

Abstract
Human urate anion transporter 1 (hURAT1) is responsible for the reabsorption of uric acid in the proximal renal tubules and is a promising therapeutic target for treating hyperuricemia. To mitigate the side effects of URAT1-targeted clinical agents such as benzbromarone, there is significant interest in discovering new URAT1 inhibitors and developing technology that can evaluate URAT1 inhibition. This review summarizes the methods for assay of URAT1 inhibition and the progress on the discovery of natural and synthetic URAT1 inhibitors in the past five years.
1. Introduction
Uric acid is the final product of purine metabolism in the human body [1, 2]. In recent years, more people suffer from an unbalanced urate metabolism [3]. Hyperuricemia is characterized by insufficient excretion and the overproduction of uric acid [4] and is defined as serum uric acid concentration of >408 μmol/L or 6.8 mg/dL [5]. The primary clinical characteristic of hyperuricemia is gout [6], which is a common and complex form of arthritis caused by the deposition of monosodium urate crystals in the peripheral joints and surrounding tissues. Gout prevalence ranges from 2.7% to 6.7% in Western countries and from 0.1% to 10% globally, and increases from 0.03% to 0.6% every year [3, 7]. Additionally, hyperuricemia is associated with other diseases, including chronic kidney dysfunction [8, 9], hyperlipidemia [10], hypertension [11], coronary heart disease [12], diabetes [13], and vascular disease [14, 15].
There are currently three treatments for hyperuricemia: reduction of urate production, increase in urate excretion, and decomposition of urate. Statistics indicate that 90% of hyperuricemia cases are due to impaired renal uric acid excretion and that only 10% are due to excessive production of uric acid [16]. The kidneys and intestines play a major role in uric acid excretion, with more than 70% of urate excretion taking place in the renal pathway. As such, the development of reagents for increasing uric acid excretion has significant therapeutic potential.
Human urate transporter 1 (hURAT1) is a transmembrane protein that consists of 555 amino acids and belongs to the organic anion transporters (OAT) family. URAT1 is expressed on the luminal side of the proximal renal tubule [17] and regulates the absorption of uric acid from the renal tubule to the epithelial cells [18]. As urate is eliminated from the kidneys, approximately 90% of the urate filtered by the glomeruli is reabsorbed back into the bloodstream and only 10% is excreted by the kidneys. The uric acid reabsorption process is primarily controlled by URAT1 and is particularly important to hyperuricemia. Inhibition of URAT1 is currently the main treatment for urate-lowering therapy, in addition to pills such as allopurinol and febuxostat, which block xanthine oxidase (XO) [19, 20]. Considering 90% of hyperuricemia cases are due to reduced renal uric acid excretion, the inhibition of URAT1 over XO could be a better approach to treating gout [21].
URAT1 inhibitory drugs currently approved for use in clinical settings include probenecid, benzbromarone, sulfinpyrazone, and lesinurad (Figure 1) [22–24]. However, these medicines have side effects and some degree of toxicity [25]. Probenecid can induce rash, gastrointestinal tract irritation, hypersensitivity, and hemolytic anemia [22]; benzbromarone results in serious hepatotoxicity and bone marrow suppression [26]; sulfinpyrazone can cause nausea, vomiting, abdominal pain, diarrhea, anemia, and skin rashes [27]; lesinurad has dose-dependent nephrotoxicity and can cause adverse cardiovascular events [28]. The use of URAT1 inhibitors is not recommended in patients with severe renal failure, urate kidney stones, or blood dyscrasias [25].
Figure 1.
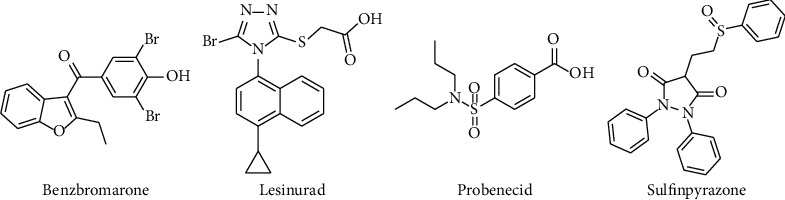
Approved URAT1 inhibitors.
While xanthine oxidase inhibition is still the primary therapy for gout, it can also have serious adverse effects, such as skin rashes, hepatitis, fever, Stevens–Johnson syndrome, nephropathy, fatal liver necrosis, allergic reactions, and cardiovascular events. As such, alternative medicines with fewer side effects are needed to treat this disorder [29–31].
Considering these challenges, there is a need for alternative medicines with fewer side effects, and new therapies are needed for patients who cannot sufficiently reduce urate with current therapies. As such, there is an urgent need to discover effective and safe compounds targeting URAT1 to lower serum uric acid [32] and to develop novel techniques validating URAT1 inhibitors. This study summarizes natural and synthetic URAT1 inhibitors in the past five years. Compared with a recently published review [33], this paper outlines the chemical structures, inhibitory effects, and evaluation methods of natural inhibitors in further detail. For synthetic URAT1 inhibitors, we demonstrate different parent structures including lesinurad, verinurad, and febuxostat derivatives, displayed with both in vitro and in vivo data. We also describe classical and recent methods for evaluating URAT1 inhibitors.
2. Materials and Methods
This review covers the literature published in the last five years and focuses on the most relevant studies assessing cell-based in vitro approaches for evaluating URAT1 inhibition and novel URAT1 inhibitors. The inhibitors are organized according to the origin of the compounds (natural and synthetic) and their chemical structures. The structures of specific compounds with promising results are presented in the figures. All studies were retrieved from the following databases: Web of Science, PubMed, Scopus, and CNKI. The following keywords were used: “URAT1 inhibitors,” “hyperuricemia,” “screening methods,” “and uric acid.” The chemical structures in this article were created by ChemDraw.
3. Results
3.1. Recent Development of URAT1 Inhibitors
3.1.1. Natural Compounds with URAT1 Inhibitory Activity
Natural products have received significant attention for their urate-lowering potential [34, 35], particularly phenolic compounds, terpenes, and fatty acids, all of which are reported to have antihyperuricemic activity.
(1) Phenolic Compounds and Analogues. Toyoda et al. [36] evaluated several flavonoids isolated from Citrus fruits and found that naringenin (compound 1) (IC50 = 16.1 μM), hesperetin (compound 2) (IC50 = 25.7 μM), and nobiletin (compound 3) (IC50 = 17.6 μM) (Figure 2) displayed substantial URAT1 inhibitory activities with negligible cytotoxicity. The three compounds have also been studied as XO inhibitors [37] and the IC50 value of hesperetin is 16.5 μM. However, naringenin and nobiletin have IC50 values exceeding 100 μM, indicating that these two affect URAT1 more than XO. Some in vivo studies also supported their urate-lowering effects [38–40]. The oral administration of the glycon of naringenin (100 mg/kg) effectively reduced serum uric acid levels by 89% in potassium oxonate (PO-) induced hyperuricemic mice, while the positive drug allopurinol showed a 156% reduction in serum uric acid [40]. The oral administration of orange juice (5 mL/kg) and hesperetin (5 mg/kg) significantly reduced the serum uric acid in PO-induced hyperuricemia rats by 38.76% and 24.93%, respectively, while allopurinol significantly decreased by 124%. The dominant flavonoids in orange juice are hesperidin and narirutin, which are hydrolyzed to hesperetin and naringenin in the colon [38]. Citrus depressa extracts (25 mL/kg) containing nobiletin also exhibited uricosuric effects in hyperuricemia model mice, with reductions in serum uric acid levels of 22.7% compared to the PO model group [39].
Figure 2.
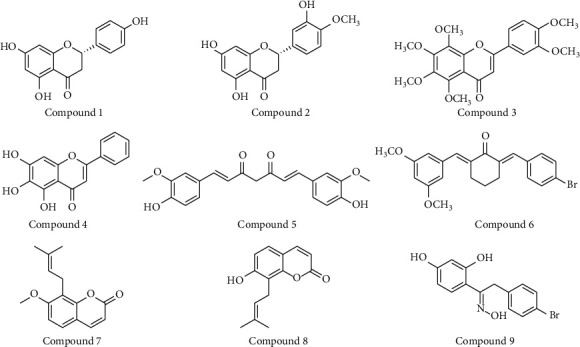
Chemical structures of phenolic compounds possessed URAT1 inhibitory activities.
Another flavonoid, baicalein (compound 4) (Figure 2), was shown by Chen et al. [41] to noncompetitively inhibit URAT1 in a dose-dependent manner with IC50 of 31.6 μM. In vivo animal studies have demonstrated that 200 mg/kg baicalein can significantly lower urate levels in PO-induced hyperuricemia mice by elevating urate excretion. Docking analysis and site mutation indicate that baicalein interacts with Ser35 and Phe241 of URAT1. Baicalein was also reported to exhibit liver and kidney protection without toxicity [42, 43].
Curcumin (compound 5) (Figure 2) is a polyphenolic compound and has been reported to have uricosuric activity and URAT1 inhibition [CN101181249]. Twenty-two α, β-unsaturated cyclohexanone and cyclopentanone analogs of curcumin were later synthesized and demonstrated a remarkable antihyperuricemia effect [44]. Among them, 4d (compound 6) (Figure 2) inhibits both URAT1 and XO. It inhibits 38.2% of URAT1 activity at 100 μM, compared with 42.5% inhibition of benzbromarone at the same concentration.
To discover novel compounds for hyperuricemia, 107 crude products were screened by uric acid uptake assays on URAT1-HEK293/PDZK1 cells [45]. Of them, the MeOH extract from Cnidii Monnieris Fructus exhibited the highest inhibitory effect. Osthol (compound 7) (Figure 2) was identified as the active compound in the extract, which noncompetitively inhibits URAT1 with an IC50 of 78.8 μM and is compatible with IC50 = 42 μM of probenecid. In vitro studies demonstrated that osthol <100 μM has no cytotoxicity in HEK293/PDZK1 cells. The authors also compared the URAT1 inhibition of different coumarins and found that only compounds with a prenyl group at the 8-position of osthol and osthenol (compound 8) displayed URAT1 inhibitory effects at 100 μM, suggesting the important role for this substitution in URAT1 inhibition.
BDEO (compound 9) (Figure 2) is a deoxybenzoins oxime analog and has a structure similar to flavonoids. Its role as a dual inhibitor for lowering urate has been studied, and it is found that it blocked the uptake of uric acid in URAT1-293T cells (Ki = 0.14 μM) in a noncompetitive manner and inhibited XO activity (IC50 = 3.3 μM). In vivo studies demonstrated that BDEO at 5 mg/kg significantly decreased serum urate in PO-induced hyperuricemia mice and exhibited a nontoxic, dose-dependent effect. The administration of 20 mg/kg BDEO has effects comparable to allopurinol or benzbromarone at 10 mg/kg [46].
(2) Terpenes. Dioscin (compound 10) (Figure 3) was discovered to have weak inhibitory activity on XO in an earlier study [47] and also shows antihyperuricemic effects in animal models [48–50]. Zhang et al. explored the metabolites of dioscin and identified tigogenin (compound 11) (Figure 3) as a URAT1 inhibitor. An in vitro uric acid uptake assay demonstrated that only 10 μM tigogenin could significantly inhibit URAT1 function and could dramatically reduce uric acid uptake by 40% at 100 μM [48].
Figure 3.
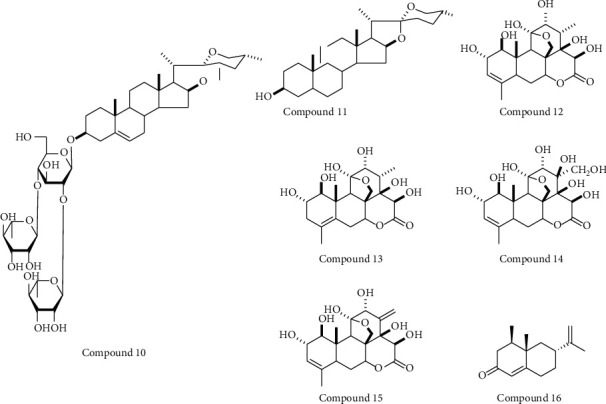
Chemical structures of terpenes compounds possessed URAT1 inhibitory activities.
Bao et al. claimed that four quassinoids (13β, 18-dihydroeurycomanol (compound 12), △4,5,14- hydroxyglaucarubol (compound 13), 13β, 21-dihydroxyeurycomanol (compound 14), and eurycomanol (compound 15)) (Figure 3) from the Eurycoma longifolia stem 70% ethanol extract (EL) possessed URAT1 inhibitory activities at 50 μM. The chemical structures of these four compounds indicate that they belong to eurycomanol type quassinoids. The EL showed the uric-lowering effect in PO-induced hyperuricemic mice and that eurycomanol significantly reduced serum uric acid levels at 20 mg/kg [51].
Additionally, alpinia oxyphylla seed ethanol extract (AE) displays strong URAT1 inhibitory activity. Its URAT1 inhibitory effect at 100 μg/mL is comparable with that of benzbromarone (100 μM), as shown by the uric uptake assay in hURAT1-expressing oocytes, in which only 1 μg/mL AE significantly inhibit URAT1 function. UPLC analysis revealed nootkatone (compound 16) (Figure 3) as the primary bioactive compound in the extract, while in vivo administration of 100 mg/kg nootkatone significantly reduced serum uric acid in PO-induced hyperuricemic rats [52].
(3) Fatty Acids. It is well established that fatty acids (FAs) influence a range of metabolic and inflammatory diseases [53, 54] and cancer [55]. A recent study demonstrated the relationship between FAs and hyperuricemia: of 25 FAs tested in vitro [56], nine unsaturated FAs exhibited URAT1 inhibitory effects at 100 μM, but no saturated FAs showed any effects. In particular, three long-chain unsaturated FAs exhibited strong URAT1 inhibitory effects: eicosapentaenoic acid (EPA) (compound 17) (Figure 4), α-linolenic acid (ALA) (compound 18) (Figure 4), and docosahexaenoic acid (DHA) (compound 19) (Figure 4), which had IC50 values of 6.0, 14.2, and 15.2 μM, respectively. This study could lead to a new kind of URAT1 inhibitor, though the interaction between FAs and URAT1 requires additional study.
Figure 4.
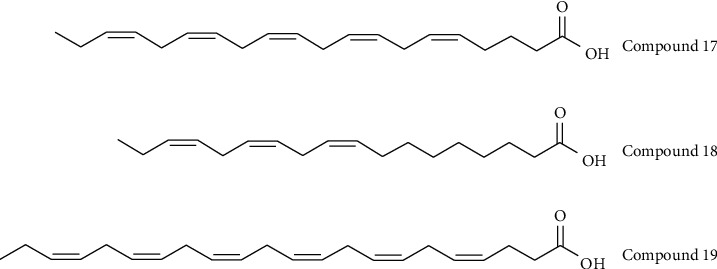
Chemical structures of fatty acids possessed URAT1 inhibitory activities.
3.1.2. Synthetic Compounds
(1) Lesinurad Analogues. Lesinurad (Figure 1) is a selective inhibitor of URAT1 and OAT4 for uric acid reabsorption, though it also displays adverse nephrotoxic effects [25]. Lesinurad is considered a scaffold compound for discovering inhibitors with reduced side effects and improved activities.
Zhao et al. [57] found that the 1-cyclopropylnaphthalene core with side chains of methyl (or dimethyl) substitution and carboxylic acid is important for lowering uric acid levels. 1H-Imidazole [4,5-b]pyridine derivative 44 (compound 20) (Figure 5) exhibited the most potent URAT1 inhibitory activity (IC50 = 1.6 μM in comparison with 13.2 μM of lesinurad). They also reported a dual inhibitor 83 (compound 21) (Figure 5) targeting both URAT1 (IC50 = 4.2 μM) and glucose transporter 9 (GLUT9) (IC50 = 31.7 μM), which could account for it having the best urate-lowering effect among all compounds tested.
Figure 5.
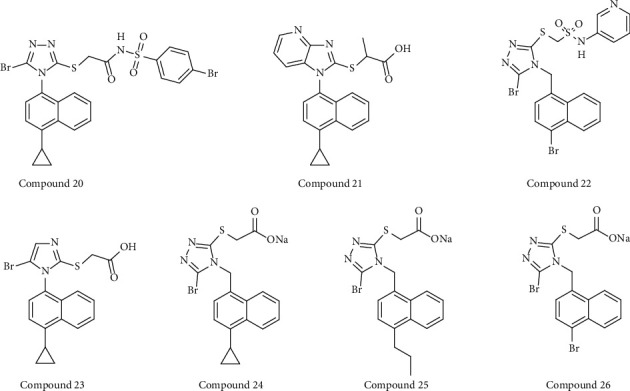
Structures of lesinurad synthetic derivatives with URAT1 inhibitory activity.
Nineteen bioisosteres of lesinurad were designed and prepared by Wu et al. [58], and two rounds of structure-activity relationship (SAR) exploration revealed a novel, highly active URAT1 inhibitor 1g (compound 22) (Figure 5), which contains an N-(pyridin-3-yl) sulfonamide moiety. URAT1-mediated urate transport assay demonstrated that 1g exhibits >200-fold URAT1 inhibition (IC50 = 0.032 μM) to lesinurad (IC50 = 7.2 μM).
Wu et al. [59] designed and synthesized a imidazole analog of lesinurad, named LUM (2(5-bromo-1(4-cyclopylnaphthalen-1H-imidazol-2-ylthio) acetate) (compound 23) (Figure 5). The IC50 value of LUM for URAT1 is 3.2 μM, which is significantly more potent than lesinurad (IC50 = 65.5 μM). Oral administration of 40 mg/kg LUM produced a urate-lowering effect comparable with that observed by 80 mg/kg lesinurad in PO-induced hyperuricemic rats, suggesting that LUM is more effective than lesinurad in in vivo experiments. However, in vitro cytotoxicity experiments found cytotoxicity beginning with 50 μM LUM, while 400 μM lesinurad significantly affected cell viability. Therefore, although LUM has better antihyperuricemia properties, its safety and tolerability must be improved.
Another active URAT1 inhibitor 1c (IC50 = 0.231 μM) (compound 24) was discovered in an earlier investigation and possesses a CH2 group between the triazole and naphthalene rings of lesinurad (Figure 5) [60]. The authors subsequently performed four rounds of SAR exploration of this flexible naphthyltriazolylmethane skeleton and discovered two compounds of 1j (sodium 2-((5-bromo-4-((4-n-propylnaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate) (compound 25) and 1m (sodium 2-((5-bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate) (compound 26) (Figure 5) as effective URAT1 inhibitors. The IC50 values of 1j and 1m against URAT1 are 0.092 μM and 0.094 μM, respectively. The authors also found several key points by the SAR analysis; first, the 5-position of the 1, 2, 4-triazole ring must be a halogen atom; second, there must be no substitution on the α-position acetate; third, the inhibitory activity of URAT1 is enhanced increase as the space volume of (cyclic) alkyl substituents increases [61].
(2) Other Synthetic Compounds. CDER167 (compound 27) is an RDEA3170 (compound 28) derivative with the insertion of methylene between the naphthalene and pyridine of RDEA3170 (Figure 6). It is described as a dual inhibitor of both URAT1 and GLUT9, with IC50 values of 2.1 μM and 91.6 μM, respectively. Compared to the parent RDEA3170, CDER167 possesses similar inhibition on URAT1, though RDEA3170 shows no inhibition of GLUT9. An in vivo study confirmed the safety of CDER167 and that it is more bioavailable than RDEA3170 [62].
Figure 6.
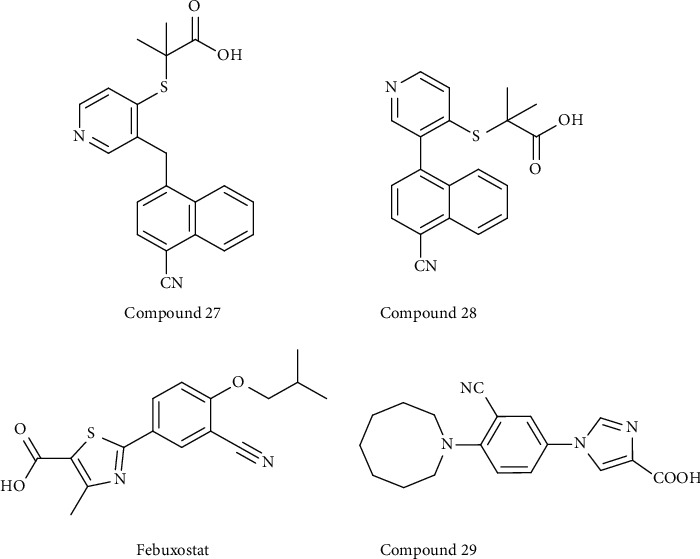
Structures of other synthetic derivatives with URAT1 inhibitory activity.
Febuxostat (Figure 6) is a nonpurine selective XO inhibitor and was approved as a first-line drug for treating hyperuricemia and gout in 2009. Zhou found that febuxostat has an inhibitory effect on URAT1 [63]. They used a fluorescence-based assay and identified the URAT1 inhibitors febuxostat and benzbromarone, which have IC50 values of 36.1 μM and 14.3 μM, respectively. As such, the authors designed and synthesized a series of “me-too” compounds using febuxostat as the lead to screen URAT1 inhibitors. They found that compound 4 (compound 29) (Figure 6) (IC50 value = 10.8 μM) exhibits a similar URAT1 inhibitory effect as benzbromarone [64].
3.2. Technical Methods for Validation of Inhibitors
Various experimental methods have been used to evaluate URAT1 inhibition. Most research uses in vitro models for cell-based approaches (Table 1). To our knowledge, there is currently no in vivo model that directly evaluates URAT1 inhibition. Many studies focus on URAT1 inhibition expression in the kidneys, but there is a lack of evidence for direct interaction. Therefore, hyperuricemia animal models are used to verify the uric-lowering effect and safety of inhibitors [65]. In this review, we focus on the in vitro approaches to evaluating URAT1 inhibition.
Table 1.
Cell-based methods for validation of URAT1 inhibitors.
| Authors | Cell model | Validation method | Compound |
|---|---|---|---|
| Toyoda et al. [36]; Saito et al. [56] |
URAT1 wild type in pEGFP-C1 vector was transiently transfected into HEK293A | Radioisotope-labeled uric acid uptake assays | 1; 2; 3; 17; 18; 19 |
| Wu et al. [58]; Zhang et al. [61] |
HEK293 cells stably expressing hURAT1 | Radioisotope-labeled uric acid uptake assays | 22; 25; 24 |
| Zhang et al. [48] | Stably hURAT1-transfected HEK293 cells | Radioisotope-labeled uric acid uptake assays | 10 |
| Yang et al. [66] | pCMV/neo-URAT1 was stably transfected into HE-K293 cells | Radioisotope-labeled uric acid uptake assays | 32; 33 |
| Ao et al. [44] | HEK293 cells were transiently transfected with URAT1 plasmid | Radioisotope-labeled uric acid uptake assays | 6 |
| Bao et al. [51] | HEK293 T cells stably expressing hURAT1 by virus transfect | UPLC | 28 |
| Tan et al. [21] | hURAT1 was subcloned into pCMV6/neo vector and transiently transfected into HEK293T cells | Radioisotope-labeled uric acid uptake assays | 28 |
| Chen et al. [41] | hURAT1 was subcloned into pcDNA3.1(+) and transiently transfected into HEK293T cells | Radioisotope-labeled uric acid uptake assays | 4 |
| Zhao et al. [57] | hURAT1 was subcloned into pcDNA3.1(+)-EGFP-vector and transiently transfected into HEK293T | Radioisotope-labeled uric acid uptake assays | 20 |
| Zhou et al. [64] | hURAT1 was subcloned into pCMV vector and stably expressed in HEK293T | Fluorescence detection method (6-CFL as the fluorescent substrate) | 29 |
| Tan et al. [67] | pCMV6/neo-hURAT1 was transiently transfected into HEK293T | URAT1 direct binding assay | — |
| Tashiro et al. [45] | hURAT1 was subcloned into pCMV-SPORT6 vector and transiently transfected into HEK293/PDZK1 cells | Radioisotope-labeled uric acid uptake assays | 7 |
| Wu et al. [59] | MDCK cells stably expressing hURAT1 | Radioisotope-labeled uric acid uptake assays | 23 |
| Shin et al. [68] | MDCK cells transfected with pcDNA3.1-hURAT1 | Radioisotope-labeled uric acid uptake assays | Benzbromarone; 30; 31 |
| Chen et al. [69] | Lentivirus vectors carrying hURAT1 were constructed and stably expressed in MDCK cells | Radioisotope-labeled uric acid uptake assays | Benzbromarone; |
| Probenecid | |||
| Sun et al. [70] | Lentivirus vectors carrying hURAT1 were constructed and stably expressed in MDCK cells | LC-MS/MS | Benzbromarone |
| Lee et al. [52] | Capped URAT1 cRNA was injected into Xenopus oocytes | Radioisotope-labeled uric acid uptake assays | Alpinia oxyphylla seed extract |
| Wempe et al. [71] | Defolliculated Xenopus oocytes were injected with capped URAT1 cRNA | Radioisotope-labeled uric acid uptake assays | 34 |
| Nakamura et al. [72] | Defolliculated Xenopus oocytes were injected with hURAT1 cRNA | Radioisotope-labeled uric acid uptake assays | Irbesartan |
| Iwanaga et al. [73] Iwanaga et al. [74] |
URAT1 cRNA was injected into Xenopus oocytes | Radioisotope-labeled uric acid uptake assays | Losartan, pratosartan, benzbromarone |
| Lee et al. [75] | Capped URAT1 cRNA was injected into Xenopus laevis oocytes | Radioisotope-labeled uric acid uptake assays | Keishibukuryogan; shakuyakukanzoto |
3.2.1. Radioisotope-Labeled Uric Acid Uptake Assays
The radioactive 14C-labeling method is the most popular method of quantitatively evaluating the URAT1 function (Figure 7A). This method directly reveals the uric acid transportation by URAT1 and was established when this protein was first identified [17]. In this assay, the following cell lines are typically used for URAT1-overexpression: Xenopus laevis Oocytes, human epithelial kidney cell, and MDCK cells. When using oocytes, hURAT1-cRNA must be synthesized, injected into the cells, and incubated for 2-3 days. The oocytes are then transferred to a Cl−-free solution containing [14C] uric acid to initiate the uptake of uric acid, while the radioactivity in the oocytes is determined by a liquid scintillation counter [71], considering that the oocyte cell model is complicated and the renal localization of URAT1, kidney cell HEK293, or MDCK cells have been used in recent years to express hURAT1 [67, 68]. In this model, the URAT1-expressing plasmid is transiently transfected into cells for 1-2 day protein expression, and the cells are then assayed for [14C] uric acid uptake at certain time points (20 s, 5 min, 10 min) [21, 76]. For example, for the inhibition of URAT1 by fatty acids, only 20 s of incubation is needed for uric acid uptake, which helps exclude the impact of fatty acids on the plasma membrane [56]. This method is easy, convenient, and quick. Some studies use hURAT1 stably expressed HEK293 or MDCK cells to obtain a persistent and stable model to evaluate URAT1 inhibitors [48, 68, 69].
Figure 7.
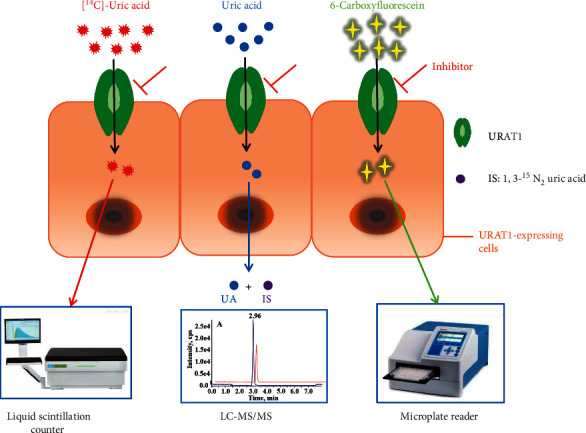
Schematic illustrations of cell-based methods for evaluation of URAT1 inhibitors. A: radioisotope-labeled uric acid uptake assay; B: chromatography-based approach, IS: internal standard; C: fluorescence detection method.
Radioisotope-labeled uric acid uptake assays have been widely used to screen URAT1 inhibitors with high sensitivity and are capable of measuring the IC50 of these inhibitors (Figure 8). However, using radioisotopes could be restricted in some laboratories and it is costly to use.
Figure 8.
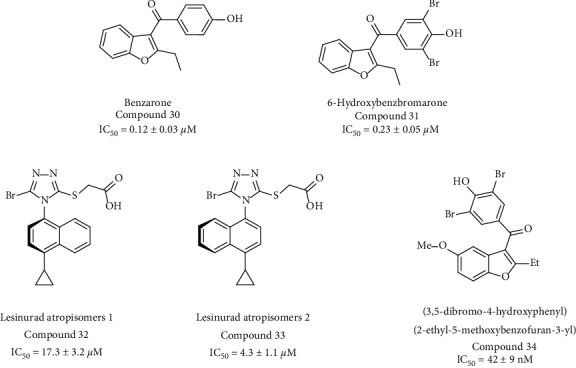
Structures of the URAT1 inhibitors shown in Table 1.
3.2.2. Chromatography-Based Approach
An ultra performance liquid chromatography (UPLC) method is used for nonradioactive uric acid transport assay to detect the uric acid contents of URAT1-expressing cells. UPLC requires a relatively large amount of uric acid (0.09 μM) and is limited to a maximum concentration of 0.18 μM [77], meaning this method depends on extracellular and intracellular concentrations of uric acid, limiting its practical use.
LC-MS/MS is an analysis used in highly selective and sensitive in vitro models of URAT1 inhibition [70] (Figure 7B). Similar to other cell-based methods, hURAT1-expressing cells are incubated with non-radioactive uric acid and the test compounds. The uric acid in the cell is then released and detected via LC-MS/MS. This method is unique in that it uses isotope-labeled 1,3-15N2 uric acid as an internal standard, which sets the limit of detection (LOD) of uric acid at 50 nM and the limit of quantitation (LOQ) at 200 nM. This approach demonstrates a highly selective and sensitive method of assessing intracellular uric acid and provides a suitable model for the in vitro evaluation of drug candidates targeting URAT1.
3.2.3. Fluorescence Method
Fluorescent substance 6-carboxyfluorescein (not uric acid) is used as the substrate for URAT1-mediated transportation [64] (Figure 7C). In stably overexpressed hURAT1-HEK293T cells, 6-carboxyfluorescein is selectively transported by URAT1, while the transport is time-dependent and saturable (Km = 239.5 μM). In this method, the cells are incubated with a fluorescent substrate for one hour, after which the cells are lysed and assayed using a microplate reader. It is an economical, environmentally friendly, and convenient approach for screening URAT1 inhibitors in vitro. However, because of the low affinity of 6-carboxyfluorescein, the IC50 values of benzbromarone and lesinurad against URAT1 measured by this method could be 100 times greater than the radioactive methods, meaning that a high concentration of compounds is required to produce the fluorescence signal. The fluorescence method has the advantages of high throughput and easy quantitative analysis and is widely used to detect in vitro activity in recent years [78–80]. The primary disadvantage of this approach is its low sensitivity, making the identification of strong fluorescence substrates a priority.
3.2.4. URAT1 Direct Binding Assay
A URAT1 binding assay was developed to identify the interactions between the proteins and inhibitors [67]. It is a similar procedure to the uric acid uptake assay, where URAT1 is first expressed in cells and URAT-enriched cell membranes are then isolated by two steps of centrifugation. The cell membranes are incubated with a radiolabeled high-affinity URAT1 inhibitor probe (e.g., 3H-RDEA3170) in the presence and absence of candidate compounds. If the candidate interacts with URAT1 at the same binding site of the radiolabeled probe, the intramembrane accumulated probes will be displaced or its binding to hURAT1 will be weakened [21]. This assay has identified some URAT1 inhibitors, including benzbromarone, sulfinpyrazone, probenecid, and lesinurad. Notably, this assay relies on the same binding site of inhibitors and probes and is ineffective for molecules that bind to other URAT1 sites. As such, this binding assay provides a tool for characterizing the molecular interactions of compounds with URAT1 and provides additional validation of new inhibitors.
4. Conclusions
URAT1 is a proven key target for promoting uric acid excretion; however, current URAT1 inhibitors have serious side effects. In recent years, many new URAT1 inhibitors have been described and this review summarizes key and novel experimental approaches that contribute to screening URAT1 inhibitors and help validate the urate-lowering effect. Radioisotope-labeled uric acid uptake assays are classical methods of validating URAT1 inhibitors. Given the disadvantages of the radioisotope method, novel identification methods have emerged in recent years. This includes nonradioactive isotope-LC-MS/MS and fluorescence detection, both of which could help explore novel URAT1 inhibitors. However, the sensitivity and efficiency of these methods must be optimized. We also summarized current progress relating to URAT1 inhibitor discovery, including natural products and synthetic compounds. Phenolic compounds are the primary category of natural URAT1 inhibitors, while synthetic inhibitors often use lesinurad as the lead scaffold. This review also demonstrates that many compounds, such as compounds 9, 21, and 27, have dual inhibitory effects on URAT1 and XO or GLUT9, which showed an excellent urate-lowering effect with more safety. Therefore, developing dual inhibitors for urate-lowering therapy is a promising area of research, though inhibition mechanisms, pharmacokinetics, and the safety of the novel URAT1 inhibitors all require further study.
Acknowledgments
The authors acknowledge Prof. Hengming Ke for reviewing the manuscript. This work was supported by the National Natural Science Foundation of China under Grant no. 31972127, the Science and Technology Program of the Beijing Municipal Education Commission under Grant no, KZ201910011013, and the Open Project Program of Beijing Advanced Innovation Center for Food Nutrition and Human Health under Grant no. 20161018.
Data Availability
The data used in the current study are included within this article.
Conflicts of Interest
The authors disclose no conflicts of interest.
Authors' Contributions
Y. C. researched the data and wrote the manuscript. R.-Y. Y. and K. W. contributed to the chemical structures drawing. Y.-S. W. contributed to the discussion and the writing of the manuscript.
References
- 1.Maiuolo J., Oppedisano F., Gratteri S., Muscoli C., Mollace V. Regulation of uric acid metabolism and excretion. International Journal of Cardiology . 2016;213:8–14. doi: 10.1016/j.ijcard.2015.08.109. [DOI] [PubMed] [Google Scholar]
- 2.James A., Ke H., Yao T., Wang Y. The role of probiotics in purine metabolism, hyperuricemia and gout: mechanisms and interventions. Food Reviews International . 2021;5:1–17. doi: 10.1080/87559129.2021.1904412. [DOI] [Google Scholar]
- 3.Dalbeth N., Choi H. K., Joosten L. A. B., et al. Gout. Nature reviews. Disease primers . 2019;5(1):p. 69. doi: 10.1038/s41572-019-0115-y. [DOI] [PubMed] [Google Scholar]
- 4.Otani N., Ouchi M., Kudo H., Tsuruoka S., Hisatome I., Anzai N. Recent approaches to gout drug discovery: an update. Expert Opinion on Drug Discovery . 2020;15(8):943–954. doi: 10.1080/17460441.2020.1755251. [DOI] [PubMed] [Google Scholar]
- 5.Dalbeth N., Merriman T. R., Stamp L. K. Gout. The Lancet . 2016;388(10055):2039–2052. doi: 10.1016/s0140-6736(16)00346-9. [DOI] [PubMed] [Google Scholar]
- 6.Burns C. M., Wortmann R. L. Gout therapeutics: new drugs for an old disease. The Lancet . 2011;377(9760):165–177. doi: 10.1016/s0140-6736(10)60665-4. [DOI] [PubMed] [Google Scholar]
- 7.Kuo C.-F., Grainge M. J., Zhang W., Doherty M. Global epidemiology of gout: prevalence, incidence and risk factors. Nature Reviews Rheumatology . 2015;11(11):649–662. doi: 10.1038/nrrheum.2015.91. [DOI] [PubMed] [Google Scholar]
- 8.Johnson R. J., Nakagawa T., Jalal D., Sanchez-Lozada L. G., Kang D.-H., Ritz E. Uric acid and chronic kidney disease: which is chasing which? Nephrology Dialysis Transplantation . 2013;28(9):2221–2228. doi: 10.1093/ndt/gft029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryu E.-S., Kim M. J., Shin H.-S., et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. American Journal of Physiology - Renal Physiology . 2013;304(5):F471–F480. doi: 10.1152/ajprenal.00560.2012. [DOI] [PubMed] [Google Scholar]
- 10.Lippi G., Montagnana M., Luca Salvagno G., Targher G., Cesare Guidi G. Epidemiological association between uric acid concentration in plasma, lipoprotein(a), and the traditional lipid profile. Clinical Cardiology . 2010;33(2):E76–E80. doi: 10.1002/clc.20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Becker B., Borghi C., Burnier M., van de Borne P. Uric acid and hypertension: a focused review and practical recommendations. Journal of Hypertension . 2019;37(5):878–883. doi: 10.1097/hjh.0000000000001980. [DOI] [PubMed] [Google Scholar]
- 12.Martínez-Quintana E., Tugores A., Rodríguez-González F. Serum uric acid levels and cardiovascular disease: the Gordian knot. Journal of Thoracic Disease . 2016;8(11):E1462–E1466. doi: 10.21037/jtd.2016.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katsiki N., Papanas N., Fonseca V., Maltezos E., Mikhailidis D. Uric acid and diabetes: is there a link? Current Pharmaceutical Design . 2013;19(27):4930–4937. doi: 10.2174/1381612811319270016. [DOI] [PubMed] [Google Scholar]
- 14.Jin M., Yang F., Yang I., et al. Uric acid, hyperuricemia and vascular diseases. Frontiers in Bioscience . 2012;17(1):656–669. doi: 10.2741/3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King C., Lanaspa M. A., Jensen T., Tolan D. R., Sánchez-Lozada L. G., Johnson R. J. Uric acid as a cause of the metabolic syndrome. Contributions to Nephrology . 2018;192:88–102. doi: 10.1159/000484283. [DOI] [PubMed] [Google Scholar]
- 16.Pascart T., Richette P. Investigational drugs for hyperuricemia, an update on recent developments. Expert Opinion on Investigational Drugs . 2018;27(5):437–444. doi: 10.1080/13543784.2018.1471133. [DOI] [PubMed] [Google Scholar]
- 17.Enomoto A., Kimura H., Chairoungdua A., et al. Molecular identification of a renal urate-anion exchanger that regulates blood urate levels. Nature . 2002;417(6887):447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 18.Xu L., Shi Y., Zhuang S., Liu N. Recent advances on uric acid transporters. Oncotarget . 2017;8(59):100852–100862. doi: 10.18632/oncotarget.20135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar R., Darpan, Sharma S., Singh R. Xanthine oxidase inhibitors: a patent survey. Expert Opinion on Therapeutic Patents . 2011;21(7):1071–1108. doi: 10.1517/13543776.2011.577417. [DOI] [PubMed] [Google Scholar]
- 20.Singh J. V., Bedi P. M. S., Singh H., Sharma S. Xanthine oxidase inhibitors: patent landscape and clinical development (2015–2020) Expert Opinion on Therapeutic Patents . 2020;30(10):769–780. doi: 10.1080/13543776.2020.1811233. [DOI] [PubMed] [Google Scholar]
- 21.Tan P. K., Liu S., Gunic E., Miner J. N. Discovery and characterization of verinurad, a potent and specific inhibitor of URAT1 for the treatment of hyperuricemia and gout. Scientific Reports . 2017;7(1):p. 665. doi: 10.1038/s41598-017-00706-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robbins N., Koch S. E., Tranter M., Rubinstein J. The history and future of probenecid. Cardiovascular Toxicology . 2012;12(1):1–9. doi: 10.1007/s12012-011-9145-8. [DOI] [PubMed] [Google Scholar]
- 23.Lee M.-H. H., Graham G. G., Williams K. M., Day R. O. A benefit-risk assessment of benzbromarone in the treatment of gout. Drug Safety . 2008;31(8):643–665. doi: 10.2165/00002018-200831080-00002. [DOI] [PubMed] [Google Scholar]
- 24.Miner J. N., Tan P. K., Hyndman D., et al. Lesinurad, a novel, oral compound for gout, acts to decrease serum uric acid through inhibition of urate transporters in the kidney. Arthritis Research and Therapy . 2016;18(1):p. 214. doi: 10.1186/s13075-016-1107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strilchuk L., Fogacci F., Cicero A. F. Safety and tolerability of available urate-lowering drugs: a critical review. Expert Opinion on Drug Safety . 2019;18(4):261–271. doi: 10.1080/14740338.2019.1594771. [DOI] [PubMed] [Google Scholar]
- 26.Hautekeete M. L., Henrion J., Naegels S., et al. Severe hepatotoxicity related to benzarone: a report of three cases with two fatalities. Liver . 1995;15:25–29. doi: 10.1111/j.1600-0676.1995.tb00102.x. [DOI] [PubMed] [Google Scholar]
- 27.Underwood M. Diagnosis and management of gout. British Medical Association . 2006;332(7553):1315–1319. doi: 10.1136/bmj.332.7553.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoy S. M. Lesinurad: first global approval. Drugs . 2016;76(4):509–516. doi: 10.1007/s40265-016-0550-y. [DOI] [PubMed] [Google Scholar]
- 29.Chien-Yi Yang B., Chi-Hua Chen M., Shin-Tarng Deng M., et al. Allopurinol use and risk of fatal hypersensitivity reactions a nationwide population-based study in Taiwan. JAMA Internal Medicine . 2015;9(175):1550–1557. doi: 10.1001/jamainternmed.2015.3536. [DOI] [PubMed] [Google Scholar]
- 30.Kumar R., Joshi G., Kler H., Kalra S., Kaur M., Arya R. Toward an understanding of structural insights of xanthine and aldehyde oxidases: an overview of their inhibitors and role in various diseases. Medicinal Research Reviews . 2017;38(4):1073–1125. doi: 10.1002/med.21457. [DOI] [PubMed] [Google Scholar]
- 31.Mehmood A., Ishaq M., Zhao L., et al. Natural compounds with xanthine oxidase inhibitory activity: a review. Chemical Biology & Drug Design . 2019;93(4):387–418. doi: 10.1111/cbdd.13437. [DOI] [PubMed] [Google Scholar]
- 32.Dong Y., Zhao T., Ai W., et al. Novel urate transporter 1 (URAT1) inhibitors: a review of recent patent literature (2016–2019) Expert Opinion on Therapeutic Patents . 2019;29(11):871–879. doi: 10.1080/13543776.2019.1676727. [DOI] [PubMed] [Google Scholar]
- 33.Song D., Zhao X., Wang F., Wang G. A brief review of urate transporter 1 (URAT1) inhibitors for the treatment of hyperuricemia and gout: current therapeutic options and potential applications. European Journal of Pharmacology . 2021;907:p. 174291. doi: 10.1016/j.ejphar.2021.174291. [DOI] [PubMed] [Google Scholar]
- 34.Jiang L. L., Gong X., Ji M. Y., Wang C. C., Wang J. H., Li M. H. Bioactive compounds from plant-based functional foods: a promising choice for the prevention and management of hyperuricemia. Foods . 2020;9(8) doi: 10.3390/foods9080973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L., Luo Z., Wang M., et al. The efficacy and mechanism of Chinese herbal medicines in lowering serum uric acid levels: a systematic review. Frontiers in Pharmacology . 2020;11 doi: 10.3389/fphar.2020.578318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toyoda Y., Takada T., Saito H., et al. Inhibitory effect of Citrus flavonoids on the in vitro transport activity of human urate transporter 1 (URAT1/SLC22A12), a renal re-absorber of urate. NPJ Science of Food . 2020;4(1):p. 3. doi: 10.1038/s41538-020-0063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu K., Wang W., Guo B.-H., et al. Chemical evidence for potent xanthine oxidase inhibitory activity of ethyl acetate extract of citrus aurantium L. dried immature fruits. Molecules . 2016;21(3):p. 302. doi: 10.3390/molecules21030302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haidari F., Ali Keshavarz S., Reza Rashidi M., Mohammad Shahi M. Orange juice and hesperetin supplementation to hyperuricemic rats alter oxidative stress markers and xanthine oxidoreductase activity. Journal of Clinical Biochemistry & Nutrition . 2009;45(3):285–291. doi: 10.3164/jcbn.09-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shunji A., Shinya I., Tadashi K. Uricosuric composition, and food/drink using the same: JP2016155825A. 2016. https://patents.google.com/patent/JP2016155825A/en .
- 40.Mo S.-F., Zhou F., Lv Y.-Z., Hu Q.-H., Zhang D.-M., Kong L.-D. Hypouricemic action of selected flavonoids in mice: structure-activity relationships. Biological and Pharmaceutical Bulletin . 2007;30(8):1551–1556. doi: 10.1248/bpb.30.1551. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y., Zhao Z., Li Y., et al. Baicalein alleviates hyperuricemia by promoting uric acid excretion and inhibiting xanthine oxidase. Phytomedicine . 2021;80:p. 153374. doi: 10.1016/j.phymed.2020.153374. [DOI] [PubMed] [Google Scholar]
- 42.Yu Z., Li Q., Wang Y., Li P. A potent protective effect of baicalein on liver injury by regulating mitochondria-related apoptosis. Apoptosis: An International Journal on Programmed Cell Death . 2020;25(5-6):412–425. doi: 10.1007/s10495-020-01608-2. [DOI] [PubMed] [Google Scholar]
- 43.Dai C., Tang S., Wang Y., Velkov T., Xiao X. Baicalein acts as a nephroprotectant that ameliorates colistin-induced nephrotoxicity by activating the antioxidant defence mechanism of the kidneys and down-regulating the inflammatory response. Journal of Antimicrobial Chemotherapy . 2017;72(9):2562–2569. doi: 10.1093/jac/dkx185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ao G.-Z., Zhou M.-Z., Li Y.-Y., et al. Discovery of novel curcumin derivatives targeting xanthine oxidase and urate transporter 1 as anti-hyperuricemic agents. Bioorganic & Medicinal Chemistry . 2017;25(1):166–174. doi: 10.1016/j.bmc.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 45.Tashiro Y., Sakai R., Hirose-Sugiura T., et al. Effects of osthol isolated from cnidium monnieri fruit on urate transporter 1. Molecules . 2018;23(11):p. 2837. doi: 10.3390/molecules23112837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu Q., Zhou M., Zhu H., et al. (E)-2-(4-bromophenyl)-1-(2, 4-dihydroxyphenyl)ethanone oxime is a potential therapeutic agent for treatment of hyperuricemia through its dual inhibitory effects on XOD and URAT1. Biomedicine & Pharmacotherapy . 2017;86:88–94. doi: 10.1016/j.biopha.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 47.Hsüch C. C., Yen Y. C. Xanthine oxidase inhibitors from the roots of eggplant (Solanum melongena L.) Taylor & Francis . 2008;7(3) doi: 10.3109/14756369309040765. [DOI] [Google Scholar]
- 48.Zhang Y., Jin L., Liu J., et al. Effect and mechanism of dioscin from Dioscorea spongiosa on uric acid excretion in animal model of hyperuricemia. Journal of Ethnopharmacology . 2018;214:29–36. doi: 10.1016/j.jep.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 49.Su J., Wei Y., Liu M., et al. Anti-hyperuricemic and nephroprotective effects of Rhizoma Dioscoreae septemlobae extracts and its main component dioscin via regulation of mOAT1, mURAT1 and mOCT2 in hypertensive mice. Archives of Pharmacal Research . 2014;37(10):1336–1344. doi: 10.1007/s12272-014-0413-6. [DOI] [PubMed] [Google Scholar]
- 50.Tan Y., Wang L., Gao J., et al. Multiomics integrative analysis for discovering the potential mechanism of dioscin against hyperuricemia mice. Journal of Proteome Research . 2021;20(1):645–660. doi: 10.1021/acs.jproteome.0c00584. [DOI] [PubMed] [Google Scholar]
- 51.Bao R., Liu M., Wang D., et al. Effect of eurycoma longifolia stem extract on uric acid excretion in hyperuricemia mice. Frontiers in Pharmacology . 2019;10:p. 1464. doi: 10.3389/fphar.2019.01464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee Y.-S., Sung Y.-Y., Yuk H. J., et al. Anti-hyperuricemic effect of Alpinia oxyphylla seed extract by enhancing uric acid excretion in the kidney. Phytomedicine . 2019;62:p. 152975. doi: 10.1016/j.phymed.2019.152975. [DOI] [PubMed] [Google Scholar]
- 53.Noels H., Lehrke M., Vanholder R., Jankowski J. Lipoproteins and fatty acids in chronic kidney disease: molecular and metabolic alterations. Nature Reviews Nephrology . 2021;17(8):528–542. doi: 10.1038/s41581-021-00423-5. [DOI] [PubMed] [Google Scholar]
- 54.Calder P. C. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochemical Society Transactions . 2017;45(5):1105–1115. doi: 10.1042/bst20160474. [DOI] [PubMed] [Google Scholar]
- 55.Currie E., Schulze A., Zechner R., Walther T. C., Farese R. V. Cellular fatty acid metabolism and cancer. Cell Metabolism . 2013;18(2):153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saito H., Toyoda Y., Takada T., et al. Omega-3 polyunsaturated fatty acids inhibit the function of human URAT1, a renal urate re-absorber. Nutrients . 2020;12(6) doi: 10.3390/nu12061601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao T., Meng Q., Sun Z., et al. Novel human urate transporter 1 inhibitors as hypouricemic drug candidates with favorable druggability. Journal of Medicinal Chemistry . 2020;63(19):10829–10854. doi: 10.1021/acs.jmedchem.0c00223. [DOI] [PubMed] [Google Scholar]
- 58.Wu J.-W., Yin L., Liu Y.-Q., et al. Synthesis, biological evaluation and 3D-QSAR studies of 1,2,4-triazole-5-substituted carboxylic acid bioisosteres as uric acid transporter 1 (URAT1) inhibitors for the treatment of hyperuricemia associated with gout. Bioorganic & Medicinal Chemistry Letters . 2019;29(3):383–388. doi: 10.1016/j.bmcl.2018.12.036. [DOI] [PubMed] [Google Scholar]
- 59.Wu T., Chen J., Dong S., et al. Identification and characterization of a potent and selective inhibitor of human urate transporter 1. Pharmacological Reports . 2017;69(5):1103–1112. doi: 10.1016/j.pharep.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 60.Tian H., Liu W., Zhou Z., et al. Discovery of a flexible triazolylbutanoic acid as a highly potent uric acid transporter 1 (URAT1) inhibitor. Molecules . 2016;21(11) doi: 10.3390/molecules21111543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang X., Wu J., Liu W., et al. Discovery of flexible naphthyltriazolylmethane-based thioacetic acids as highly active uric acid transporter 1 (URAT1) inhibitors for the treatment of hyperuricemia of gout. Medicinal Chemistry . 2017;13(3):260–281. doi: 10.2174/1573406412666160915163002. [DOI] [PubMed] [Google Scholar]
- 62.Zhao Z. A., Jiang Y., Chen Y. Y., et al. CDER167, a dual inhibitor of URAT1 and GLUT9, is a novel and potent uricosuric candidate for the treatment of hyperuricemia. Acta Pharmacologica Sinica . 2021;1 doi: 10.1038/s41401-021-00640-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou H. Y. Establishment and Evaluation of Cell Screening Model for URAT1 Inhibitors Based on Fluorescence Method . Guangzhou, China: South China University of Technology; 2020. [Google Scholar]
- 64.Zhou H., Zhong G., Bai J., et al. Development of a fluorescence-based assay for screening of urate transporter 1 inhibitors using 6-carboxyfluorescein. Analytical Biochemistry . 2021;626:p. 114246. doi: 10.1016/j.ab.2021.114246. [DOI] [PubMed] [Google Scholar]
- 65.Lu J., Dalbeth N., Yin H., Li C., Merriman T. R., Wei W.-H. Mouse models for human hyperuricaemia: a critical review. Nature Reviews Rheumatology . 2019;15(7):413–426. doi: 10.1038/s41584-019-0222-x. [DOI] [PubMed] [Google Scholar]
- 66.Yang C., Zhou D., Shen Z., et al. Characterization of stereoselective metabolism, inhibitory effect on uric acid uptake transporters, and pharmacokinetics of lesinurad atropisomers. Drug Metabolism and Disposition . 2019;47(2):104–113. doi: 10.1124/dmd.118.080549. [DOI] [PubMed] [Google Scholar]
- 67.Tan P. K., Ostertag T. M., Miner J. N. Mechanism of high affinity inhibition of the human urate transporter URAT1. Scientific Reports . 2016;6(1):p. 34995. doi: 10.1038/srep34995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shin H. J., Takeda M., Enomoto A., et al. Interactions of urate transporter URAT1 in human kidney with uricosuric drugs. Nephrology . 2011;16(2):156–162. doi: 10.1111/j.1440-1797.2010.01368.x. [DOI] [PubMed] [Google Scholar]
- 69.Chen J. S., Wu T., Qiu Y. C., Cao Y., Pang J. X. A cell line stably expressing lentivirus-mediated hURAT1 as an in vitro model of screening uricosuric agents. Life Sciences . 2016;20(3):248–254. [Google Scholar]
- 70.Sun C., Zhang M., Zhao Y., Pang J., Peng Y., Zheng J. An LC-MS/MS- and hURAT1 cell-based approach for screening of uricosuric agents. Journal of Chromatography B . 2020;1159:p. 122336. doi: 10.1016/j.jchromb.2020.122336. [DOI] [PubMed] [Google Scholar]
- 71.Wempe M. F., Jutabha P., Quade B., et al. Developing potent human uric acid transporter 1 (hURAT1) inhibitors. Journal of Medicinal Chemistry . 2011;54(8):2701–2713. doi: 10.1021/jm1015022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakamura M., Anzai N., Jutabha P., Sato H., Sakurai H., Ichida K. Concentration-dependent inhibitory effect of Irbesartan on renal uric acid transporters. Journal of Pharmacological Sciences . 2010;114(1):115–118. doi: 10.1254/jphs.10064sc. [DOI] [PubMed] [Google Scholar]
- 73.Iwanaga T., Kobayashi D., Hirayama M., Maeda T., Tamai I. Involvement of uric acid transporter in increased renal clearance of the xanthine oxidase inhibitor oxypurinol induced by a uricosuric agent, benzbromarone. Drug Metabolism and Disposition . 2005;33(12):1791–1795. doi: 10.1124/dmd.105.006056. [DOI] [PubMed] [Google Scholar]
- 74.Iwanaga T., Sato M., Maeda T., Ogihara T., Tamai I. Concentration-dependent mode of interaction of angiotensin II receptor blockers with uric acid transporter. Journal of Pharmacology and Experimental Therapeutics . 2006;320(1):211–217. doi: 10.1124/jpet.106.112755. [DOI] [PubMed] [Google Scholar]
- 75.Lee H.-S., Shin H. J., Cho M., Lee S. H., Oh D.-S. Inhibitory effects of Kampo medicines, Keishibukuryogan and Shakuyakukanzoto, on the substrate uptake activities of solute carrier organic anion transporters. Journal of Pharmacological Sciences . 2018;138(4):279–283. doi: 10.1016/j.jphs.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 76.Miyata H., Takada T., Toyoda Y., Matsuo H., Ichida K., Suzuki H. Identification of febuxostat as a new strong ABCG2 inhibitor: potential applications and risks in clinical situations. Frontiers in Pharmacology . 2016;7:p. 518. doi: 10.3389/fphar.2016.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wen S., Zhang Z., Chen X., et al. An improved UPLC method for determining uric acid in rat serum and comparison study with commercial colorimetric kits. Acta Chromatographica . 2019;31(3):201–205. doi: 10.1556/1326.2018.00449. [DOI] [Google Scholar]
- 78.Raudszus R., Nowotny R., Gertzen C. G. W., et al. Fluorescent analogs of peptoid-based HDAC inhibitors: synthesis, biological activity and cellular uptake kinetics. Bioorganic & Medicinal Chemistry . 2019;27(19):p. 115039. doi: 10.1016/j.bmc.2019.07.055. [DOI] [PubMed] [Google Scholar]
- 79.Bibic L., Herzig V., King G. F., Stokes L. Development of high-throughput fluorescent-based screens to accelerate discovery of P2X inhibitors from animal venoms. Journal of Natural Products . 2019;82(9):2559–2567. doi: 10.1021/acs.jnatprod.9b00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen Y. Recent progress in natural product-based inhibitor screening with enzymatic fluorescent probes. Analytical Methods . 2021;13(15):1778–1787. doi: 10.1039/d1ay00245g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used in the current study are included within this article.