

Abstract
Inflammation contributes centrally to cardiovascular diseases, and anti-inflammatory treatments can reduce cardiovascular events. The JAK–STAT pathway is an emerging target in inflammation, mainly in rheumatoid arthritis (RA) and chronic myeloproliferative neoplasms (MPNs), disorders that heighten cardiovascular risk. The aim of this study was to review the international literature on the relationship between dysregulation of the JAK–STAT pathway in RA/MPNs and cardiovascular risk and on the potential cardiovascular effects of JAK–STAT inhibitors. The JAK–STAT pathway sustains inflammatory and thrombotic events in autoimmune disorders such as RA and MPNs. Here, an imbalance exists between pro- and anti-inflammatory cytokines [increased levels of interleukin (IL)-6, IL-1-β, tumour necrosis factor-α, decreased levels of IL-10] and the over-expression of some prothrombotic proteins, such as protein kinase Cε, on the surface of activated platelets. This pathway also operates in atherosclerotic cardiovascular disease. JAK–STAT inhibitors may reduce cardiovascular events and related deaths in such conditions, but the potential of these agents requires more studies, especially with regard to cardiovascular safety, and particularly for potential prothrombotic effects. JAK–STAT inhibitors merit consideration to curb heightened cardiovascular risk in patients with RA and MPNs, with rigorous assessment of the potential benefits and risks.
Keywords: Cardiovascular disease, Rheumatoid arthritis, Inflammation, Atherosclerosis, Myeloproliferative neoplasm, JAK–STAT pathway, Tofacitinib, Baricitinib, Ruxolitinib
Graphical Abstract
Introduction
Cardiovascular diseases cause nearly one‐third of deaths worldwide.1 Atherosclerosis is the most important substrate for cardiovascular diseases, with acute myocardial infarction (MI), stroke, and ischaemic cardiomyopathy being major causes of death and disability.2 The participation of inflammation in cardiovascular diseases has come into focus as clinical trials have validated a large body of biomarkers and experimental work. Immune-inflammatory cells foster the initiation and subsequent development of atherosclerosis. Individuals at risk of developing cardiovascular diseases have increased levels of inflammatory cytokines or biomarkers of their activation, such as C-reactive protein (CRP).3,4 These findings have encouraged clinical research in targeting inflammatory molecules, with the aim of reducing cardiovascular risk.5–10
From this perspective, the JAK–STAT inflammatory pathway has recently attracted interest. The JAK–STAT pathway participates in the pathogenesis of several autoimmune diseases including rheumatoid arthritis (RA)11–13 and myeloproliferative neoplasms (MPNs),14 but it also operates in cardiovascular diseases.15 Recently, several JAK–STAT inhibitors have become available for the treatment of RA16 and MPNs17 and are under evaluation for other rheumatological disorders including systemic lupus erythematosus,18,19 autoinflammatory diseases and giant cell arteritis.12 Considering that both rheumatological and haematological disorders are themselves characterized by increased cardiovascular risk11–15,20,21 the use of JAK inhibitors in these conditions affords a unique opportunity to understand their therapeutic role in atherosclerosis.
We here therefore review the evidence for the hypothesis that these inhibitors benefit cardiovascular diseases in the two specific conditions, RA and MPNs, where they have been most studied.
The JAK–STAT pathway in physiology and disease
Janus kinases (JAKs) comprise a family of intracellular non-receptor tyrosine kinases. Originally named ‘just another kinase’, these enzymes were subsequently renamed ‘Janus kinases’ from the name of Janus, the two-faced Roman god of beginnings, endings and duality. This is because JAKs possess two nearly identical phosphate-transferring domains: one exhibiting kinase activity and the other one inhibiting the kinase activity of the former. There are four currently listed JAK proteins—JAK1, JAK2, JAK3, and TYK2—involved in signalling activated by various members of cytokine receptor families.22
Members of the signal transducer and activator of transcription (STAT) protein family are intracellular transcription factors that mediate many aspects of cellular immunity, proliferation, apoptosis, and differentiation. There are seven STAT proteins—STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6—activated by different combinations of cytokines or growth factors in addition to the JAK proteins.22 STATs require activation by the recruitment of a receptor complex, consisting of two JAK combinations and several cytokines, growth hormones, or growth factors.
Signalling through the JAK–STAT pathway (Figure 1) begins when a cytokine binds to its corresponding receptor. This ligation leads to conformational changes in the cytoplasmic moiety of the receptor, activating receptor-associated members of the JAK family. These JAK proteins, in turn, mediate the phosphorylation at specific tyrosine residues of the receptor’s cytoplasmic domains, which then serve as docking sites for STATs and other signalling molecules. Once recruited to the receptor, STATs also become phosphorylated by JAKs on a single tyrosine residue. Activated STATs then dissociate from the receptor, dimerize, translocate to the nucleus, and bind to members of the gamma-activated site family of enhancers. This event allows the transcription of different portions of DNA-targeted genes that encode molecules involved in processes of proliferation, differentiation, and apoptosis.23 Modulators of the signal transduction to the nucleus include inhibitory molecules, such as suppressors of cytokine signalling (SOCSs) and protein tyrosine phosphatases (PTPs). SOCSs act by negative feedback, binding to JAK proteins or to the cytokine receptors, and limiting STAT phosphorylation. PTPs remove phosphate groups from phosphorylated tyrosine residues on JAKs and STATs.24
Figure 1.
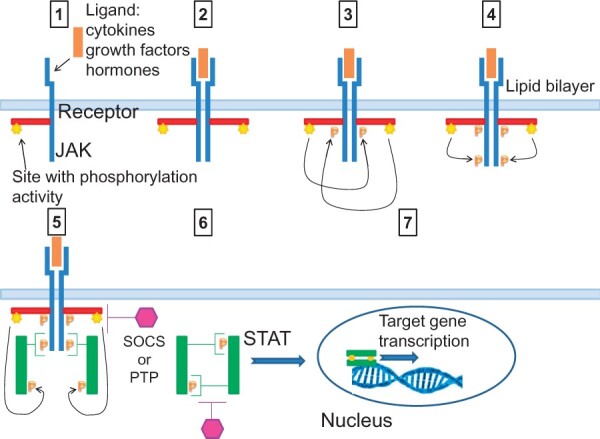
Signalling through the JAK–STAT pathway. (1) The signal through the JAK–STAT pathway is initiated when a ligand (e.g. cytokines, growth factors, hormones) binds to its corresponding transmembrane receptor. (2) Ligand binding causes receptor dimerization. (3) This leads to the activation of the receptor-associated members of JAKs through proximity‐mediated trans‐phosphorylation. (4) JAKs are endowed with a phosphorylation site. This mediates the phosphorylation of a specific tyrosine residue of the receptor, which then becomes a docking site for latent cytoplasmic STAT proteins. (5) Inactive STAT proteins associate with the phosphorylated receptor, allowing the JAK protein to phosphorylate its C‐terminus on a single tyrosine residue. (6) Activated STATs then dissociate from the receptor, dimerize, and translocate to the nucleus. (7) Here, they bind to nuclear DNA. This allows the transcription of specific DNA-targeted genes that encode for molecules involved in processes of proliferation, differentiation, and apoptosis. (8) The signal transduction to the nucleus is turned-off by inhibiting molecules, such as suppressors of cytokine signalling and protein tyrosine phosphatases.
In several pathological states, the JAK–STAT pathway does not operate in a physiological manner. This is because of mutations—sporadic or inherited—of the signalling elements involved directly in the pathway or in regulatory molecules. Different clinical manifestations may then occur, depending on which combination of ligands, JAK and STAT proteins, DNA sequences, or regulatory proteins are defective compared with the physiological situation. Consequently, derangements of the pathway may contribute to rheumatological diseases or to MPNs.25
Several aspects of the JAK–STAT pathway still require full elucidation. These include cell-intrinsic and -extrinsic factors that govern its activity, molecular, and genomic mechanisms with epigenetic and transcriptional effects, causes that induce and maintain JAK mutations, and the correlation between biology and clinical manifestations of dysregulated JAK–STAT signalling. Nevertheless, the rapidly accumulating knowledge on these aspects should lead to development of novel possible pathway-targeting drugs and their practical implementation in disease states.23
Atherosclerosis, inflammation, and the JAK–STAT pathway
The relationship between cardiovascular diseases, atherosclerosis, and inflammation has gained wide recognition over the past 20 years26 and provides a background for the role of the JAK–STAT pathway in modulating cardiovascular risk. Inflammatory activation of the arterial endothelium occurs early in the development of atherosclerosis. Inflammatory cytokines, such as interleukin (IL)-1, IL-6, and tumour necrosis factor (TNF)-α, which orchestrate the clinical course of RA and other inflammatory diseases, play a pivotal role in endothelial activation and dysfunction, inhibiting the production of nitric oxide and cyclooxygenase-1.27 Endothelial activation in turn increases vascular permeability, leucocyte adhesion, and propensity to thrombosis. After monocyte adhesion to the activated, ‘inflamed’ endothelium, these cells migrate into the intima, mature into macrophages, and accumulate lipids, forming foam cells.28 Smooth muscle cells can undergo metaplasia to acquire characteristics of foam cells as well.29 Platelets can also adhere to the endothelium, leading to the release of several mediators. These stimuli, in concert with molecules derived from macrophages and activated T lymphocytes, induce smooth muscle cell migration, proliferation, and increased production of extracellular matrix.30 These processes result in the formation of atheromata, dynamic lesions consisting of dysfunctional endothelial cells, modulated smooth muscle cells, and admixed T lymphocytes and macrophages of various subtypes.31 These various leukocytes can each release mediators that promote further changes in the vessel wall. As lesions progress, cell death releases lipids and necrotic debris.26 The net result of macrophage and T-cell activation is the local production of cytokines and chemokines that further activate inflammatory cells, such as polymorphonuclear neutrophils. In advanced or disrupted human atheromata, neutrophils may contribute to lesion inflammation through release of numerous mediators such as myeloperoxidase, calgranulins, and formation of neutrophil extracellular traps (NETs).32–37 Activated macrophages actively produce reactive oxygen species that enhance low-density lipoprotein (LDL) oxidation and elaborate growth factors driving smooth muscle cell proliferation and extracellular matrix production.38 This connective tissue typically forms a fibrous cap beneath the luminal surface that overlies a central core of lipid-laden cells and fatty debris that may then accumulate calcium mineral. The accumulation of cholesterol crystals and free fatty acids in macrophages and other cells also leads to activation of pattern recognition receptors and cytosolic innate immune receptors. The latter include components of inflammasomes, multiprotein oligomers of the innate immune system that mediate inflammatory responses.39,40 Inflammasome activation leads to the maturation of IL-1β and IL-18, pro-inflammatory mediators that promote further leucocyte recruitment, including monocytes, and activated T lymphocytes.
The intimal plaque grows initially abluminally, preserving the lumen calibre, but may progressively encroach on the arterial lumen often accompanied by a thinning of the underlying tunica media. Rupture of the fibrous cap that separates the necrotic core from the flowing blood or superficial erosion may eventually cause an acute thrombotic event41–43 due to the sudden exposure of the highly thrombogenic material in the plaque to the blood, with the release of subendothelial von Willebrand factor, collagen, tissue factor (TF), as well as an array of mediators including platelet-derived thromboxane A2.44 Locally produced and circulating plasminogen activator inhibitor-1 can limit endogenous thrombolysis, favouring thrombus accumulation.
Inflammatory biomarkers to assess cardiovascular risk
Elevated plasma concentrations of pro-inflammatory mediators, such as cytokines and chemokines, adhesion molecules (intercellular adhesion molecule-1, vascular cell adhesion molecule-1), and acute-phase reactants (CRP, fibrinogen), have undergone extensive study, delineating them as markers of different phases of atherosclerosis and overt cardiovascular diseases. Many cell types can produce cytokines that mediate and regulate immune and inflammatory reactions, not only leukocytes but also endothelial, epithelial, and mesenchymal cells. These molecules circulate in the bloodstream and can affect distant tissues.45 Their plasma concentrations can serve, therefore, to predict their peripheral action and the severity of ensuing tissue injury. For example, CRP is a biomarker that—alone or in combination with clinical and other serum markers—can predict the occurrence of a first acute MI and ischaemic stroke in healthy humans.46,47 CRP derives largely from the liver in response to IL-6. This cytokine contributes causally to atherosclerotic events, while CRP is a downstream readout of IL-1β/IL-6 production and of the activation of other inflammatory pathways and most likely does not contribute directly to atherosclerotic disease.48,49 CRP detected by a high-sensitivity assay (high-sensitivity CRP) has emerged as the benchmark biomarker for inflammatory risk in atherosclerotic disease.50,51
Interleukin-6 and the JAK–STAT pathway: a two-faced Janus cornerstone in atherosclerosis and cardiovascular diseases
Among the atherosclerosis-related stimuli of JAK–STAT signalling, interferons and cytokines figure prominently. Interferon gamma (IFN-γ) augments atherosclerosis and signals through JAK1 and JAK2.52–54 IL-6 participates in many biological processes, including the activation of B cells, haematopoiesis and oncogenesis, as well as in the regulation of cell growth, gene activation, proliferation, survival, and differentiation.55 IL-6 may have direct pro- and anti-atherogenic effects and contributes causally to human atherosclerotic events.56 IL-6 signalling is complex:57–59 classical signalling involves engagement of the canonical IL-6 surface receptor (CD126) found primarily on hepatocytes and leukocytes by the soluble ligand IL-6. After binding IL-6, CD126 associates with gp130 that signals to JAK–STAT components downstream. Cleavage of CD126 by proteinases of the ADAM (short for a disintegrin and metalloproteinase) family generates a circulating soluble form of CD126 that can bind IL-6 and partner promiscuously with gp130, expressed by many cells, a pathway designated trans signalling. The roles of trans- and classical IL-6 signalling in atherosclerosis have engendered controversy.59,60 Recent human data, however, support a net pro-inflammatory effect of IL-6 signalling in humans.61
Binding of IL-6 to its receptor initiates several cellular events, including the activation of JAK- and Ras-mediated signalling. Activated JAKs phosphorylate and activate STAT transcription factors, particularly STAT3 and Src homology-2 domain-containing tyrosine phosphatase (SHP2).62 Phosphorylated STAT3 then forms a dimer and translocates to the nucleus to induce the transcription of genes containing STAT3-responding elements, dealing with cell survival and cell-cycle transitions.63 In addition to eliciting acute phase protein production in hepatocytes, IL-6 induces lipolysis and fat oxidation57,63 (Figure 1).
Many cell types involved in atherogenesis or myocardial diseases express STAT family members including cardiac myocytes, fibroblasts, and endothelial cells. Various stimuli that activate hypertrophic growth of cardiac myocytes or provide cardioprotection, as well as those occurring after mechanical stretch, pressure overload or MI, can activate the JAK–STAT signalling.64 Profound alterations in the JAK–STAT signalling pathways can occur in patients with end-stage dilated cardiomyopathy, where tyrosine phosphorylation of JAK2 is diminished, while increased gp130 phosphorylation occurs, indicating impaired downstream activation of this critical pathway.65 Activation of JAK leads to phosphorylation and activation of STAT substrates. JAK2 phosphorylates STAT3 or STAT6, which then activate the angiotensinogen gene promoter in cardiac myocytes. Angiotensinogen eventually leads to the production of angiotensin II that in turn induces the production of cardiotrophin-1, a member of the IL-6 family, ultimately producing cardiac hypertrophy.
On the other hand, several studies have demonstrated that STAT3 also mediates cardioprotective signalling when the pathway is activated by the anti-inflammatory cytokine IL-10.66,67 This activation may exert protective effects in the heart, such as compensatory hypertrophy or a reduction in apoptosis.15 Thus, the JAK–STAT pathway, in keeping with the reminiscence of Janus, can either promote or protect diseases of the myocardium and the coronary arteries.
Anti-inflammatory therapies and cardiovascular diseases
Several recent studies have highlighted the central role of inflammation in provoking cardiovascular events by targeting inflammatory pathways. Although statins exert anti-inflammatory activity independent of LDL lowering, analyses cannot rigorously deconvolute the relative contribution of these two actions. Trials of agents that act primarily as anti-inflammatory agents in cardiovascular disease have studied the anti-IL-1β monoclonal antibody canakinumab, as well as methotrexate and colchicine and the anti-IL-6 receptor monoclonal antibody tocilizumab (Table 1).
Table 1.
Main trials of anti-inflammatory strategies to avert cardiovascular events
| First author and reference | Study name | Year of start and follow-up duration | Study population | Intervention | End points | Results |
|---|---|---|---|---|---|---|
| Ridker et al.,68 USA | JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosurvastatin) | 2003, median follow-up of 1.9 years (maximum, 5.0) | Healthy people with high levels of hsCRP (≥2 mg/L), but LDL-C 130 mg/dL (3.4 mmol/L) | Rosuvastatin 20 mg daily | MI, stroke, arterial revascularization, hospitalization for unstable angina, or death from CV causes | Rosuvastatin reduced LDL-C levels, hsCRP levels, and the main CV outcome |
| Ridker et al.,69 USA | CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcomes Study) | 2011, median follow-up of 3.7 years | Patients with previous MI and hsCRP ≥2 mg/L | Canakinumab 50 or 150 or 300 mg every 3 months | Nonfatal MI, nonfatal stroke, or CV death | The 150 mg dose of canakinumab every 3 months led to a rate of recurrent CV events significantly lower than placebo |
| Kleveland et al.,70 Norway | Effect of a single dose of the IL-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with NSTEMI | 2011, follow-up of 6 months | Patients with NSTEMI | Single dose of 280 mg of tocilizumab | hsCRP and hsTnT | Tocilizumab attenuated the inflammatory response and TnT release |
| Ridker et al.,71 USA | CIRT (CV Inflammation Reduction Trial) | 2013, median follow-up of 2.3 years | Patients with previous MI or multivessel coronary disease who additionally had either type 2 diabetes or the metabolic syndrome | Methotrexate 15–20 mg weekly | Composite of MI, stroke, CV death or hospitalization for unstable angina that led to urgent revascularization | Methotrexate did not reduce levels of IL-1β, IL-6, or hsCRP and did not result in fewer CV events than placebo |
| Nidorf et al.,6 USA | LoDoCo (Low-Dose Colchicine) | 2013, median follow-up of 3 years | Patients with stable coronary disease receiving aspirin and/or clopidogrel and statins | Colchicine 0.5 mg daily | A composite of acute coronary syndrome, cardiac arrest or ischaemic stroke | Colchicine 0.5 mg daily was safe and effective in reducing the risk of CV events |
| Nidorf et al.,9 USA | LoDoCo2 | 2014, 30-Day run-in phase | Patients with stable coronary disease receiving aspirin and/or clopidogrel and statins | Colchicine 0.5 mg daily | Reduce the incidence of composite acute coronary syndrome, cardiac arrest or ischaemic stroke | Confirmation of the safety and benefit of colchicine 0.5 mg daily for secondary prevention of CV events |
| Hennessy et al.,72 Australia | LoDoCo-MI (Low-Dose Colchicine after Myocardial Infarction) | 2015, follow-up of 30 days | Patients with acute MI | Colchicine 0.5 mg daily | Proportion of patients with residual hsCRP level ≥2 mg/L after 30 days of treatment | The treatment was safe and well tolerated, but it was not associated with a significant reduction of hsCRP level |
| Tardif et al.,8 Canada | COLCOT (Colchicine CV Outcomes Trial) | 2015, median follow-up of 22.6 months | Patients within 30 days after an MI | Colchicine 0.5 mg daily | A composite of death from CV causes, resuscitated cardiac arrest, MI, stroke, or urgent hospitalization for angina leading to coronary revascularization | Colchicine produced a 23% lower composite efficacy endpoint but did not lower hsCRP level compared to placebo |
| Giles et al.,73 USA | ENTRACTE (A Clinical Outcomes Study to Evaluate the Effects of IL-6 Receptor Blockade With Tocilizumab in Comparison With Etanercept on the Rate of CV Events in Patients With Moderate to Severe Rheumatoid Arthritis) | 2016, follow-up of 4.9 years | Patients with active RA who had inadequate response to conventional synthetic disease-modifying antirheumatic drugs and who had at least 1 CV risk factor | Tocilizumab 8 mg/kg/month or Etanercept 50 mg/week | Comparison of time to first occurrence of a major adverse CV event | Tocilizumab was associated with a relative risk of 1.43 for the occurrence of major CV events compared to etanercept |
CV, cardiovascular; hsCRP, high-sensitivity C-reactive protein; IL, interleukin; LDL-C, low-density lipoprotein cholesterol; MI, myocardial infarction; NSTEMI, non-ST elevation MI; RA, rheumatoid arthritis.
Particularly, it is still debated whether tocilizumab, an IL-6 receptor inhibitor used in active RA, exerts any influence on cardiovascular risk. Current literature has not yet proven a clinically relevant cardioprotective effect for tocilizumab in RA. However, tocilizumab appears to reduce ApoLp(a) within LDL particles despite increasing their serum levels and increases protective HDL.74 Tocilizumab also decreases leptin levels in RA patients, another potentially beneficial cardiovascular effect in such patients.75
Rheumatoid arthritis: a main emerging arena for JAK–STAT inhibitors
RA exemplifies a disease in which inflammation, and in particular the JAK–STAT pathway, plays a central role, and for which agents, such as JAK–STAT inhibitors, may play a role in curbing the associated cardiovascular risk. RA is a chronic inflammatory autoimmune disorder characterized by a chronic synovitis of the joints, but also fostering significant cardiovascular morbidity and mortality.21,76–78 The threads that link joint damage and increased cardiovascular risk in RA include the participation of multiple cytokines (i.e. IL-6, IL-1, TNF), the downstream signalling of which crucially involves JAK–STAT.79 In RA, the JAK–STAT pathway appears to be constitutively activated, thus leading to the progression of joint damage through a higher expression of matrix metalloproteinase genes, chondrocyte apoptosis, and apoptosis resistance in the synovial tissue.80 In particular, among inhibitors of the JAK–STAT pathway, SOCS proteins appear deficient in RA,81,82 promoting the pathway constitutive activation. The same inflammatory cascade that affects the joints can mediate RA-associated endothelial inflammation, contributing to the development of cardiovascular complications.83,84
Blockade of JAK–STAT signalling can improve the clinical manifestations and progression of RA.85 Accordingly, the US Food and Drug Administration (FDA) already in 2014 approved the first JAK1/3-selective small molecule inhibitor (SMI) tofacitinib for the treatment of RA.86 Currently, three oral JAK inhibitors (tofacitinib, baricitinib, and upadacitinib) have received FDA approval for the treatment of RA. Baricitinib is a selective oral JAK1/2 inhibitor with moderate activity against TYK2 and significantly less inhibition of JAK3,87 whereas upadacitinib is an oral JAK1-selective inhibitor.88,89 In Japan, peficitinib, another pan-JAK inhibitor, has received approval,90 and the pipeline now lists several other molecules, such as filgotinib and decernotib.91 Overall, phase II and III randomized controlled trials (RCTs) have demonstrated the efficacy of JAK inhibitors in both RA patients with inadequate response to conventional synthetic drugs or disease-modifying antirheumatic drugs (DMARDs) and in patients with inadequate response or intolerance to TNF inhibitors.85 According to the European League Against Rheumatism (EULAR) recommendations, JAK inhibitors, either combined with a conventional synthetic DMARD or in monotherapy, are now indicated as second-line treatment for moderate-to-severe active RA in patients with an inadequate response or intolerance to methotrexate.92
Cardiovascular risk in patients with rheumatoid arthritis
Patients with RA have an incidence of cardiovascular events threefold higher than the general population.93 RA patients suffer about the double of cardiovascular disease-associated deaths compared with control subjects.83,94 Traditional cardiovascular disease risk factors, including smoking, hypertension, insulin resistance, obesity and physical inactivity, play an important role in explaining the higher cardiovascular disease risk in RA.95 The lipid profile in RA patients is characterized by the so called ‘lipid paradox’: indeed, RA patients with active disease feature lower levels of total cholesterol (TC), LDL-cholesterol, and HDL-cholesterol (HDL-C) than treated patients with normal acute-phase reactants.96 The reduction in HDL-C levels occurring in RA patients leads to a high atherogenic index of TC/HDL-C ratio, that in turn may contribute to cardiovascular risk.97 However, conventional cardiovascular risk factors do not completely explain cardiovascular morbidity in this disease.98 The increased cardiovascular risk extends beyond patients with established RA, as rheumatoid factor-positive patients with early inflammatory arthritis also have increased cardiovascular mortality.99 Elevated concentrations of cytokines, such as TNF-α, IL-6, and IL-1β, can mediate activation of both synovial and vascular cells and contribute to both pannus formation in the joints and associated higher carotid artery intima-media thickness or increased prevalence of carotid plaques in comparison with controls.100 Pro-inflammatory IL-6 and TNF-α are significantly involved in the ‘lipid paradox’ up-regulating LDL receptors on hepatocytes, thus decreasing circulating LDL. Interestingly, recent studies have shown a decrease in LDL catabolic clearance in patients treated with tofacitinib or anti-IL-6 inhibitor.101–103
Myeloproliferative neoplasms as disorders of JAK/STAT pathway dysfunction
Activation of the JAK–STAT pathway also plays a pivotal pathogenetic in chronic Philadelphia-chromosome-negative MPNs, through the presence of mutated, constitutively activated tyrosine kinases, such as JAK2, the thrombopoietin receptor (monophosphoryl lipid A) and calreticulin.104–109 Under physiological conditions, haematopoietic growth factors act on progenitor cells by binding their surface receptors and activating tyrosine kinases in a controlled manner. In contrast, in MPNs the mutated tyrosine kinases circumvent the physiological control of haematopoiesis, thus leading to continued proliferation and survival of progenitors.110 This phenomenon gives rise to polycythemia vera (PV), characterized by excess production of erythrocytes; essential thrombocythemia (ET), where platelet number exceeds 400 × 109/L; or myelofibrosis (MF), where collagen overproduction in the bone marrow associates with cytopenia in various lineages and progressive splenomegaly.111,112 Gain-of-function mutations in the JAK2 kinase-like domain are frequent in MPNs, the most common being the V617F mutation—a valine-to-phenylalanine substitution at residue 617 of the JAK2 gene113—that characterizes more than 95% of cases of PV,114 and about 50% of cases of ET and MF.110 When mutated, JAK2 binds to homodimeric cytokine receptors,115 unrestricted signalling occurs via the STAT 3/5, the phosphatidylinositol-3-kinase/AKT, and the RAS/mitogen-activated protein kinases pathways, with consequent uncontrolled expansion of myeloid lineages. STAT proteins are differentially activated in MPNs: increased STAT5 and STAT3 phosphorylation occurs in PV, increased STAT3—but reduced STAT5—phosphorylation are reported in ET, and reduced phosphorylation of both STAT5 and STAT3 characterizes MF.116 JAK V617F can also trigger the absent in melanoma 2 (Aim2) inflammasome and generate mature IL-1β.117
Activation of the JAK–STAT pathway also increases the formation of NETs, which can aggravate thrombosis. Indeed, mice with the JAK2 V617F mutation overexpress PAD4, a protein that contributes to NET formation. Administration of the JAK1/2 inhibitor ruxolitinib limits NET formation and reduces venous thrombus formation, as seen in mice.118 NETs link thrombosis119 to inflammation120 observed in MPNs. Indeed, high levels of pro-inflammatory cytokines (IL-6, IL-8, TNF-α, IFN-α, transforming growth factor-β) might contribute to the endothelial–mesenchymal transition,121 thus favouring the evolution of ET/PV into secondary MF.122 Indeed, high levels of IL-6 correlate with a higher incidence of cardiovascular events also in other chronic myeloid neoplasms.123 Especially in MF, ‘inflammatory’ symptoms are very relevant, with significant impairment of patients’ quality of life: in a survey involving 904 patients, half of responders reported a decline in the ability to work caused either by symptoms, such as itching, or the occurrence of thrombotic events.124JAK2 mutations can also increase circulating levels of the coagulation initiator TF, promoting a prothrombotic milieu,125 and augment venous thrombosis through the increased adhesion of granulocytes to endothelium via β1 and β2 integrins.126 Such findings help explain the well-known high risk of thrombosis and thrombosis recurrence that characterizes MPNs.20 In a series of 494 patients affected by PV or ET, thrombosis recurred in about one-third, especially involving the central nervous system and the heart. Leukocytosis and age (>60 years) predict thrombosis recurrence, the incidence of which decreases after starting cytoreductive therapy.127 Moreover, in about 15% of cases, portal or mesenteric vein thrombosis precedes the diagnosis of MPN.128
Many studies have now investigated the relationship between JAK2 mutations and the occurrence of thrombotic events in MPNs, offering some interesting pathogenetic hypotheses.129–131 For example, the protein kinase Cε, which is also over-expressed in platelets from patients with acute MI and cardiac hypertrophy, appears to be abundant on platelets from patients affected by MF, with significant correlation with a history of cardiovascular events and disease aggressiveness.132 It has been clearly demonstrated that hyper-activation of the JAK–STAT pathway induces significant biological changes in megakaryocytes and platelets that could contribute to the increased thrombotic risk observed in MPNs.133 In particular, two different transgenic murine models were used to demonstrate that megakaryocytes from JAK2 mutated animals show increased ploidy and mobility, that JAK–STAT activation leads to increased formation of more active pro-platelets, and that platelets from JAK2 mutated mice show increased aggregation, spreading, and propensity to thrombus formation. For a better understanding of the cause for these observations, the authors performed gene expression profiling experiments, confirming that in JAK2 mutated mice several pathways known to be important in thrombosis are upregulated.134,135 Translated in the human context, it has been reported that platelets from subjects affected by JAK2-mutated ET or with a previous history of thrombosis show higher P-selectin expression136 and higher levels of TF-positive platelets.137
All these data clearly suggest that JAK–STAT pathway hyperactivity might significantly affect different cellular pathways potentially involved in the activation of the hemostatic system, which might contribute to explain the increased risk and cardiovascular events observed in patients with MPNs.
Cardiovascular risk in patients with myeloproliferative neoplasms
Many studies available from the literature clearly show that MPNs are significantly associated with a heightened cardiovascular risk and with a higher incidence of cardiovascular thrombotic events.138 The incidence of cardiovascular events in MPN patients is about 10 times higher than in the general population, as reported by the Swedish group that matched 9429 MPN patients with 35 820 controls. Here, in the MPN cohort, arterial thrombotic events were thrice higher than in the control population, while the ratio of occurrence of venous thrombosis was even 9.7, and across all age groups.139 Moreover, the hearts of MPNs patients harbouring JAK2 mutations accumulate collagen fibres, and cardiac myocytes become hypertrophic.140
In PV, the European Collaboration on Low-dose Aspirin in Polycytemia Vera (ECLAP) study reported that cardiovascular mortality accounted for 41% of all deaths, mainly due to coronary heart disease (15%), congestive heart failure (8%), non-hemorrhagic stroke (8%), and pulmonary embolism (PE) (8%).141 The maintenance of a haematocrit level <45% significantly reduced the occurrence of cardiovascular events.142 PV is the MPN subtype more frequently associated with arterial and venous thrombosis,143 abnormal coronary flow, and myocardial ischaemia.144,145
In ET, the rate of thrombotic events ranges from 2% to 4% patient/year, with arterial events being 2–3 times higher than venous ones.146 In a series of 891 patients with a median follow-up of 6 years, 12% experienced arterial or venous thrombosis. At multivariable analysis, arterial thrombosis was predicted by age >60 years, history of previous thrombosis, ‘conventional’ cardiovascular risk factors (tobacco use, hypertension, diabetes mellitus), leukocytes >11 × 109/L, platelet count >1000 × 109/L and presence of the JAK2V617F mutation.146 Patients with ET also experience a higher rate of ischaemic strokes, attributed to increased platelet activation, coagulation changes, endothelial and leucocyte activation.147,148
In MF, the overall cumulative rate of cardiovascular death and nonfatal thrombotic complications was 2.2%/year. Here age >60 years, hyperleukocytosis and the presence of JAK2 mutations were significantly associated with thrombosis occurrence. When JAK2 mutation was present along with leukocytosis, the rate of thrombosis increased up to 3.9% person/years.20 Serum uric acid levels have also been reported to be associated with shorter times to thrombotic events, independent of the Dynamic International Prognostic Staging System score or age.149
Finally, in about 15% of cases the diagnosis of MPN is made following that of a thrombotic event, especially when occurring at an ‘atypical’ site: for example in a study involving 1062 patients with the Budd–Chiari syndrome and 855 patients with portal vein thrombosis, the prevalence of MPNs resulted to be 40.9% and 31.5%, respectively.128
Available data on the cardiovascular safety of JAK–STAT inhibitors
Pathophysiological considerations illustrated above suggest viability for the hypothesis of favourable effects of JAK–STAT inhibitors on cardiovascular disease affecting RA and MPNs. Safety concerns with the use of any new class of drugs, however, require careful consideration. This is even more so because, despite a rather solid rationale—and perhaps paradoxically—some observations have raised concern on possible prothrombotic effects of JAK inhibitors. We therefore searched and analysed PubMed-indexed studies, the EULAR documents reporting on treatments with JAK–STAT inhibitory drugs and adverse reactions connected with their use, as well as post-marketing safety documents specifically focusing on three currently used such agents—tofacitinib, baricitinib, and ruxolitinib—to identify cardiovascular events connected with their extended use in patients with RA or MPNs.
Rheumatoid arthritis
Data regarding the cardiovascular safety of JAK inhibitors in RA remain inconclusive, as they derive primarily from short-term RCTs. Observational studies without randomized allocation of treatments and those without rigorous adjudication of cardiovascular events can prove misleading (Table 2). Data from six phase III studies and two open-label long-term extension (LTE) studies of tofacitinib in patients with RA and inadequate response to DMARDs show that tofacitinib treatment associates with a low incidence of cardiovascular events.150 In a post hoc analysis of six phase III and two LTE studies over 7 years including 4076 patients, the same authors found that, after 24 weeks of tofacitinib treatment, increased HDL-C, but not higher LDL or total cholesterol, appeared to be associated with a lower rate of subsequent cardiovascular events.151 Tofacitinib may also reduce carotid intima-media thickness after 54 weeks of treatment in patients with increased carotid intima-media thickness at baseline.152 Similarly, pooling data from nine clinical trials with baricitinib, no difference in the incidence of major adverse cardiovascular events was found in patients treated with baricitinib in comparison with placebo; six events of deep vein thrombosis (DVT)/PE occurred in patients treated with 4 mg baricitinib, but no cases of DVT/PE were reported in the placebo group. During longer‐term evaluation, the incidence of DVT/PE was similar between the baricitinib dose groups.153 Finally, in a recent meta-analysis of 26 RCTs, no significant differences regarding the risk for cardiovascular events in RA patients treated with JAK inhibitors were found in general, and specifically with tofacitinib, baricitinib, upadacitinib, peficitinib, or decernotinib. The authors did not observe any significant difference in the occurrence of major adverse cardiovascular events in patients receiving JAK inhibitors with respect to placebo.154 This meta-analysis, which evaluated the risk of thromboembolic events comparing tofacitinib 5 mg against 10 mg or upadacitinib 15 mg against 30 mg, showed no significant overall risk of thromboembolic events. Conversely, however, baricitinib at dose of 2 mg appeared to be safer than 4 mg.154
Table 2.
Main studies on JAK-inhibitors and cardiovascular safety in patients with rheumatoid arthritis
| First author and reference | Objectives | Methods | Results |
|---|---|---|---|
| Charles-Schoeman et al.150 | To evaluate the incidence of CV events, including major adverse CV events (including CV death, as coronary, cerebrovascular, cardiac, and non-cardiac vascular events, and non-fatal CV events, as MI and cerebrovascular events) and CHF | Data from six phase III studies and two LTE studies of tofacitinib (administered alone or with non-biological DMARDs) in patients with RA and inadequate response to DMARDs | Lower incidence of major adverse CV events and CHF; lipid levels increased in the first 3 months and stabilized thereafter; blood pressure remained stable |
| Charles-Schoeman et al.151 | To evaluate the incidence of major adverse CV events (including MI, stroke, CV death) | Post hoc analysis of six phase III and two LTE studies over 7 years, in patients with RA and inadequate response to DMARDs, after a 24-week treatment with tofacitinib | Lower incidence of major adverse CV events; HDL-scholesterol increased, but not LDL-cholesterol or total cholesterol |
| Kume et al.152 | To analyse the effects of tofacitinib on atherosclerosis, comparing the CIMT at the baseline and 54 weeks after | Open-label prospective study in which patients with RA received tofacitinib 10 mg/day for 54 weeks | The CIMT had a signifcant decrease after the observation period |
| Taylor et al.153 | To evaluate the incidence of major adverse CV events (including MI, stroke, and CV death) and other CV events (hospitalization for unstable angina, hospitalization for heart failure, serious arrhythmia, resuscitated sudden death, cardiogenic shock, or coronary revascularizations); ATE (MI and ischaemic stroke); DVT/PE; CHF | Data from nine studies, placebo comparison up to 24 weeks included data from six studies. Randomized dose comparison between baricitinib doses of 2 and 4 mg used data from four studies and from the associated long‐term extension study | No association between baricitinib and the incidence of major adverse CV events, ATE or CHF. DVT/PE occurred in patients treated with 4 mg baricitinib, but there were no cases of DVT/PE in the placebo group |
| Xie et al.154 | To evaluate the incidence of major adverse CV events (including MI, ischaemic, and haemorrhagic strokes) or cardiovascular death, and DVT/PE | Systematic review and meta-analysis of 26 randomised controlled trials comparing tofacitinib 5 mg against 10 mg, baricitinib 2 mg against 4 mg, upadacitinib 15 mg against 30 mg | No significant differences regarding the risk for major adverse CV events were found; baricitinib at the dose of 2 mg appeared to be safer than 4 mg |
ATE, arterial thrombotic events; CHF, congestive heart failure; CIMT, carotid intima-media thickness; DVT/PE, deep vein thrombosis/pulmonary embolism; DMARDs, disease-modifying antirheumatic drugs; LTE studies, open-label long-term extension studies; MI, myocardial infarction.
There are indeed some concerns about the occurrence of thromboembolic events attributable to JAK inhibitors at higher dosage. A recent post-marketing ongoing safety trial (study A3921133) aroused concerns regarding increased risk of PE and death in RA patients older than 50 years with at least one cardiovascular risk factor when treated with high-dose tofacitinib (10 mg twice daily). Current recommendations suggest avoiding such high doses continuously in patients with augmented risk of thrombosis (older or obese subjects, patients undergoing hormone replacement therapy, patients with previous history of DVT or PE).155 More reliable results should soon become available. Similar data have been reported for baricitinib in Europe,156 and the US FDA did not approve the 4-mg dose once daily of baricitinib due to unclear additional benefit vs. the 2-mg dose once daily, and also due to concern of a dose-related effect on safety outcomes, particularly thromboembolism.87
In summary, existing data raise concern regarding the safety of high-dose JAK inhibitors with respect to thromboembolism, but further randomized and well-controlled studies are required to determine if this is a class effect and if a threshold and a dose–effect relationship exists.157 Such studies should definitely also clarify if the anti-atherosclerotic effects hypothesized above might counterbalance such potential risks.
Myeloproliferative neoplasms
The JAK1/2 inhibitor ruxolitinib was initially approved for treating patients with MPNs of intermediate-2 or high risk, according to the International Prognostic Scoring System.158 Since 2015, expanded indications include treatment of patients with PV resistant or intolerant to hydroxyurea. Two phase III pivotal trials (COMFORT-I159 and COMFORT-II160) have compared ruxolitinib to placebo or to the best available therapy, respectively. For the first time in the history of MF, a drug prolonged overall survival reducing the risk of death by 30% and proving to be quite safe also from the cardiovascular standpoint. Indeed, the 5-year update of results from these trials reported an incidence of only 0.9% of MI at 2 years, and of 2.7% at 4 years.161 More recently, a post-marketing safety study confirmed the occurrence of heart failure in only 1.6% of treated subjects.162 Other authors have evaluated the possible adverse effects of ruxolitinib on lipid metabolism. This inhibitor apparently increases cholesterol levels163 and alters the lipid structure of the cell membrane, but without damaging cell membrane integrity.164 A compilation of studies reporting on the cardiovascular safety of ruxolitinib is provided in Table 3.
Table 3.
Studies on ruxolitinib reporting on cardiovascular risk
| First author and Reference | Title | Type of study | Results |
|---|---|---|---|
| Verstovsek et al.159 | Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial | 5-year update of COMFORT I phase-III trial: ruxolitinib (n = 155) vs. placebo (n = 154) | Myocardial infarction 2.7% |
| Verstovsek et al.161 | Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses | Pooled analysis | The update of results at 5 years reported 0.9% of myocardial infarction at 2 years and 2.7% at 4 years. Overall, by 48 months of therapy 6.2% of patients receiving ruxolitinib presented cardiac failure |
| Harrison et al.160 | Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs. best available therapy for myelofibrosis | 5-year update of COMFORT II phase-III trial: ruxolitinib (n = 146) vs best available therapy (n = 118). | Cardiac failure 2% |
| Barraco et al.162 | Real-world non-interventional long-term post-authorisation safety study of ruxolitinib in myelofibrosis | Real-world experience | Occurrence of cardiac failure in only 1.6% of treated subjects |
| Samuelson et al.165 | The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: a meta-analysis | Meta-analysis | Rates of thrombosis were significantly lower among patients treated with ruxolitinib (risk ratio 0.45, 95% confidence interval 0.23–0.88) |
| Masciulli et al.166 | Ruxolitinib for the prevention of thrombosis in polycythemia vera: a systematic review and meta-analysis | Meta-analysis involving four randomized trials with 663 patients | Thrombosis risk ratio of 0.56 for ruxolitinib vs. best available therapy, corresponding to an incidence of 3.09% and 5.51% patients per year |
| Saliba et al.167 | Association between myelofibrosis and thromboembolism: a population-based retrospective cohort study | Population-based retrospective study; 1 469 790 adults were followed from 2007 until 2016 for the occurrence of myelofibrosis | One-third of myelofibrosis patients received ruxolitinib: no significant association was found between JAK-2 inhibitor treatment and the risk of venous or arterial thromboembolism, indirectly suggesting a favourable role of this drug from the CV |
| Colafigli et al.168 | The advantages and risks of ruxolitinib for the treatment of polycythemia vera | Expert opinion: authors searched Medline, Embase, archives from the European Hematology Association and the American Society of Hematology annual congresses from 2014 to 2020 about ruxolitinib treatment in polycythemia vera patients and safety | Thromboembolic events per 100 patient-years of 1.8 in the ruxolitinib vs. 8.2 in the best available therapy arm and 2.7 in the cross-over cohort |
Concerning the possible cardiovascular ‘protective’ effect of ruxolitinib, one meta-analysis reported the halving of arterial and venous thrombosis in MF and PV.165 A more recent meta-analysis including more than 600 PV patients has reported an annual incidence of thrombosis of 5.5% in the control arm vs. 3% in the ruxolitinib arm.166 Although this difference was not statistically significant, both meta-analyses suggest that ruxolitinib protects MPN patients from cardiovascular events. Another study matched 642 MF patients with 2568 controls and followed them for 10 years for thrombotic events. As expected, the MF cohort had an increased risk of DVT and PE, especially in subjects with a history of previous venous thromboembolism and atrial fibrillation. Interestingly, atypical sites of thrombosis occurred in these patients compared with controls (26.1% vs. 4.0%). In this study one-third of patients received ruxolitinib: those receiving this JAK2 inhibitor showed no significantly higher risk of venous or arterial thromboembolism, although one has to recognize that non-randomized observational studies are subject to multiple sources of confounding.167
In PV as well, ruxolitinib has so far appeared safe and protective: the pivotal phase III RESPONSE trial, which included 110 patients followed for a median of 80 weeks, reported a rate of thromboembolic events of 1.8% patient-years in the ruxolitinib vs. 8.2% in the best available therapy arm and 2.7% in the crossover cohort, again suggesting that this JAK inhibitor may play a role in reducing cardiovascular and thrombotic events in MPN patients.169 The JAK2 V617F mutation can cause clonal haematopoiesis, associated with increased cardiovascular risk, particular in younger individuals.170 Targeting JAK2 inhibitors to bearers of JAK2 V617F clonal haematopoiesis thus has a strong rationale.118
Conclusions
Patients affected by rheumatological and haematological diseases, such as RA and MPNs, share a pro-inflammatory state and concomitantly higher cardiovascular risk and increased prevalence of cardiovascular disease-related deaths than the general population.171,172 In these conditions, the JAK–STAT pathway contributes fundamentally, as demonstrated by the efficacy of JAK inhibitors, now extensively used in clinical practice.
Several research groups are now assessing the cardiovascular safety of JAK inhibitors. Treatment with tofacitinib seems to be associated with a lower incidence of cardiovascular events.150 There are some concerns about the occurrence of thromboembolic events attributable to JAK inhibitors at higher dosage (study A3921133). There appears to be an increased risk of PE and death in RA patients older than 50 years with at least one cardiovascular risk factor when treated with a high dose of tofacitinib (10 mg twice daily). Current recommendations suggest avoiding such high doses in patients with high risk of thrombosis. Similar concerns have been reported for baricitinib.153 Baricitinib at dose of 2 mg appeared to be safer than 4 mg.154
The use of tofacitinib and baricitinib in patients with autoimmune diseases and a high thromboembolic risk requires caution. Favourable observations in treatment of COVID-19173,174 support the possibility of using these drugs proactively to reduce cardiovascular events in patients at high thrombotic risk by reducing the underlying chronic inflammatory state.
Treatment with ruxolitinib improved the quality of life of MPN patients, but further studies are necessary to definitively ascertain if the use of these drugs in early disease prevents the occurrence of cardiovascular events. Results from trials on this drug reported a lower incidence of MI and heart failure in treated subjects.159,162 There are also possible adverse effects on lipid metabolism, with an increase in cholesterol levels.163 Other studies showed a lower incidence of thrombosis in patients treated with ruxolitinib, but these data are not statistically significant.166,167 In a study on PV, ruxolitinib appeared safe and protective with a lower incidence of thromboembolic events in patients treated with ruxolitinib vs. the best available therapy.169
Thus, with a word of caution, and recollecting the troublesome story of two previous drug strategies also aimed at reducing atherosclerosis—hormone replacement therapy175 and cyclooxygenase 2 inhibitors (coxibs)176—and then found harmful because of the increased thrombotic risk, currently available data support the hypothesis of a net clinical cardiovascular benefit of JAK–STAT inhibitors due to anti-atherosclerotic effects (Graphical abstract). This possibility requires rigorous investigation in well-conducted, randomized, and adequately powered trials to balance potential benefit and harm.

Clinical effects of JAK–STAT pathway activations and JAK–STAT inhibitors. (A) Stimuli, such as mechanical stretch, pressure overload and myocardial infarction, activate the JAK2/JAK2-STAT3/STAT6 pathway. This way allows the expression of the angiotensinogen gene promoter in cardiac myocytes. Angiotensinogen eventually leads to the production of angiotensin II that in turn induces the production of cardiothropin-1, a member of the IL-6 family, ultimately producing cardiac hypertrophy. The clinical effects of this pathway are cardiac hypertrophy or myocardial infarction (both could result in heart failure). Cardiotrophin-1 in turn allows the activation of the B-pathway. (B) Cytokines, such as IL-6, could activate the JAK2/JAK2-STAT3/STAT3 pathway in fibroblasts and endothelial cells. They are responsible for the endothelial damage that leads to atherosclerosis, with the formation of atheroma. There are several events linked to atherosclerotic cardiovascular disease, such as myocardial infarction, deep vein thrombosis and pulmonary embolism, cerebrovascular events (transient ischaemic attack, stroke), peripheral artery disease, and the formation of aortic aneurysms. JAK–STAT inhibitors, through the inhibition of the JAK–STAT pathway, may represent a way to prevent these adverse cardiovascular events. Baricitinib (JAK1/JAK2 inhibitor), tofacitinib (JAK1/JAK2/JAK3 inhibitor) and ruxolitinib (JAK1/JAK2 inhibitor) are treatment approved in RA and in MPNs that are hypothesized to have potential cardiovascular benefit.
Conflict of interest: F.R.M. and C.B. report no disclosures. S.G. participated in clinical trials and meetings supported by Novartis. P.L. is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Merck, Novartis, Pfizer, and Sanofi-Regeneron. P.L. is a member of scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, and XBiotech, Inc. P.L.’s laboratory has received research funding in the last 2 years from Novartis. P.L. is on the Board of Directors of XBiotech, Inc. and has a financial interest in Xbiotech, a company developing therapeutic human antibodies. P.L.'s interests were reviewed and are managed by Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies. P.L. receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund. R.D.C. has served as a co-author of several ESC Guidelines including those on acute coronary syndromes, has been Steering Committee member, National Coordinator for Italy, a co-author of the APPRAISE-2, ARISTOTLE, AVERROES, ENGAGE AF-TIMI 38, and Re-DUAL PCI trials investigating anticoagulants in atrial fibrillation and acute coronary syndromes, and has received fees, honoraria, and research funding from Sanofi-Aventis, Boehringer Ingelheim, Bayer, BMS/Pfizer, Daiichi-Sankyo, Novartis, Merck, Portola, Roche, AstraZeneca, Menarini, and Guidotti.
References
- 1. Rana JS, Khan SS, Lloyd-Jones DM, Sidney S.. Changes in mortality in top 10 causes of death from 2011 to 2018. J Gen Intern Med 2020;1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amini M, Zayeri F, Salehi M.. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: results from global burden of disease study 2017. BMC Public Health 2021;21:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramji DP, Davies TS.. Cytokines in atherosclerosis: key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev 2015;26:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fiordelisi A, Iaccarino G, Morisco C, Coscioni E, Sorriento D.. NFkappaB is a key player in the crosstalk between inflammation and cardiovascular diseases. Int J Mol Sci 2019;20:1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ridker PM, Thuren T, Zalewski A, Libby P.. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J 2011;162:597–605. [DOI] [PubMed] [Google Scholar]
- 6. Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL.. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol 2013;61:404–410. [DOI] [PubMed] [Google Scholar]
- 7. O'Donoghue ML, Glaser R, Cavender MA, Aylward PE, Bonaca MP, Budaj A, Davies RY, Dellborg M, Fox KA, Gutierrez JA, Hamm C, Kiss RG, Kovar F, Kuder JF, Im KA, Lepore JJ, Lopez-Sendon JL, Ophuis TO, Parkhomenko A, Shannon JB, Spinar J, Tanguay JF, Ruda M, Steg PG, Theroux P, Wiviott SD, Laws I, Sabatine MS, Morrow DA; LATITUDE-TIMI 60 Investigators. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: a randomized clinical trial. JAMA 2016;315:1591–1599. [DOI] [PubMed] [Google Scholar]
- 8. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, López-Sendón J, Ostadal P, Koenig W, Angoulvant D, Grégoire JC, Lavoie MA, Dubé MP, Rhainds D, Provencher M, Blondeau L, Orfanos A, L'Allier PL, Guertin MC, Roubille F.. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 2019;381:2497–2505. [DOI] [PubMed] [Google Scholar]
- 9. Nidorf SM, Fiolet ATL, Eikelboom JW, Schut A, Opstal TSJ, Bax WA, Budgeon CA, Tijssen JGP, Mosterd A, Cornel JH, Thompson PL; LoDoCo2 Investigators. The effect of low-dose colchicine in patients with stable coronary artery disease: the LoDoCo2 trial rationale, design, and baseline characteristics. Am Heart J 2019;218:46–56. [DOI] [PubMed] [Google Scholar]
- 10. Lawler PR, Bhatt DL, Godoy LC, Luscher TF, Bonow RO, Verma S, Ridker PM.. Targeting cardiovascular inflammation: next steps in clinical translation. Eur Heart J 2021;42:113–131. [DOI] [PubMed] [Google Scholar]
- 11. Fragoulis GE, McInnes IB, Siebert S.. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford) 2019;58:i43–i54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jamilloux Y, El Jammal T, Vuitton L, Gerfaud-Valentin M, Kerever S, Sève P.. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun Rev 2019;18:102390. [DOI] [PubMed] [Google Scholar]
- 13. El Jammal T, Gerfaud-Valentin M, Sève P, Jamilloux Y.. Inhibition of JAK/STAT signaling in rheumatologic disorders: the expanding spectrum. Joint Bone Spine 2020;87:119–129. [DOI] [PubMed] [Google Scholar]
- 14. O'Sullivan JM, Harrison CN.. JAK-STAT signaling in the therapeutic landscape of myeloproliferative neoplasms. Mol Cell Endocrinol 2017;451:71–79. [DOI] [PubMed] [Google Scholar]
- 15. Kishore R, Verma SK.. Roles of STATs signaling in cardiovascular diseases. Jakstat 2012;1:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moura RA, Fonseca JE.. JAK inhibitors and modulation of B cell immune responses in rheumatoid arthritis. Front Med (Lausanne) 2020;7:607725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee SE. Disease modifying agents of myeloproliferative neoplasms: a review. Blood Res 2021;56:S26–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aringer M, Leuchten N, Dörner T.. Biologicals and small molecules for systemic lupus erythematosus. Z Rheumatol 2020;79:232–240. ]. [DOI] [PubMed] [Google Scholar]
- 19. You H, Xu D, Zhao J, Li J, Wang Q, Tian X, Li M, Zeng X.. JAK inhibitors: prospects in connective tissue diseases. Clin Rev Allergy Immunol 2020;59:334–351. [DOI] [PubMed] [Google Scholar]
- 20. Barbui T, Carobbio A, Cervantes F, Vannucchi AM, Guglielmelli P, Antonioli E, Alvarez-Larrán A, Rambaldi A, Finazzi G, Barosi G.. Thrombosis in primary myelofibrosis: incidence and risk factors. Blood 2010;115:778–782. [DOI] [PubMed] [Google Scholar]
- 21. Schieir O, Tosevski C, Glazier RH, Hogg-Johnson S, Badley EM.. Incident myocardial infarction associated with major types of arthritis in the general population: a systematic review and meta-analysis. Ann Rheum Dis 2017;76:1396–1404. [DOI] [PubMed] [Google Scholar]
- 22. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM.. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 2017;77:521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Villarino AV, Kanno Y, O'Shea JJ.. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol 2017;18:374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Valentino L, Pierre J.. JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol 2006;71:713–721. [DOI] [PubMed] [Google Scholar]
- 25. Venugopal S, Mascarenhas J.. Novel therapeutics in myeloproliferative neoplasms. J Hematol Oncol 2020;13:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgözoğlu L, Lewis EF.. Atherosclerosis. Nat Rev Dis Primers 2019;5:56. [DOI] [PubMed] [Google Scholar]
- 27. Khanna NN, Jamthikar AD, Gupta D, Piga M, Saba L, Carcassi C, Giannopoulos AA, Nicolaides A, Laird JR, Suri HS, Mavrogeni S, Protogerou AD, Sfikakis P, Kitas GD, Suri JS.. Rheumatoid arthritis: atherosclerosis imaging and cardiovascular risk assessment using machine and deep learning-based tissue characterization. Curr Atheroscler Rep 2019;21:7. [DOI] [PubMed] [Google Scholar]
- 28. Chen PY, Schwartz MA, Simons M.. Endothelial-to-mesenchymal transition, vascular inflammation, and atherosclerosis. Front Cardiovasc Med 2020;7: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, Howe KL, Jarr K-U, Flores AM, Tsantilas P, Tsao N, Rao A, Newman AAC, Eberhard AV, Priest JR, Ruusalepp A, Pasterkamp G, Maegdefessel L, Miller CL, Lind L, Koplev S, Björkegren JLM, Owens GK, Ingelsson E, Weissman IL, Leeper NJ.. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Natl Acad Sci USA 2020;117:15818–15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Borén J, Williams KJ.. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol 2016;27:473–483. [DOI] [PubMed] [Google Scholar]
- 31. Williams JW, Winkels H, Durant CP, Zaitsev K, Ghosheh Y, Ley K.. Single cell RNA sequencing in atherosclerosis research. Circ Res 2020;126:1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Döring Y, Drechsler M, Soehnlein O, Weber C.. Neutrophils in atherosclerosis: from mice to man. Arterioscler Thromb Vasc Biol 2015;35:288–295. [DOI] [PubMed] [Google Scholar]
- 33. Gaul DS, Stein S, Matter CM.. Neutrophils in cardiovascular disease. Eur Heart J 2017;38:1702–1704. [DOI] [PubMed] [Google Scholar]
- 34. Cassatella MA, Östberg NK, Tamassia N, Soehnlein O.. Biological roles of neutrophil-derived granule proteins and cytokines. Trends Immunol 2019;40:648–664. [DOI] [PubMed] [Google Scholar]
- 35. Bonaventura A, Montecucco F, Dallegri F, Carbone F, Lüscher TF, Camici GG, Liberale L.. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc Res 2019;115:1266–1285. [DOI] [PubMed] [Google Scholar]
- 36. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O.. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol 2020;17:327–340. [DOI] [PubMed] [Google Scholar]
- 37. Döring Y, Libby P, Soehnlein O.. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res 2020;126:1228–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Badimon L, Vilahur G.. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med 2014;276:618–632. [DOI] [PubMed] [Google Scholar]
- 39. Próchnicki T, Latz E.. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab 2017;26:71–93. [DOI] [PubMed] [Google Scholar]
- 40. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA.. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res 2020;126:1260–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med 2013;368:2004–2013. [DOI] [PubMed] [Google Scholar]
- 42. Libby P, Pasterkamp G, Crea F, Jang IK.. Reassessing the mechanisms of acute coronary syndromes. Circ Res 2019;124:150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kolte D, Libby P, Jang IK.. New insights into plaque erosion as a mechanism of acute coronary syndromes. JAMA 2021;325:1043–1044. [DOI] [PubMed] [Google Scholar]
- 44. De Caterina R, D'Ugo E, Libby P.. Inflammation and thrombosis—testing the hypothesis with anti-inflammatory drug trials. Thromb Haemost 2016;116:1012–1021. [DOI] [PubMed] [Google Scholar]
- 45. Grebe A, Hoss F, Latz E.. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res 2018;122:1722–1740. [DOI] [PubMed] [Google Scholar]
- 46. Wang J, Tan GJ, Han LN, Bai YY, He M, Liu HB.. Novel biomarkers for cardiovascular risk prediction. J Geriatr Cardiol 2017;14:135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lawler PR. Glycomics and cardiovascular disease: advancing down the path towards precision. Circ Res 2018;122:1488–1490. [DOI] [PubMed] [Google Scholar]
- 48. Maier W, Altwegg LA, Corti R, Gay S, Hersberger M, Maly FE, SüTsch G, Roffi M, Neidhart M, Eberli FR, Tanner FC, Gobbi S, von Eckardstein A, LüScher TF, Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation 2005;111:1355–1361. [DOI] [PubMed] [Google Scholar]
- 49. Wyss CA, Neidhart M, Altwegg L, Spanaus KS, Yonekawa K, Wischnewsky MB, Corti R, Kucher N, Roffi M, Eberli FR, Amann-Vesti B, Gay S, von Eckardstein A, Lüscher TF, Maier W.. Cellular actors, Toll-like receptors, and local cytokine profile in acute coronary syndromes. Eur Heart J 2010;31:1457–1469. [DOI] [PubMed] [Google Scholar]
- 50. Koenig W. High-sensitivity C-reactive protein and atherosclerotic disease: from improved risk prediction to risk-guided therapy. Int J Cardiol 2013;168:5126–5134. [DOI] [PubMed] [Google Scholar]
- 51. Ridker PM. A test in context: high-sensitivity C-reactive protein. J Am Coll Cardiol 2016;67:712–723. [DOI] [PubMed] [Google Scholar]
- 52. Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, Lorber MI, Pober JS.. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature 2000;403:207–211. [DOI] [PubMed] [Google Scholar]
- 53. Horvath CM. The Jak-STAT pathway stimulated by interferon gamma. Sci STKE 2004;2004:tr8. [DOI] [PubMed] [Google Scholar]
- 54. Boshuizen MC, de Winther MP.. Interferons as essential modulators of atherosclerosis. Arterioscler Thromb Vasc Biol 2015;35:1579–1588. [DOI] [PubMed] [Google Scholar]
- 55. Zhao J, Turpin-Nolan S, Febbraio MA.. IL-6 family cytokines as potential therapeutic strategies to treat metabolic diseases. Cytokine 2021;144:155549. [DOI] [PubMed] [Google Scholar]
- 56. Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN, Gao P, Saleheen D, Rendon A, Nelson CP, Braund PS, Hall AS, Chasman DI, Tybjærg-Hansen A, Chambers JC, Benjamin EJ, Franks PW, Clarke R, Wilde AA, Trip MD, Steri M, Witteman JC, Qi L, van der Schoot CE, de Faire U, Erdmann J, Stringham HM, Koenig W, Rader DJ, Melzer D, Reich D, Psaty BM, Kleber ME, Panagiotakos DB, Willeit J, Wennberg P, Woodward M, Adamovic S, Rimm EB, Meade TW, Gillum RF, Shaffer JA, Hofman A, Onat A, Sundström J, Wassertheil-Smoller S, Mellström D, Gallacher J, Cushman M, Tracy RP, Kauhanen J, Karlsson M, Salonen JT, Wilhelmsen L, Amouyel P, Cantin B, Best LG, Ben-Shlomo Y, Manson JE, Davey-Smith G, de Bakker PI, O'Donnell CJ, Wilson JF, Wilson AG, Assimes TL, Jansson JO, Ohlsson C, Tivesten Å, Ljunggren Ö, Reilly MP, Hamsten A, Ingelsson E, Cambien F, Hung J, Thomas GN, Boehnke M, Schunkert H, Asselbergs FW, Kastelein JJ, Gudnason V, Salomaa V, Harris TB, Kooner JS, Allin KH, Nordestgaard BG, Hopewell JC, Goodall AH, Ridker PM, Hólm H, Watkins H, Ouwehand WH, Samani NJ, Kaptoge S, Di Angelantonio E, Harari O, Danesh J; IL6R Genetics Consortium Emerging Risk Factors Collaboration. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 2012;379:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Libby P, Rocha VZ.. All roads lead to IL-6: a central hub of cardiometabolic signaling. Int J Cardiol 2018;259:213–215. [DOI] [PubMed] [Google Scholar]
- 58. Kang S, Narazaki M, Metwally H, Kishimoto T.. Historical overview of the interleukin-6 family cytokine. J Exp Med 2020;217:e20190347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Libby P. Targeting inflammatory pathways in cardiovascular disease: the inflammasome, interleukin-1, interleukin-6 and beyond. Cells 2021;10:951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim KW, Ivanov S, Williams JW.. Monocyte recruitment, specification, and function in atherosclerosis. Cells 2020;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ridker PM. Inhibiting interleukin-6 to reduce cardiovascular event rates: a next step for atherothrombosis treatment and prevention. J Am Coll Cardiol 2021;77:1856–1858. [DOI] [PubMed] [Google Scholar]
- 62. Ni CW, Hsieh HJ, Chao YJ, Wang DL.. Interleukin-6-induced JAK2/STAT3 signaling pathway in endothelial cells is suppressed by hemodynamic flow. Am J Physiol Cell Physiol 2004;287:C771–80. [DOI] [PubMed] [Google Scholar]
- 63. Schust J, Sperl B, Hollis A, Mayer TU, Berg T.. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 2006;13:1235–1242. [DOI] [PubMed] [Google Scholar]
- 64. Booz GW, Day JN, Baker KM.. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol 2002;34:1443–1453. [DOI] [PubMed] [Google Scholar]
- 65. Podewski EK, Hilfiker-Kleiner D, Hilfiker A, Morawietz H, Lichtenberg A, Wollert KC, Drexler H.. Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation 2003;107:798–802. [DOI] [PubMed] [Google Scholar]
- 66. Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R.. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res 2009;104:e9-18–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bolli R, Stein AB, Guo Y, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu Y, Xuan YT.. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol 2011;50:589–597. [DOI] [PubMed] [Google Scholar]
- 68. Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 69. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ; CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 70. Kleveland O, Kunszt G, Bratlie M, Ueland T, Broch K, Holte E, Michelsen AE, Bendz B, Amundsen BH, Espevik T, Aakhus S, Damås JK, Aukrust P, Wiseth R, Gullestad L.. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur Heart J 2016;37:2406–2413. [DOI] [PubMed] [Google Scholar]
- 71. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, Mam V, Hasan A, Rosenberg Y, Iturriaga E, Gupta M, Tsigoulis M, Verma S, Clearfield M, Libby P, Goldhaber SZ, Seagle R, Ofori C, Saklayen M, Butman S, Singh N, Le May M, Bertrand O, Johnston J, Paynter NP, Glynn RJ; CIRT Investigators. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med 2019;380:752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hennessy T, Soh L, Bowman M, Kurup R, Schultz C, Patel S, Hillis GS.. The low dose colchicine after myocardial infarction (LoDoCo-MI) study: a pilot randomized placebo controlled trial of colchicine following acute myocardial infarction. Am Heart J 2019;215:62–69. [DOI] [PubMed] [Google Scholar]
- 73. Giles JT, Sattar N, Gabriel S, Ridker PM, Gay S, Warne C, Musselman D, Brockwell L, Shittu E, Klearman M, Fleming TR.. Cardiovascular safety of tocilizumab versus etanercept in rheumatoid arthritis: a randomized controlled trial. Arthritis Rheumatol 2020;72:31–40. [DOI] [PubMed] [Google Scholar]
- 74. Greco D, Gualtierotti R, Agosti P, Adorni MP, Ingegnoli F, Rota M, Bernini F, Meroni PL, Ronda N.. Anti-atherogenic modification of serum lipoprotein function in patients with rheumatoid arthritis after tocilizumab treatment, a pilot study. J Clin Med 2020;9:2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pulito-Cueto V, Remuzgo-Martínez S, Genre F, Calvo-Alén J, Aurrecoechea E, Llorente I, Triguero-Martinez A, Blanco R, Llorca J, Ruiz-Lucea E, Rivera-García N, Gualillo O, López-Mejías R, Castañeda S, González-Gay MA.. Anti-IL-6 therapy reduces leptin serum levels in patients with rheumatoid arthritis. Clin Exp Rheumatol 2020;38:1201–1205. [PubMed] [Google Scholar]
- 76. Lévy L, Fautrel B, Barnetche T, Schaeverbeke T.. Incidence and risk of fatal myocardial infarction and stroke events in rheumatoid arthritis patients. A systematic review of the literature. Clin Exp Rheumatol 2008;26:673–679. [PubMed] [Google Scholar]
- 77. Meune C, Touzé E, Trinquart L, Allanore Y.. High risk of clinical cardiovascular events in rheumatoid arthritis: levels of associations of myocardial infarction and stroke through a systematic review and meta-analysis. Arch Cardiovasc Dis 2010;103:253–261. [DOI] [PubMed] [Google Scholar]
- 78. Mason JC, Libby P.. Cardiovascular disease in patients with chronic inflammation: mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur Heart J 2015;36:482–49c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McInnes IB, Schett G.. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017;389:2328–2337. [DOI] [PubMed] [Google Scholar]
- 80. Malemud CJ. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskelet Dis 2018;10:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lao M, Shi M, Zou Y, Huang M, Ye Y, Qiu Q, Xiao Y, Zeng S, Liang L, Yang X, Xu H.. Protein inhibitor of activated STAT3 regulates migration, invasion, and activation of fibroblast-like synoviocytes in rheumatoid arthritis. J Immunol 2016;196:596–606. [DOI] [PubMed] [Google Scholar]
- 82. Malemud CJ. Negative regulators of JAK/STAT signaling in rheumatoid arthritis and osteoarthritis. Int J Mol Sci 2017;18:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. England BR, Thiele GM, Anderson DR, Mikuls TR.. Increased cardiovascular risk in rheumatoid arthritis: mechanisms and implications. BMJ 2018;361:k1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Provan SA, Lillegraven S, Sexton J, Angel K, Austad C, Haavardsholm EA, Kvien TK, Uhlig T.. Trends in all-cause and cardiovascular mortality in patients with incident rheumatoid arthritis: a 20-year follow-up matched case-cohort study. Rheumatology (Oxford) 2020;59:505–512. [DOI] [PubMed] [Google Scholar]
- 85. Wang F, Sun L, Wang S, Davis JM 3rd, Matteson EL, Murad MH, Luo F, Vassallo R.. Efficacy and safety of tofacitinib, baricitinib, and upadacitinib for rheumatoid arthritis: a systematic review and meta-analysis. Mayo Clin Proc 2020;95:1404–1419. [DOI] [PubMed] [Google Scholar]
- 86. Kaur K, Kalra S, Kaushal S.. Systematic review of tofacitinib: a new drug for the management of rheumatoid arthritis. Clin Ther 2014;36:1074–1086. [DOI] [PubMed] [Google Scholar]
- 87. Mogul A, Corsi K, McAuliffe L.. Baricitinib: the second FDA-approved JAK inhibitor for the treatment of rheumatoid arthritis. Ann Pharmacother 2019;53:947–953. [DOI] [PubMed] [Google Scholar]
- 88. Duggan S, Keam SJ.. Upadacitinib: first approval. Drugs 2019;79:1819–1828. [DOI] [PubMed] [Google Scholar]
- 89. Burja B, Mertelj T, Frank-Bertoncelj M.. Hi-JAKi-ng synovial fibroblasts in inflammatory arthritis with JAK inhibitors. Front Med (Lausanne) 2020;7: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Markham A, Keam SJ.. Peficitinib: first global approval. Drugs 2019;79:887–891. [DOI] [PubMed] [Google Scholar]
- 91. Angelini J, Talotta R, Roncato R, Fornasier G, Barbiero G, Dal Cin L, Brancati S, Scaglione F.. JAK-inhibitors for the treatment of rheumatoid arthritis: a focus on the present and an outlook on the future. Biomolecules 2020;10:1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, McInnes IB, Sepriano A, van Vollenhoven RF, de Wit M, Aletaha D, Aringer M, Askling J, Balsa A, Boers M, den Broeder AA, Buch MH, Buttgereit F, Caporali R, Cardiel MH, De Cock D, Codreanu C, Cutolo M, Edwards CJ, van Eijk-Hustings Y, Emery P, Finckh A, Gossec L, Gottenberg J-E, Hetland ML, Huizinga TWJ, Koloumas M, Li Z, Mariette X, Müller-Ladner U, Mysler EF, da Silva JAP, Poór G, Pope JE, Rubbert-Roth A, Ruyssen-Witrand A, Saag KG, Strangfeld A, Takeuchi T, Voshaar M, Westhovens R, van der Heijde D.. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis 2020;79:685–699. [DOI] [PubMed] [Google Scholar]
- 93. Del Rincon ID, Williams K, Stern MP, Freeman GL, Escalante A.. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum 2001;44:2737–2745. [DOI] [PubMed] [Google Scholar]
- 94. Avina-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D.. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Rheum 2008;59:1690–1697. [DOI] [PubMed] [Google Scholar]
- 95. Jagpal A, Navarro-Millán I.. Cardiovascular co-morbidity in patients with rheumatoid arthritis: a narrative review of risk factors, cardiovascular risk assessment and treatment. BMC Rheumatol 2018;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Venetsanopoulou AI, Pelechas E, Voulgari PV, Drosos AA.. The lipid paradox in rheumatoid arthritis: the dark horse of the augmented cardiovascular risk. Rheumatol Int 2020;40:1181–1191. [DOI] [PubMed] [Google Scholar]
- 97. Robertson J, Peters MJ, McInnes IB, Sattar N.. Changes in lipid levels with inflammation and therapy in RA: a maturing paradigm. Nat Rev Rheumatol 2013;9:513–523. [DOI] [PubMed] [Google Scholar]
- 98. Amaya-Amaya J, Sarmiento-Monroy JC, Mantilla RD, Pineda-Tamayo R, Rojas-Villarraga A, Anaya JM.. Novel risk factors for cardiovascular disease in rheumatoid arthritis. Immunol Res 2013;56:267–286. [DOI] [PubMed] [Google Scholar]
- 99. Kaplan MJ. Cardiovascular complications of rheumatoid arthritis: assessment, prevention, and treatment. Rheum Dis Clin North Am 2010;36:405–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ambrosino P, Lupoli R, Di Minno A, Tasso M, Peluso R, Di Minno MN.. Subclinical atherosclerosis in patients with rheumatoid arthritis. A meta-analysis of literature studies. Thromb Haemost 2015;113:916–930. [DOI] [PubMed] [Google Scholar]
- 101. Strang AC, Bisoendial RJ, Kootte RS, Schulte DM, Dallinga-Thie GM, Levels JHM, Kok M, Vos K, Tas SW, Tietge UJF, Müller N, Laudes M, Gerlag DM, Stroes ESG, Tak PP.. Pro-atherogenic lipid changes and decreased hepatic LDL receptor expression by tocilizumab in rheumatoid arthritis. Atherosclerosis 2013;229:174–181. [DOI] [PubMed] [Google Scholar]
- 102. Charles-Schoeman C, Fleischmann R, Davignon J, Schwartz H, Turner SM, Beysen C, Milad M, Hellerstein MK, Luo Z, Kaplan IV, Riese R, Zuckerman A, McInnes IB.. Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol 2015;67:616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Robertson J, Porter D, Sattar N, Packard CJ, Caslake M, McInnes I, McCarey D.. Interleukin-6 blockade raises LDL via reduced catabolism rather than via increased synthesis: a cytokine-specific mechanism for cholesterol changes in rheumatoid arthritis. Ann Rheum Dis 2017;76:1949–1952. [DOI] [PubMed] [Google Scholar]
- 104. Tefferi A. JAK2 mutations and clinical practice in myeloproliferative neoplasms. Cancer J 2007;13:366–371. [DOI] [PubMed] [Google Scholar]
- 105. Kilpivaara O, Levine RL.. JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia 2008;22:1813–1817. [DOI] [PubMed] [Google Scholar]
- 106. Jäger R, Kralovics R.. Molecular pathogenesis of Philadelphia chromosome negative chronic myeloproliferative neoplasms. Curr Cancer Drug Targets 2011;11:20–30. [DOI] [PubMed] [Google Scholar]
- 107. Tibes R, Bogenberger JM, Benson KL, Mesa RA.. Current outlook on molecular pathogenesis and treatment of myeloproliferative neoplasms. Mol Diagn Ther 2012;16:269–283. [DOI] [PubMed] [Google Scholar]
- 108. Viny AD, Levine RL.. Genetics of myeloproliferative neoplasms. Cancer J 2014;20:61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bose P, Gotlib J, Harrison CN, Verstovsek S.. SOHO state-of-the-art update and next questions: MPN. Clin Lymphoma Myeloma Leuk 2018;18:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Marneth AE, Mullally A.. The molecular genetics of myeloproliferative neoplasms. Cold Spring Harb Perspect Med 2020;10:a034876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rumi E, Baratè C, Benevolo G, Maffioli M, Ricco A, Sant'Antonio E.. Myeloproliferative and lymphoproliferative disorders: state of the art. Hematol Oncol 2020;38:121–128. [DOI] [PubMed] [Google Scholar]
- 112. Wan Z, Han B.. Comparison and implications of mutational profiles of myelodysplastic syndromes, myeloproliferative neoplasms, and myelodysplastic/myeloproliferative neoplasms: a meta-analysis. Front Oncol 2020;10:579221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC.. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352:1779–1790. [DOI] [PubMed] [Google Scholar]
- 114. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W.. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–1148. [DOI] [PubMed] [Google Scholar]
- 115. Lu X, Huang LJ, Lodish HF.. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J Biol Chem 2008;283:5258–5266. [DOI] [PubMed] [Google Scholar]
- 116. Teofili L, Martini M, Cenci T, Petrucci G, Torti L, Storti S, Guidi F, Leone G, Larocca LM.. Different STAT-3 and STAT-5 phosphorylation discriminates among Ph-negative chronic myeloproliferative diseases and is independent of the V617F JAK-2 mutation. Blood 2007;110:354–359. [DOI] [PubMed] [Google Scholar]
- 117. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, Silvestre-Roig C, Kotini AG, Luchsinger LL, Wei Y, Westerterp M, Snoeck HW, Papapetrou EP, Schulz C, Massberg S, Soehnlein O, Ebert B, Levine RL, Reilly MP, Libby P, Wang N, Tall AR.. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021;592:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, Silver AJ, Adams D, Castellano CA, Schneider RK, Padera RF, DeAngelo DJ, Wadleigh M, Steensma DP, Galinsky I, Stone RM, Genovese G, McCarroll SA, Iliadou B, Hultman C, Neuberg D, Mullally A, Wagner DD, Ebert BL.. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med 2018;10:eaan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Martinod K, Wagner DD.. Thrombosis: tangled up in NETs. Blood 2014;123:2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jariwala MP, Laxer RM.. NETosis in rheumatic diseases. Curr Rheumatol Rep 2021;23: 9. [DOI] [PubMed] [Google Scholar]
- 121. Fazzi R, Pacini S, Testi R, Azzarà A, Galimberti S, Testi C, Trombi L, Metelli MR, Petrini M.. Carboxy-terminal fragment of osteogenic growth peptide in vitro increases bone marrow cell density in idiopathic myelofibrosis. Br J Haematol 2003;121:76–85. [DOI] [PubMed] [Google Scholar]
- 122. Koschmieder S, Chatain N.. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev 2020;42:100711. [DOI] [PubMed] [Google Scholar]
- 123. Bocchia M, Galimberti S, Aprile L, Sicuranza A, Gozzini A, Santilli F, Abruzzese E, Baratè C, Scappini B, Fontanelli G, Trawinska MM, Defina M, Gozzetti A, Bosi A, Petrini M, Puccetti L.. Genetic predisposition and induced pro-inflammatory/pro-oxidative status may play a role in increased atherothrombotic events in nilotinib treated chronic myeloid leukemia patients. Oncotarget 2016;7:72311–72321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yu J, Parasuraman S, Paranagama D, Bai A, Naim A, Dubinski D, Mesa R.. Impact of Myeloproliferative neoplasms on patients' employment status and work productivity in the United States: results from the living with MPNs survey. BMC Cancer 2018;18:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kogan I, Chap D, Hoffman R, Axelman E, Brenner B, Nadir Y.. JAK-2 V617F mutation increases heparanase procoagulant activity. Thromb Haemost 2016;115:73–80. [DOI] [PubMed] [Google Scholar]
- 126. Edelmann B, Gupta N, Schnoeder TM, Oelschlegel AM, Shahzad K, Goldschmidt J, Philipsen L, Weinert S, Ghosh A, Saalfeld FC, Nimmagadda SC, Müller P, Braun-Dullaeus R, Mohr J, Wolleschak D, Kliche S, Amthauer H, Heidel FH, Schraven B, Isermann B, Müller AJ, Fischer T.. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J Clin Invest 2018;128:4359–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. De Stefano V, Za T, Rossi E, Vannucchi AM, Ruggeri M, Elli E, Mico C, Tieghi A, Cacciola RR, Santoro C, Gerli G, Vianelli N, Guglielmelli P, Pieri L, Scognamiglio F, Rodeghiero F, Pogliani EM, Finazzi G, Gugliotta L, Marchioli R, Leone G, Barbui T, for the GIMEMA CMD-Working PartyT. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–380. [DOI] [PubMed] [Google Scholar]
- 128. Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW.. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–4928. [DOI] [PubMed] [Google Scholar]
- 129. Patrono C, Rocca B, tefano V.. Platelet activation and inhibition in polycythemia vera and essential thrombocythemia. Blood 2013;121:1701–1711. [DOI] [PubMed] [Google Scholar]
- 130. Tefferi A, Barbui T.. Personalized management of essential thrombocythemia-application of recent evidence to clinical practice. Leukemia 2013;27:1617–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Vianello F, Cella G, Osto E, Ballin A, Famoso G, Tellatin S, Iliceto S, Cucchini U, Saggiorato G, Omenetto E, Tona F.. Coronary microvascular dysfunction due to essential thrombocythemia and policythemia vera: the missing piece in the puzzle of their increased cardiovascular risk? Am J Hematol 2015;90:109–113. [DOI] [PubMed] [Google Scholar]
- 132. Masselli E, Carubbi C, Pozzi G, Martini S, Aversa F, Galli D, Gobbi G, Mirandola P, Vitale M.. Platelet expression of PKCepsilon oncoprotein in myelofibrosis is associated with disease severity and thrombotic risk. Ann Transl Med 2017;5:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Fleischman AG, Tyner JW.. Causal role for JAK2 V617F in thrombosis. Blood 2013;122:3705–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Veninga A, De Simone I, Heemskerk JWM, Cate HT, van der Meijden PEJ.. Clonal hematopoietic mutations linked to platelet traits and the risk of thrombosis or bleeding. Haematologica 2020;105:2020–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hobbs CM, Manning H, Bennett C, Vasquez L, Severin S, Brain L, Mazharian A, Guerrero JA, Li J, Soranzo N, Green AR, Watson SP, Ghevaert C.. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood 2013;122:3787–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Villamor N, Colomer D, Cervantes F.. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica 2006;91:169–175. [PubMed] [Google Scholar]
- 137. Falanga A, Marchetti M, Vignoli A, Balducci D, Russo L, Guerini V, Barbui T.. V617F JAK-2 mutation in patients with essential thrombocythemia: relation to platelet, granulocyte, and plasma hemostatic and inflammatory molecules. Exp Hematol 2007;35:702–711. [DOI] [PubMed] [Google Scholar]
- 138. Reeves BN, Beckman JD.. Novel pathophysiological mechanisms of thrombosis in myeloproliferative neoplasms. Curr Hematol Malig Rep 2021;16:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Hultcrantz M, Björkholm M, Dickman PW, Landgren O, Derolf ÅR, Kristinsson SY, Andersson TML.. Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: a population-based cohort study. Ann Intern Med 2018;168:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ciuffetti G, Schillaci G, Lombardini R, Pirro M, Vaudo G, Mannarino E.. Prognostic impact of low-shear whole blood viscosity in hypertensive men. Eur J Clin Invest 2005;35:93–98. [DOI] [PubMed] [Google Scholar]
- 141. Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, Marilus R, Villegas A, Tognoni G, Barbui T.. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–2232. [DOI] [PubMed] [Google Scholar]
- 142. Marchioli R, Finazzi G, Specchia G, Masciulli A, Mennitto MR, Barbui T.. The CYTO-PV: a large-scale trial testing the intensity of CYTOreductive therapy to prevent cardiovascular events in patients with polycythemia vera. Thrombosis 2011;2011:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Vannucchi AM, Antonioli E, Guglielmelli P, Longo G, Pancrazzi A, Ponziani V, Bogani C, Ferrini PR, Rambaldi A, Guerini V, Bosi A, Barbui T; for the MPD Research Consortium. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007;21:1952–1959. [DOI] [PubMed] [Google Scholar]
- 144. Yang M, Bai XY, Ni JJ, Xu JC, Tang JY, Zeng Y, Fang Q.. Recurrent coronary arterial thromboses as the tip of the iceberg: the JAK2 mutation-related disease. Int J Cardiol 2015;182:48–49. [DOI] [PubMed] [Google Scholar]
- 145. Inami T, Okabe M, Matsushita M, Kobayashi N, Inokuchi K, Hata N, Seino Y, Shimizu W.. JAK2 mutation and acute coronary syndrome complicated with stent thrombosis. Heart Vessels 2016;31:1714–1716. [DOI] [PubMed] [Google Scholar]
- 146. Carobbio A, Thiele J, Passamonti F, Rumi E, Ruggeri M, Rodeghiero F, Randi ML, Bertozzi I, Vannucchi AM, Antonioli E, Gisslinger H, Buxhofer-Ausch V, Finazzi G, Gangat N, Tefferi A, Barbui T.. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–5859. [DOI] [PubMed] [Google Scholar]
- 147. Lata K, Madiraju N, Levitt L.. JAK2 mutations and coronary ischemia. N Engl J Med 2010;363:396–397. [DOI] [PubMed] [Google Scholar]
- 148. Nazha B, Garcia G, Kandov R, Odaimi M.. Calreticulin mutated essential thrombocythemia presenting as acute coronary syndrome. Case Rep Hematol 2015;2015:161764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lucijanic M, Krecak I, Galusic D, Sedinic M, Holik H, Perisa V, Moric Peric M, Zekanovic I, Stoos-Veic T, Pejsa V, Kusec R.. Higher serum uric acid is associated with higher risks of thrombosis and death in patients with primary myelofibrosis. Wien Klin Wochenschr 2021. doi:10.1007/s00508-020-01802-x. [DOI] [PubMed] [Google Scholar]
- 150. Charles-Schoeman C, Wicker P, Gonzalez-Gay MA, Boy M, Zuckerman A, Soma K, Geier J, Kwok K, Riese R.. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Semin Arthritis Rheum 2016;46:261–271. [DOI] [PubMed] [Google Scholar]
- 151. Charles-Schoeman C, DeMasi R, Valdez H, Soma K, Hwang LJ, Boy MG, Biswas P, McInnes IB.. Risk factors for major adverse cardiovascular events in phase III and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol 2019;71:1450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kume K, Amano K, Yamada S, Kanazawa T, Ohta H, Hatta K, Amano K, Kuwaba N.. Tofacitinib improves atherosclerosis despite up-regulating serum cholesterol in patients with active rheumatoid arthritis: a cohort study. Rheumatol Int 2017;37:2079–2085. [DOI] [PubMed] [Google Scholar]
- 153. Taylor PC, Weinblatt ME, Burmester GR, Rooney TP, Witt S, Walls CD, Issa M, Salinas CA, Saifan C, Zhang X, Cardoso A, Gonzalez-Gay MA, Takeuchi T.. Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol 2019;71:1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Xie W, Huang Y, Xiao S, Sun X, Fan Y, Zhang Z.. Impact of Janus kinase inhibitors on risk of cardiovascular events in patients with rheumatoid arthritis: systematic review and meta-analysis of randomised controlled trials. Ann Rheum Dis 2019;78:1048–1054. [DOI] [PubMed] [Google Scholar]
- 155. Aschenbrenner DS. Tofacitinib receives new boxed safety warning. Am J Nurs 2019;119:20. [DOI] [PubMed] [Google Scholar]
- 156. Taylor PC. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology (Oxford) 2019;58:i17–i26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Vallejo-Yagüe E, Weiler S, Micheroli R, Burden AM.. Thromboembolic safety reporting of tofacitinib and baricitinib: an analysis of the WHO VigiBase. Drug Saf 2020;43:881–891. [DOI] [PubMed] [Google Scholar]
- 158. Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, Vannucchi AM, Mesa RA, Demory JL, Barosi G, Rumi E, Tefferi A.. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009;113:2895–2901. [DOI] [PubMed] [Google Scholar]
- 159. Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano JV, Deininger MW, Miller CB, Silver RT, Talpaz M, Winton EF, Harvey JH Jr., Arcasoy MO, Hexner EO, Lyons RM, Paquette R, Raza A, Jones M, Kornacki D, Sun K, Kantarjian H; for the COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol 2017;10:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Harrison CN, Vannucchi AM, Kiladjian J-J, Al-Ali HK, Gisslinger H, Knoops L, Cervantes F, Jones MM, Sun K, McQuitty M, Stalbovskaya V, Gopalakrishna P, Barbui T; on behalf of the COMFORT-II Investigators. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016;30:1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Verstovsek S, Gotlib J, Mesa RA, Vannucchi AM, Kiladjian JJ, Cervantes F, Harrison CN, Paquette R, Sun W, Naim A, Langmuir P, Dong T, Gopalakrishna P, Gupta V.. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol 2017;10:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Barraco F, Greil R, Herbrecht R, Schmidt B, Reiter A, Willenbacher W, Raymakers R, Liersch R, Wroclawska M, Pack R, Burock K, Karumanchi D, Gisslinger H.. Real-world non-interventional long-term post-authorisation safety study of ruxolitinib in myelofibrosis. Br J Haematol 2020;191:764–774. [DOI] [PubMed] [Google Scholar]
- 163. Mesa RA, Verstovsek S, Gupta V, Mascarenhas JO, Atallah E, Burn T, Sun W, Sandor V, Gotlib J.. Effects of ruxolitinib treatment on metabolic and nutritional parameters in patients with myelofibrosis from COMFORT-I. Clin Lymphoma Myeloma Leuk 2015;15:214–221. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Fischer M, Luck M, Werle M, Scheidt HA, Müller P.. Binding of the small-molecule kinase inhibitor ruxolitinib to membranes does not disturb membrane integrity. Biochem Biophys Rep 2020;24: 100838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Samuelson BT, Vesely SK, Chai-Adisaksopha C, Scott BL, Crowther M, Garcia D.. The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: a meta-analysis. Blood Coagul Fibrinolysis 2016;27:648–652. [DOI] [PubMed] [Google Scholar]
- 166. Masciulli A, Ferrari A, Carobbio A, Ghirardi A, Barbui T.. Ruxolitinib for the prevention of thrombosis in polycythemia vera: a systematic review and meta-analysis. Blood Adv 2020;4:380–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Saliba W, Mishchenko E, Cohen S, Rennert G, Preis M.. Association between myelofibrosis and thromboembolism: a population-based retrospective cohort study. J Thromb Haemost 2020;18:916–925. [DOI] [PubMed] [Google Scholar]
- 168. Colafigli G, Scalzulli E, Pepe S, Di Prima A, Efficace F, Martelli M, Foà R, Breccia M.. The advantages and risks of ruxolitinib for the treatment of polycythemia vera. Expert Rev Hematol 2020;13:1067–1072. [DOI] [PubMed] [Google Scholar]
- 169. Verstovsek S, Vannucchi AM, Griesshammer M, Masszi T, Durrant S, Passamonti F, Harrison CN, Pane F, Zachee P, Kirito K, Besses C, Hino M, Moiraghi B, Miller CB, Cazzola M, Rosti V, Blau I, Mesa R, Jones MM, Zhen H, Li J, Francillard N, Habr D, Kiladjian JJ.. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL.. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, Bhagat G, Pernis AB.. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest 2010;120:3280–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Galimberti S, Baldini C, Baratè C, Fornili M, Balducci S, Ricci F, Ferro F, Elefante E, Di Paolo A, Baglietto L, Donati V, Petrini M.. Myeloid neoplasms and autoimmune diseases: markers of association. Clin Exp Rheumatol 2021. [DOI] [PubMed] [Google Scholar]
- 173. Calabrese LH, Calabrese C.. Baricitinib and dexamethasone for hospitalized patients with COVID-19. Cleve Clin J Med 2021. doi:10.3949/ccjm.88a.ccc073. [DOI] [PubMed] [Google Scholar]
- 174. Moreno-González G, Mussetti A, Albasanz-Puig A, Salvador I, Sureda A, Gudiol C, Salazar R, Marin M, Garcia M, Navarro V, de la Haba Vaca I, Coma E, Sanz-Linares G, Dura X, Fontanals S, Serrano G, Cruz C, Mañez R.. A phase I/II clinical trial to evaluate the efficacy of baricitinib to prevent respiratory insufficiency progression in onco-hematological patients affected with COVID19: a structured summary of a study protocol for a randomised controlled trial. Trials 2021;22: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Lobo RA. Hormone-replacement therapy: current thinking. Nat Rev Endocrinol 2017;13:220–231. [DOI] [PubMed] [Google Scholar]
- 176. Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med 2004;351:1709–1711. [DOI] [PubMed] [Google Scholar]