedestal with dH2O. Apply 2 μL in the bottom pedestal arm, then lower the upper pedestal to form a liquid column. Wait for a couple of minutes. Wipe away the water from both pedestals using a clean wipe (i.e., Kimtech ScienceTM KimwipeTM).
-
ii.Select the Protein A280 option and perform the blank reading using 1–2 μL of the respective storage buffer.Note: Make sure that your buffer does not contain high concentrations of any reagent that reports a considerable absorbance at 280 nm (i.e., Triton X-100 that is used to store the TEV protease).
-
iii.When the measurement is complete, raise the upper pedestal arm and clean the buffer from both upper and bottom measurement surfaces using a clean dry paper wipe.
-
iv.Enter the sample name and select “E and MW” as sample type option to use the extinction coefficient and molecular weight of the protein to calculate the sample concentration.Note: For LbCas12a use 167780 M−1 cm−1 extinction coefficient, with a MW of 130 kDa; for Taq DNA polymerase use 112760 M−1 cm−1 extinction coefficient, with a MW of 94 kDa for and for M-MLV reverse transcriptase use 79780 M−1 cm−1 extinction coefficient, with a MW of 71 kDa.
-
v.Place 2 μL of your sample and measure in triplicates. Calculate the average and the standard deviation. It is recommended to measure at least two different dilutions of the protein stock.
-
vi.Clean both pedestal arms with a dry wipe. Then, continue with the next sample.
-
i.
-
b.Gel quantificationNote: For gel quantification, a protein standard is required. BSA could be used as standard (i.e., BSA protein standard, Cat. No.: P0834, Sigma-Aldrich).
-
i.Prepare at least five dilutions of the standard protein. We recommend using a range between 1 – 10 μg/sample (20 μL). Then mix each dilution with the SDS sample buffer.
-
ii.Prepare serial dilutions of your protein, at least three sample dilutions. Then mix the diluted samples with the SDS sample buffer.
-
iii.Heat the diluted standard and protein samples at 95°C for 5 min. Spin briefly.
-
iv.Prepare the SDS-PAGE gel according to the percentage required for each protein.
-
v.Load the standard and samples (100 V for 15 min followed by 200 V for 40 min).
-
vi.Stain the gel with Coomassie blue dye or another method available. Destain the gel matrix as described above and visualize the protein bands.
-
vii.Gel images can be analyzed with ImageJ software. A step-by-step protocol for using ImageJ is available in the following link: https://imagejdocu.tudor.lu/gui/analyze/gels
-
viii.Plot the band density (Y-axis) of the standard curve versus BSA concentration (X-axis) (Syrový and Hodný, 1991).
-
ix.Determine the unknown sample concentration of the protein using linear regression of your standard curve using the function:With (m) being the slope and (b) the background. [X] = (Y – b)/m
-
x.Calculate the sample concentration by interpolating the corresponding density with the linear regression. Multiply the resulting concentrations by the dilution factor applied at the (b.ii) step. Average and calculate your standard deviation.
-
i.
-
c.Bradford quantificationNote: For Bradford quantification, a protein standard is required. BSA could be used as standard (i.e., BSA protein standard, Cat. No.: P0834, Sigma-Aldrich).Note: The procedure could be performed in a 96-well plate if a plate reader is available (e.g., SynergyTM H1 Hybrid Multi-Mode plate reader) or in microcentrifuge tubes if a spectrophotometer is available.
-
i.Prepare five dilutions (use storage buffer as diluent) of the protein standard. We recommend using a range between 15 – 250 μg/mL in 100 μL. Prepare a blank (storage buffer only) as well. The volume can be scaled depending on the spectrophotometer that is available.
-
ii.Prepare three 10 μL serial dilutions of the proteins (1/10- 1 is recommended).
-
iii.Add 90 μL of Bradford (Bio-Rad) reagent to each dilution in a well or microcentrifuge tube.
-
iv.Mix gently 3 to 5 times.
-
v.Incubate at 22°C–25°C for 5 min in the dark.
-
vi.Read absorbance at 595 nm.
-
vii.Normalize the raw data by subtracting the blank absorbance (storage buffer only).
-
viii.Plot the normalized absorbance (Y-axis) of the standard curve versus BSA concentration (X-axis) (Bradford, 1976).
-
ix.Determine the unknown concentration of the protein, using linear regression of your standard curve using the formula:With (m) being the slope and (b) the background. [X] = (Y – b)/m
-
x.Calculate the sample concentration by interpolating the corresponding Abs595 (blank subtracted) with the linear regression. Multiply the resulting concentrations by the dilution factor applied at the (c.ii) step. Average and calculate your standard deviation.
-
i.
-
a.
Enzyme activity evaluation
-
9.
Enzyme activity tests
An RT-PCR assay can be performed to confirm that the Taq DNA polymerase and the M-MLV reverse transcriptase are in an active state (Figure 9A). Here, we describe a one-step RT-PCR assay. For protocol details, please refer to (Alcántara et al., 2021b). For buffer composition and a recipe for enzyme dilution and RT-PCR reaction see the Data S1.Alternatives: A two-step RT-PCR reaction can also be performed. Additionally, a commercial buffer can be used to evaluate enzymatic activity. We tested the buffers and components of the RevertAid First Strand cDNA Synthesis Kit (Cat. No. K1622, ThermoFisher) for M-MLV reverse transcriptase; and Standard Taq Buffer (Cat. No. M0273S, NEB) for Taq DNA polymerase.-
a.Prepare a 20X working stock of each enzyme as follows:
-
i.20X working stock of M-MLV reverse transcriptase: Dilute the enzyme stock in MB6 buffer to a final concentration of 34 ng/μL.
-
ii.20X working stock of Taq DNA polymerase: Dilute the enzyme stock in MB6 buffer to a final concentration of 32 ng/μL.
-
i.
-
b.Prepare the master mix according to the following recipe, scale the required volume of each reagent according to the number of samples to test. It is recommended to prepare the master mix for 1-2 additional reactions to account for volume loss during pipetting.Note: We recommend performing the RT-PCR using positive and negative standardized controls.
Reagent Initial concentration Final concentration Volume (μL) per reaction RPB4X 4X 1X 5 M-MLV RT 20X 1X 1 Taq polymerase 20X 1X 1 Reverse primer 5 μM 0.2 μM 0.8 Forward primer 5 μM 0.2 μM 0.8 Nuclease-free water - - 9.4 Sample/controls - - 2 Final volume 20 -
c.To monitor potential reaction contaminations, a non-template negative control (NTC) should be included in each assay. For the NTC, nuclease-free water must be added to the respective PCR tube
-
d.Run the following RT-PCR program:
PCR cycling conditions
Step Temperature (°C) Time Cycles Reverse transcription 50 20 min 1 Initial denaturation 95 5 min 1 Denaturation 95 5 s 45 Annealing/Extension 55 30 s Hold 10 ∞ Note: Total run time 1:40 hours -
e.The RT-PCR products can be visualized by agarose gel electrophoresis (Figure 9B). Use a percentage of agarose according to the expected RT-PCR product size (3–5%).
-
a.
Figure 9.

Enzyme activity evaluation and quantitative and qualitative analysis
A RT-PCR assay was performed to confirm that the Taq DNA polymerase and the M-MLV reverse transcriptase were in an active state.
(A) Purified enzyme stocks were analyzed by SDS-PAGE gel electrophoresis and visualized by Coomassie blue staining. Average purity ranged between 90% and 99% for each enzyme.
(B) RT-PCR reaction for functional enzyme confirmation. Results with commercial and in-house produced enzymes using a positive control (+) and a non-template negative control (-, NTC) were analyzed by 1.5% agarose gel electrophoresis and visualized with a blue light transilluminator.
(C) CRISPR/Cas reaction for functional enzymes confirmation. The fluorescent target detection was based on the indicated RT-PCR products on (B). The dark and light blue lines represent the normalized fluorescence intensity (RFU) over time (min) of commercial and in-house LbCas12a proteins, respectively. The full lines represent the positive control reactions, while the dotted lines the negative NTC controls. The fluorescence was measured on a SynergyTM H1 Hybrid Multi-Mode plate reader (Biotek).
(D) The bar graph shows the mean of the normalized fluorescence intensity ratio of the positive control over the NTC controls. The bars represent the mean of five measurements, and the error bars the standard deviation of the mean. Bar colors as on (C).
(E) The CRISPR/Cas reaction was also assessed qualitatively by mixing the reaction mixture in a tube, without exposure to light for 30 min and visualization with a blue light transilluminator.
(F) The same reaction and visualization method as in (E) was used for positive samples with different levels of SARS-CoV-2 RNA viral load. This panel has been elaborated from (Alcántara et al., 2021a)(Figure 5).
-
10.
CRISPR/Cas reaction for functional enzymes confirmation
A CRISPR/Cas assay can be performed to confirm that the LbCas12a is in an active state. Here, we describe a fluorescent detection assay using the target-specific RT-PCR products as input DNA (Figures 9C and 9D). For methodology details for SARS-CoV-2 detection, please refer to (Alcántara et al., 2021a). For buffer composition and a recipe for the CRISPR/Cas reaction see the Data S1.Note: A crRNA sequence and synthetic dsDNA target or the corresponding primers to amplify the target region are necessary before performing the CRISPR/Cas assay. For a quick guide for crRNA design, please see https://international.neb.com/faqs/2018/05/03/how-do-i-design-a-guide-rna-for-use-with-engen-lba-cas12a.Alternatives: The 1X NEBufferTM r2.1 (Cat. No. B6002S, NEB) can be used as an alternative for reaction buffer. Local production of the crRNA used here can be found in (Alcántara et al., 2021b).-
a.Perform the crRNA re-folding by incubation at 65°C for 10 min followed by cooling at 22°C–25°C for 10 min.
-
b.Prepare a 10X CRISPR-Cas12a complex according to the following recipe:
Reagent Final concentration per reaction LbCas12a 0.1μM Re-folded crRNA 0.15 μM Dual-labeled single-stranded DNA (ssDNA) reporter probe 3 μM 1X CrB1∗ - Final volume 10 μL ∗1X CRISPR/Cas buffer 1, see Data S1. -
c.Incubate the 10X CRISPR-Cas12a complex at 22°C–25°C for 10 min. Protect it from light.
-
d.Add MgCl2 to the 1X CrB1 to get a final concentration of 17.65 mM. This is labeled 1X CrB2.
-
e.The CRISPR/Cas assay is prepared according to the following scheme:
Pipetting
OrderReagent Initial concentration Final concentration Final volume (μL) per assay Notes 1 1X CrB2 (17.65 mM MgCl2) 1X 1X 85 Mix separately 2 RT-PCR product or 1 μM synthetic template - - 5 3 CRISPR-Cas12a complex 10X 1X 10 Final volume - - 100 Note: You can prepare the CRISPR/Cas assay in separate tubes, or PCR strips. -
f.Once the CRISPR/Cas reaction is prepared, incubate it without light exposure at 22°C–25°C for 30 min.
-
g.Transfer the tubes to a blue transilluminator and close the filter tap (Figures 9E and 9F).
-
h.Register the results of direct fluorescence visualization using a photo camera or a smartphone.Note: We recommend using a black cabinet, box, or darkroom to improve direct visualization of the results.
-
i.Alternatively, a fluorescence 96-well plate reader was used. Fluorescence was excited at 491 nm and measured at 525 nm on a SynergyTM H1 Hybrid Multi-Mode plate reader (Biotek) (Figures 9C and 9D).
-
a.
Expected outcomes
Each required enzyme is intended to be produced to high purity following a four-day scheme. Typically, 3–5 g of dry cells are obtained from 1 L of cell culture. After chromatography steps, 6–12 mL of protein are obtained and stored. The standardized protocols show high purity and yields of the enzymes. Average purity ranged between 90% and 99%. Regarding protein production, Cas12a shows the highest yield with 2 mg/L of cell culture, followed by the Taq DNA polymerase with 1.2 mg/L of cell culture. M-MLV reverse transcriptase shows the lowest yield with 0.6 mg/L of cell culture. Concerning the total number of reactions, 1 L of cell culture can produce enzymes for 20 to 40 thousand one-step RT-PCR reactions, and around 20 thousand CRISPR/Cas detection reactions.
Limitations
The described protocol is not intended to be used for commercial purposes. Batch-to-batch variation in expression and purification of recombinant enzymes could result in subtle changes of activity. Therefore, each new batch of recombinant enzyme must be evaluated to guarantee the quality of each preparation. In vitro assays such as RT-PCR and CRISPR/Cas reactions have been described here to evaluate the functionality of the recombinant enzymes. However, long-term enzyme stability has not been assessed. The enzyme activity has been validated in a molecular diagnostic pipeline (Alcántara et al., 2021a). However, we recommend local validation with the pertinent samples of interest. Check your national and/or local regulatory guidelines. A series of technical guides for in vitro diagnostic validation is available at the WHO website (https://extranet.who.int/pqweb/vitro-diagnostics/technical-guidance-series). The enzymes described here are the wild-type version. Modifications on their amino acid sequences have been reported to increase activity and genome-wide optimization of E. coli could improve production yields (Mahalik et al., 2014). The yield of protein expression can vary depending on the E. coli strain used, type of medium, and culture conditions for expression.
Troubleshooting
Problem 1
Transformed bacteria did not grow on LB plate + antibiotics (obtaining enzyme-expressing E. coli section, step 2b). Competent cells transformed with the correct plasmid must grow on an LB agar plate or LB broth supplemented with the appropriate antibiotic (i.e., for the plasmids used in this protocol: kanamycin, ampicillin, or chloramphenicol).
Potential solution
It is recommended to use fresh competent cells. Usually, incubation at 37°C for 16–18 h is sufficient to see colonies on LB plate or turbidity in LB broth. The efficiency of transformation may decrease when using long-stored competent cells. Check the procedure used for competent cells preparation. It is important to maintain the cold chain along the total process to reduce the loss of cell viability. Check that culture media is supplemented with the adequate antibiotic concentration. If other transformation techniques or a modified version was performed, we recommend assessing the methodology described in literature (Chan et al., 2013; Inoue et al., 1990)
Problem 2
No or few isolated bacterial colonies are observed on plates (obtaining enzyme-expressing E. coli section, step 2b). Isolated colonies are required to identify recombinant bacteria harboring the right plasmid by colony PCR. Although the antibiotic restrains the growth of bacteria without the plasmid, not all observed colonies may contain the plasmid.
Potential solution
If there are few isolated colonies, you can re-plate one colony using a streak technique in a new plate. One important tip before the plate streak is to check that the culture plate is already dry. Always incubate the culture plates at 37°C for a few minutes before the plate streak (i.e., particularly if using stored culture plates). Finally, check that the antibiotic concentration is adequate for the bacterial selection. If it is necessary, prepare new plates and/or antibiotic stocks.
Problem 3
Lack of protein induction as monitored by SDS-PAGE gel (expression in E. coli section, step 5f). Induction monitoring is an essential step to verify that proteins are being expressed correctly. SDS-PAGE gel evaluation can give insights if it is necessary to optimize expression conditions.
Potential solution
Verify the time and temperature used for induction and the concentration of IPTG. Review colony PCR confirmation. Verify and change IPTG stocks. If the protein band is not seen, the expression protocol must be optimized.
Problem 4
Lack of proteins in the elution fractions as monitored by SDS-PAGE gel (protein purification section, steps 7a–d). Protein purification monitoring is crucial to determine which aliquots will be pooled for downstream steps (i.e., concentration, quantification, or secondary purification). Depending on the volume of expression culture, the number of aliquots that contain the protein of interest may vary. Usually, after all the purification steps, 6–12 mL of protein are obtained from 1 L of expression culture. It is recommended to keep all the purification fractions until the SDS-PAGE is done.
Potential solution
First, identify in which fraction (flow-through or washing) is the protein of interest by SDS-PAGE electrophoresis. If protein bands are seen in other fractions than the elution, we recommend to (1) check the pH of the used buffers, since low or high pH could affect the performance of the purification; especially when an ion exchange column is used. (2) If the elution is done with a unique concentration elution buffer, consider using a gradient to improve/optimize the elution step. (3) In the case of using an affinity column, check that the column is re-equilibrated and freshly reconstituted before the chromatography. If protein bands are not seen in any purification fraction, check the conditions of the previous cell lysis step. Optimization could be necessary, or the expressed protein could be in the insoluble cell fraction.
Problem 5
Protein is precipitating after the dialysis step (protein purification section, steps 7a–d). A protein pellet after centrifugation may indicate that the protein is precipitating. Small pellets are commonly found in many protein preparations.
Potential solution
If the protein pellet is large, evaluate and revise the dialysis buffer composition. If the equipment is available (e.g., Tycho or Prometheus NT48, Nanotemper), evaluate the stability of the protein in the selected buffer.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pohl Milón (pmilon@upc.pe).
Materials availability
This study did not generate new unique reagents. All the plasmids used in this study are available in the Addgene catalog (See key resources table).
Acknowledgments
We are very thankful to Dr. Marcos Milla for donating equipment that was used in this study and others. We would also like to thank all lab members of the Adaui and Milón groups for their help, support, and great working atmosphere. This work was supported by grants from the Peruvian Fondo Nacional de Desarrollo Científico, Tecnológico y de Innovación Tecnológica [070-2020-FONDECYT] to VA and [036-2019-FONDECYT-BM-INC.INV] and [154-2017-FONDECYT] to PM.
Author contributions
Conceptualization and methodology, R.A., K.P., J.A.N., L.C.-S., and P.M.; investigation, G.M.-R., A.S.-C., V.S.-V., L.T., K.P., and R.A.; formal analysis, R.A., K.P., and P.M., writing – original draft, V.S.-V, A.S.-C., K.P., R.A., and P.M., writing – review & editing, V.S.-V., A.S.-C., G.M.-R., R.A., L.T., K.P., J.A.N., L.C.-S., V.A., and P.M.; visualization, V.S.-V. and A.S.-C.; supervision, R.A. and P.M.; project administration, P.M. and K.P.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100899.
Contributor Information
Roberto Alcántara, Email: pcmeralc@upc.edu.pe.
Pohl Milón, Email: pmilon@upc.edu.pe.
Supplemental Information
Data and code availability
Original data have been deposited to Mendeley Data: .1 https://doi.org/10.17632/wynyf35xf3.2.
References
- Alcántara R., Peñaranda K., Mendoza G., Nakamoto J.A., Martins-Luna J., Valle J. del, Adaui V., Pohl M. Unlocking SARS-CoV-2 detection in low- and middle-income. Cell Rep. Methods. 2021 doi: 10.1016/j.crmeth.2021.100093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcántara R., Peñaranda K., Mendoza-Rojas G., Nakamoto J.A., Dueñas E., Alvarez D., Adaui V., Milón P. UnCovid: a versatile, low-cost, and open-source protocol for SARS-CoV-2 RNA detection. STAR Protoc. 2021 doi: 10.1016/j.xpro.2021.100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chan W.-T., Verma C.S., Lane D.P., Gan S.K.-E. A comparison and optimization of methods and factors affecting the transformation of Escherichia coli. Biosci. Rep. 2013;33:e00086. doi: 10.1042/BSR20130098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.S., Ma E., Harrington L.B., Costa M.D., Tian X., Palefsky J.M., Doudna J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360:eaar6245. doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien A., Edgar D.B., Trela J.M. Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J. Bacteriol. 1976;127:1550–1557. doi: 10.1128/jb.127.3.1550-1557.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham T.G.W., Dugast-Darzacq C., Dailey G.M., Nguyenla X.H., Dis E.V., Esbin M.N., Abidi A., Stanley S.A., Darzacq X., Tjian R. Open-source RNA extraction and RT-qPCR methods for SARS-CoV-2 detection. PLoS One. 2021;16:e0246647. doi: 10.1371/journal.pone.0246647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H., Nojima H., Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- Kapust R.B., Tözsér J., Fox J.D., Anderson D.E., Cherry S., Copeland T.D., Waugh D.S. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. Des. Sel. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- Kotewicz M.L., D’Alessio J.M., Driftmier K.M., Blodgett K.P., Gerard G.F. Cloning and overexpression of Moloney murine leukemia virus reverse transcriptase in Escherichia coli. Gene. 1985;35:249–258. doi: 10.1016/0378-1119(85)90003-4. [DOI] [PubMed] [Google Scholar]
- Kotewicz M.L., Sampson C.M., D’Alessio J.M., Gerard G.F. Isolation of cloned Moloney murine leukemia virus reverse transcriptase lacking ribonuclease H activity. Nucleic Acids Res. 1988;16:265–277. doi: 10.1093/nar/16.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawyer F.C., Stoffel S., Saiki R.K., Myambo K., Drummond R., Gelfand D.H. Isolation, characterization, and expression in Escherichia coli of the DNA polymerase gene from Thermus aquaticus. J. Biol. Chem. 1989;264:6427–6437. [PubMed] [Google Scholar]
- Madadlou A., O’Sullivan S., Sheehan D. Protein chromatography, methods and protocols. Methods Mol. Biol. 2016;1485:365–373. doi: 10.1007/978-1-4939-6412-3_19. [DOI] [PubMed] [Google Scholar]
- Mahalik S., Sharma A.K., Mukherjee K.J. Genome engineering for improved recombinant protein expression in Escherichia coli. Microb. Cell Fact. 2014;13:177. doi: 10.1186/s12934-014-0177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynal B., Lenormand P., Baron B., Hoos S., England P. Quality assessment and optimization of purified protein samples: why and how? Microb. Cell Fact. 2014;13:180. doi: 10.1186/s12934-014-0180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reischl U., Polayes D.A., Parks T.D., Johnston S.A., Dougherty W.G. Molecular diagnosis of infectious diseases. Methods Mol. Med. 1997;13:169–183. doi: 10.1385/0-89603-485-2:169. [DOI] [PubMed] [Google Scholar]
- Sasse J., Gallagher S. Staining Proteins in Gels. Current Protocols in Molecular Biology. 2009;85(1) doi: 10.1002/0471142727.mb1006s85. [DOI] [PubMed] [Google Scholar]
- Schneider C., Rasband W., Eliceiri K. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrový I., Hodný Z. Staining and quantification of proteins separated by polyacrylamide gel electrophoresis. J. Chromatogr. B Biomed. Sci. Appl. 1991;569:175–196. doi: 10.1016/0378-4347(91)80229-6. [DOI] [PubMed] [Google Scholar]
- Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data have been deposited to Mendeley Data: .1 https://doi.org/10.17632/wynyf35xf3.2.