

Abstract
The nature of the terminal oxidant in oxidation reactions is an important reaction variable that can profoundly impact the mechanism, efficiency, and practicality of a synthetic protocol. One might reasonably categorize catalyitic oxidation reactions into either “oxygenase” type reactions, in which the oxidant serves as an atom- or group-transfer reagent, or “oxidase” type reactions, where the oxidant is involved in catalyst turnover but does not become structurally incorporated into the product. As the field of photoredox catalysis has matured over the past decade, many successful oxygenase type photoreactions have been reported. The development of photocatalytic oxidase reactions, on the other hand, has been somewhat slower. This tutorial review presents selected examples of some of the key classes of terminal oxidants that have been used in the design of photoredox oxidase transformations, along with the mechanistic features and benefits of each.
1. Introduction
The realization that many visible light-absorbing chromophores can efficiently convert photonic energy into synthetically useful chemical potential has motivated a recent renaissance in organic photochemistry.1,2,3 Photocatalysis offers a uniquely facile strategy for the generation of a wide variety of open-shell intermediates, and the development of new photoredox transformations based upon their reactivity has been a major theme of research in the past decade. An important aspect of these reactions is the involvement of balanced photocatalyst-mediated electron transfer steps. That is, the initiating step of a photoredox reaction is a photoinduced one-electron oxidation or reduction. Turnover of the photocatalytic cycle, therefore, requires a complementary redox step to regenerate the photocatalyst. This mechanistic requirement may explain why many of the most successful early applications of photoredox catalysis were net redox-neutral in nature.
Recently, significant efforts have led to the development of complementary net-oxidation and reduction reactions that leverage the unique reactivity accessible from photoredox catalysis. These transformations have been somewhat more difficult to develop, however, because photocatalyst regeneration requires the use of stoichiometric terminal oxidants or reductants. Appropriate redox reagents must be compatible with the photocatalyst, the organic substrates, any photogenerated intermediates, and the products in each transformation. Thus, the identification of compatible terminal oxidants and reductants is a key consideration in the development of this class of reactions. Tertiary amines and dihydropyridines have long been recognized as practical, inexpensive terminal reductants in photocatalytic solar energy applications. This insight enabled the early, rapid development of a wide range of photocatalytic reduction reactions.4
The development of efficient photocatalytic oxidation reactions has been somewhat slower. The most widely utilized terminal oxidants have been group transfer reagents in which a portion of the terminal oxidant becomes structurally incorporated into the final product. An excellent example of this strategy is the photocatalytic decarboxylative fluorination strategy developed initially by Sammis5 and later expanded by MacMillan6 that uses Selectfluor as a terminal oxidant (Scheme 1). Photooxidation of the carboxylate of acid 1 affords organoradical 2, which can be intercepted by Selectfluor to produce the desired C–F bond. Aminyl radical 3, generated during C–F bond formation, serves to either turn over the photocatalyst or oxidize another equivalent of 1 in a chain propagation pathway. This mechanistic manifold has proven quite powerful, enabling the transfer of a variety of functional groups under photoredox conditions (Figure 1).
Scheme 1.
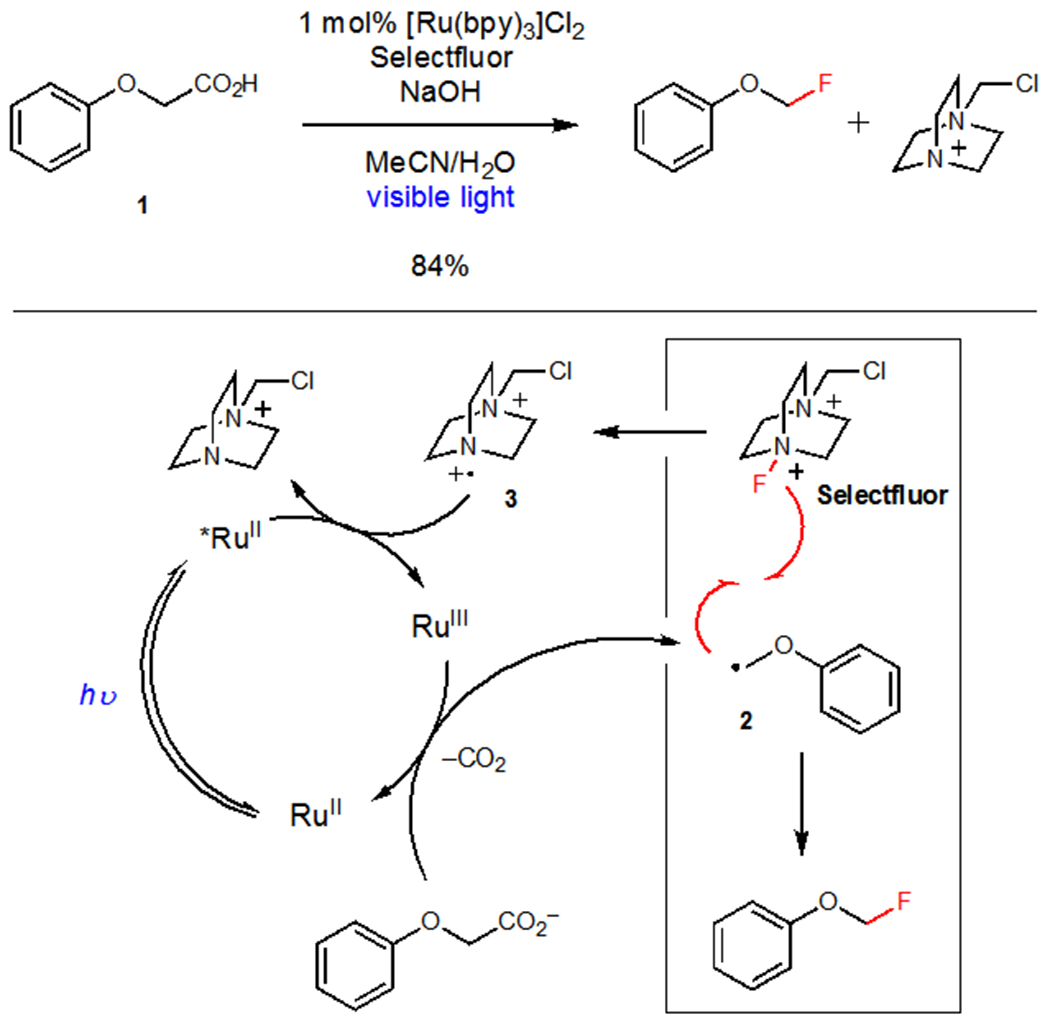
Decarboxylative Fluorination using Selectfluor as a Group Transfer Oxidant
Figure 1.
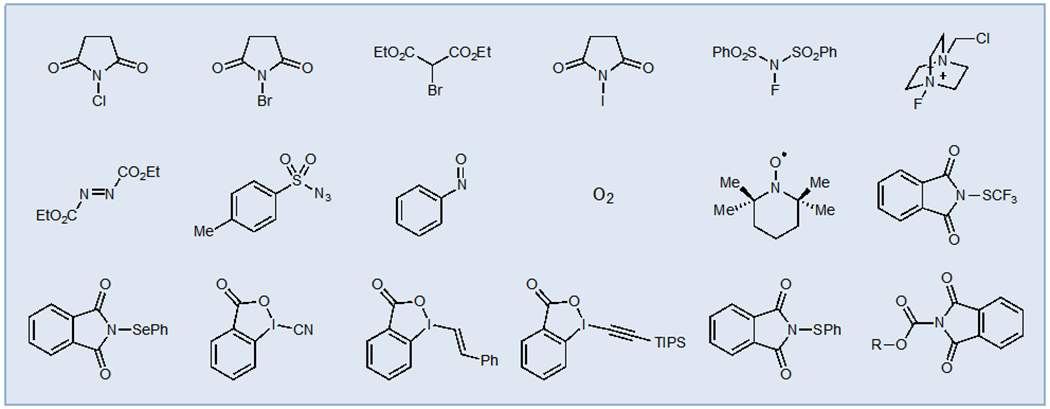
Representative Terminal Oxidants Commonly Used for Atom- or Group-transfer Photoredox Reactions
Stahl has drawn an analogy between transition metal-catalyzed group-transfer oxidation reactions and biological “oxygenase” reactions, in which metalloenzymes use molecular oxygen to install oxygen-containing functionalities.7 Aerobic oxidation reactions in which oxygen is simply used as an electron acceptor to turn over a transition metal catalyst, in contrast, are comparable to enzymatic “oxidase” reactions. The generalization of the chemical oxidase paradigm has informed synthetic methods development by enabling the coupling of O2 reduction to a variety of oxidative transformations, including those that do not introduce any new atoms at all (e.g., dehydrogenation). The extension of the oxidase/oxygenase analogy to other classes of redox transformations can also be conceptually quite useful.
In the context of photoredox catalysis, the group-transfer reactions that might be categorized as oxygenase-type reactions are relatively well developed (vide supra). In contrast, oxidase reactions have been much more difficult to develop. This observation could be due to the fact that photoredox catalysis introduces several unique challenges related to the choice of terminal oxidant. For example, many terminal oxidants that are commonly used in ground-state transition metal catalysis react rapidly and unproductively with photogenerated radical intermediates and can quench the excited states of many photocatalysts. Furthermore, these oxidants can react through multiple, diverse mechanistic pathways that can be difficult to deconvolute and rationalize. Thus, the identification of an appropriate terminal oxidant for a given photocatalytic reaction is often the result of laborious empirical screening. This review is designed to present a targeted survey of the major classes of terminal oxidants that have been used in photocatalytic oxidase chemistry. As this review is intended to be tutorial rather than comprehensive, examples have been chosen to illustrate unifying themes and common pitfalls that we hope could aid in future reaction development. Protocols that rely on the preactivation of an organic substrate in a separate, prior step (e.g. the conversion of a carboxylic acid to a redox-active ester) will not be discussed because the photocatalytic process would then be oxygenase in nature.
2. Molecular Oxygen
Molecular oxygen is often considered an ideal terminal oxidant due to its abundance, low toxicity, and environmentally benign byproducts.8 Its use in photocatalytic oxidase reactions, however, presents significant challenges. Molecular oxygen exists as a triplet in its ground state and efficiently quenches the excited state of many common photocatalysts.9 Furthermore, it reacts rapidly with the radical and radical ion intermediates that typify modern photoredox catalysis.10 Nevertheless, its successful use has been reported in a variety of photocatalytic transformations operating via several distinct mechanisms. Photoreduction of O2 generates superoxide anion, a potent hydrogen atom transfer reagent.11 Molecular oxygen can also mediate photocatalyst quenching or turnover by serving as an electron/proton acceptor.
Stephenson and coworkers reported an oxidative aza-Henry reaction of tetrahydroisoquinolines using an aerobic photoredox catalyst system (Scheme 2).12 Tetrahydroisoquinoline 4 was oxidized by photoexcited [Ir(ppy)2(dtbbpy)]PF6 to afford radical cation 5 and an Ir(II) complex. Photocatalyst turnover generated superoxide anion, which can abstract a hydrogen atom from radical cation 5 to generate iminium 6. Interception of 6 by nitromethane enolate delivered 7. Mechanistic experiments strongly suggested that photocatalyst turnover was mediated primarily by molecular oxygen and not nitromethane. These conclusions were subsequently confirmed by König, Gshwind, and coworkers who found that O2 was integral to both efficient hydrogen atom transfer and photocatalyst turnover.13 The success of this system hinged on the highly selective hydrogen atom transfer between photogenerated superoxide anion and radical cation 5, a result of severe weakening of its α-amino C–H bonds.
Scheme 2.
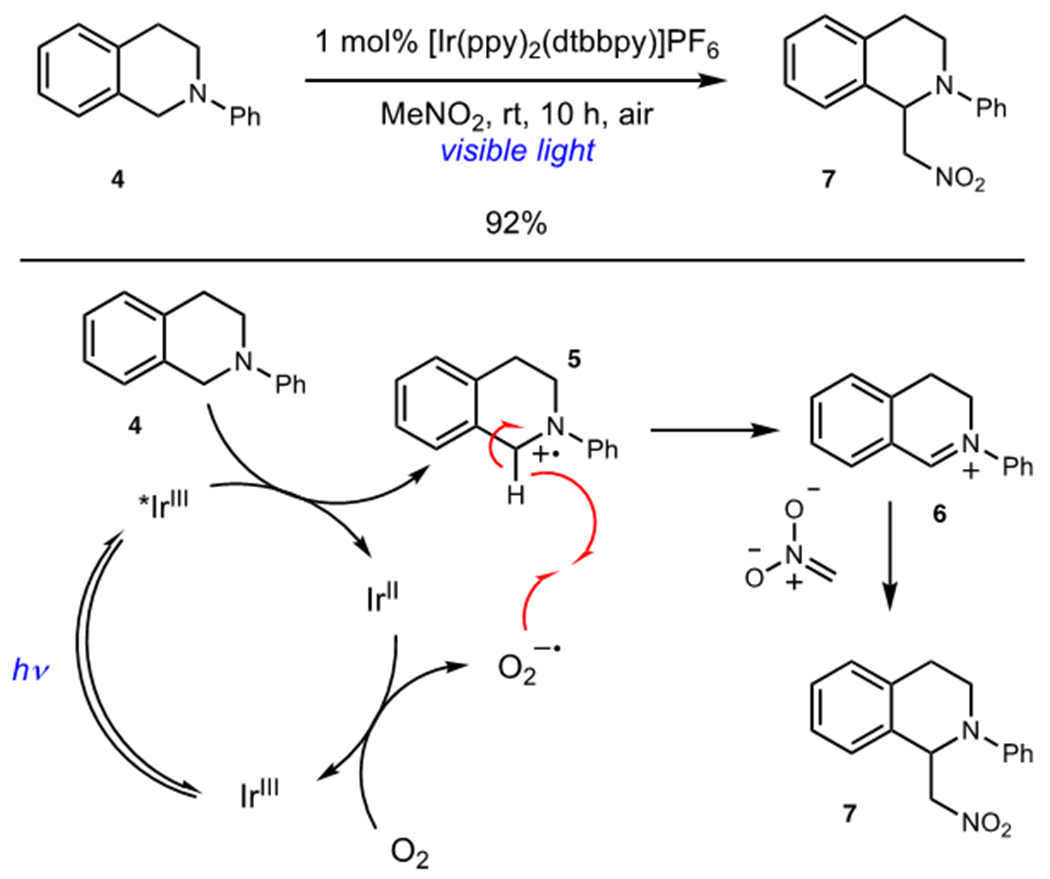
Photocatalytic Oxidative Aza-Henry Reaction
Reactive oxygen species such as superoxide anion react with many common organic functional groups and can result in undesired decomposition pathways. The oxidative amination of arenes developed by Nicewicz and coworkers offers an instructive case study (Scheme 3).14 Photooxidation of anisole by acridinium 8 afforded an electrophilic arene radical cation (9) which could be intercepted by N–H heterocycles. Subsequent oxidation generated aryl amine 11 via the intermediacy of delocalized radical 10. Turnover of the photocatalyst can occur via reduction of molecular O2. However, Nicewicz found that the resulting superoxide decomposed both acridinium 8 and the anisole starting materials. The addition of catalytic TEMPO was found to prevent decomposition and resulted in significantly higher isolated yields of aminated products. TEMPO was proposed to sequester reactive oxygen species via hydrogen atom transfer. Subsequent reports have found that the addition of radical scavengers is not necessary in all cases, presumably because the rates of catalyst turnover and reduction of hydroperoxyl radical are sufficiently matched to avoid these decomposition pathways entirely.15,16 These studies also highlight a key limitation of using molecular oxygen as a terminal oxidant in photocatalytic reactions: the presence of weak C–H bonds can result in significant oxidative decomposition and often requires the identification of alternate terminal oxidants to achieve the desired reactivity.
Scheme 3.
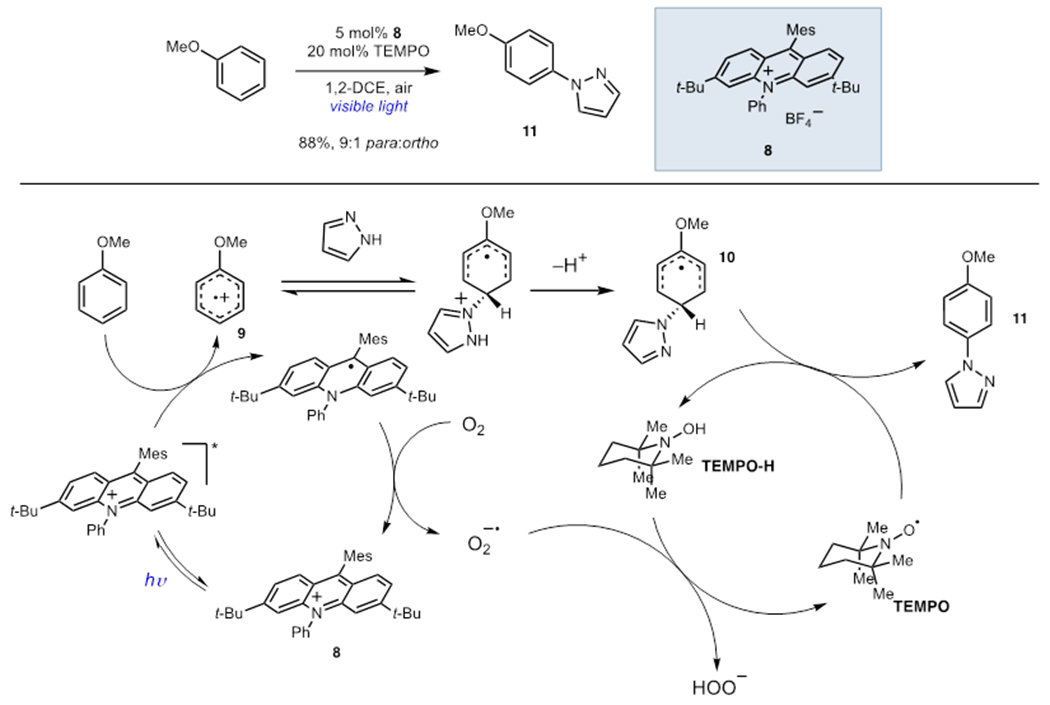
Arene Amination via Organic Photoredox Catalysis
Reoxidation of a transition metal catalyst can often be the rate-limiting step in catalytic oxidation reactions. Photoredox catalysis offers a particularly attractive solution to this problem because it can rapidly oxidize organometallic complexes via photoinduced electron transfer, and these processes can be readily coupled to the reduction of O2 to regenerate the photocatalyst. Kobayashi and coworkers reported a photocatalytic Chan–Evans–Lam coupling that utilized a dual photoredox/Cu(II) system (Scheme 4).17 Irradiation of a solution of catalytic Ir(ppy)3 under Chan–Evans–Lam conditions rapidly afforded high yields of N,N-diaryl amine products. Kobayashi proposed that photooxidation of an intermediate Cu(I)–aryl complex to regenerate their Cu(II) catalyst was responsible for the significantly enhanced rates of reaction. Rapid oxidative quenching of *Ir(ppy)3 by molecular oxygen generated the Ir(IV) complex necessary to oxidize the intermediate Cu(I)–aryl species.
Scheme 4.
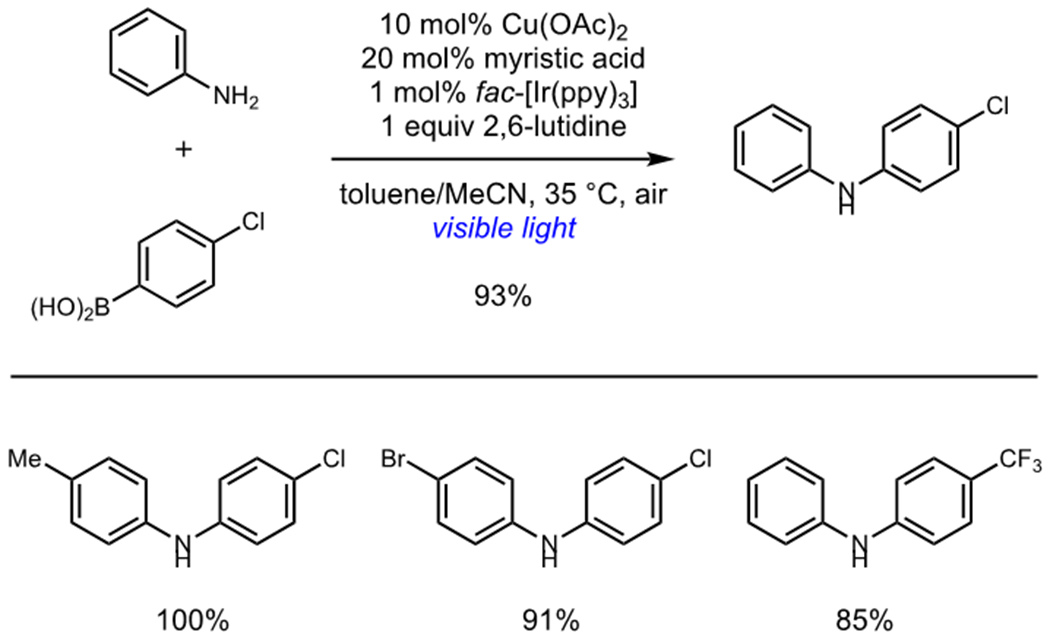
Photocatalytic Chan–Evans–Lam Coupling
3. Persulfates
Persulfate salts are among the most potent chemical oxidants that are commercially available, inexpensive, and conveniently handled. As such, they have found extensive use as radical initiators, bleaching agents, and chemical oxidants.18 In the context of photocatalytic oxidase chemistry, persulfate oxidants have primarily been used to generate sulfate radical anion, a potent hydrogen atom transfer reagent. Unlike superoxide, sulfate radical anion does not readily engage in atom transfer radical addition and generates only innocuous sulfate byproducts. One distinctive limitation of persulfate oxidants, however, is their limited solubility in most organic solvents, which can lead to slow rates of reaction. This practical consideration often dominates the design of new photocatalytic reactions using this class of oxidants. In fact, some of the most successful early applications of these oxidants (e.g. water oxidation19 and protein-protein cross-linking20) use H2O as the reaction medium.
MacMillan and coworkers leveraged the high reactivity of persulfate oxidants in the development of a photocatalytic Minisci reaction (Scheme 5).21 The direct oxidation of ethers occurs at high positive reduction potentials and can be difficult to achieve using most photocatalysts. Oxidative quenching of the photocatalyst by Na2S2O8 generated sulfate radical anion, which can abstract a hydrogen atom from the weak C–H bonds of tetrahydropyran to generate radical 12. The high reactivity of sulfate radical anion allowed for a range of cyclic and acyclic ethers to be used as radical precursors. Radical 12 underwent addition to protonated heteroarenes such as 13 in Minisci fashion to generate α-amino radicals (14). Photocatalyst turnover was accomplished by oxidation of 14, furnishing α-arylation product 15. In contrast to molecular oxygen, persulfate oxidants generate benign sulfate byproducts that cannot promote further oxidative side reactions.
Scheme 5.
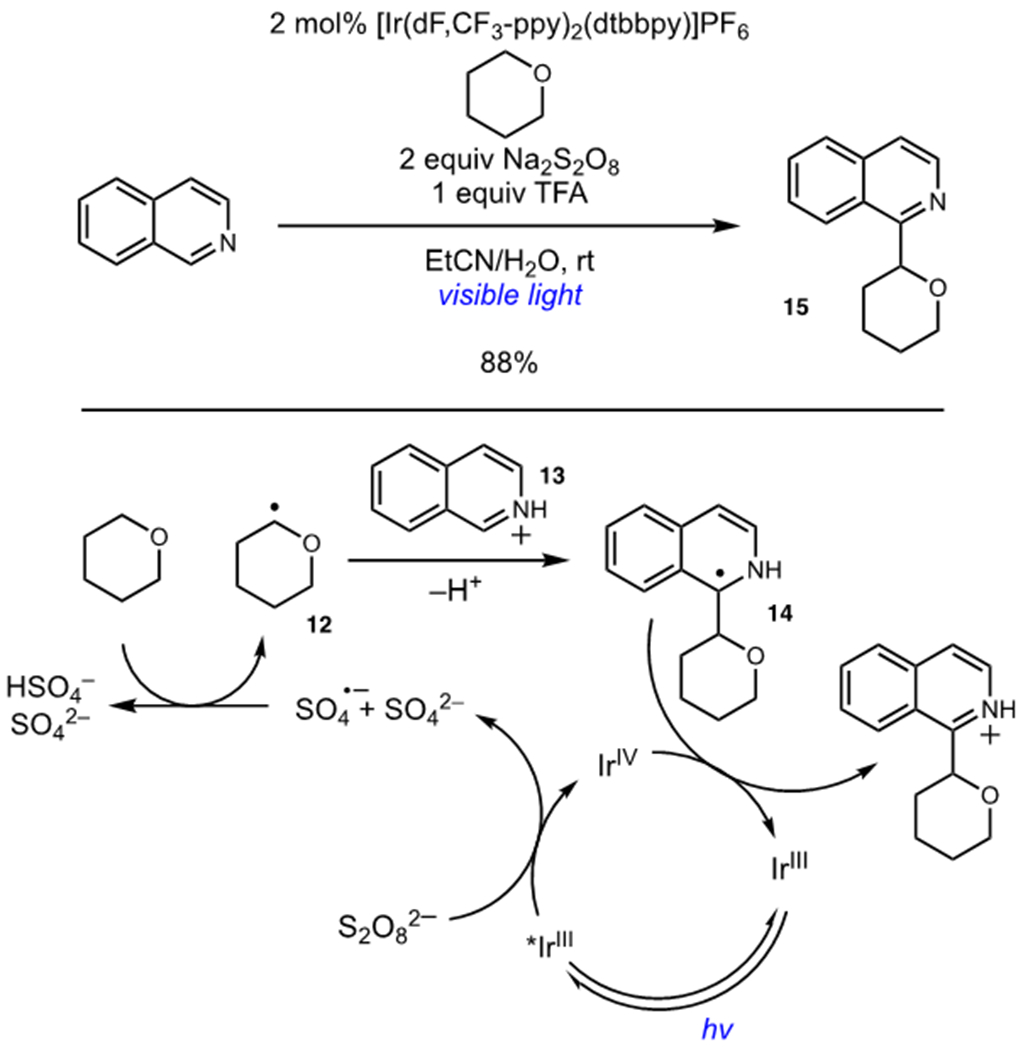
Arylation of Ethers via C–H Abstraction and Minisci-Type Addition
A common method of taming the high reactivity of sulfate radical anion is to generate a highly activated hydrogen atom donor via photooxidation as a coupled process. Using this strategy, Woo and coworkers reported a photocatalytic deprotection of para-methoxybenzyl (PMB) ethers utilizing (NH4)2S2O8 as a terminal oxidant (Scheme 6).22 Woo found that the use of aqueous MeCN was necessary to ensure high solubility of the persulfate oxidant and consequently attain high yields of deprotected alcohol products. Photooxidation of arene 16 generated radical cation 17 while subsequent photocatalyst turnover generated sulfate radical anion. Hydrogen atom transfer between 17 and sulfate radical anion afforded oxocarbenium 18 which underwent hydrolysis to reveal alcohol 19. By coupling generation of sulfate radical anion to the generation of an activated hydrogen atom donor, these processes minimize unproductive side reactions.
Scheme 6.
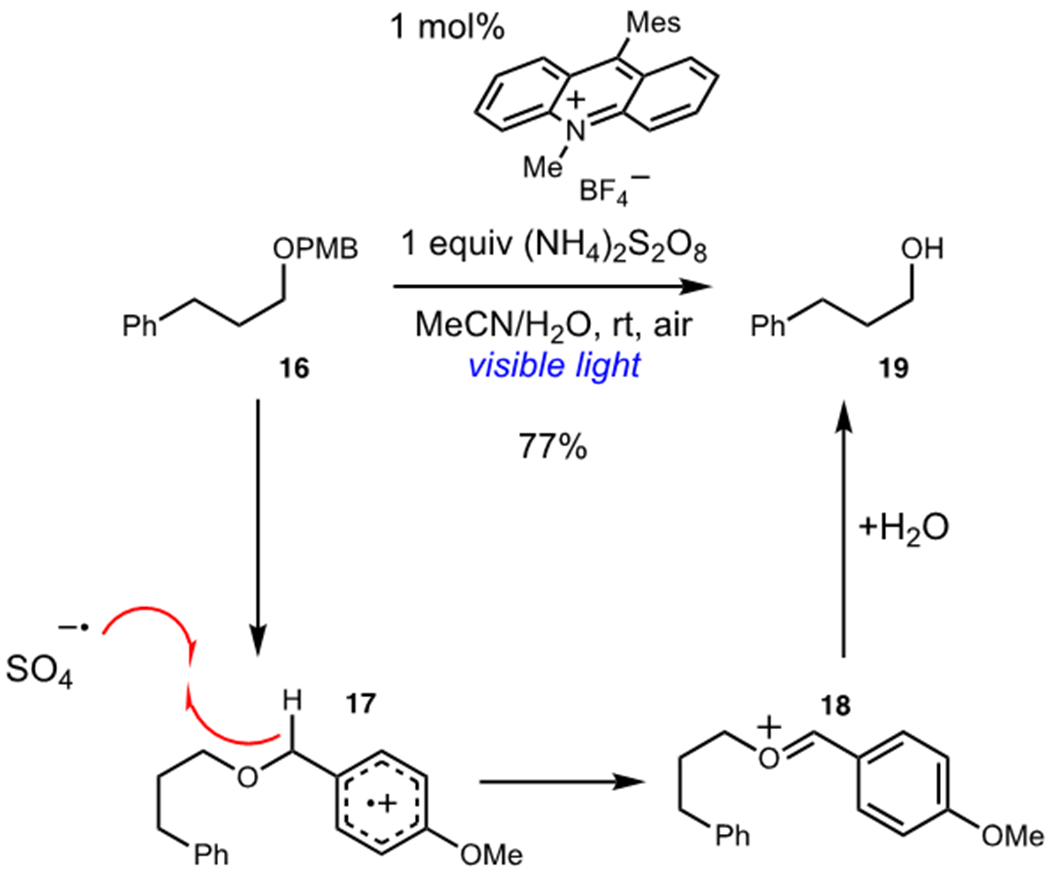
Photocatalytic Deprotection of para-Methoxybenzyl Ethers
While the use of aqueous cosolvents can often be beneficial, Yoon and coworkers provided an alternative strategy in the development of a photocatalytic [3+2] cycloaddition between phenols and styrenic olefins using persulfate as a terminal oxidant (Scheme 7).23 The photooxidation of electron-rich phenols and their subsequent cycloadditions with styrenic olefins was found to proceed in high yields and diastereoselectivity using catalytic [Ru(bpz)3](PF6)2 and (NH4)2S2O8 as a terminal oxidant. Yoon found that these reactions exhibited a significant induction period and rationalized this observation by proposing a salt metathesis between [Ru(bpz)3](PF6)2 and (NH4)2S2O8 to generate catalytically active [Ru(bpz)3](S2O8). Upon photoexcitation, [Ru(bpz)3](S2O8) underwent oxidative quenching within the ion pair to generate the highly oxidizing [Ru(bpz)3](SO4)+ complex ([Ru]3+/2+ = +1.98 V vs. SCE), which was capable of oxidizing the phenolic starting materials. Evidence for this pathway was obtained via independent synthesis of [Ru(bpz)3](S2O8), which proved to be both competent as a catalyst for this reaction and to exhibit no induction period. Thus, even though [Ru(bpz)3](S2O8) was only sparingly soluble in MeCN, the generation of this salt as the active photocatalyst in situ meant that oxidative quenching occurred rapidly within the ion pair upon irradiation.
Scheme 7.
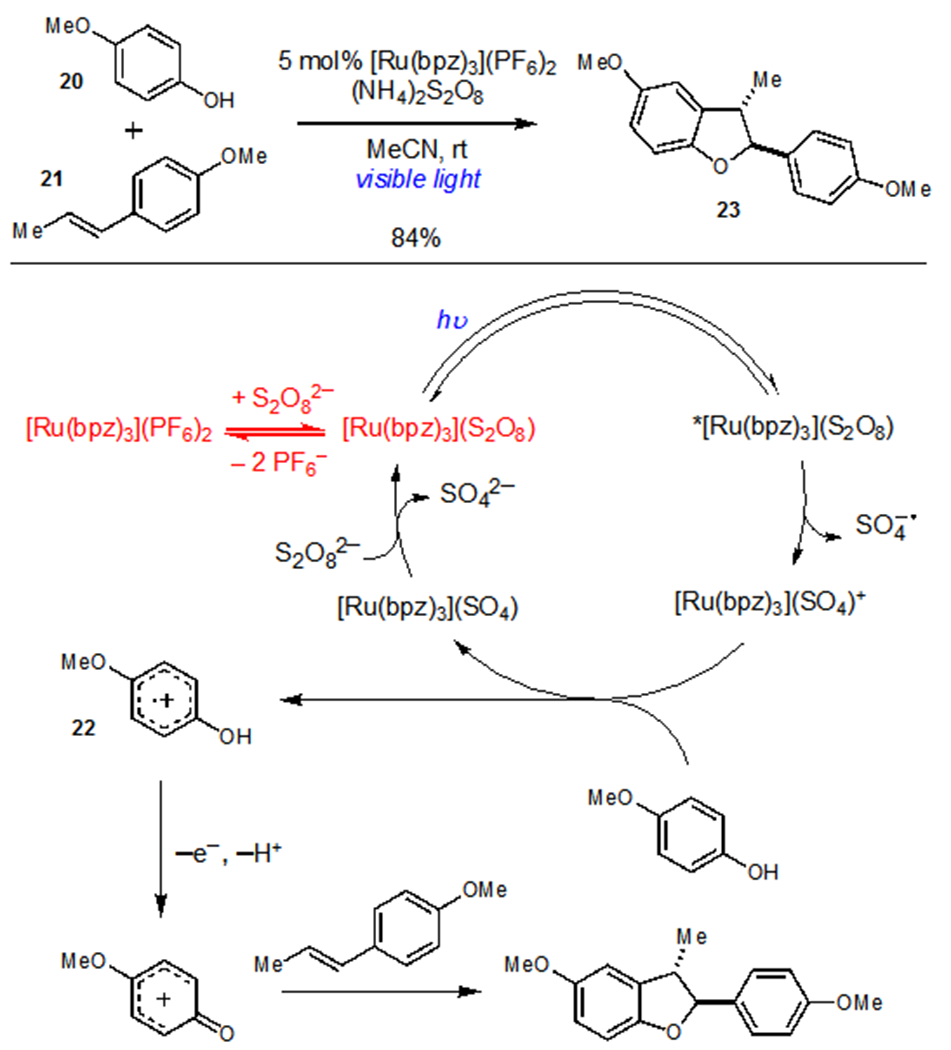
Photocatalytic [3+2] Cycloaddition of Phenols
4. Peroxides and Peresters
Peroxides and peresters have been used relatively infrequently as terminal oxidants in photocatalytic oxidase reactions because the intermediate oxyradicals are highly reactive and exhibit limited functional group tolerance. They do, however, undergo single-electron reduction to afford oxygen-centered radical intermediates that are capable of hydrogen atom transfer with organic substrates. Given the relative scarcity and limited mechanistic diversity of peroxides in photocatalytic oxidase chemistry, an extensive treatment of the available synthetic methods would not likely be instructive. Instead, we present a single illustrative example in the synthesis of elbasvir, a collaboration between the Merck Research Laboratories and the Knowles group at Princeton University (Scheme 8).24
Scheme 8.
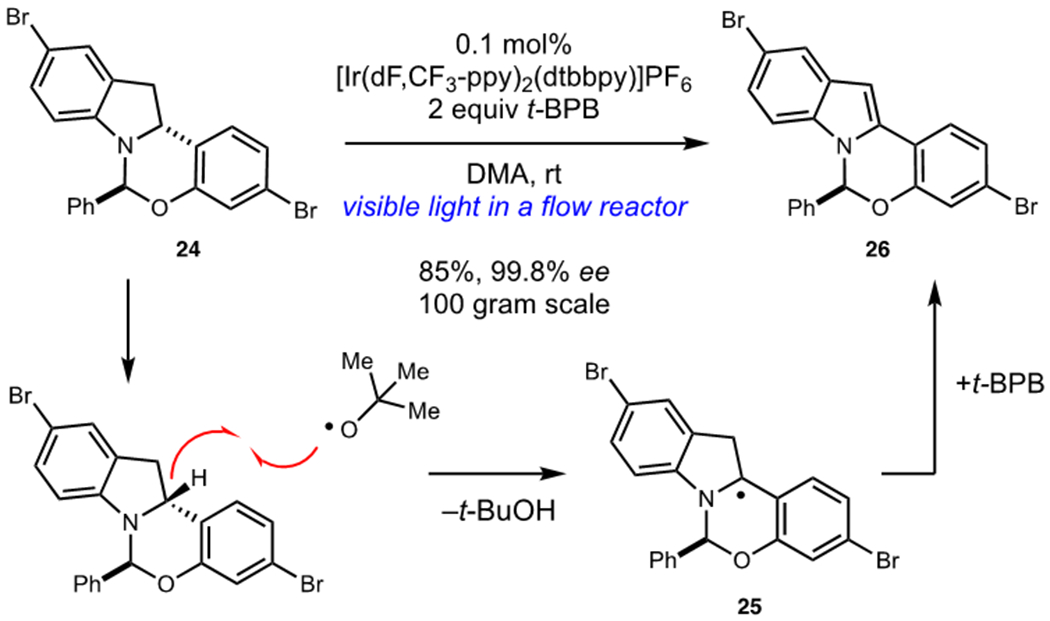
Photocatalytic Indoline Dehydrogenation in the Synthesis of Elbasvir
During the development of a process route to elbasvir, an inhibitor of heptatitis C virus NS5A protein, dehydrogenation of indoline 24 proved troublesome due to facile epimerization of the hemiaminal stereocenter. An exhaustive screen of oxidation protocols revealed that photocatalytic dehydrogenation using [Ir(dF,CF3-ppy)2(dtbbpy)]PF6 as a photocatalyst and tert-butylperbenzoate (t-BPB) as a terminal oxidant enabled selective oxidation of 24 to indole 26 without epimerization. This transformation proved highly scalable, and a continuous flow method enabled the synthesis of 100 g of this key intermediate. Detailed mechanistic investigations revealed that selective hydrogen atom transfer between photogenerated t-butoxy radical and indoline 24 was the key step responsible for maintaining stereochemical fidelity. Fortuitously, the indoline α-amino hydrogen atom was found to have the lowest bond dissociation free energy and other oxidation methods that proceeded via oxidation of the nitrogen atom resulted in significant epimerization of the adjacent stereocenter. This reaction was found to be a radical chain process, and propagation was proposed to occur via benzoyl transfer between t-BPB and radical 25.
5. Halocarbons
Halocarbon oxidants undergo facile single-electron reduction under photoredox conditions to generate highly electrophilic carbon-centered radicals that react rapidly with a variety of unsaturated organic functionality.25 In the presence of photogenerated organoradical intermediates, however, these halocarbon oxidants react primarily by halogen atom transfer to generate stable, closed shell organohalides which can then undergo elimination or substitution in a subsequent step. Furthermore, the halomethyl radical byproducts do not readily participate in hydrogen atom transfer reactions with organic substrates. For these reasons, reactions that utilize these oxidants typically display expanded scope and functional group tolerance compared to their aerobic or persulfate-mediated counterparts. Additionally, halocarbon oxidants are freely soluble in most organic solvents, rendering issues of mass transport or gas diffusion inconsequential. The main drawback of these reagents is that they are quite toxic and environmentally problematic on scale. Although many halocarbons might be used as terminal oxidants, in practice, carbon tetrabromide (CBr4), carbon tetrachloride (CCl4), and bromotrichloromethane (BrCCl3) have been the most frequently employed for photoredox applications (Figure 3).
Figure 3.

Common Halocarbon Oxidants
Stephenson and coworkers dramatically expanded the scope of viable nucleophiles in their photocatalytic aza-Henry reaction (see Section 1.2) by utilizing BrCCl3 as a terminal oxidant instead of molecular oxygen (Scheme 9).26 The aerobic conditions suffered from slow photocatalyst turnover, and Stephenson found that the use of BrCCl3 as a terminal oxidant afforded high yields of aza-Henry adduct 30. Stephenson proposed that amine radical cation 28, generated by photooxidation of 27, afforded highly reactive bromide 29 after reaction with BrCCl3. Photocatalyst turnover was accomplished via reduction of trichloromethyl radical by the reduced photocatalyst. Bromide 29 could then be subjected to nucleophilic substitution with a variety of nucleophiles to afford high yields of functionalized tetrahydroisoquinoline products. A diverse range of nucleophiles, including allyl silanes (32), silyl enol ethers (33, 35), malonates (34), and aza-heterocycles (36), could be used.
Scheme 9.
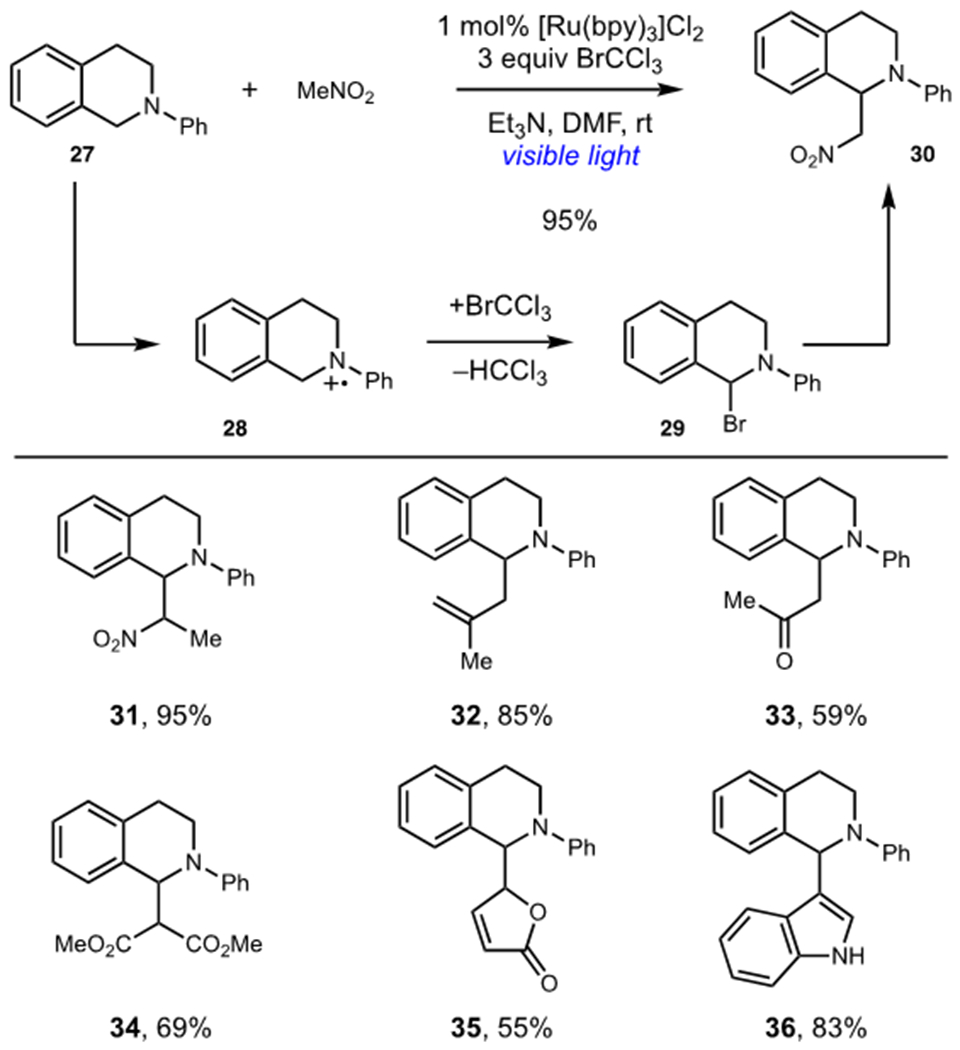
Nucleophilic Trapping of Photogenerated Iminium Ions
In a subsequent report, Stephenson and Jacobsen reported that these photogenerated α-aminohalide intermediates serve as excellent iminium ion precursors in the presence of chiral anion-binding catalysts (Scheme 10).27 The generation of a stable, closed shell intermediate (38) from 37 was crucial to the success of this method as the photochemical oxidation and alkylation steps required drastically different solvents to achieve both high yields and enantioselectivities. This method clearly demonstrates a key advantage of using this class of terminal oxidants: the organohalide intermediates are relatively stable and can generally be isolated. Thus, they can be utilized in subsequent transformations much more readily than photogenerated radical intermediates, which are rarely stable at high concentrations.
Scheme 10.
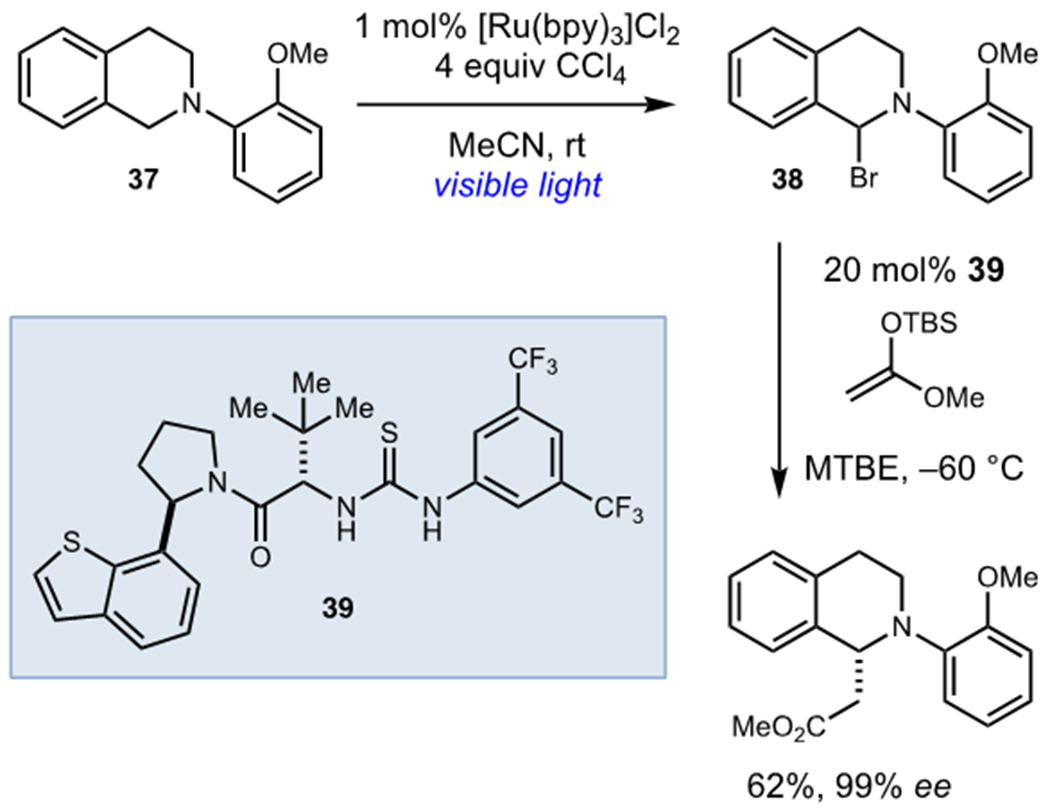
Enantioselective Oxidative Alkylation of Tetrahydroisoquinolines
6. Hypervalent Iodine Reagents
Hypervalent iodine reagents have been used extensively in organic synthesis as mild oxidants with diverse chemical reactivity.28 Hypervalent iodine reagents are most often utilized as group transfer oxidants in photoredox catalysis due to their proclivity to undergo facile single electron reduction and subsequent fragmentation, which offers a valuable route to diverse organoradical intermediates.29 The most widely studied members of this class are the cyclic benziodoxole and periodinane reagents due to their greater stability (Figure 4). In contrast, their use as terminal oxidants in photocatalytic oxidase reactions has only recently been reported. Under photoredox conditions, they typically serve to activate an organic substrate in situ towards single-electron reduction and fragmentation. Hypervalent iodine reagents can also mediate radical generation via a wide range of unconventional mechanistic manifolds, however, and optimization of the oxidant structure can often reveal new modes of reactivity.
Figure 4.
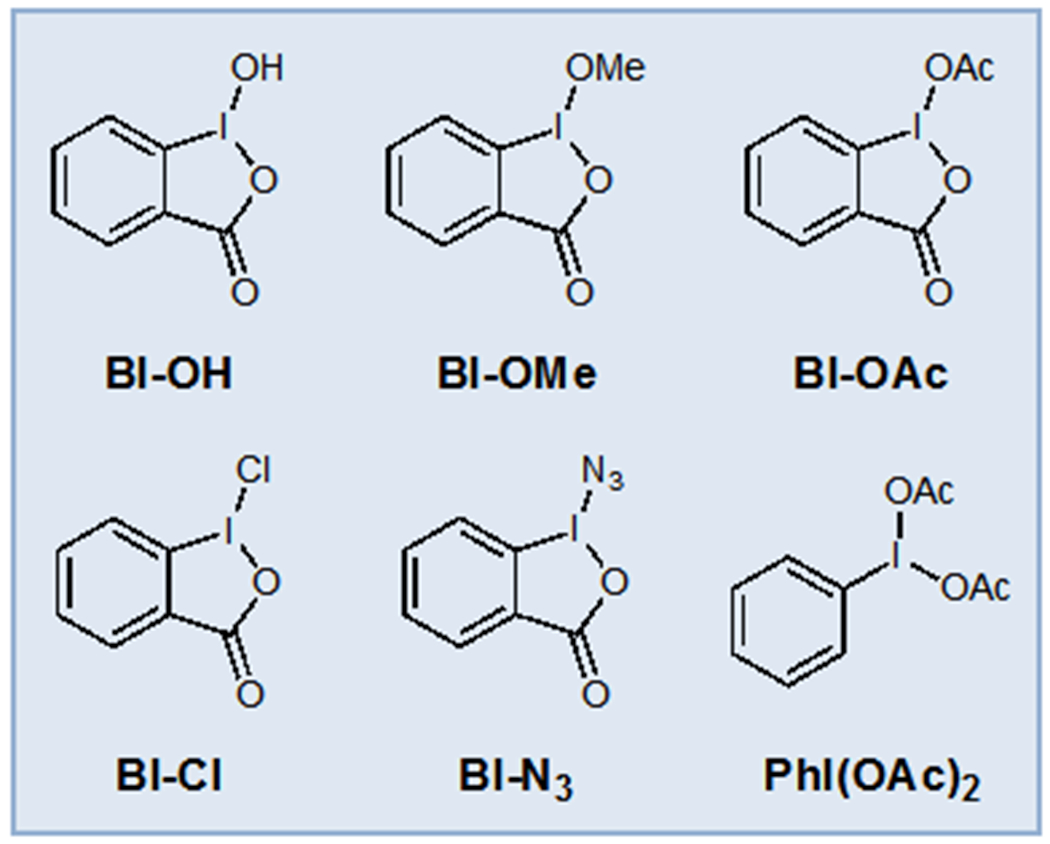
Common Hypervalent Iodine Oxidants
In 2015, Y. Chen and coworkers demonstrated that the oxidative coupling of acrylic acids and organotrifluoroborate salts could be accomplished using benziodoxole terminal oxidants under photoredox conditions (Scheme 11).30 Chen proposed that BI-OAc served to activate acrylic acid 40 towards reduction by Ru(bpy)32+ via condensation to initiate the reaction. Turnover of the photocatalyst was accomplished by oxidation of the organotrifluoroborate salt to liberate alkyl radical 42, which underwent addition to 41 to give radical 43. Fragmentation of 43 released carboxyl radical 44 and alkenylation product 45. Carboxyl radical 44 could then be reduced by Ru(bpy)3+ to turn over the photocatalyst and continue the reaction. This strategy allowed for the direct use of a wide array of carboxylic acids without the need for preactivation.
Scheme 11.
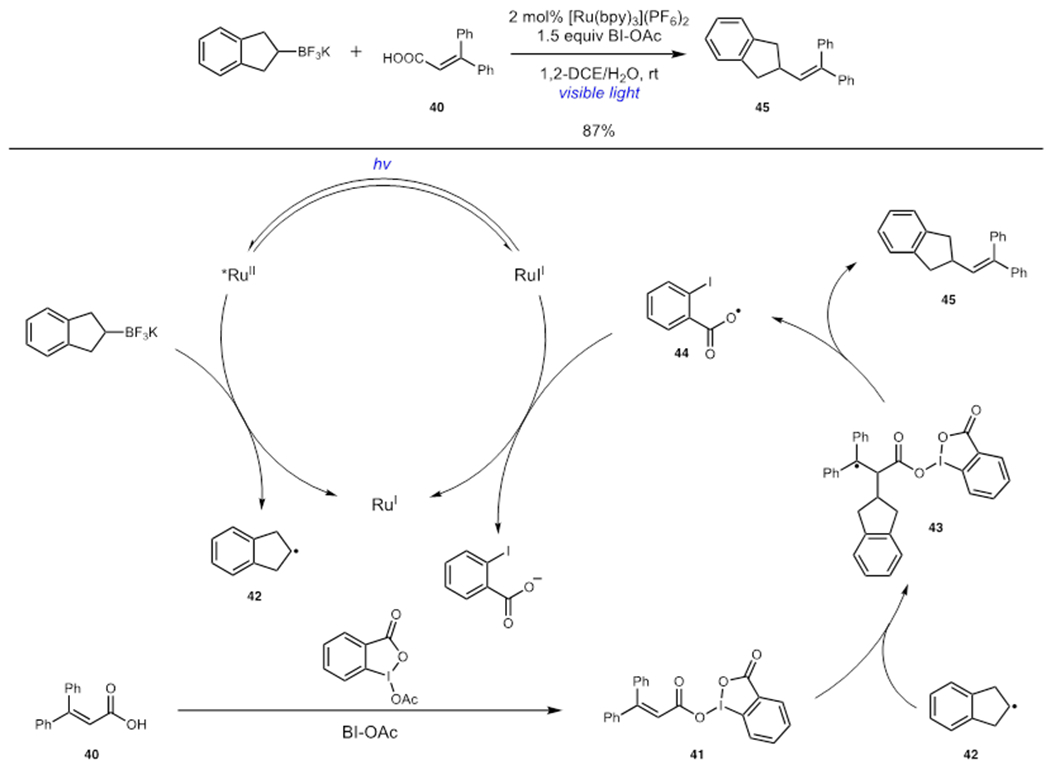
Decarboxylative Alkenylation via Photoredox Catalysis
G. Chen and coworkers have probed the mechanistic diversity available to this class of terminal oxidants. Chen utilized BI-OAc as a terminal oxidant in a photocatalytic alkylation of aza-arenes, finding that BI-OAc generated organoradical intermediates from trifluoroborate salts via an unusual radical “ate” mechanism (Scheme 12).31 Oxidative quenching of [Ru(bpy)3]Cl2 by BI-OAc resulted in formation of the corresponding carboxyl radical, which formed metastable complex 46 with n-butyl boronic acid, generated by hydrolysis of the corresponding trifluoroborate salt. This complex decomposed to afford n-butyl radical. Addition of the alkyl radical to heteroarene 47 and subsequent oxidation by the photocatalyst afforded the desired alkylated heterocycle 48. This unusual radical “ate” pathway allowed for the use of a wide variety of alkyl trifluoroborate salts as radical precursors.
Scheme 12.
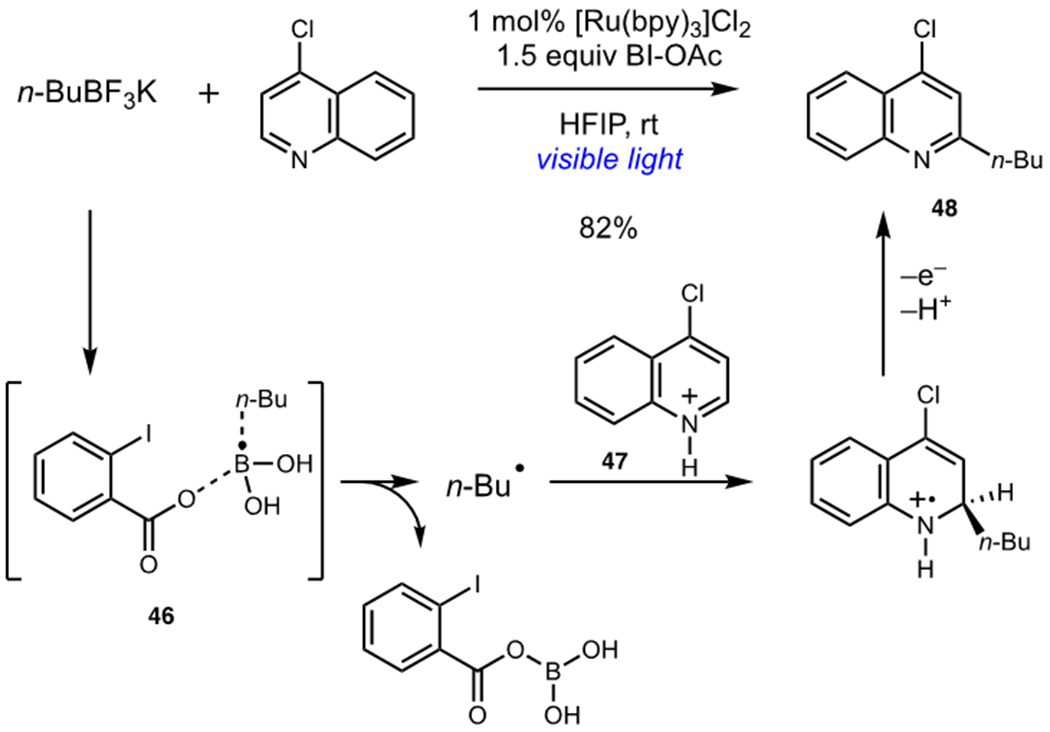
Photocatalytic Alkylation of Aza-Arenes Using Trifluoroborate Salts
The same group found that highly electrophilic hypervalent iodine oxidants could also mediate hydrogen atom transfer reactions (Table 1).32 Reasoning that the increased stability of the benziodoxole oxidant was responsible for their poor hydrogen atom transfer reactivity, a series of increasingly electrophilic benziodoxole reagents were examined. Chen found that 49 cleanly effected hydrogen atom transfer to afford organoradical intermediates, which delivered hydroxylated or amidated products in excellent yields upon oxidation and solvolysis.
Table 1.
Effect of Benziodoxole Structure on Photocatalytic Hydroxylation
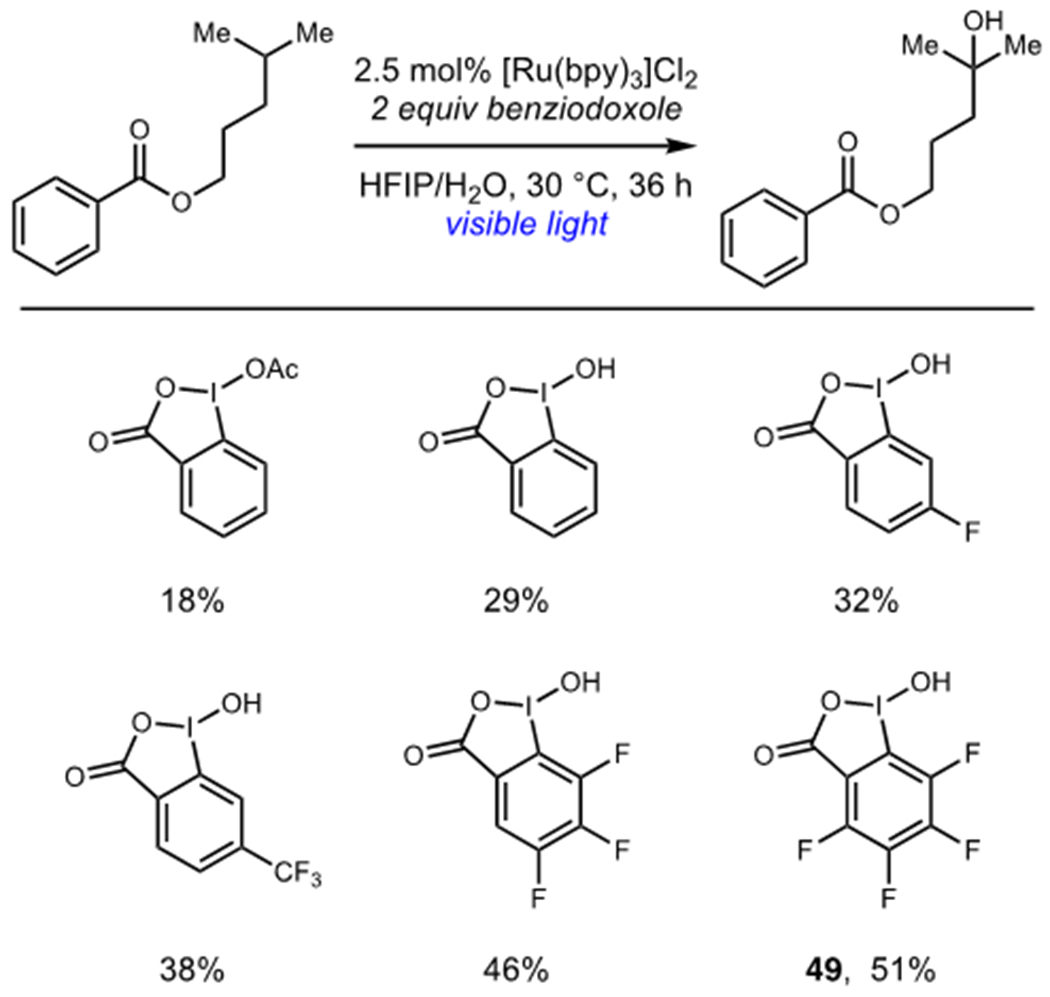
|
7. Transition Metals
Transition metals have most often been used as co-catalysts in photoredox catalysis33 while their use as stoichiometric terminal oxidants has been much less common. This is surprising given the fact that many transition metals undergo facile reduction via outer-sphere photoinduced electron transfer, making them especially well-suited to mediate photocatalyst turnover. Furthermore, the reduced metal by-products are relatively unreactive and avoid many of the decomposition pathways that can occur using other classes of terminal oxidants (e.g. hydrogen atom transfer). The use of transition metal complexes as terminal oxidants does impose several practical limitations, however. Many transition metals are prohibitively expensive and can require complicated ligand scaffolds for good reactivity. Transition metal terminal oxidants should ideally be commercially available or easily synthesized from commodity chemicals, air- and moisture-stable, and non-toxic. For these reasons, they are generally limited to simple, first row transition metal salts.
A seminal example comes from the work of Okhubo and coworkers who utilized chiral-at-metal ruthenium catalysts in an oxidative dimerization of naphthols (Scheme 13).34 The photocatalyst, a menthol derivative of Ru(bpy)32+, underwent oxidative quenching by Co(acac)3 upon excitation and the resulting Ru(III) complex was capable of oxidizing naphthol 50 to radical 51. Radical 51 underwent dimerization and oxidation with another equivalent of naphthol 50 to furnish biaryl 52. The terminal oxidant was found to have a negligible impact on enantioinduction as enantiopure Λ- or Δ-Co(acac)3 did not impart significant enantioselectivity. This is consistent with the fact that the Co(III) oxidant is not intimately involved in the oxidation of the organoradical intermediates and serves only to mediate redox state changes of the photocatalyst.
Scheme 13.
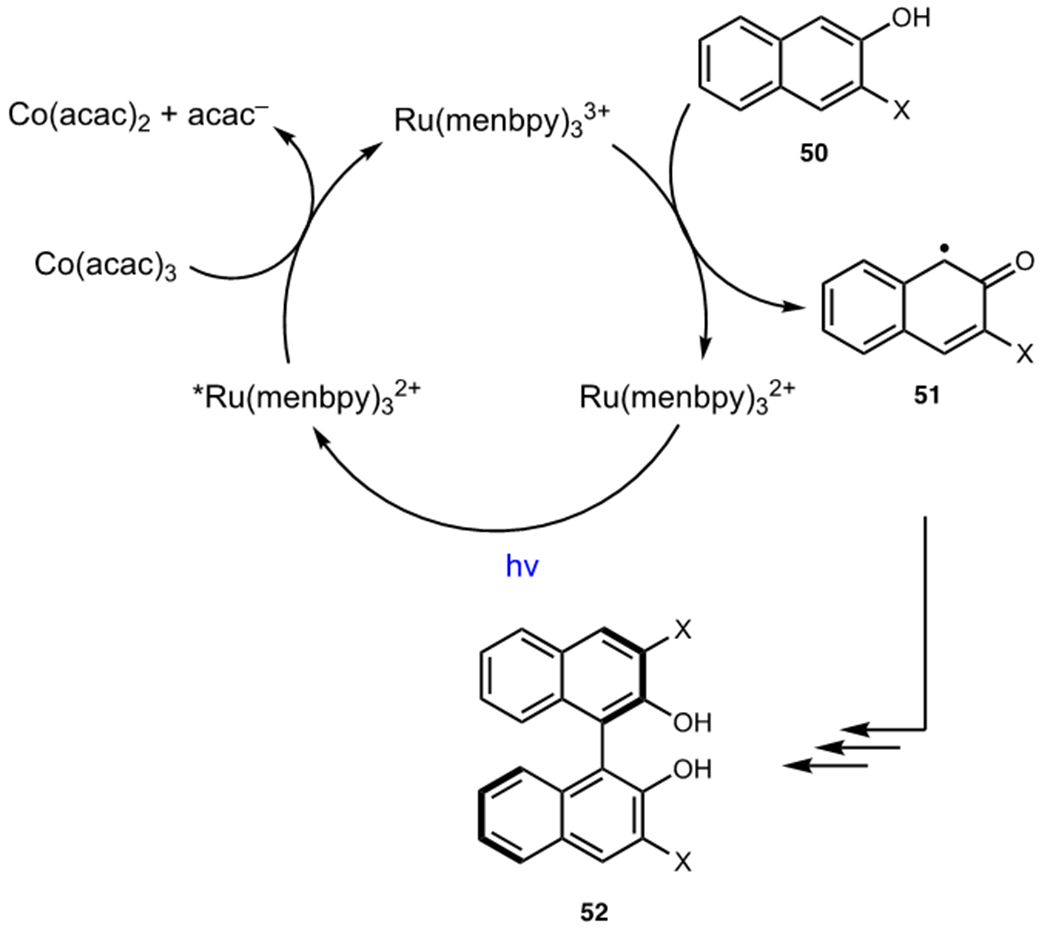
Okhubo’s Mechanism for the Dimerization of Naphthols using Ru(menbpy)32+
Cu(II) has found use as a terminal oxidant in thermal catalysis, but its utilization in photoredox catalysis has only recently gained attention. Cu(II) readily oxidizes organoradical intermediates to their corresponding carbocations, which can then be intercepted by a wide variety of nucleophilic functionality. The reduction of Cu(II) also provides a strong thermodynamic driving force to drive a wide array of oxidative transformations. Additionally, the reduced metal byproducts are innocuous and easily removed at the end of a reaction by aqueous washing or filtration.
Yoon and coworkers developed an approach to photocatalytic alkene difunctionalization that utilized triphenyl pyrylium tetrafluoroborate (TPPT) as a photocatalyst and Cu(II) salts as terminal oxidants (Scheme 14).35 Initial photooxidation of 53 resulted in formation of styrene radical cation 54 which underwent cyclization with tethered carbamates to afford radical 55. The role of Cu(II) in this reaction was proposed to be twofold: (1) to oxidize 55 and (2) to turn over the photocatalyst. Cyclization of the carbamoyl oxygen and loss of tert-butyl cation upon oxidation of 55 afforded oxazolidone 56. Interestingly, Yoon found that one equivalent of Cu(II) was able to effect both oxidations, resulting in formation of Cu(0) which could be observed precipitating from solution. Due to the oxidase nature of this reaction, alkene diamination and dioxygenation could also be accomplished under very similar conditions by changing the identity of the heteroatomic nucleophile.
Scheme 14.
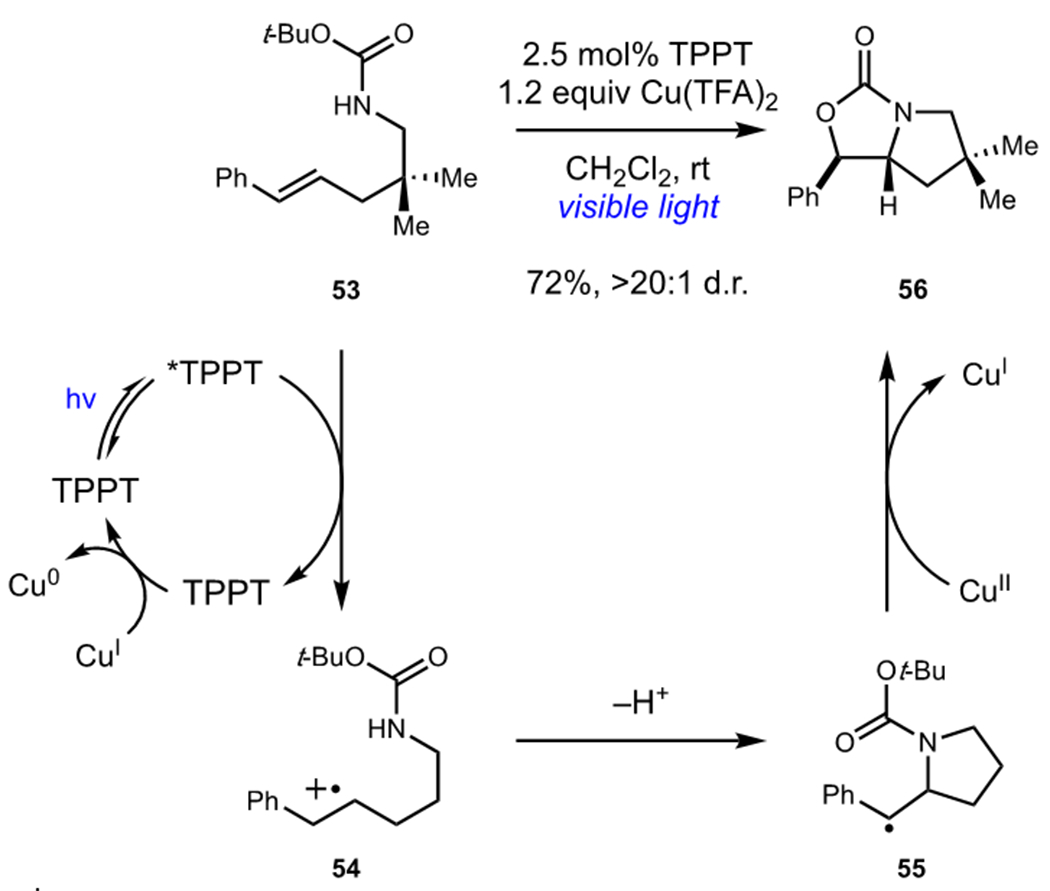
Photocatalytic Oxidative Alkene Difunctionalization
Realizing that the oxidation of radical intermediates by Cu(II) should be agnostic to the method of radical generation, Yoon subsequently developed a direct benzylic alkoxylation reaction between electron-rich arenes and alcohols (Scheme 15).36 Oxidation and subsequent deprotonation of 57 generated radical 58. Oxidation of 58 by Cu(II) afforded quinone methide cation 59 which was then intercepted by oxygen nucleophiles to generate benzylic functionalization products (60). Disproportionation of Cu(I) generated Cu(II), which effected photocatalyst turnover. The mild reaction conditions enabled the late stage functionalization of several arene natural products with complex alcohols. In conceptually related work, Tunge and coworkers demonstrated that the outcome of organoradical oxidation can be diverted towards alkene formation to accomplish the decarboxylative alkenylation of amino acid derivatives.37
Scheme 15.
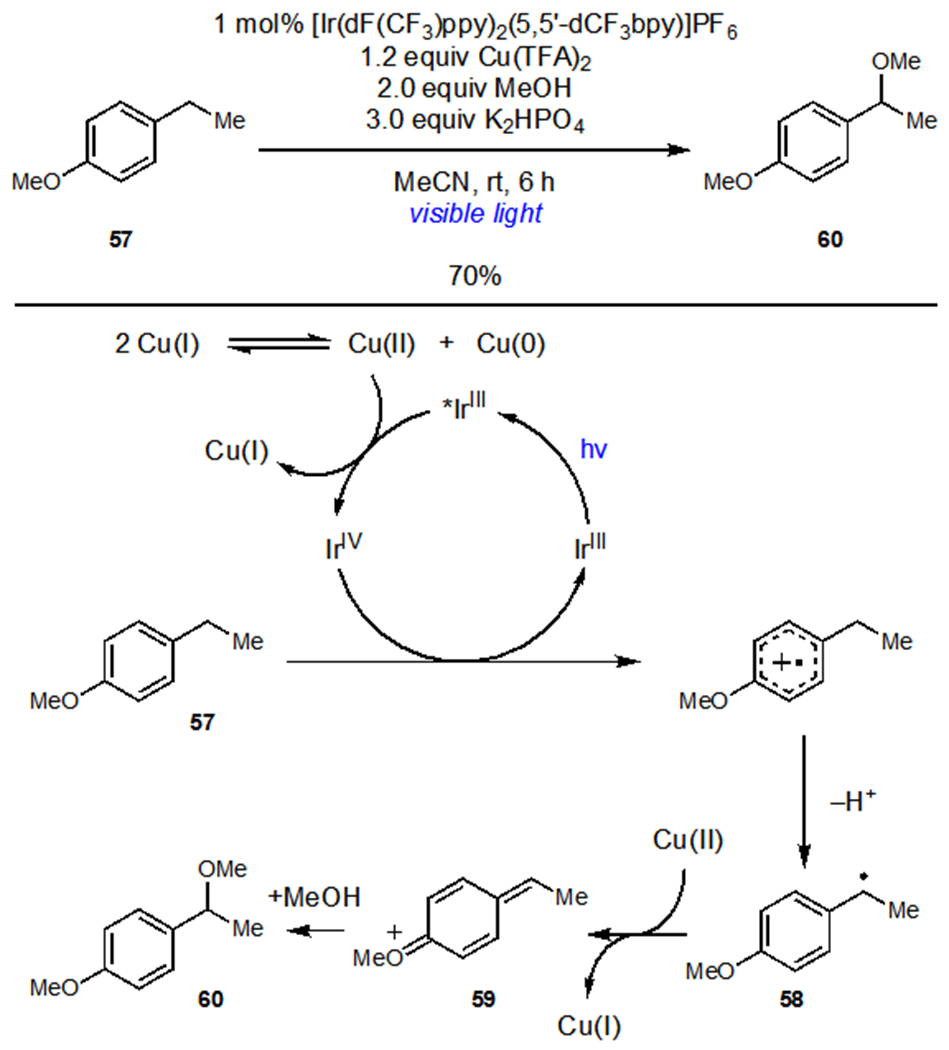
Photocatalytic Benzylic Alkoxylation
8. Hydrogen Evolution Catalysis
One particularly attractive approach to the development of photocatalytic oxidase reactions is to couple the oxidation of an organic substrate to the reduction of H+ and generation of H2 gas. The use of hydrogen evolution catalysis in combination with photoredox catalysis has allowed for the development of a wide variety of “external oxidant-free” reactions in which the protons and electrons necessary for hydrogen evolution come from the organic substrate upon photooxidation. The most commonly utilized catalysts for this purpose are the Co(III) diglyoxime complexes first reported by Connolly and Espenson (Figure 5)38 whose synthesis, reactivity, and applications have been examined in detail elsewhere.39 The main advantage of these systems is that they eliminate the need for stoichiometric bases and oxidants and generate H2 as the byproduct. While the mechanism of hydrogen evolution can be quite complex, a simplified mechanism under photoredox conditions is presented in Scheme 16. Reduction of a Co(III) complex by two sequential single-electron transfers gives rise to an intermediate Co(I) complex. Protonation affords a Co(III)–H which, after an additional proton transfer, liberates H2 to regenerate the starting Co(III) catalyst.
Fig. 5.
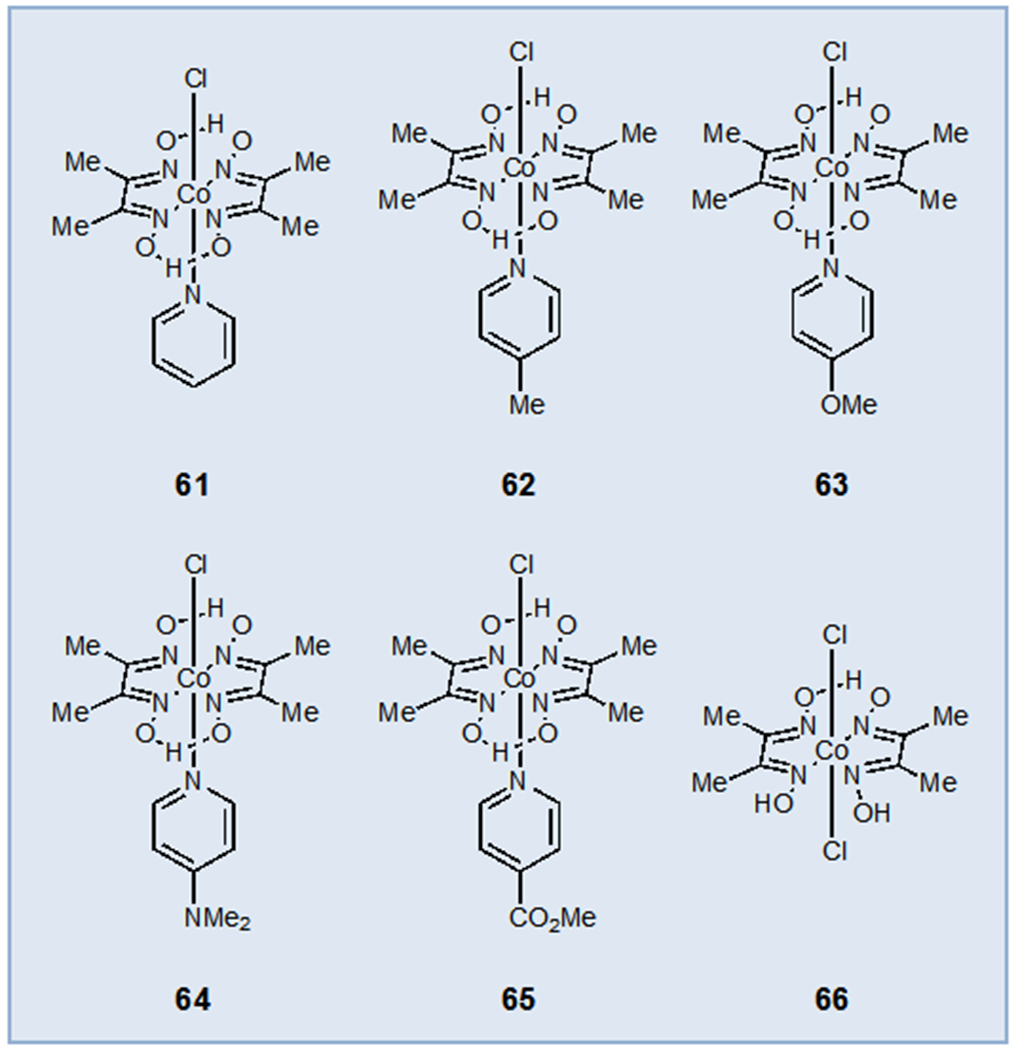
Common Hydrogen Evolution Catalysts
Scheme 16.
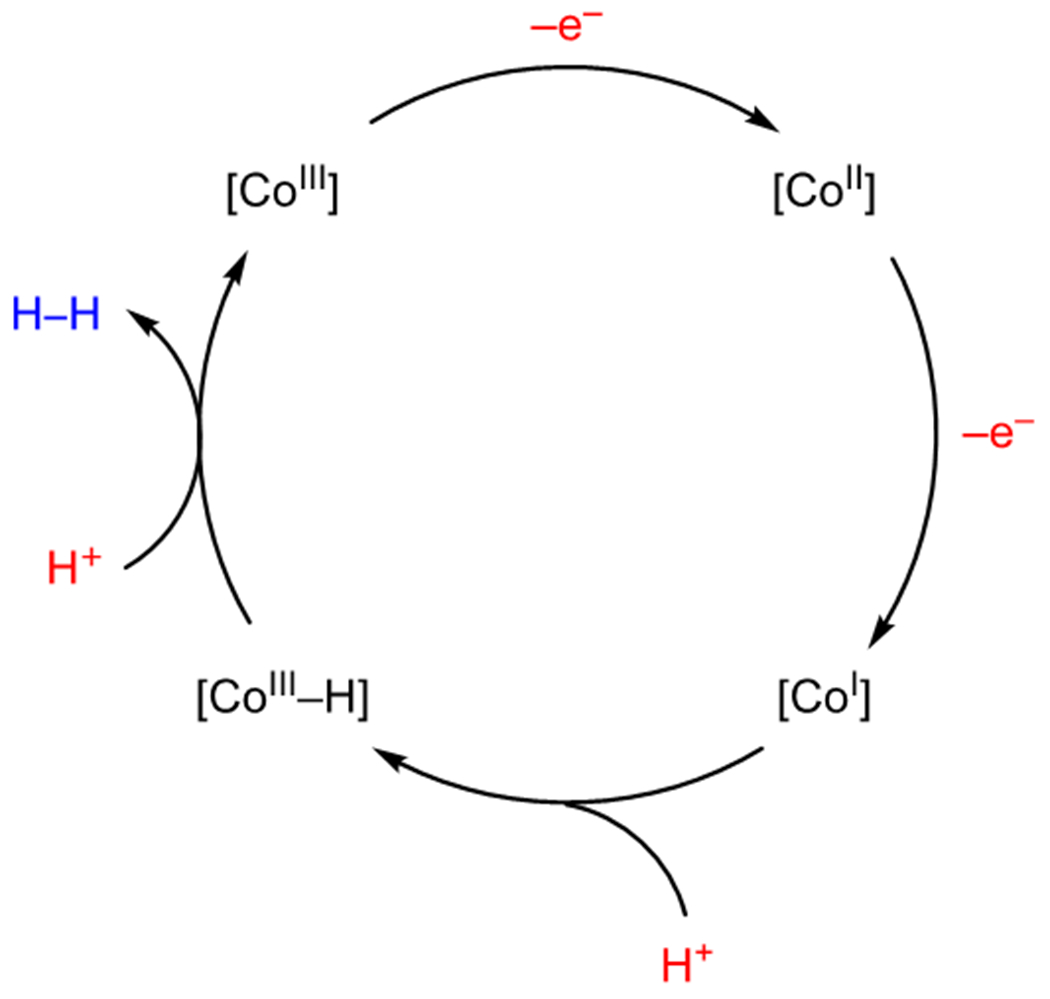
Common Mechanism for Hydrogen Evolution by Cobalt Diglyoxime Catalysts under Photoredox Conditions
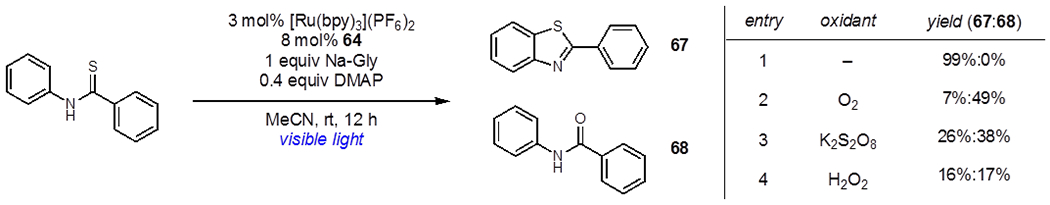
Lei and coworkers used a dual photoredox/hydrogen evolution strategy to develop a photocatalytic C–H thiolation reaction (Table 2).40 Using catalytic Ru(bpy)32+ and 64 Lei observed quantitative yields of benzothiazole 67. They found these conditions applicable to the synthesis of a wide variety of substituted benzothiazoles and found no traces of overoxidation. To demonstrate the advantages of this strategy, Lei used other common terminal oxidants in place of hydrogen evolution catalyst 64 and found that they resulted in mixtures of 67 and amide 68, with 68 favored in all cases. Thus, the use of hydrogen evolution catalysis avoids this deleterious pathway entirely. Lei has since demonstrated the broad generality of this approach, developing a wide variety of oxidative transformations, including hydration, phosphonylation, arene amination, dehydrogenative lactonization, and oxidative annulation reactions.41 In all cases, the protons and electrons needed for hydrogen evolution are provided by the organic substrate upon photooxidation. Sorenson and coworkers reported that this strategy can also be used to effect the dehydrogenation of simple alkanes and alcohols when used in combination with a decatungstate photocatalyst.42
Table 2.
Effect of the Oxidant on Photocatalytic Thiolation
|
Nearly simultaneous reports by the groups of Ritter43 and Tunge44 described the application of dual photoredox/hydrogen evolution catalysis approaches to decarboxylative olefination. Ritter’s system converted alkyl carboxylic acids to alkenes using an iridium photocatalyst and 63 as a hydrogen evolution catalyst (Scheme 17a) and olefination proceeded in excellent yields using a wide variety of carboxylic acids. Interestingly, stoichiometric experiments of isolated alkyl–Co(III) complexes showed that alkene formation occurs from discrete organocobalt complexes upon irradiation with visible light. Tunge and coworkers reported a similar strategy for the decarboxylative olefination of N-acyl amino acid derivatives (Scheme 17b). Tunge found that enamides and enecarbamates could be readily synthesized upon irradiation of the corresponding amino acids in the presence of an acridinium photocatalyst and 61. This method greatly expanded the scope of decarboxylative elimination as initially described by Kochi by eliminating the need for stoichiometric Pb(OAc)4 and Cu(OAc)2, which afforded only low yields of the desired products and significant decomposition.
Scheme 17.
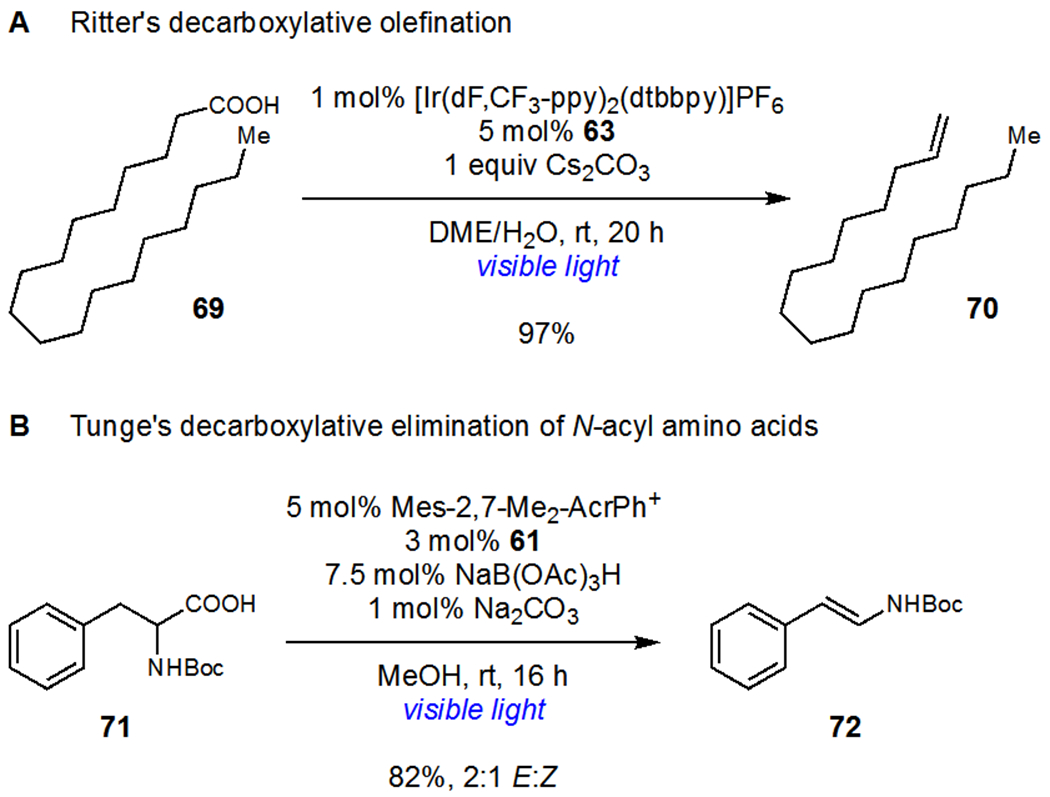
(a) Ritter’s Decarboxylative Olefination and (b) Tunge’s Decarboxylative Elimination of N-Acyl Amino Acids
9. Electrochemistry
There has been a significant renewal of interest in synthetic organic electrochemistry over the last two decades. While the combination of photoredox catalysis and electrochemistry has only recently been explored in depth, it represents an exciting new frontier in photocatalytic reaction development. The main advantage of this strategy is that anodic oxidation can be coupled to the cathodic reduction of an innocuous terminal oxidant (e.g., H+). Consequently, these reactions typically require acidic additives although they afford H2 gas as the sole by-product. There are, however, several limitations that must be considered in the development of such a reaction. Anodic oxidation of organic compounds typically requires high electrode potentials, which can lead to decomposition, and the resulting organoradical intermediates can undergo further oxidation at the electrode surface, leading to electrode passivation. Finally, electrochemical synthesis comes with significant barriers to entry in terms of equipment and experimental expertise and has long been viewed as a “specialist” branch of synthetic organic chemistry.
These barriers have been lowered substantially through the publication of tutorial reviews and the introduction of standardized instruments for electrochemical synthesis.45,46 Methods that combine electrochemical and photochemical activation of organic substrates have been enabled primarily by indirect electrolysis strategies in which a redox mediator, often the photocatalyst, is oxidized at the anode surface, diffuses into the bulk solution, and oxidizes an organic substrate (Scheme 18).47 Turnover of the electrochemical system is most often coupled to cathodic generation of H2 from H+. This strategy allows for many oxidative transformations to be performed at low applied potentials (ca. <1 V versus SCE), limiting overoxidation and electrode passivation. Indirect electrolysis also allows for efficient irradiation of commonly used electrochemical cells, significantly simplifying the experimental intricacies associated with the development of these methods.
Scheme 18.
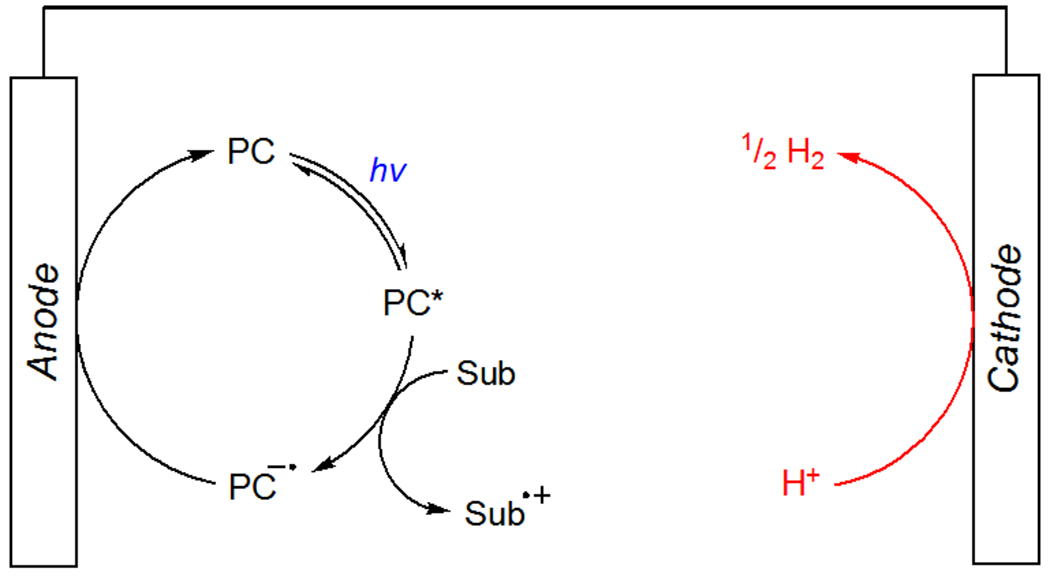
General Mechanism for Dual Photoredox/Electrochemical Oxidase Reactions
Several recent pioneering studies have clearly demonstrated the power and unique reactivity accessible by combining these two modes of activation. Xu and coworkers demonstrated that electrochemical oxidation could be used in place of persulfate oxidants during the development of a Minisci reaction of heteroarenes (Scheme 19).48 Their proposed mechanism involved photooxidation of a trifluoroborate salt (74) by MesAcrMe+ to generate an alkyl radical intermediate that subsequently adds to an aza-arene (73). Subsequent oxidation by either the photocatalyst or at the anode surface afforded the desired functionalized heteroarenes (75–78). Photocatalyst turnover was accomplished via anodic oxidation and was coupled to cathodic reduction of H+. Lin and coworkers recently reported an elegant study on the oxidation of unactivated alcohols using riboflavin tetraacetate (RFT) as a photocatalyst (Scheme 20).49 Unactivated alcohols could not be directly oxidized by the excited state photocatalyst but Lin found that addition of thiourea 79 as a catalytic redox mediator afforded high yields of aldehyde and ketone products. These reactions were proposed to proceed via hydrogen atom transfer between photogenerated thiourea radical 80 and the alcohol substrates to afford an electron-rich α-oxy radical which undergoes subsequent oxidation at the anode surface.
Scheme 19.
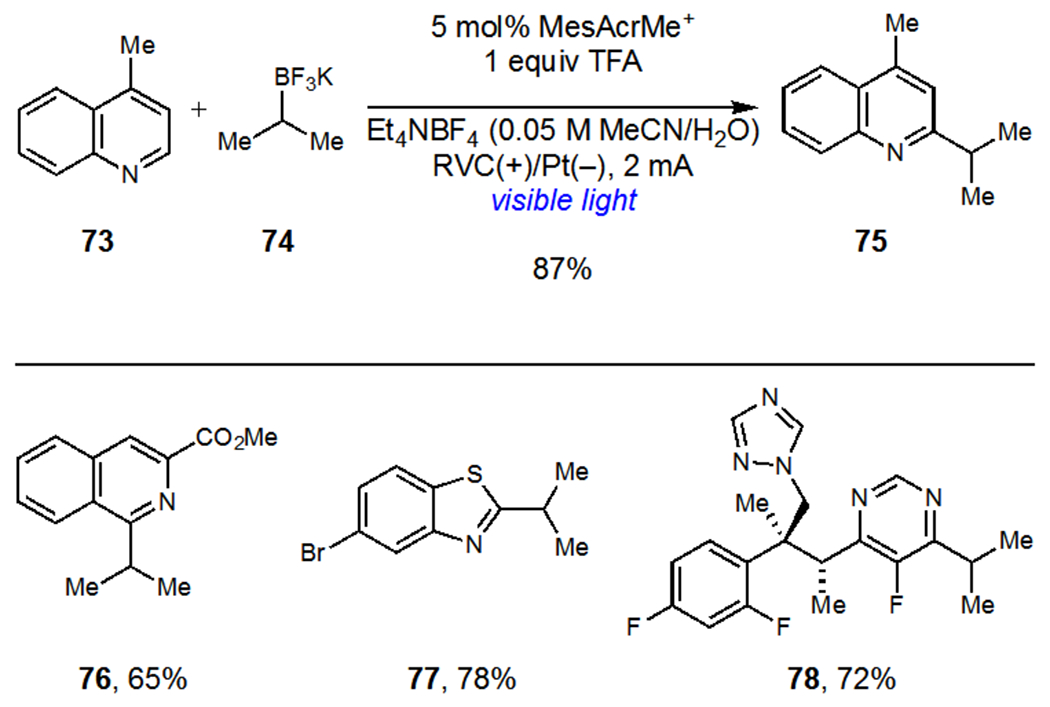
Xu’s Alkylation of Heteroarenes
Scheme 20.
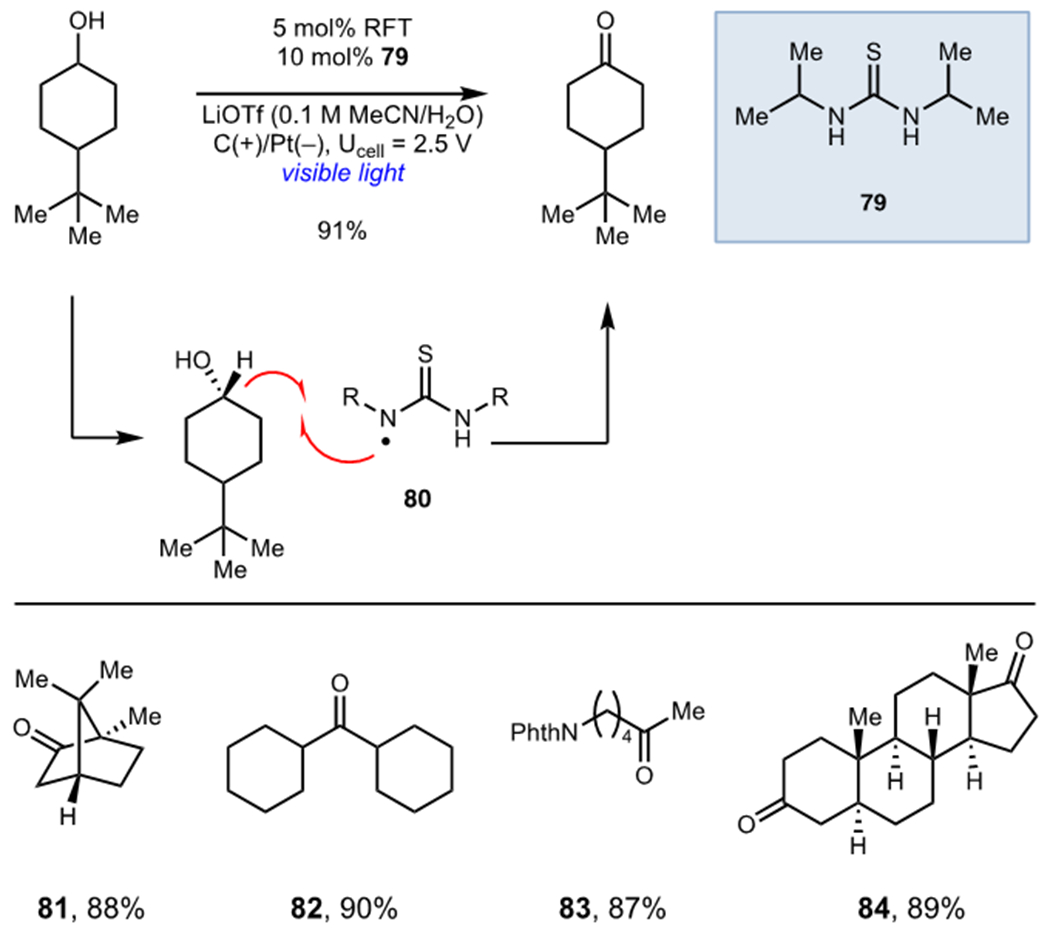
Lin’s Oxidation of Unactivated Alcohols
Stahl and Wang greatly expanded the scope of the traditional Hoffman-Löffler-Freytag (HLF) reaction by merging the photochemical cleavage of N–I bonds with the electrochemical oxidation of an iodide catalyst (Scheme 21).50 Stahl found that tetrabutylammonium iodide (TBAI) could be oxidized at low applied potentials to generate catalytic I2. Subsequent reaction with sulfonamide or imidate nucleophiles generated iodoamine intermediates, like 86, which underwent homolysis to generate nitrogen-centered radical 87 upon irradiation with visible light. The reaction then proceeded via 1,5-HAT, iodination, and base-promoted nucleophilic ring closure to deliver aza-heterocycle 88 and this method proved applicable to the synthesis of a wide range of aza-heterocyclic scaffolds (89–92). Photochemical cleavage of the N–I bond proved crucial to the success of this method as β-hydride elimination of the iodoamine intermediates predominated under thermal conditions.
Scheme 21.
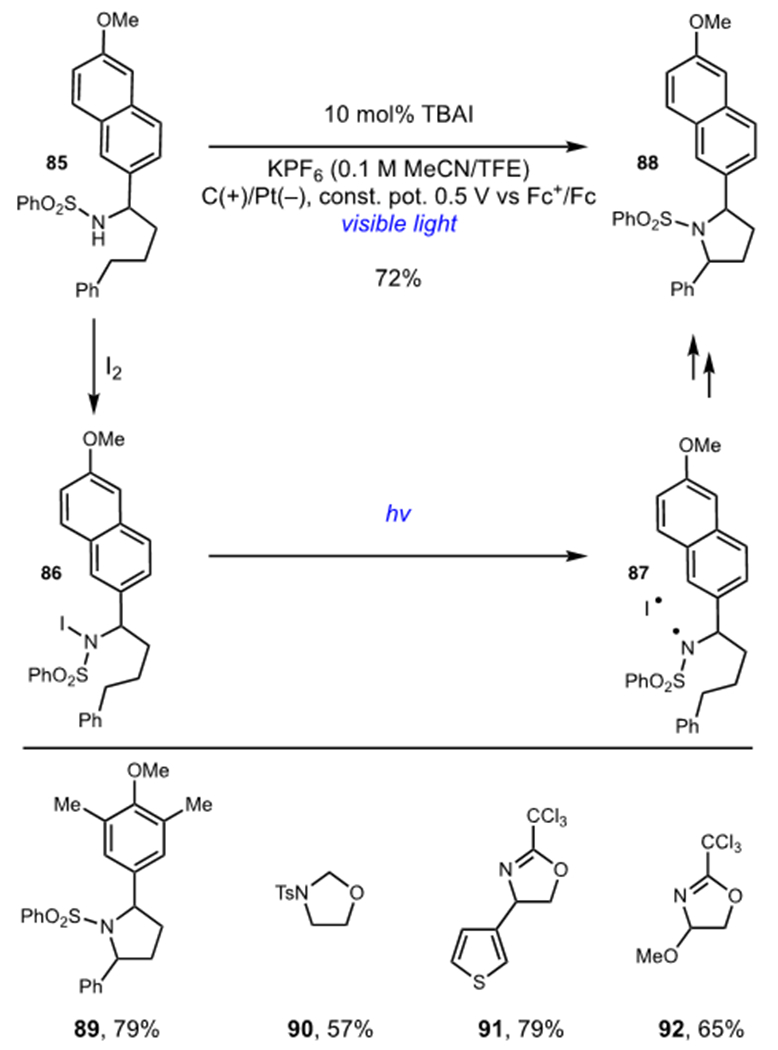
Stahl’s Hoffman-Löffler-Freytag Reaction
Lambert and coworkers reported the trisaminocyclopropenium ion (TAC) as a privileged catalyst for dual photoredox/electrochemical applications. The relevant mechanism for oxidase chemistry is outlined in Scheme 22. Anodic oxidation of TAC generates a radical dication (TAC+), which, upon excitation with visible light, affords an extremely oxidizing excited state (*TAC+, Eox = +3.33 V vs SCE). Oxidation of an organic substrate regenerates the ground state monocationic catalyst. Lambert initially applied this catalyst system to the oxidative amination of arenes using nitrogen heterocycles which was proposed to proceed via the intermediacy of arene radical cations (Scheme 23a).51 In comparison to Nicewicz’s arene amination (see Scheme 3),14 Lambert’s conditions displayed expanded scope due to the highly oxidizing nature of the excited state radical dication and, as such, even electron-deficient arenes were viable reaction partners. The use of TAC as a photochemical redox mediator was crucial as bulk electrolysis led to polymerization and poor yields of aminated product.
Scheme 22.
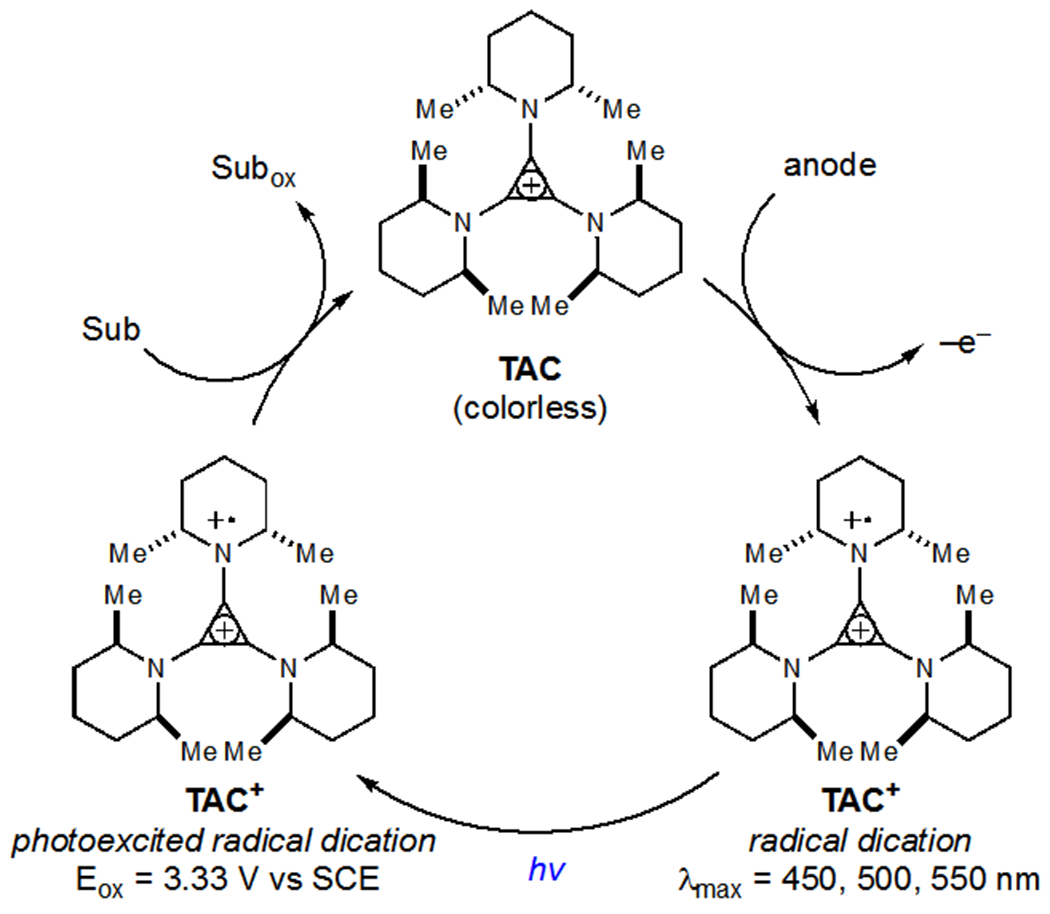
Photophysical and Electrochemical Properties of TAC
Scheme 23.
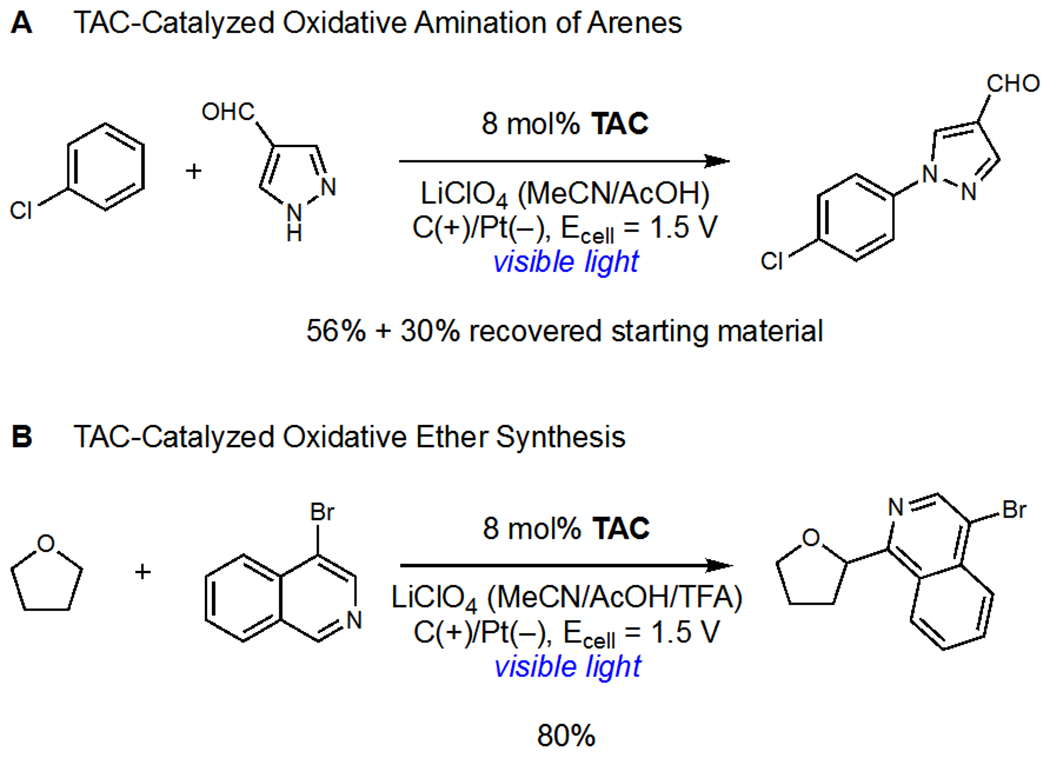
Lambert’s (a) Oxidative Amination of Arenes and (b) Oxidative Ether Synthesis
Lambert demonstrated that the excited state radical dication can mediate hydrogen atom transfer to generate organoradical intermediates (Scheme 23b).52 Using this new mode of reactivity, a Minisci reaction between alkyl ethers and heteroarenes was developed. The oxidase nature of this process allowed electron deficient olefins and N–H heterocycles to be used in place of the heteroarene reaction partner with no change to the catalytic conditions. This process is noteworthy because the hydrogen atom transfer reagent is generated catalytically as turnover of the electrochemical system is coupled to cathodic reduction of H+. Cathodic generation of H2 also turns over the TAC catalyst and allows the photocatalytic cycle to continue. This contrasts most photocatalytic oxidase reactions which use stoichiometric terminal oxidants to generate hydrogen atom transfer reagents upon photoreduction (see Sections 2–4).
10. Other Oxidants
A variety of organic compounds have also been used as terminal oxidants in photocatalytic oxidase reactions. A unifying theme among these reagents is that the reduced oxidant (often an organoradical species itself) must not promote deleterious side reactions. This is most commonly accomplished by a subsequent reaction in which the reduced oxidant generates a closed shell organic molecule. An early example is the seminal work of Cano-Yelo and Deronzier, who reported a photocatalytic oxidation of benzyl alcohols to benzaldehydes using aryl diazonium salts as terminal oxidants (Scheme 24).53 Diazonium salt 93 oxidatively quenches the excited state photocatalyst, generating aryl radical 94 as a byproduct. Aryl radicals are normally quite reactive and might be expected to engage in unproductive side reactions, but 94 underwent Pschorr cyclization to yield fluorenone 95 as a stable byproduct instead. A more recent example comes from DiRocco and Rovis who reported the asymmetric α-acylation of tertiary amines using a combination of photoredox and N-heterocyclic carbene catalysis.54 They showed that meta-dinitrobenzene acts as an excellent terminal oxidant for this process; its absence results in decomposition and low yields. Other oxidants, like bromotrichloromethane, also resulted in significant decomposition of both the amine substrate and N-heterocyclic carbene catalyst presumably due to the highly reactive nature of the trichloromethyl radical intermediate.
Scheme 24.
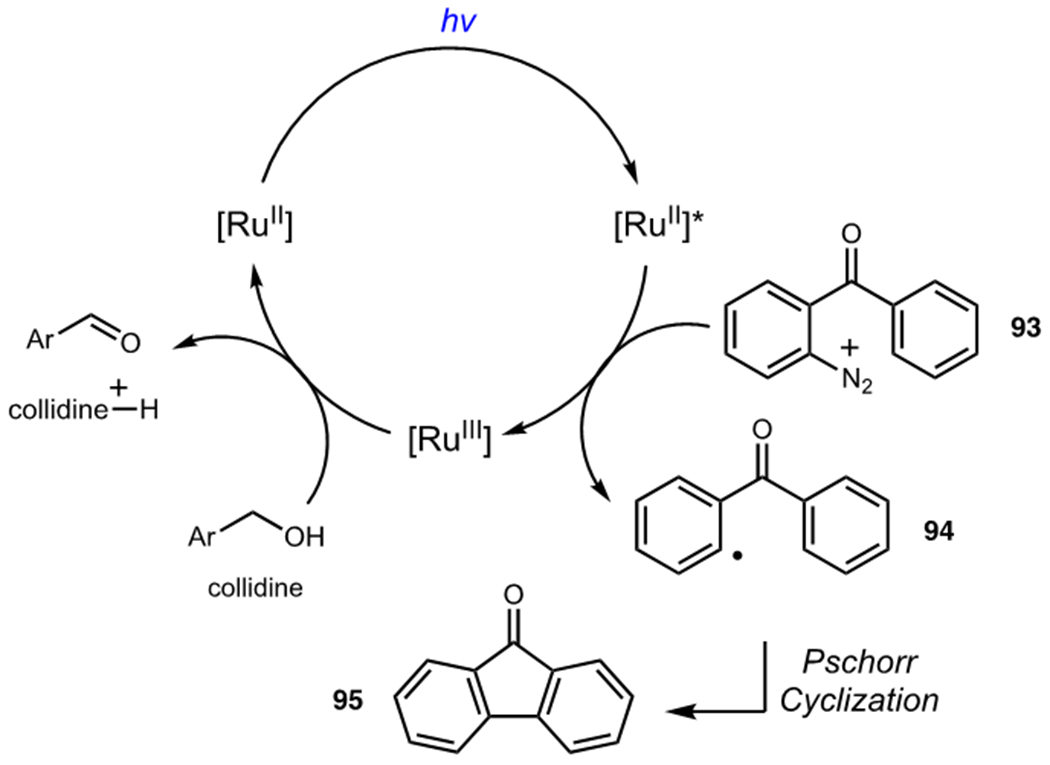
Mechanism for Photocatalytic Oxidation of Carbinols
Conclusions
The identification of terminal oxidants for photoredox applications has resulted in the development of a variety of synthetically useful oxidative transformations. While the majority of these proceed via group-transfer pathways, there has been an increasing appreciation for the power of oxidase-type processes where the identity of the terminal oxidant is critical to the success of the methods. Under this paradigm, the terminal oxidants can react through a diverse array of mechanistic pathways enabled by photoredox catalysis. They can quench the photocatalyst to generate highly reactive intermediates; they can activate an organic substrate towards reaction with an excited state photocatalyst; they can directly oxidize photogenerated radical intermediates; or they can mediate photocatalyst turnover by outer sphere electron transfer. In most cases, several of these processes are performed in concert to accomplish a single photocatalytic transformation.
Our purpose in crafting this tutorial review has been to survey the major classes of terminal oxidants that have been utilized in photocatalytic oxidase chemistry and to provide instructive guidance using selected examples. The most important insight that arises from this analysis is that terminal oxidants that are ideal for thermal catalysis are often not ideal for photoredox applications, and it is not uncommon to see highly unconventional terminal oxidants (e.g. diazonium salts) employed under photoredox conditions. Thus, the design of new photocatalytic oxidase reactions requires a high degree of flexibility and creativity in the selection of appropriate terminal oxidants. While this might seem like a daunting task at first, it also affords exciting opportunities to develop fundamentally new strategies in catalytic oxidation chemistry. Our hope is that this review will provide an appreciation for the critical role of the terminal oxidant in these processes and aid in the future development of novel photocatalytic transformations.
Fig. 2.
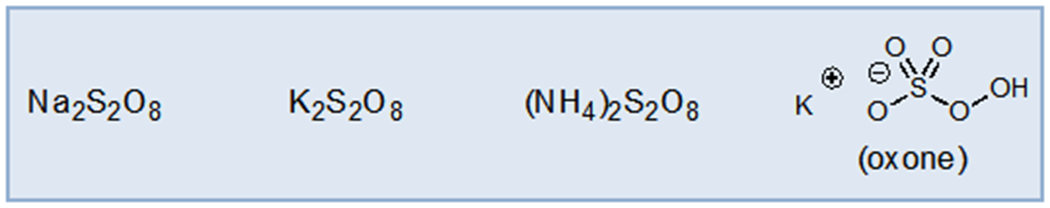
Common Persulfate Oxidants
Acknowledgements
Funding for our laboratory’s research in oxidative photoredox catalysis is provided by the NIH (GM095666).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.See: Narayanam JMR and Stephenson CRJ, Chem. Soc. Rev, 2011, 40, 102–113 [DOI] [PubMed] [Google Scholar]
- 2.See:Prier CK, Rankic DA, and MacMillan DWC, Chem. Rev, 2013, 113, 5322–5363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.See:Romero NA and Nicewicz DA, Chem. Rev, 2016, 116, 10075–10166 [DOI] [PubMed] [Google Scholar]
- 4.For a leading modern example, see: Narayanam JMR, Tucker JW, and Stephenson CRJ, J. Am. Chem. Soc. 2009, 131, 8756–8757. [DOI] [PubMed] [Google Scholar]
- 5.Rueda-Becerril M, Mahé O, Drouin M, Majewski MB, West JG, Wolf MO, Sammis GM, and Pacquin J-F, J. Am. Chem. Soc, 2014, 136, 2637–2641. [DOI] [PubMed] [Google Scholar]
- 6.Ventre S, Petronijevic FR, and MacMillan DWC, J. Am. Chem. Soc, 2015, 137, 5654–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.See:Stahl SS, Angew. Chem., Int. Ed, 2004, 43, 3400–3420 [DOI] [PubMed] [Google Scholar]
- 8.See:Stahl SS, Science, 2005, 309, 1824–1826 [DOI] [PubMed] [Google Scholar]
- 9.Winterle JS, Kliger DS, and Hammond GS, J. Am. Chem. Soc, 1976, 98, 3719–3721. [Google Scholar]
- 10.Maillard B, Ingold KU, and Scaiano JC, J. Am. Chem. Soc, 1983, 105, 5095–5099. [Google Scholar]
- 11.See:Hayyan M, Hashim MA, and AlNashef IM, Chem. Rev, 2016, 116, 3029–3085 [DOI] [PubMed] [Google Scholar]
- 12.Condie AG, González-Goméz JC, and Stephenson CRJ, J. Am. Chem. Soc, 2010, 132, 1464–1465. [DOI] [PubMed] [Google Scholar]
- 13.Bartling H, Eisenhofer A, König B, and Gschwind RM, J. Am. Chem. Soc, 2016, 138, 11860–11871. [DOI] [PubMed] [Google Scholar]
- 14.Romero NA, Margrey KA, Tay NE, and Nicewicz DA, Science, 2015, 349, 1326–1330. [DOI] [PubMed] [Google Scholar]
- 15.McManus JB and Nicewicz DA, J. Am. Chem. Soc, 2017, 139, 2880–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margrey KA, Levens A, and Nicewicz DA, Angew. Chem., Int. Ed, 2017, 56, 15644–15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo W-J, Tsukamoto T, and Kobayashi S, Angew. Chem., Int. Ed, 2015, 54, 6587–6590. [DOI] [PubMed] [Google Scholar]
- 18.See:Mandal S, Bera T, Dubey G, Saha J, and Laha JK, ACS Catal, 2018, 8, 5085–5144 [Google Scholar]
- 19.See:Yagi M and Kaneko M, Chem. Rev, 2001, 101, 21–35 [DOI] [PubMed] [Google Scholar]
- 20.Fancy DA and Kodadek T, Proc. Natl. Acad. Sci. U.S.A, 1999, 96, 6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin J and MacMillan DWC, Angew. Chem., Int. Ed, 2015, 54, 1565–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahn DK, Kang YW, and Woo SK, J. Org. Chem, 2019, 84, 3612–3623. [DOI] [PubMed] [Google Scholar]
- 23.Blum TR, Zhu Y, Nordeen SA, and Yoon TP, Angew. Chem., Int. Ed, 2014, 53, 11056–11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yayla HG, Peng F, Mangion IK, McLaughlin M, Campeau L-C, Davies IW, DiRocco DA, and Knowles RR, Chem. Sci, 2016, 7, 2066–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.For a leading modern example, see: Nguyen JD, Tucker JW, Konieczynska MD, and Stephenson CRJ, J. Am. Chem. Soc, 2011, 133, 4160–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freeman DB, Furst L, Condie AG, and Stephenson CRJ, Org. Lett, 2012, 14, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergonzini G, Schindler CS, Wallentin C-J, Jacobsen EN, and Stephenson CRJ, Chem. Sci, 2014, 5, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.See: Yoshimura A and Zhdankin VV, Chem. Rev. 2016, 116, 3328–3435 [DOI] [PubMed] [Google Scholar]
- 29.See: Wang L and Liu J, Eur. J. Org. Chem, 2016, 10, 1813–1824 [Google Scholar]
- 30.Huang H, Jia K, and Chen Y, Angew. Chem., Int. Ed, 2015, 54, 1881–1884. [DOI] [PubMed] [Google Scholar]
- 31.Li G-X, Morales-Rivera CA, Wang Y, Gao F, He G, Liu P, and Chen G, Chem. Sci, 2016, 7, 6407–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li G-X, Morales-Rivera CA, Gao F, Wang Y, He G, Liu P, and Chen G, Chem. Sci, 2017, 8, 7180–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.See: Twilton J, Le C, Zhang P, Shaw MH, Evans RW, and MacMillan DWC, Nat. Rev. Chem, 2017, 1, 0052 [Google Scholar]
- 34.Hamada T, Ishida H, Usui S, Watanabe Y, Tsumura K, and Ohkubo K, J. Chem. Soc., Chem. Commun, 1993, 909–911. [Google Scholar]
- 35.Reed NL, Herman MI, Miltchev VP, and Yoon TP, Org. Lett, 2018, 20, 7345–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee BJ, DeGlopper KS, and Yoon TP, Angew. Chem., Int. Ed, 2019, 58, 1–7. [Google Scholar]
- 37.Cartwright KC, Lang SB, and Tunge JA, J. Org. Chem, 2019, 84, 2933–2940. [DOI] [PubMed] [Google Scholar]
- 38.Connolly P, and Espenson JH, Inorg. Chem, 1986, 25, 2684–2688. [Google Scholar]
- 39.See:Dempsey JL, Brunschwig BS, Winkler JR, and Gray HB, Acc. Chem. Res, 2009, 42, 1995–2004 [DOI] [PubMed] [Google Scholar]
- 40.Zhang G, Liu C, Yi H, Meng Q, Bian C, Chen H, Jian J-X, Wu L-Z, and Lei A, J. Am. Chem. Soc, 2015, 137, 9273–9280. [DOI] [PubMed] [Google Scholar]
- 41.See: Wang H, Goa X, Lv Z, Abdelilah T, and Lei A, Chem. Rev, 2019, 119, 6769–6787 [DOI] [PubMed] [Google Scholar]
- 42.West JG, Huang D, and Sorensen EJ, Nat. Commun, 2015, 6, 10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun X, Chen J, and Ritter T, Nat. Chem, 2018, 10, 1229–1233. [DOI] [PubMed] [Google Scholar]
- 44.Cartwright KC and Tunge JA, ACS Catal, 2018, 8, 11801–11806. [Google Scholar]
- 45.See: Feng R, Smith JA, and Moeller KD, Acc. Chem. Res, 2017, 50, 2346–2352 [DOI] [PubMed] [Google Scholar]
- 46.See: Moeller KD, Chem. Rev, 2018, 118, 4817–4833 [DOI] [PubMed] [Google Scholar]
- 47.For a recent review on the combination of electrochemistry and photoredox catalysis, see:; Liu J, Lu L, Wood D, and Lin S, ACS Cent. Sci, 2020, 6, 1317–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan H, Hou Z-W, and Xu H-C, Angew. Chem., Int. Ed, 2019, 58, 4592–4595. [DOI] [PubMed] [Google Scholar]
- 49.Zhang W, Carpenter KL, and Lin S, Angew. Chem., Int. Ed, 2020, 59, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang F and Stahl SS, Angew. Chem., Int. Ed, 2019, 58, 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang H, Strater ZM, Rauch M, Shee J, Sisto TJ, Nuckolls C, and Lambert TH, Angew. Chem., Int. Ed, 2019, 58, 13318–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang H, Strater ZM, and Lambert TH, J. Am. Chem. Soc, 2020, 142, 1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cano-Yelo H and Deronzier A, Tetrahedron Lett, 1984, 25, 5517–5520. [Google Scholar]
- 54.DiRocco DA and Rovis T, J. Am. Chem. Soc, 2012, 134, 8094–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]