

Abstract
Cancer stem cells (CSCs), a group of tumour cells with stem cell characteristics, have the ability of self‐renewal, multi‐lineage differentiation and tumour formation. Since CSCs are resistant to conventional radiotherapy and chemotherapy, their existence may be one of the root causes of cancer treatment failure and tumour progression. The elimination of CSCs may be effective for eventual tumour eradication. Because of the good therapeutic effects without major histocompatibility complex (MHC) restriction and the unique characteristics of CSCs, chimeric antigen receptor T‐cell (CAR‐T) therapy is expected to be an important method to eliminate CSCs. In this review, we have discussed the feasibility of CSCs‐targeted CAR‐T therapy for cancer treatment, summarized current research and clinical trials of targeting CSCs with CAR‐T cells and forecasted the challenges and future direction from the perspectives of toxicity, persistence and potency, trafficking, infiltration, immunosuppressive tumour microenvironment, and tumour heterogeneity.
Keywords: cancer stem cells, CAR‐T, immunotherapy, targeted therapy
1. INTRODUCTION
The hypothesis that tumours originated from “stem cells” was first proposed about 150 years ago. However, relevant research had been progressing slowly until a growing number of experimental data strongly supported the tumour stem cell hypothesis which has changed views on tumourigenesis and tumour cell biology. 1 , 2 In cancer stem cell theory, tumours originate from a small portion of cancer stem cells (CSCs), and they have the capacity of immortal proliferation and multi‐lineage differentiation, which drives tumour formation, growth, recurrence, metastasis, drug resistance, chemo/radio‐resistance and other malignant phenotypic characteristics. 3 In this context, while CSCs are resistant to radiotherapy, chemotherapy and certain targeted therapies, the key to cancer treatment remains in CSCs. In addition, because of the special tumour‐killing mechanism, cancer stem cell may be more sensitive to immunotherapy, and in‐depth study of CSCs characteristics may also significantly promote the development of tumour immunotherapy. 4
Recently, the clinical application of chimeric antigen receptor T (CAR‐T)‐cell therapy has made an unprecedented breakthrough in the treatment of haematological diseases. 5 The safety and feasibility of CAR‐T therapy in the treatment of solid tumours have also been confirmed. 6 For CAR‐T therapy, T cells from the patients will be genetically engineered to express chimeric antigen receptors (CAR), and then be adoptively transferred back to patients. The genetically engineered CAR‐T cells will recognize the surface antigen of tumour cells and selectively target and kill those tumour cells. 7 CAR‐T cells can recognize the target antigen independently of MHC restrictions. 8 After the recognition, CAR‐T cells fix their position specifically in the tumour site and can have sustained persistence for a while. The successful application of CAR‐T cells in cancer treatment represents a milestone in anticancer therapy.
Cancer stem cells have some unique characteristics, such as slow rate of division, high expression of drug efflux pumps, 9 heightened activation of DNA repair mechanisms 10 and microenvironment characteristics: hypoxia and acidosis, 11 which is due to the expression of specific surface markers. These surface markers can be used as specific targets for CAR‐T therapy to eliminate CSCs. In addition, the expression of MHC molecules on the surface of CSCs is low, which causes MHC restriction when immunotherapy is used to target CSCs. 8 However, in CAR‐T therapy, CAR‐T cells can recognize the target antigen with no MHC restrictions, 8 which endows some advantages for the application of CAR‐T therapy to eliminate CSCs. In this review, we analysed the feasibility of targeting CSCs by using CAR‐T cells, summarized published studies on CSCs‐targeted CAR‐T therapy, pointed out the challenges of targeting CSCs by CAR‐T cells related to toxicity, persistence and potency, trafficking, infiltration, immunosuppressive tumour microenvironment, tumour heterogeneity and purposed promising strategies, such as novel CAR containing a JAK‐STAT signalling domain, modulation of chemokine signalling, directing CAR‐T cell to target vascular endothelial growth factor receptor 2(VEGFR2), combining CSCs‐targeted therapy with FDA‐approved PD‐1/PD‐L1 checkpoint inhibitors, multi‐target CAR‐T cell therapies and transgenic modification of the CAR structure, for the future development of CSCs‐targeted therapy.
2. GENERATION OF CAR‐T CELLS FOR CANCER IMMUNOTHERAPY
Chimeric antigen receptor T‐cell therapy has emerged as a novel therapeutic T‐cell engineering practice, in which T cells derived from patient blood were engineered in vitro to express artificial receptors to target a specific tumour antigen, then the modified T cells will be adoptively infused back to the patient's body to fight against cancer. 12 For the preparation of CAR‐T cells, activated T cells were infected with retroviruses or lentiviruses loaded with CAR sequences to express receptors, and these modified T cells can recognize tumour‐associated antigens and express the tandem co‐stimulation molecular signal transduction fragments which were related to T‐cell activation. 13 The engineered CAR‐T cells were expanded in vitro and then infused into patients to fight against tumours. In 1989, Gross et al. first proposed the concept of CAR‐T cell therapy. 14 At present, this therapy has made breakthroughs in clinical trials for the treatment of leukaemia, and has gradually extended to the clinical treatment of solid tumours. 15
The development of CAR‐T structure has gone through four generations. Each generation of CAR‐T structure is modified by adding more components in the intracellular space to make it more specific, efficient and durable (Table 1). The first generation of CAR‐T cells composed of the single‐chain variable fragment (scFv) and an intracellular CD3ζ signalling domain for T‐cell activation mainly solved the problem of targeting, but it lacked complete costimulatory signals and cannot fully activate T cells which limited its antitumour activity. 16 Subsequently, second‐ and third‐generation CARs were invented, which included one or two costimulatory domains respectively. The second‐generation CAR‐T cells had one costimulatory domain – CD28 or 4‐1BB, which effectively improves the tumour‐killing effect. 17 The third‐generation CAR‐T cells carried two costimulatory molecular domains, which significantly increased the proliferation activity of CAR‐T cells and enhanced cytokines release, which improves the in vivo persistence of CAR‐T cells and results in a stronger cytotoxic activity. 18 More recently, the fourth‐generation CAR‐T cells, also called TRUCK‐T (CAR redirected T cells that deliver a transgenic product to the targeted tumour tissue) cells, were engineered to secrete specific cytokines, such as IL‐12, IL15, IL‐18, CCL19 and IL‐7, so as to overcome the suppression from the tumour immune microenvironment, recruit and activate the second wave of immune cells to produce an immune response. 19
TABLE 1.
Architectural evolution of CAR‐T cell design
| Generation | The evolution of chimeric antigen receptors (CARs) |
|---|---|
| 1st generation | First‐generation CARs contain the CD3ζ chain of the T‐cell receptor complex |
| 2nd generation | Second‐generation synthetic antigen receptors differ from the first generation by the addition of a costimulatory domain (either CD28 or 4‐1BB). |
| 3rd generation | Third‐generation CARs contain two costimulatory domains, respectively, such as CD28 and OX40. |
| 4th generation | Fourth‐generation CARs, the so‐called TRUCKs or armoured CARs which are additionally modified with a constitutive or inducible expression cassette for a transgenic protein, which is released by the CAR‐T cell to modulate the T‐cell response. |
| Other evolution | Introducing some regulatory elements into CAR‐T cells, which include suicide‐initiated, negative‐regulatory and switch‐initiated components, or using dual antigen‐targeting CARs and inhibitory CARs. |
In addition to the evolution of CAR designs outlined above, some elements with regulatory functions can be added with the expression of an “armour” protein, by introducing suicide‐initiated, negative‐regulatory and switch‐initiated components into CAR‐T cells to have better control of the cytotoxic response of engineered CAR‐T cells on tumours. 20 In recent years, some new types of CAR‐T cells have been developed to increase the safety and therapeutic effect of CAR‐T cell therapy, such as dual antigen‐targeting CARs which improved specificity through targeting multiple antigens, and inhibitory CARs which were engineered to inhibit T‐cell activation upon binding to an antigen expressed on non‐malignant cells instead of tumour cells 21 , 22 (Table 1).
The CAR‐T cell therapy currently used in clinical practice is based on the second‐generation CAR‐T cells and mainly targets B‐cell‐related diseases, while the clinical application of the third‐ or fourth‐generation CAR‐T cells is at the early stages. 23 Since the infusion of allogeneic T cells is prone to cause human immune rejection, CAR‐T treatment currently uses patient's own T cells. As shown in Figure 1, the general treatment process can be divided into five steps: (1) Separation: isolate and purify T cells from the peripheral blood mononuclear cell (PBMC) of tumour patients through leucapheresis; (2) Modification: genetic engineering technology is applied to prepare CAR‐T cells through adding a chimeric antibody to recognize tumour cells and activate T cells at the same time; (3) Expansion: culture CAR‐T cells in vitro to the required dose for treatment, generally at the level of one billion to ten billion; (4) Infusion: infuse CAR‐T cells into patients with cancer; (5) Monitoring: observe the efficacy and closely monitor the adverse reactions. 24 The advantages of CAR‐T cell therapy include potential in vivo proliferation, long‐term in vivo persistence and efficient homing of CAR‐T cells to the tumour site. 25 Some of these patients have achieved durable complete remissions (CRs), which standard cytotoxic chemotherapy seldom achieves. 26 The advantages and encouraging results of CAR‐T therapy mentioned above make us optimistic about its clinical prospects.
FIGURE 1.
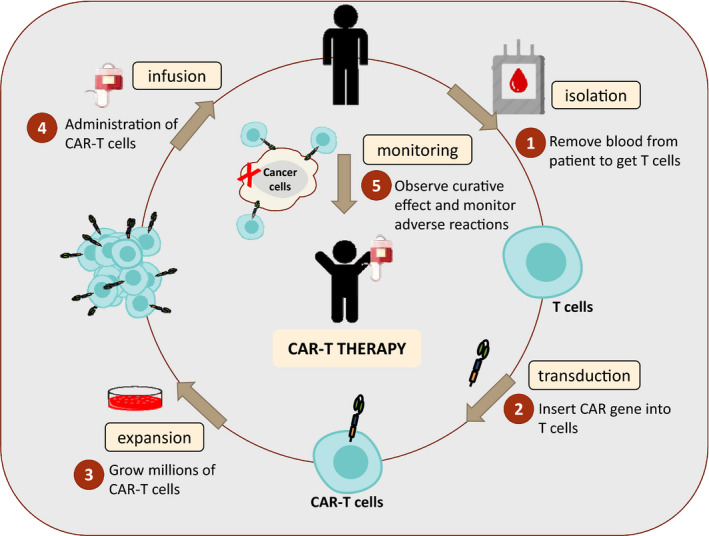
Diagram of CAR‐T treatment process. CAR‐T cell therapy can be defined as a treatment in which a patient's T cells are genetically modified in laboratory to express chimeric antigen receptors (CARs) and attack cancer cells. The process of adoptive CAR‐T cell therapy includes the following steps:(1) isolation of T cells from the peripheral blood sample by the process of leukapheresis; (2) transduction of cells by a viral vector encoding the CAR gene; (3) expansion of in vitro CAR‐T cells; (4) administration into the patient to kill cancer; (5) monitoring: observe curative effect and monitor adverse reactions
3. ANALYSES OF THE FEASIBILITY AND ADVANTAGES OF TARGETING CSCS WITH CAR‐T THERAPY FOR CANCER TREATMENT
Among many anticancer therapies, the primary problem impediment against cancer curability is tumour recurrence, which is mainly caused by the presence of CSCs. 27 Recently, cell therapy represented by CAR‐T cells has shown strong curative effects in tumour treatment and has the tendency to become an essential tumour treatment strategy. 28 Based on the characteristics of CAR‐T cells and CSCs, we proposed that targeting CSCs with CAR‐T therapy is feasible, and analysed the advantages and feasibility of this promising therapy.
3.1. Boost the body's own immune system to eliminate CSCs
Cancer stem cells play an important role in tumour occurrence, drug resistance, cancer recurrence, invasion and metastasis. Therefore, the elimination of CSCs improves tumour‐killing efficacy and even achieves radically curing tumours. Current strategies of targeting CSCs mainly include: (1) Targeting surface biomarkers of CSCs, mainly by developing monoclonal antibodies that specifically target the CSCs biomarkers 29 ; (2) Targeting key intracellular signal transduction pathways, such as NOTCH, Hedgehog (Hh) or WNT/ß‐Catenin signalling pathway 30 , 31 , 32 ; (3) Inhibiting ATP‐binding cassette transporters, for example, use Verapamil, Vardenafil and Laniquidar to inhibit the ABC transporters or use RNAi for ABC transporter gene silencing 33 ; (4) Induces CSCs differentiation and changes the microenvironment of CSCs. 34 Although a variety of strategies mentioned above have been used to target CSCs, these treatments are currently only in the laboratory stage. Up to now, no effective therapies have been verified by clinical trials, and there are still no drugs specifically used for targeting CSCs in clinical application. 35
Harnessing the power of the immune system to target CSCs is a promising therapeutic approach. For instance, J.C. Sun et al. (2010) applied dendritic cell‐based vaccines, which were treated with antigens from CD133+ hepatocellular carcinoma cells to activate specific cytotoxic lymphocytes, and therefore destroyed hepatocellular carcinoma CSCs. 36 In the last decade, cell‐based immunotherapy represented by CAR‐T therapy is being considered as an efficient approach for the treatment of cancer. 6 This is a way to eliminate tumour stem cells using the power of the body's own immune system, based upon the principle of targeting the surface markers of CSCs. CAR‐T cells eliminate CSCs relying on body's own immune system, and the infusion of millions of exogenous modified T cells can highly enhance the body's immune function.
3.2. The unique characteristics of CSCs are suitable for CAR‐T therapy
Targeting CSCs may realize tumour radical eradication, while the CSCs are protected by their unique characteristics, such as infrequent replication, enhanced drug resistance and heightened activation of DNA repair mechanisms as we mentioned in part 2. However, some of the CSCs characteristics may be harnessed to eradicate CSCs. For example, ATP‐binding cassette subfamily B member 5 (ABCB5), a marker of CSCs in a number of malignancies and a drug efflux transporter which associates multidrug resistance, tumour progression and recurrence, can be used in the tumour eradication. 37 , 38 Treatment with anti‐ABCB5 monoclonal antibodies has been shown to inhibit tumour growth in xeno‐transplantation models which prove that ABCB5 could be a good target for CSCs eradication. 39 Furthermore, as ABCB5‐reactive CD8+ T cells are present in the peripheral blood of melanoma patients and an ABCB5‐specific response can be induced in vitro in naive donors, which implicate that ABCB5 could be a potential target for cancer immunotherapy. 40 The subcellular location for ABCB5 expression is on cell membrane, which creates conditions for CAR‐T therapy targeting at ABCB5. 41 Interaction between CAR and ABCB5 helps to the formation of immune synapse, through which the contact‐dependent cytotoxicity may occur.
3.3. CAR‐T is an MHC‐independent adoptive cellular immunotherapy
CAR‐T therapy stands out among many CSCs‐targeted therapies for it is an MHC‐independent adoptive cellular immunotherapy. In 1975, Doherty and Zinkernagel first proposed the phenomenon of “MHC restriction” – viral peptides can only be recognized by T cells when combined with specific MHC molecules. 42 MHC is an important component of the immune system and plays a key role in antigen presentation, enabling specific T lymphocytes detect foreign antigen. 43 However, the expression of MHC molecules on CSCs is lower, which may prevent the body from boosting immune system to eliminate CSCs. 44 Fortunately, CAR‐T therapy is a MHC‐independent adoptive cellular immunotherapy for the unique structure of single‐chain variable fragment (scFv), mainly formed by variable regions of heavy and light chains, which can recognize cell surface antigens directly and specifically, instead of being restricted by the down‐regulation of MHC molecules. 45 Therefore, although CSCs are not easily eliminated by the immune cells from the immune system of patients, it is feasible to eliminate CSCs through CAR‐T therapy.
3.4. The existence surface antigens of CSCs that can be targeted by CAR‐T cells
Chimeric antigen receptor T‐cell cells can specifically recognize the surface antigens of tumour cells and effectively inhibit their growth and proliferation, which suggests that molecular markers on CSCs may be used as targets for CAR‐T cell immunotherapy. The discovery of surface markers of CSCs provides specific therapeutic targets for the treatment of CSCs. A large number of previous experiments have identified the expression of CD133, CD90, ALDH and EpCAM in CSCs of many types of cancers. 46 These markers can be used as an important molecular target for CAR‐T cells to kill CSCs to achieve the therapeutic effect of eliminating CSCs and inhibiting tumour recurrence and metastasis. In addition, certain molecular markers expressed in common tumour cells, such as epidermal growth factor receptor variant III (EGFRvIII), chondroitin sulphate proteoglycan 4 (CSPG4), human epidermal growth factor receptor 2(HER2) and NKG2D ligands (NKG2DLs), etc., are also expressed on the surface of tumour stem cells. 47 , 48 , 49 The construction of CAR‐T cells with molecular markers of CSCs as targets has a certain theoretical effect in the elimination of CSCs.
4. REPORTED LABORATORY RESEARCH OF CSCS‐TARGETED CAR‐T THERAPY
It is necessary that CSCs‐targeted CAR‐T cells efficiency, cytolytic activities and CAR molecule expression must be evaluated in preclinical setting before utilizing these cells as a therapy. In the following part, after carefully investigating laboratory studies on targeting CSCs with CAR‐T therapies from very limited number of reports, we proposed that the existing laboratory research on CSCs‐targeted CAR‐T therapies can be divided into two categories (Table 2): (1) the first category is targeting specific antigen molecules of CSCs, such as CD133, EpCAM or ALDH and designing corresponding CAR‐T cells for carrying out in vitro killing experiments and in vivo animal experiments for verification and (2) the second category is targeting “general” antigens (antigens also expressed in cancer cells) on CSCs.
TABLE 2.
Current studies on CSCs‐targeted CAR‐T therapy
| Antigen | Tumour target | Type of CARs | Target cells | Animal experiment | Main findings | Specific target |
|---|---|---|---|---|---|---|
| CD133 | Glioblastoma | 3rd generation CAR | AC133+ patient‐derived NCH421k GBM‐SCs | Orthotopic NMRI mouse model of GBM | CSCs isolated from glioblastoma patients were successfully killed by anti‐CD133 CAR‐T cells both in vitro and in vivo models of orthotopic tumour. 50 | Yes |
| EpCAM | Human prostate cancer | 2nd generation CAR | PC3M cells and PC cells | NOD/SCID mice (injected with PC3M‐luc cells) | Anti‐EpCAM CAR‐T cells were able to eliminate PC3M cells which express high levels of EpCAM in vivo and in vitro, as well as able to inhibit the tumour growth of PC3 cells that express low levels of EpCAM and prolong mouse survival in a PC3 metastasis model. 51 | Yes |
| EpCAM | Human ovarian and colorectal cancer | 3rd generation CAR | EpCAM‐positive ovarian cancer cell lines | NSG mouse xenograft model of human ovarian and colorectal carcinoma | CAR‐T cells targeting EpCAM on human ovarian and colorectal cancer cells are capable of killing the cancer cells in vitro, and the adoptive transfer of these CAR‐T cells resulted in prolonged animal survival by eradicating the established ovarian xenografts 52 | Yes |
| HER2 | Glioblastoma | 2nd generation CAR | Glioblastoma stem cells | Orthotopic xenogeneic SCID mouse model of GBM | Patients' HER2‐specific CAR‐T cells killed CD133‐positive and CD133‐negative cells derived from primary HER2‐positive glioblastomas. These HER2‐specific T cells had a potent antitumor activity against autologous tumours in an orthotopic xenogeneic SCID mouse model 48 | No |
| EGFRvIII | Glioblastoma | 2nd and 3rd generation CAR | Glioma stem cells(GSCs): GSCs (0308, 1228, and 0822) | NA | Anti‐EGFRvIII CAR‐engineered T cells produced the effector cytokine, IFN‐γ and lysed antigen expressing target cells. 49 | No |
| CSPG4 | Glioblastoma | 2nd generation CAR | Glioblastoma stem cells | NA | It is reported for the first time that CSPG4 is expressed on glioblastoma cancer stem cells (GSC) and demonstrate that anti‐CSPG4 CAR‐transduced T cells recognize and kill these GSC. 55 | No |
| NKG2DLs | Glioblastoma | 2nd generation CAR | Patient‐derived GSC‐3# | B‐NDG mice bearing U251MG xenografts | NKG2D ligands‐targeted CAR‐T cells efficiently lysed glioblastoma cells and cancer stem cells in vitro and produced high levels of cytokines, perforin and granzyme B. In vivo, the CAR‐T cells markedly eliminated xenograft tumours and did not exhibit significant toxicity in the treated mice 47 | No |
In the experiment of co‐culturing glioma stem cells with CAR‐T cells, Zhu et al. proved that anti‐CD133 CAR‐T cells can kill tumour patients‐derived glioma stem cells in vitro and have also shown therapeutic effects in animal models of orthotopic tumours. 50 However, in this study, when CAR‐T cells have an indirect contact with CD57‐positive target cells, the expression of CD57, which is a marker to abrogate T‐cell function, on CAR‐T cells is rapidly up‐regulated, leading to function impairment of CAR‐T cells. In another study by Deng et al., it is proved that EpCAM‐specific CARs can inhibit the growth of the human prostate cancer cell line PC3, a kind of cell line with low expression levels of EpCAM or kill PC3M (a highly metastatic clone of PC3 with high expression of EpCAM) in vitro and in vivo. Interestingly, despite the low expression level of EpCAM on PC3 cells, lymphocytes targeting EpCAM can cause significant tumour‐killing effects and inhibit the metastasis of PC3 cells in NOD/SCID mice. 51 For further study, Ang et al. reported that anti‐EpCAM CAR‐T cells exhibited specific in vitro killing activity against EpCAM‐positive human ovarian and colorectal cancer cells, and successfully treated local peritoneal cancer in xenograft mice with anti‐EpCAM CAR‐T cells, which show the feasibility of this therapy for curing clinical gastrointestinal and gynaecological malignancies. 52
Compared with targeting the specific surface antigens, engineering the corresponding CAR‐T cells to target “general” CSCs markers is another way to eliminate CSCs with CAR‐T therapy. General antigen‐targeted CAR‐T cells may not be initially designed to specifically kill CSCs. Due to the expression of these markers was also detected on the surface of CSCs, when such CAR‐T cells were co‐cultured with corresponding CSCs, they also have a cancer‐killing effect (Table 2). HER2 is a tumour‐associated antigen that is expressed by up to 80% of glioblastomas (GBMs) but not by normal postnatal neurons or glia. 53 Ahmed et al. generated HER2‐specific T cells from 10 consecutive glioblastoma patients with a retroviral vector encoding a HER2‐specific CAR to produce effector cells. They showed that these effector cells recognized autologous HER2‐positive GBMs including their CD133‐positive stem cells in vitro and had potent antitumor activity in an orthotopic, xenograft model. 48 EGFRvIII is one of the few tumour‐specific antigens and thus an attractive candidate for the development of immunotherapy for glioblastoma patients. Richard A. Morgan et al. sought to develop immune‐based approaches targeting GCS as a potential treatment for glioblastoma, and report for the first time that EGFRvIII is expressed in glioblastoma cancer stem cells (GSC) lines and EGFRvIII CAR‐engineered T cells effectively target these lines. 49 Moreover, CSPG4, a highly immunogenic cell surface proteoglycan identified on melanoma cells, has been shown to facilitate the progression from radial to vertical growth in melanoma tumours. 54 Rachel E Beard et al. report for the first time that CSPG4 is also expressed on GSC and demonstrate that anti‐CSPG4 CAR‐transduced T cells recognize and kill these GSC. 55 Similarly, the expression of NKG2DLs is usually expressed in most epithelial‐derived tumour cells, such as ovarian cancer, colon cancer and leukaemia, while it is rarely detected in healthy adult tissues. 56 Yang et al. confirmed the high expression of NKG2DLs in GSCs and verified anti‐NKG2DLs CAR‐T cells efficiently lysed glioblastoma cells and cancer stem cells in vitro, and obviously eliminated xenograft tumours and did not exhibit significant toxicity in the mice model. 47
The above experimental results indicated that adoptive cellular immunotherapy with CSCs‐targeted CAR‐T cells is expected to become a promising cancer treatment method. For other common markers of CSCs, such as CD90 and ALDH that could be theoretically ideal targets for CAR‐T therapy, unfortunately, there is no reported studies using CD90 or ALDH‐specific CAR‐T cell for cancer treatment.
5. CLINICAL APPLICATION OF CSCS‐TARGETED CAR‐T THERAPY
To investigate the clinical application of CSCs‐targeted CAR‐T therapy, we have searched the ClinicalTrials.gov website and summarized the latest registered clinical trials of CAR‐T therapy using surface markers of CSCs (Table 3). These trials are within Phase I or Phase II, most of which are carried out in China and performed in recruiting stage. Twenty‐one trials are specific to haematological tumours treatment, in which acute myelocytic leukaemia (AML) treatment accounts for the majority; seven trials are for treating solid tumours, such as nasopharyngeal carcinoma (NCT02915445), breast cancer (NCT02915445), stomach neoplasms (NCT02725125), liver neoplasms (NCT02729493) and small cell lung cancer (NCT03392064).
TABLE 3.
Registered clinical trials of CSCs‐targeted CAR‐T therapy
| Target | Tumour target | Sponsor | NCT number | Phase | Status | Start date |
|---|---|---|---|---|---|---|
| EpCAM | EpCAM‐positive cancer | First Affiliated Hospital of Chengdu Medical College | NCT03013712 | Phase I/II | Recruiting/unknown | 2017 |
| Nasopharyngeal carcinoma or breast cancer | Sichuan University | NCT0avb2915445 | Phase I | Recruiting | 2016 | |
| Advanced gastric cancer with peritoneal metastasis | West China Hospital, Sichuan University | NCT03563326 | Phase I | Recruiting | 2018 | |
| Advanced solid tumour neoplasms | Tang‐Du Hospital | NCT04151186 | NA | Not yet recruiting | 2019 | |
| Stomach neoplasms | Sinobioway Cell Therapy Co., Ltd. | NCT02725125 | Phase II | Unknown | 2015 | |
| Liver neoplasms | Sinobioway Cell Therapy Co., Ltd. | NCT02729493 | Phase II | Unknown | 2015 | |
| CD133 | Relapsed and/or chemotherapy refractory advanced malignancies | Chinese PLA General Hospital | NCT02541370 | Phase I/II | Completed | 2015 |
| Relapsed/refractory acute myeloid leukaemia | Zhujiang Hospital | NCT03473457 | NA | Recruiting | 2018 | |
| CD123 | Acute myeloid leukaemia (AML) | Hebei Senlang Biotechnology Inc., Ltd. | NCT03796390 | Phase I | Recruiting | 2018 |
| AML | University of Pennsylvania | NCT03766126 | Phase I | Active, not recruiting | 2018 | |
| CD123+ AML | Beijing Immunochina Medical Science & Technology Co., Ltd. | NCT03585517 | Phase I | Recruiting/unknown | 2018 | |
| Relapsed/refractory AML | Cellectis S.A. | NCT03190278 | Phase I | Recruiting | 2017 | |
| Relapsed/refractory AML | The Affiliated Hospital of the Chinese Academy of Military Medical Sciences | NCT03556982 | Phase I/II | Recruiting/unknown | 2018 | |
| Relapsed/refractory AML | Chongqing Precision Biotech Co., Ltd | NCT04265963/NCT04272125 | Phase I/II | Recruiting | 2020 | |
| Relapsed/refractory AML | Zhujiang Hospital | NCT03473457 | NA | Recruiting | 2018 | |
| Relapsed/refractory AML | Shenzhen Geno‐Immune Medical Institute | NCT03222674 | Phase I/II | Unknown | 2017 | |
| CD123+ acute myeloid leukaemia | Wuhan Union Hospital, China | NCT04014881 | Phase I | Recruiting | 2019 | |
| Acute myeloid leukaemia or blastic plasmacytoid dendritic cell neoplasm | City of Hope Medical Center | NCT02159495 | Phase I | Recruiting | 2015 | |
| Myeloid malignancies | Southwest Hospital, China | NCT02937103 | Phase I/II | Recruiting | 2016 | |
| Adult acute myeloid leukaemia | Affiliated Hospital to Academy of Military Medical Sciences | NCT03114670 | Phase I | Recruiting | 2017 | |
| B‐cell malignancies | Shenzhen Geno‐Immune Medical Institute | NCT03125577 | Phase I/II | Recruiting | 2019 | |
| B‐cell leukaemia | Shenzhen Geno‐Immune Medical Institute | NCT04016129 | Phase I/II | Recruiting | 2019 | |
| CD19‐Negative B‐cell malignancies | Shenzhen Geno‐Immune Medical Institute | NCT04430530 | Phase I/II | Recruiting | 2020 | |
| AML | St. Jude Children's Research Hospital | NCT04318678 | Phase I | Recruiting | 2020 | |
| Refractory/relapsed acute leukaemia | Second Affiliated Hospital of Xi'an Jiaotong University | NCT03672851 | Phase I | Terminated (adverse effect) | 2019 | |
| paediatric subjects with relapsed/refractory AML | University of Pennsylvania | NCT04678336 | Phase I | Recruiting | 2020 | |
| Relapsed/refractory AML | Fujian Medical University | NCT03631576 | Phase II/III | Recruiting | 2018 | |
| DLL3 | Small cell lung cancer (SCLC) | Amgen | NCT03392064 | Phase I | Suspended | 2018 |
| CD44v6 | AML or multiple myeloma (MM) | AGC Biologics S.p.A. | NCT04097301 | Phase I/II | recruiting | 2019 |
| Cancers which are CD44v6 positive | Shenzhen Geno‐Immune Medical Institute | NCT04427449 | Phase I/II | Recruiting | 2020 | |
| Breast cancer | Shenzhen Geno‐Immune Medical Institute | NCT04430595 | Phase I/II | Recruiting | 2020 |
Among the clinical trials, a CD133‐targeted Phase I/II clinical study at Chinese PLA General Hospital for treating the relapsed and/or chemotherapy refractory advanced malignancies is the world's first successful clinical trial of CSCs‐targeted CAR‐T therapy for cancer treatment (NCT02541370). 57 CD133 is a marker of CSCs and endothelial progenitor cells (EPCs) which had been scientifically proven to be involving in tumour metastasis and recurrence. 58 In this clinical trial, 23 patients were enrolled, 14 of which are with hepatocellular carcinoma [HCC], seven of which pancreatic carcinomas and two of which colorectal carcinomas. Finally, three subjects achieved partial remission (PR) and 14 subjects' condition became stable with no serious adverse events. However, a side effect, namely, “on‐target, off‐tumour” effects, manifested as haematopoietic system toxicities were observed in most subjects in the study. This might be caused by the expression of CD133 on the surface of CD34+ progenitor cells in adult bone marrow and peripheral blood. 59 Moreover, cells with expressed non‐targeted antigen might give rise to tumour recurrences though CD133‐targeted CAR‐T cells eliminate target‐express cells. The process indicates antigen escape. Therefore, avoiding rapid growth of antigen‐negative cells after the antigen‐positive cell clearance is of great importance for improving clinical application of CSCs‐targeted CAR‐T therapy.
Side effects in patients treated with CSCs surface antigen‐targeted CAR‐T therapy were also shown in several literature. In a pilot trial for studying the safety of anti‐CD123 CAR‐T cell product, the fourth‐generation, apoptosis‐inducible lentiviral CAR‐T cells targeting CD123 (4SCAR123 T cells) were used to treat a patient with AML‐M2, 60 who was administered with cyclophosphamide (CTX) as conditioning regimen for 3 consecutive days and was subsequently infused with 1.8 × 106/kg anti‐CAR123 T cell. One day after the infusion, the patient had rigorous chills and fevers, low blood pressure and hypoxaemia. The subject also suffered from severe cytokine release syndrome (CRS) 4 days after the infusion, because of the controlling effects from administration of one dose tocilizumab. Fortunately, in this first human experiment of CD123‐specific CAR‐T cells for AML treatment, obvious off‐target cytotoxicity from the 4SCAR123 T cells were not found except for a decrease of blasts 20 days after CAR‐T therapy. 60 Apart from that, one side effect caused by CSCs‐targeted CAR‐T therapy was shown in a clinical trial registered in ClinicalTrials.gov (NCT03672851). The trial was a Phase I study designed to determine the safety and efficacy of anti‐CD123 CAR‐T cells in treating patients diagnosed with refractory/relapsed acute leukaemia and conducted in a dose‐escalation administration pattern. Unfortunately, it was terminated due to side effects while the experiment results was not released. Based on the experimental design, only two subjects were enrolled. The situation might be caused by individual differences of the included cases or the unreasonable design of the CAR structure. With limited subjects, the study was ill‐grounded in proving that CD123 was unsuitable for CAR‐T cell targeting.
6. CHALLENGES AND FUTURE DIRECTIONS
Although some successful animal experiments conducted with CSCs‐targeted CAR‐T cells have been reported and some ongoing CSCs‐targeted CAR‐T therapy clinical trials have shown good tumour treatment prospects, many challenges in clinical application exist. (Table 4).
TABLE 4.
Challenges and overcoming strategies of CSCs‐targeted CAR‐T therapy
| Challenges | Overcoming strategies |
|---|---|
| Toxicity |
|
| Persistence and potency |
|
| Trafficking | |
| Infiltration |
|
| Immunosuppressive tumour microenvironment |
|
| Heterogeneity |
|
6.1. Toxicity
One of the limitations in CAR‐T therapy is on‐target off‐tumour toxicity, caused by the direct attack on normal cells which have the shared expression of the targeted antigen. 61 On‐target/off‐tumour toxicity becomes a major hindrance of CSCs‐targeted CAR‐T therapy, because in normal cells, some CSCs markers are found, such as CD133 expressed in normal neural stem cells or ALDH expressed in hematopoietic stem cells. 62 , 63 To reduce the toxicity, selecting a safer antigen of CAR‐T cells is needed or designing dual‐targeted CARs to enhance the tumour specificity of CAR‐T cells may work. 64 Toxicity can also be minimized by local (intratumoural) delivery of CSCs‐targeted CAR‐T cells. 65 Moreover, introducing suicidal genes as a “safety switch” in CAR‐T cells when adverse reactions are uncontrollable may limit on‐target, off‐tumour toxicities. 66 Similarly, modifying CAR‐T cells and enabling them to express an inhibitory chimeric antigen receptor, such as CTLA‐4 or PD‐1, can achieve antigen‐specific suppression of T‐cell cytotoxicity, cytokine release and proliferation. 21
Adverse events other than on‐target/off‐tumour toxicity include cytokine release syndrome (CRS) and immune effector cell‐associated neurotoxicity syndrome (ICANS), which were common toxicities of CAR‐T cells in treating tumours. 67 In the first several days after CAR‐T cell infusion, CRS is mostly found and patients may have fever, hypotension and tachycardia which might lead to haemodynamic instability, causing end‐organ injury; after the onset of CRS, neurotoxicity syndrome, which manifests as subtle cognitive decline, may occur. 12 CRS is associated with elevated IL‐6 levels in patients receiving CAR‐T therapy and anti‐IL‐6 receptor antagonist tocilizumab is, thus, used to treat CRS. For instance, FDA sanctified the use of the drug in the treatment of CRS when the first CAR‐T cell product was approved. 68 Corticosteroids, which suppress immune responses, are also commonly used in the management of the toxicity once the patient does not have a rapid response to IL‐6 receptor blockade. 69 Alternatively, therapeutic options for ICANS are corticosteroids, antiepileptics and care measures with intensive care unit (ICU) monitoring. 12
6.2. Persistence and potency
A large number of clinical trials of CAR‐T cell therapy show the poor persistence of infused T cells, especially in solid tumours. 70 Therefore, improving the persistence and efficacy of CAR‐T cells has become one of the focuses in current research on CSCs‐targeted CAR‐T therapy. CAR structure and T cells exhaustion determine the cells persistence, which can be enhanced through improving the costimulatory domain of CAR‐T cells. 71 CAR‐T cell proliferation, persistence and potency can be elevated when one or more costimulatory signal domains were incorporated, such as 4‐1BB, ICOS, OX40 or CD27, which has been made clear in preclinical studies. 72 , 73 , 74 Moreover, several studies have proven that CAR‐T cell persistence was better maintained through incorporation of immune checkpoint blockade into CAR‐T cells, and such immune checkpoint inhibitors include anti‐programmed cell death protein 1 (PD‐1), anti‐cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4), anti‐mucin domain‐containing protein 3 (TIM3), anti‐Lymphocyte activation gene 3 (LAG3) and anti‐adenosine 2A receptor (A2AR). 74 In addition, optimizing T‐cell activation, expansion and persistence require not only antigen participation (signal1) or costimulatory signals (signal2), but also cytokine support (signal3). 75 Providing cytokine signals promotes the activation and proliferation of CAR‐T cells. However, some clinical trials found adverse reactions when patients were directly administered with exogenous cytokines. 76 , 77 In order to minimize systemic toxicity and induce the accumulation of high cytokine concentrations at the tumour site, CAR‐T cells were modified to produce IL‐12, IL‐18, IL‐7, IL‐15 and IL‐21 cytokines, and activation and persistence of CAR‐T cells were, therefore, enhanced in vivo. 78 Similar to the forced expression of cytokine genes in CAR‐T cells, constructing CAR‐T cells with the capability of inducing cytokine signalling upon antigen stimulation can also provide cytokine support. Based upon that, a novel CAR containing a JAK‐STAT signalling domain has been developed by Kagoya et al. 79 This invention incorporated cytoplasmic domain of IL‐2 receptor β between the cytoplasmic domains of CD28 and CD3z for JAK‐STAT pathway activation and a YXXQ motif at the C‐terminus of CD3z for STAT3 recruitment. The novel CAR demonstrated superior in vivo persistence and antitumour effects in models of liquid and solid tumours. All the approaches mentioned above to improve the persistence and potency of CAR‐T cells are good references in CSCs‐targeted therapy.
6.3. Trafficking
Apart from effectively treating haematological tumours, the CSCs‐targeted CAR‐T therapy is applicable to kill cancer stem cells in solid tumours in radical treatment. A major challenge in solid tumour treatment is less trafficking of CAR‐T cells into these sites, since CSCs in solid tumours are less possible to access immune cells and are usually surrounded by compact stroma and tumour cells. 80 We proposed the following methods currently used to improve the trafficking or homing ability of CAR‐T cells so as to overcome the challenge. The most straightforward method is to directly infuse the CSCs‐targeted CAR‐T cells into tumour sites. In a previous report, the local infusion of CAR‐T cells resulted in significant regression of glioblastoma. Nevertheless, localized therapy is not suitable for many metastatic solid tumours. 81 Modulation of chemokine signalling would be another choice to elevate the trafficking ability of CSCs, as numerous chemokines mediate immune cell trafficking. Several preclinical models demonstrated that the forced expression of a chemokine receptor of CCR4, CCR2b or CXCR2 improved the homing ability of CAR‐T cells. 82 , 83 , 84 Since CSCs are encompassed by a bulk of tumour cells, we proposed that combining CSCs‐specific CAR‐T and conventional anticancer therapies may be effective. Specifically, it is recommended that traditional therapies, including radiotherapy or chemotherapy are used to remove most of the tumour cells for better exposure of CSCs, and CSCs‐targeted CAR‐T cells are subsequently infused for full eradication of the tumour. 85
6.4. Infiltration
After CAR‐T cells reach the tumour site, an issue needs to be considered how the cells should approach CSCs expressing target antigens to form immune synapses and destroy tumour stem cells. Due to the physical and biochemical barriers established by the extracellular matrix (ECM) around CSCs, the infiltration of CAR‐T cells has become one challenge. 80 , 86 Fibroblast activation protein (FAP) is a surface marker of cancer‐associated stromal cells (CASCs), and has a role in remodelling ECM. FAP‐targeted CAR‐T cells have the ability to break through the physical barrier established by ECM, which is achieved by targeting PAF+CASCs to inhibit matrix production and angiogenesis. 87 Directing CAR‐T cell to target vascular endothelial growth factor receptor 2(VEGFR2) is another way to improve penetration and antitumor response, which is achieved by destroying tumour vascular endothelial cells. 88 In addition, Caruana et al. reported that engineering CAR‐T cells to express ECM‐degrading enzyme heparanase improved the infiltration of T cells in tumours. 89
6.5. Immunosuppressive tumour microenvironment
Cancer stem cells survive in an immunosuppressive tumour microenvironment (TME) composed of vascular niches, cancer‐associated fibroblasts, cancer‐associated mesenchymal stem cells, hypoxia, tumour‐associated macrophages and extracellular matrix, which hinders the direct killing of tumour stem cells by one's own immune cells and the adoptive CAR‐T cells. 90 Therefore, combining CSCs‐targeted CAR‐T therapy and the strategy of targeting the immunosuppressive TME of CSCs may help improve the efficiency of CSCs removal. As we mentioned in “persistence and potency” section in this review, cytokine support served as one of the important signals for optimal T‐cell activation and proliferation. However, this signal was lacking in the TME of CSCs. 91 Constructing CSCs‐targeted CAR‐T cells which overexpress IL‐12, IL‐18, IL‐7, IL‐15 and IL‐21 cytokines may be an effective way to provide support for the activation, proliferation and killing of CSCs of CAR‐T cells in immunosuppressive TME. 78
Several studies have proved that CSCs have the ability to evade the immune system, because these cells secrete several substances into the TME, such as TGFβ, IL‐10, IL‐4 and IL‐13, which exert inhibitory effects on an array of immune cells. 3 Neutralization of immunosuppressive mediators within the TME is another way to enhance the potency of CSCs‐targeted cells. For example, Takahashi has proved that the neutralization of TGF‐β, IL‐10 and arginase I with anti‐TGF‐β mAb, anti‐IL‐10 mAb and the arginase I inhibitor Noha, or L‐arginine significantly restored T‐cell proliferation. 92
Several other studies showed that the immunosuppressive TME can be triggered by CSCs through the mechanism of up‐regulated expression of PD‐L1 on the CSCs' surfaces. 93 , 94 PD‐L1 suppresses CAR‐T cells' functions and induces their exhaustion upon binding to PD‐1 on activated T cells. 95 Therefore, combining CSCs‐targeted therapy with FDA‐approved PD‐1/PD‐L1 checkpoint inhibitors, including three for PD‐1 (pembrolizumab, nivolumab and cemiplimab) and three for PD‐L1 (atezolizumab, avelumab and durvalumab), may be a choice to mitigate the immunosuppressive microenvironment. 96 Fang Zheng et al. have observed significant antitumor effects and dramatic ALDHhigh CSCs elimination, following the triple therapy of the dual blockade of PD‐L1 and CTLA‐4 and CSC‐DC vaccine which were accompanied by significantly enhanced T‐cell expansion, suppressed TGF‐β secretion, enhanced IFN‐γ secretion and significantly enhanced host‐specific CD8+ T cell response against CSCs. 97 Based upon all the results from the literature, administration of a‐PD‐L1 and a‐CTLA‐4 blockade combined with CSCs‐targeted CAR‐T cells may be an effective immunotherapeutic strategy for cancer patients.
6.6. Heterogeneity
One great challenge in cancer therapy is intratumour heterogeneity, while CSCs are one of the determining factors causing the problem. 98 Therefore, eradication of CSCs by CAR‐T therapy is promising for overcoming the heterogeneity. However, accumulating evidence suggests that CSCs represent phenotypically and functionally heterogeneous populations, which has been found in colorectal, 99 colon, 100 hepatocellular 101 and breast cancer stem cells, 102 leading to antigen loss or escape when applying CSCs‐targeted CAR‐T therapy.
Given that CD19/CD22 bispecific CAR‐T cells have demonstrated clinical efficacy in patients with B‐cell malignancies, 103 bispecific CAR‐T cells can be bio‐engineered by designing a single CAR molecule with two (or more) distinct binding domains of CSCs‐specific markers, so as to overcome antigen escape caused by CSCs heterogeneity. Furthermore, multi‐target CAR‐T cell therapies can be created to overcome the limitation of antigen loss by mixing different CAR‐T cell products targeting single antigens prior to infusion or by transducing T cells with multiple CAR constructs. 13
Transgenic modification of the CAR structure to elicit an endogenous immune response through recruiting additional effector cells is an alternative approach to avoid the heterogeneity. To recruit bystander T cells against a second tumour‐associated surface antigen, CAR‐T cell targeting can be combined with the release of bispecific T‐cell engagers (BiTEs). 104 Iwahori et al. first reported the generation of T cells which can secrete a bispecific T‐cell engager specific both for CD3 and the tumour‐associated antigen, erythropoietin‐producing hepatocellular carcinoma A2 (EphA2), for bystander T‐cell‐mediated in vitro cytolysis. 105 More recently, EGFRvIII‐targeted CAR‐T cells were constructed to secrete engagers against wild‐type EGFR for local recruitment of bystander T cells against EGFRvIII‐negative tumour cell subpopulations in glioblastoma, so as to overcome the limitation of antigen escape. 106
7. CONCLUSION
Due to CSC's characteristics of self‐renewal, multi‐lineage differentiation, tumour‐formation ability and chemo‐radio‐resistance, the CSCs existence is the key factor causing cancer treatment failure. The advantages of CAR‐T therapies are recognizing specific surface antigen, activating T cells in an MHC‐unrestricted manner, long‐term in vivo persistence and proliferation, and efficient homing of CAR‐T cells to the tumour site, which are believed to be a potential method to eliminate CSCs. Apart from the detailed analysis of the feasibility and advantages of targeting CSCs with the bio‐modified CAR‐T cells, the paper also reviewed the few existing reported laboratory results and summarized the registered clinical trials about this method. Finally, we discussed the current challenges of this therapy and the solutions that can be adopted for the future development of CSCs‐targeted therapy. As the first therapy mentioned in the review has the potential to cure cancers and is currently on the market, harnessing CAR‐T cells to target CSCs is believed to achieve greater success in treating tumours.
CONFLICT OF INTEREST
Conflict of interest relevant to this article was not reported.
AUTHOR CONTRIBUTIONS
Xiaoyue Cui: Conceptualization (lead); Data curation (lead); Formal analysis (lead); Investigation (lead); Resources (lead); Software (lead); Writing‐original draft (lead); Writing‐review & editing (lead). Rui Liu: Data curation (equal); Supervision (supporting); Writing‐review & editing (equal). Lian Duan: Investigation (equal); Writing‐review & editing (supporting). Dan Cao: Data curation (supporting); Investigation (supporting). Qiaoling Zhang: Data curation (supporting); Writing‐review & editing (supporting). Aijie Zhang: Resources (equal); Writing‐review & editing (equal).
ACKNOWLEDGEMENTS
We thank members in Zhao laboratory for providing the platform to conduct research related to CAR‐T therapy, and we are grateful to Dr. Yang Dong and Dr. Sun Bin for their previous technical support.
Cui X, Liu R, Duan L, Cao D, Zhang Q, Zhang A. CAR‐T therapy: Prospects in targeting cancer stem cells. J Cell Mol Med. 2021;25:9891–9904. doi: 10.1111/jcmm.16939
Funding information
This work was supported by the fund of Sichuan Science and Technology Department (2018JY0665).
REFERENCES
- 1. Cohnheim J. Ueber entzündung und eiterung. Arch Pathol Anat Physiol Klin Med. 1867;40(1):1‐79. [Google Scholar]
- 2. D'Andrea V, Guarino S, Matteo FMD, Maugeri Sacca M, Maria RD. Cancer stem cells in surgery. G Chir. 2014;35(11‐12):257‐259. [PMC free article] [PubMed] [Google Scholar]
- 3. Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells — a clinical update. Nat Rev Clin Oncol. 2020;17(4):204‐232. [DOI] [PubMed] [Google Scholar]
- 4. Quaglino E, Conti L, Cavallo F. Breast cancer stem cell antigens as targets for immunotherapy. Semin Immunol. 2020;47:101386. [DOI] [PubMed] [Google Scholar]
- 5. Lee DW, Kochenderfer JN, Stetler‐Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet. 2015;385(9967):517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stern LA, Jonsson VD, Priceman SJ. CAR T cell therapy progress and challenges for solid tumors, in Tumor Microenvironment. Springer; 2020:297‐326. [DOI] [PubMed] [Google Scholar]
- 7. Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T‐cell immunotherapy for human cancer. Am J Hematol. 2019;94(S1):S3‐s9. [DOI] [PubMed] [Google Scholar]
- 8. He Q, Jiang X, Zhou X, Weng J. Targeting cancers through TCR‐peptide/MHC interactions. J Hematol Oncol. 2019;12(1):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene. 2007;26(9):1357‐1360. [DOI] [PubMed] [Google Scholar]
- 10. Skvortsov S, Debbage P, Lukas P, Skvortsova I. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin Cancer Biol. 2015;31:36‐42. [DOI] [PubMed] [Google Scholar]
- 11. Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu‐Economescu M, et al. Therapies targeting cancer stem cells: Current trends and future challenges. World J Stem Cells. 2015;7(9):1185‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020;21(3):e168‐e178. [DOI] [PubMed] [Google Scholar]
- 13. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin‐T‐cell receptor chimeric molecules as functional receptors with antibody‐type specificity. Proc Natl Acad Sci USA. 1989;86(24):10024‐10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mohanty R, Chowdhury C, Arega S, Sen P, Ganguly P, Ganguly N. CAR T cell therapy: a new era for cancer treatment (Review). Oncol Rep. 2019;42(6):2183‐2195. [DOI] [PubMed] [Google Scholar]
- 16. Brocker T, Karjalainen K. Signals through T cell receptor‐zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181(5):1653‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abate‐Daga D, Davila ML. CAR models: next‐generation CAR modifications for enhanced T‐cell function. Mol Ther Oncolytics. 2016;3:16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ramos CA, Rouce R, Robertson CS, et al. In vivo fate and activity of second‐ versus third‐generation CD19‐specific CAR‐T cells in B cell Non‐Hodgkin's lymphomas. Mol Ther. 2018;26(12):2727‐2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hong M, Clubb JD, Chen YY. Engineering CAR‐T cells for next‐generation cancer therapy. Cancer Cell. 2020;38(4):473‐488. [DOI] [PubMed] [Google Scholar]
- 20. Duong MT, Collinson‐Pautz MR, Morschl E, et al. Two‐dimensional regulation of CAR‐T cell therapy with orthogonal switches. Mol Ther Oncolytics. 2019;12:124‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fedorov VD, Themeli M, Sadelain M. PD‐1‐ and CTLA‐4‐based inhibitory chimeric antigen receptors (iCARs) divert off‐target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van der Schans JJ, van de Donk N, Mutis T. Dual targeting to overcome current challenges in multiple myeloma CAR T‐cell treatment. Front Oncol. 2020;10:1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seif M, Einsele H, Löffler J. CAR T cells beyond cancer: hope for immunomodulatory therapy of infectious diseases. Front Immunol. 2019;10:2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang M, Munoz J, Goy A, et al. KTE‐X19 CAR T‐cell therapy in relapsed or refractory mantle‐cell lymphoma. N Engl J Med. 2020;382(14):1331‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off‐the‐shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185‐199. [DOI] [PubMed] [Google Scholar]
- 26. Magnani CF, Gaipa G, Lussana F, et al. Sleeping Beauty‐engineered CAR T cells achieve antileukemic activity without severe toxicities. J Clin Invest. 2020;130(11):6021‐6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rossi F, Noren H, Jove R, Beljanski V, Grinnemo KH. Differences and similarities between cancer and somatic stem cells: therapeutic implications. Stem Cell Res Ther. 2020;11(1):489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weber EW, Maus MV, Mackall CL. The emerging landscape of immune cell therapies. Cell. 2020;181(1):46‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stratford EW, Bostad M, Castro R, et al. Photochemical internalization of CD133‐targeting immunotoxins efficiently depletes sarcoma cells with stem‐like properties and reduces tumorigenicity. Biochim Biophys Acta. 2013;1830(8):4235‐4243. [DOI] [PubMed] [Google Scholar]
- 30. Harrison H, Farnie G, Howell SJ, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70(2):709‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Katoh M. Canonical and non‐canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. 2017;51(5):1357‐1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zinzi L, Contino M, Cantore M, Capparelli E, Leopoldo M, Colabufo NA. ABC transporters in CSCs membranes as a novel target for treating tumor relapse. Front Pharmacol. 2014;5:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dzobo K, Senthebane DA, Ganz C, Thomford NE, Wonkam A, Dandara C. Advances in therapeutic targeting of cancer stem cells within the tumor microenvironment: an updated review. Cells. 2020;9(8):1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li Y, Wang Z, Ajani JA, Song S. Drug resistance and Cancer stem cells. Cell Commun Signal. 2021;19(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun JC, Pan K, Chen MS, et al. Dendritic cells‐mediated CTLs targeting hepatocellular carcinoma stem cells. Cancer Biol Ther. 2010;10(4):368‐375. [DOI] [PubMed] [Google Scholar]
- 37. Wilson BJ, Saab KR, Ma J, et al. ABCB5 maintains melanoma‐initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res. 2014;74(15):4196‐4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang M, Li W, Fan D, et al. Expression of ABCB5 gene in hematological malignances and its significance. Leuk Lymphoma. 2012;53(6):1211‐1215. [DOI] [PubMed] [Google Scholar]
- 39. Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature. 2008;451(7176):345‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Borchers S, Mablo C, Muller CA, et al. Detection of ABCB5 tumour antigen‐specific CD8(+) T cells in melanoma patients and implications for immunotherapy. Clin Exp Immunol. 2018;191(1):74‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Frank NY, Pendse SS, Lapchak PH, et al. Regulation of progenitor cell fusion by ABCB5 P‐glycoprotein, a novel human ATP‐binding cassette transporter. J Biol Chem. 2003;278(47):47156‐47165. [DOI] [PubMed] [Google Scholar]
- 42. Doherty PC, Zinkernagel RM. Enhanced immunological surveillance in mice heterozygous at the H‐2 gene complex. Nature. 1975;256(5512):50‐52. [DOI] [PubMed] [Google Scholar]
- 43. Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823‐836. [DOI] [PubMed] [Google Scholar]
- 44. Volonté A, Di Tomaso T, Spinelli M, et al. Cancer‐initiating cells from colorectal cancer patients escape from T cell‐mediated immunosurveillance in vitro through membrane‐bound IL‐4. J Immunol. 2014;192(1):523‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Srivastava S, Riddell SR. Engineering CAR‐T cells: design concepts. Trends Immunol. 2015;36(8):494‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gzil A, Zarębska I, Bursiewicz W, Antosik P, Grzanka D, Szylberg Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol Biol Rep. 2019;46(6):6629‐6645. [DOI] [PubMed] [Google Scholar]
- 47. Yang D, Sun B, Dai H, et al. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J Immunother Cancer. 2019;7(1):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ahmed N, Salsman VS, Kew Y, et al. HER2‐specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morgan RA, Johnson LA, Davis JL, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23(10):1043‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient‐derived glioblastoma stem cells are killed by CD133‐specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6(1):171‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Deng Z, Wu Y, Ma W, Zhang S, Zhang Y‐Q. Adoptive T‐cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015;16(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ang WX, Li Z, Chi Z, et al. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti‐EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget. 2017;8(8):13545‐13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER‐2, gp100, and MAGE‐1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64(14):4980‐4986. [DOI] [PubMed] [Google Scholar]
- 54. Price MA, Colvin Wanshura LE, Yang J, et al. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 2011;24(6):1148‐1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beard RE, Zheng Z, Lagisetty KH, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer. 2014;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu H, Wang S, Xin J, Wang J, Yao C, Zhang Z. Role of NKG2D and its ligands in cancer immunotherapy. Am J Cancer Res. 2019;9(10):2064‐2078. [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Y, Chen M, Wu Z, et al. CD133‐directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. 2018;7(7):e1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liou GY. CD133 as a regulator of cancer metastasis through the cancer stem cells. Int J Biochem Cell Biol. 2019;106:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Aghajani M, Mansoori B, Mohammadi A, Asadzadeh Z, Baradaran B. New emerging roles of CD133 in cancer stem cell: Signaling pathway and miRNA regulation. J Cell Physiol. 2019;234(12):21642‐21661. [DOI] [PubMed] [Google Scholar]
- 60. Luo Y, Chang LJ, Hu Y, Dong L, Wei G, Huang H. First‐in‐man CD123‐specific chimeric antigen receptor‐modified t cells for the treatment of refractory acute myeloid leukemia. Blood. 2015;126(23):3778. [Google Scholar]
- 61. Sun S, Hao H, Yang G, Zhang Y, Fu Y. Immunotherapy with CAR‐modified T cells: toxicities and overcoming strategies. J Immunol Res. 2018;2018:2386187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Peh GSL, Lang RJ, Pera MF, Hawes SM. CD133 expression by neural progenitors derived from human embryonic stem cells and its use for their prospective isolation. Stem Cells Dev. 2008;18(2):269‐282. [DOI] [PubMed] [Google Scholar]
- 63. Schuurhuis GJ, Meel MH, Wouters F, et al. Normal hematopoietic stem cells within the AML bone marrow have a distinct and higher ALDH activity level than co‐existing leukemic stem cells. PLoS ONE. 2013;8(11):e78897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang E, Yang P, Gu J, et al. Recombination of a dual‐CAR‐modified T lymphocyte to accurately eliminate pancreatic malignancy. J Hematol Oncol. 2018;11(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Theruvath J, Sotillo E, Mount CW, et al. Locoregionally administered B7‐H3‐targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med. 2020;26(5):712‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gargett T, Brown MP. The inducible caspase‐9 suicide gene system as a "safety switch" to limit on‐target, off‐tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chou CK, Turtle CJ. Assessment and management of cytokine release syndrome and neurotoxicity following CD19 CAR‐T cell therapy. Expert Opin Biol Ther. 2020;20(6):653‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Si S, Teachey DT. Spotlight on tocilizumab in the treatment of CAR‐T‐Cell‐Induced cytokine release syndrome: clinical evidence to date. Ther Clin Risk Manag. 2020;16:705‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T‐cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433‐444. [DOI] [PubMed] [Google Scholar]
- 70. McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol. 2019;97(7):664‐674. [DOI] [PubMed] [Google Scholar]
- 71. Jayaraman J, Mellody MP, Hou AJ, et al. CAR‐T design: elements and their synergistic function. EBioMedicine. 2020;58:102931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Guedan S, Posey AD, Shaw C, et al. Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI Insight. 2018;3(1):e96976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119(3):696‐706. [DOI] [PubMed] [Google Scholar]
- 74. Zhang H, Li F, Cao J, et al. A chimeric antigen receptor with antigen‐independent OX40 signaling mediates potent antitumor activity. Sci Transl Med. 2021;13(578):eaba7308. [DOI] [PubMed] [Google Scholar]
- 75. Kershaw MH, Westwood JA, Darcy PK. Gene‐engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13(8):525‐541. [DOI] [PubMed] [Google Scholar]
- 76. Pachella LA, Madsen LT, Dains JE. The toxicity and benefit of various dosing strategies for interleukin‐2 in metastatic melanoma and renal cell carcinoma. J Adv Pract Oncol. 2015;6(3):212. [PMC free article] [PubMed] [Google Scholar]
- 77. Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first‐in‐human clinical trial of recombinant human interleukin‐15 in patients with cancer. J Clin Oncol. 2015;33(1):74‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next‐generation CAR T cells against solid cancers. J Hematol Oncol. 2020;13(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kagoya Y, Tanaka S, Guo T, et al. A novel chimeric antigen receptor containing a JAK‐STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nallanthighal S, Heiserman JP, Cheon D‐J. The role of the extracellular matrix in cancer stemness. Front Cell Dev Biol. 2019;7:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T‐cell therapy. N Engl J Med. 2016;375(26):2561‐2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu G, Rui W, Zheng H, et al. CXCR2‐modified CAR‐T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. 2020;50(5):712‐724. [DOI] [PubMed] [Google Scholar]
- 83. Craddock JA, Lu A, Bear A, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33(8):780‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Di Stasi A, De Angelis B, Rooney CM, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392‐6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yu WD, Sun G, Li J, Xu J, Wang X. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett. 2019;452:66‐70. [DOI] [PubMed] [Google Scholar]
- 86. Begum A, Ewachiw T, Jung C, et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE. 2017;12(7):e0180181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang LC, Lo A, Scholler J, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2(2):154‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chinnasamy D, Yu Z, Theoret MR, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR‐2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest. 2010;120(11):3953‐3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR‐redirected T lymphocytes. Nat Med. 2015;21(5):524‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Badrinath N, Yoo SY. Recent advances in cancer stem cell‐targeted immunotherapy. Cancers (Basel). 2019;11(3):310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019;234:116781. [DOI] [PubMed] [Google Scholar]
- 92. Takahashi H, Sakakura K, Kudo T, et al. Cancer‐associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget. 2017;8(5):8633‐8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Polónia A, Pinto R, Cameselle‐Teijeiro JF, Schmitt FC, Paredes J. Prognostic value of stromal tumour infiltrating lymphocytes and programmed cell death‐ligand 1 expression in breast cancer. J Clin Pathol. 2017;70(10):860‐867. [DOI] [PubMed] [Google Scholar]
- 94. Inaguma S, Lasota J, Wang Z, et al. Expression of ALCAM (CD166) and PD‐L1 (CD274) independently predicts shorter survival in malignant pleural mesothelioma. Hum Pathol. 2018;71:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hu W, Zi Z, Jin Y, et al. CRISPR/Cas9‐mediated PD‐1 disruption enhances human mesothelin‐targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68(3):365‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ai L, Chen J, Yan H, et al. Research status and outlook of PD‐1/PD‐L1 inhibitors for cancer therapy. Drug Des Devel Ther. 2020;14:3625‐3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zheng F, Dang J, Zhang H, et al. Cancer stem cell vaccination with PD‐L1 and CTLA‐4 blockades enhances the eradication of melanoma stem cells in a mouse tumor model. J Immunother. 2018;41(8):361‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Prasetyanti PR, Medema JP. Intra‐tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hirata A, Hatano Y, Niwa M, Hara A, Tomita H. Heterogeneity in colorectal cancer stem cells. Cancer Prev Res (Phila). 2019;12(7):413‐420. [DOI] [PubMed] [Google Scholar]
- 100. Hirata A, Hatano Y, Niwa M, Hara A, Tomita H. Heterogeneity of colon cancer stem cells. Adv Exp Med Biol. 2019;1139:115‐126. [DOI] [PubMed] [Google Scholar]
- 101. Zheng H, Pomyen Y, Hernandez MO, et al. Single‐cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology. 2018;68(1):127‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sousa B, Ribeiro AS, Paredes J. Heterogeneity and plasticity of breast cancer stem cells. Adv Exp Med Biol. 2019;1139:83‐103. [DOI] [PubMed] [Google Scholar]
- 103. Dai H, Wu Z, Jia H, et al. Bispecific CAR‐T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol. 2020;13(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kailayangiri S, Altvater B, Wiebel M, Jamitzky S, Rossig C. Overcoming heterogeneity of antigen expression for effective CAR T cell targeting of cancers. Cancers. 2020;12(5):1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Iwahori K, Kakarla S, Velasquez MP, et al. Engager T cells: a new class of antigen‐specific T cells that redirect bystander T cells. Mol Ther. 2015;23(1):171‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Choi BD, Yu X, Castano AP, et al. CAR‐T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37(9):1049‐1058. [DOI] [PubMed] [Google Scholar]