

Abstract
Background
IgG4-related disease (IgG4-RD) is a fibro-inflammatory condition involving loss of B cell tolerance and production of autoantibodies. However, the relevant targets and role of these aberrant humoral immune responses are not defined.
Objective
To identify novel autoantibodies and autoantigen targets that promote pathogenic responses in IgG4-RD.
Methods
We sequenced plasmablast antibody repertoires in patients with IgG4-RD. Representative monoclonal antibodies (mAb) were expressed and their specificities characterized using cytokine microarrays. The role of anti-interleukin-1 receptor-antagonist (IL-1RA) autoantibodies was investigated using in vitro assays.
Results
We identified strong reactivity against human IL-1RA using a clonally-expanded plasmablast-derived mAb from a patient with IgG4-RD. IgG4-RD patient plasma exhibited elevated levels of reactivity against IL-1RA compared to controls and neutralized IL-1RA activity, resulting in inflammatory and fibrotic mediator production in vitro. IL-1RA was detected in lesional tissues from IgG4-RD patients. Patients with anti-IL-1RA autoantibodies of the IgG4 subclass had greater numbers of organs affected than those without anti-IL-1RA autoantibodies. Peptide analyses identified IL-1RA epitopes targeted by anti-IL-1RA antibodies at sites near the IL-1RA/IL-1R interface. Serum from patients with systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) also had elevated levels of anti-IL-1RA autoantibodies compared to controls.
Conclusion
A subset of patients with IgG4-RD have anti-IL-1RA autoantibodies, which promote pro-inflammatory and pro-fibrotic meditator production via IL-1RA neutralization. These findings support a novel immunological mechanism underlying the pathogenesis of IgG4-RD. Anti-IL-1RA autoantibodies are also present in a subset of patients with SLE and RA, suggesting a potential common pathway in multiple autoimmune diseases.
Keywords: IL-1 receptor-antagonist, cytokine, IgG4-related disease, lupus, rheumatoid arthritis, autoimmune disease, autoantibodies, plasmablast, sequencing
CAPSULE SUMMARY
Anti-IL-1RA autoantibodies in patients with IgG4-RD are associated with the number of affected organs, promote pro-inflammatory and pro-fibrotic meditator production in vitro, and may represent a novel immunological mechanism promoting inflammation and fibrosis in B cell-mediated autoimmune diseases.
INTRODUCTION
IgG4-related disease (IgG4-RD) is a fibroinflammatory condition characterized by multi-organ involvement, tissue infiltration by IgG4-expressing plasma cells, and storiform fibrosis.1, 2 Autoantibodies against various self-antigens including a ubiquitin ligase-associated protein,3 carbonic anhydrase IV,4 annexin A11,5 amylaseα−2A,6 and galectin-3 7 were previously described in the context of IgG4-RD. Furthermore, passive transfer of purified human IgG and IgG4 antibodies derived from patients with IgG4-RD induced organ manifestations resembling IgG4-RD in recipient mice.8 Nevertheless, the specific autoantibodies that promote inflammation and fibrosis contributing to the diverse organ manifestations in this disease, and their unique targets, are yet to be identified.1 Further, questions remain about the contribution of B cells and autoantibodies to the pathogenesis of IgG4-RD.
Given the broad heterogeneity of organ involvement observed in IgG4-RD, we hypothesized that a subset of autoreactive B cells and autoantibodies target self-antigens that are ubiquitously expressed across multiple inflamed tissues in IgG4-RD. Cytokines are broadly expressed, and anti-cytokine autoantibodies have been demonstrated in 38% of SLE, 42% of Sjögren’s syndrome, and 20% of RA patients.9 Here we describe the discovery of a novel target of anti-cytokine autoantibodies, interleukin-1 receptor-antagonist (IL-1RA), in IgG4-RD and the expression of IL-1RA in the lesional tissue of patients with this disease. We show that anti-IL-1RA autoantibodies can promote expression of inflammatory and fibrotic mediators by neutralizing IL-1RA binding to the IL-1 receptor. Moreover, we also find elevated levels of anti-IL-1RA autoantibodies in patients with SLE and RA. Together these findings suggest a central mechanism by which anti-cytokine autoantibodies targeting IL-1RA could promote inflammation and tissue damage in multiple autoimmune conditions.
METHODS
Human Subjects
Human samples were collected under protocols approved by the Institutional Review Boards at Stanford University, Massachusetts General Hospital (MGH), and the University Hospital Heidelberg (UHH), Germany. Patients provided written informed consent. IgG4-RD (n = 14), multiple sclerosis (MS), RA (n = 55), and SLE (n = 64) patients were recruited from Stanford Hospital. Additional samples from IgG4-RD patients (n = 147) and MS patients (n = 36) were collected at MGH and UHH, respectively.
IgG4-RD diagnosis was made as described by Umehara et al.10, and defined patients as having definite (positive for 3 diagnostic criteria) or probable (positive for criteria 1 and criteria 2 or 3) IgG4-RD. Control groups included age- and sex-matched healthy donors and subjects with other immune-mediated diseases (Sjögren’s syndrome or gout).
Antibody sequencing, expression, and analysis
Flow cytometry was used to isolate CD19+CD20−CD27+CD38hiIgA−IgM− (IgG+) plasmablasts. As described previously,11 immunoglobulin heavy- (HC) and light-chain (LC) genes were cell-barcoded, PCR amplified, and sequenced using MiSeq (Illumina). Bioinformatic analysis was used to assign cell barcodes, obtain error-corrected consensus sequences, and pair the HC and LC sequences expressed by individual plasmablasts. Clonal family antibodies were identified based on sharing HC and LC V- and J-genes and possessing >60% CDR3 region identity, as described.12 Recombinant mAbs were expressed 12 and characterized using antigen microarrays 13 containing 335 cytokines, chemokines, and growth factors.14
In vitro blocking assays
40pg/uL human IL-1RA (PeproTech, US) was pre-incubated with 5ng/uL antibody (G4–21 or isotype control. IL-1RA and antibody were applied to HEK-Blue IL-1β cells (InvivoGen, US) for 2 hours and stimulated with 2pg/uL human IL-1β (PeproTech, US) for 36 hours at 37°C. Alternatively, cells were treated with 8–10 nM IL-1RA and commercial monoclonal (mAbs) or polyclonal antibodies (pAbs) at 40–2000nM, or with patient plasma at 1:10 and 1:20 dilutions, and subsequently stimulated with 0.1–1 nM IL-1β for 24 hours. Cells were treated with antibodies in the presence of IL-1α and IL-1RA, IL-1RA alone, or media, and supernatants assayed using the QUANTI-Blue assay (InvivoGen, hkb-il1b, US).
Human A549 lung epithelial cells and MRC-5 lung fibroblasts (ATCC, US) were treated with 10nM IL-1RA and patient plasma and stimulated with 0.5nM human IL-1α (PeproTech, US) for 24 hours at 37°C. RNA was isolated, cDNA prepared, and samples analyzed using TaqMan FAM-conjugated primer sets. Supernatants were collected and assayed for human IL-6 (R&D, US), IL-8 (R&D, US), and G-CSF (R&D, US) via ELISA.
Immunofluorescence Staining
FFPE tissue sample sections were deparaffinized and incubated with diluted primary antibody followed by washing in PBS-Tween and incubation with a secondary antibody and detected using the SuperPicTure Polymer Detection Kit (Invitrogren, US). Images were acquired using the TissueFAXS platform and quantified with TissueQuest software (TissueGnostics, Austria).
Statistical Analysis
Comparisons of IL-1RA reactivities in diseased subjects and healthy controls were performed using the Kruskal-Wallis (>2 group comparisons) or Mann-Whitney U-tests (2 groups comparison). Unpaired Student ’s t-test was used for comparison when ≤ three replicates were performed. The Fisher’s exact test was used for groups with and without detectable levels of anti-IL-1RA autoantibodies in the validation cohort. P < 0.05 was considered statistically significance.
RESULTS
Identification of IL-1RA autoantigen in IgG4-RD
We performed barcode-enabled single cell sequencing of the heavy and light chain variable regions of IgG plasmablasts from four individuals with IgG4-RD. Using bioinformatics analysis, we identified and selected immunoglobulin sequences representative of 42 clonal family plasmablasts (shared heavy chain and light chain variable and joining region gene usage and greater than 60% sequence identity in their CDR3 regions), across these four patients for recombinant expression as monoclonal antibodies (mAbs). Analysis of these mAbs for binding on a protein microarray containing 335 unique human cytokines, chemokines, and growth factors revealed that one mAb, G4–21, robustly reacted with interleukin-1 receptor-antagonist cytokine (IL-1RA) (Fig. 1, A–C, S. Figs. 2 and 3). We also observed significantly higher levels of G4–21 reactivity against IL-1RA as compared to related cytokines, IL-1α and IL-1β when analyzed via ELISA (Fig. 1, D). We did not detect reactivity to IL-1RA in mAbs derived from unrelated plasmablast clonal families from the same patient (G4–05), an additional IgG4-RD patient (G4–25), or the isotype control mAb (Fig. 1, E).
FIG 1.
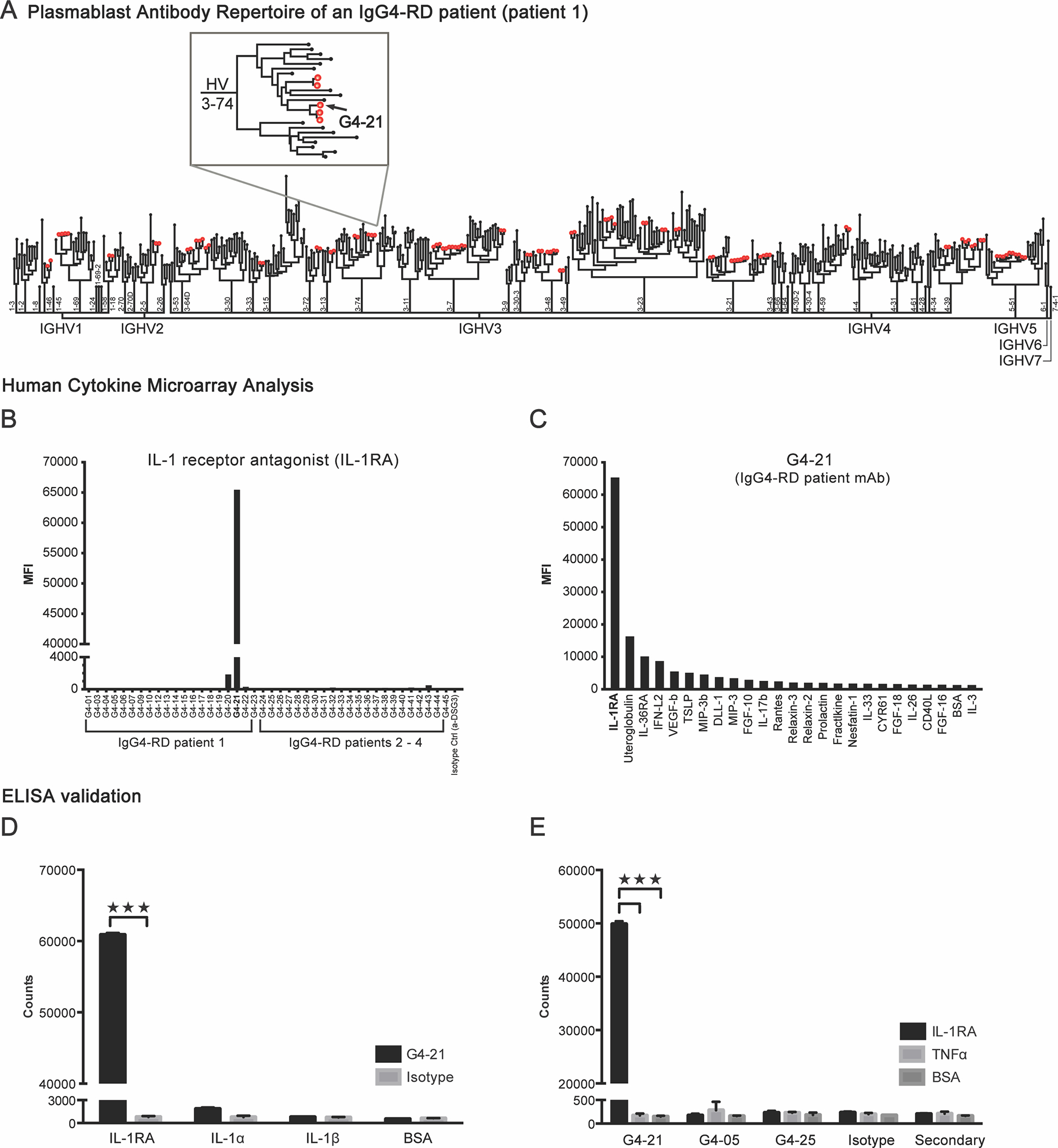
Identification of IL-1 receptor antagonist (IL-1RA) as a target of a plasmablast-derived IgG antibody in a patient with IgG4-related disease (IgG4-RD). A, Phylogenetic tree of the plasmablast antibody repertoire from one patient with IgG4-RD. Each peripheral node represents an antibody expressed by a single plasmablast. Red dots indicate clonal family antibodies. Inset: magnification of the branch containing the plasmablast-derived monoclonal antibody (mAb), G4–21. B, Quantification of the binding reactivities of 42 recombinantly expressed mAbs, derived from the plasmablast clonal families of four IgG4-RD patients, against IL-1RA from the human cytokine microarray analysis. C, Quantification of G4–21 binding to the top 25 reactive antigens on the human cytokine microarray. D, ELISA quantification of G4–21 binding to recombinant human IL-1RA, IL-1α or IL-1β and E, additional mAbs G4–05 and G4–25, to IL-1RA. Recombinant human TNFα and BSA, and reactivity of a human IgG secondary antibody alone (E) and an anti-desmoglein 3 (Isotype, α-DSG3) antibody served as a negative control (B – E). Data represent means ± SD of the median fluorescent intensities (MFI) of four replicate spots of each antigen (B and C) or of europium counts of triplicate wells (D and E). ***P<0.001 by two-tailed unpaired Student’s t tests.
Anti-IL-1RA autoantibodies are elevated in patients with IgG4-RD
We evaluated the prevalence of anti-IL-1RA autoantibodies in patients with IgG4-RD by performing ELISAs on plasma from patients in a discovery cohort of IgG4-RD (n = 13), disease controls (DC) consisting of patients with Sjögren’s syndrome (n = 6) and gout (n = 6), and healthy controls (HC; n = 10) (Fig. 2, A). We observed significantly higher levels of reactivities to IL-1RA in plasma from patients with IgG4-RD as compared to disease controls or healthy controls (Fig. 2, A), with 5 out of 13 patients (38%) positive for reactivity against IL-1RA. Findings were confirmed via detection of significantly higher levels of IgG reactivity to IL-1RA in serum from a large, independent cohort of patients with IgG4-RD (n = 122) as compared to healthy controls (n = 50) (Fig. 2, B, S. Table 1), with 19 of 122 IgG4-RD patients (15.6%) positive for reactivity against IL-1RA.
FIG 2.
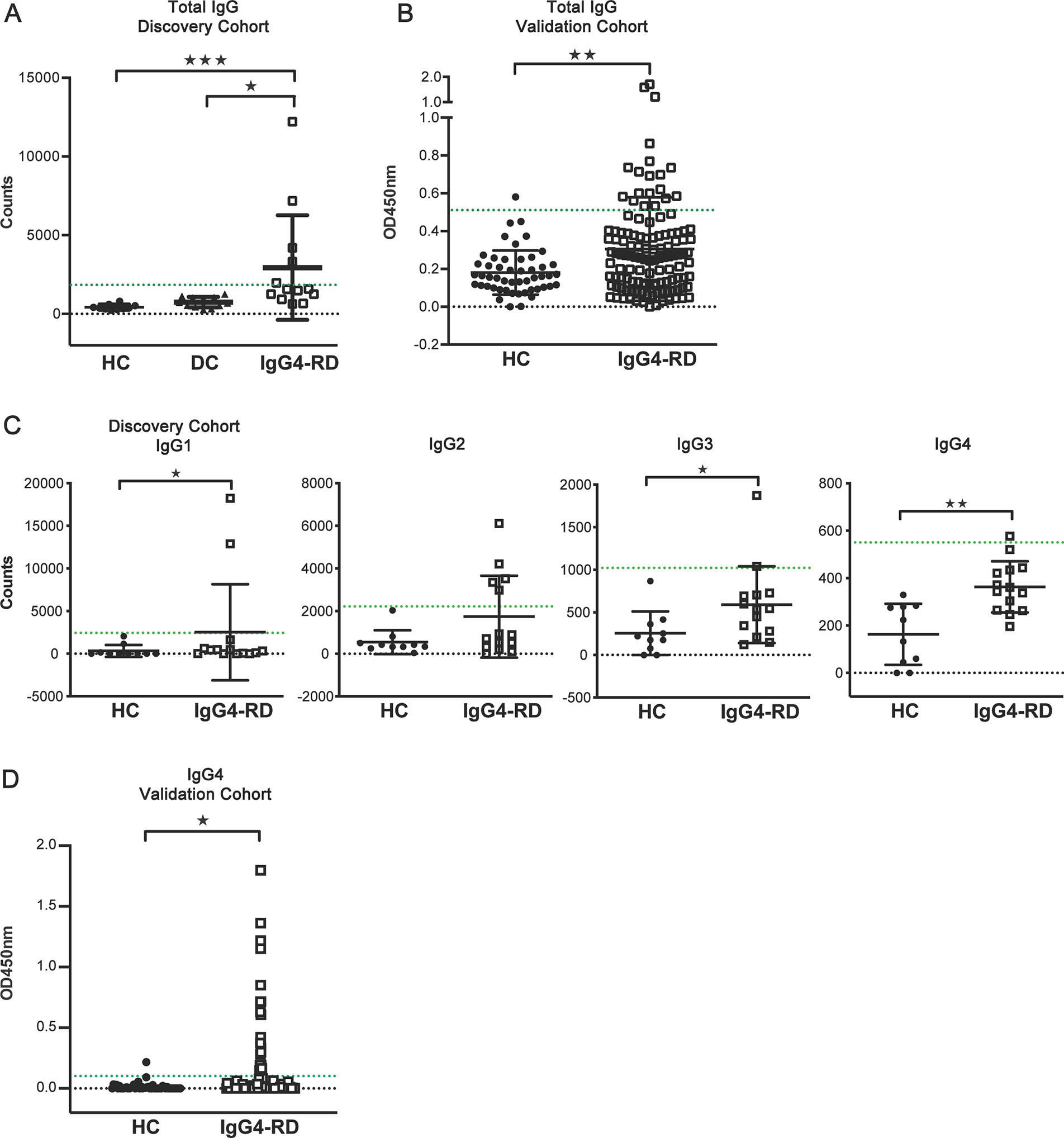
IgG and IgG4 reactivity to IL-1RA is elevated in plasma and serum of subgroups of patients with IgG4-RD. A and B, ELISA quantification of IgG reactivities to recombinant human IL-1RA in plasma from a discovery cohort consisting of healthy controls (HC; n 10), disease controls (subjects with Sjögren’s syndrome or gout; DC; n = 12), and patients with IgG4-RD (n = 13) (A) and in serum from a validation cohort consisting of age-matched healthy controls (HC; n = 50) and IgG4-RD patients (n = 122) (B). C, ELISA quantification of reactivities of individual IgG subclass (IgG1–4) antibodies to recombinant human IL-1RA in plasma from patients in the discovery cohort (HC, n = 10; IgG4-RD, n = 14). D, ELISA quantification of IgG4 reactivities to recombinant human IL-1RA in serum from a validation cohort (HC, n = 50; IgG4-RD, n = 147). Green lines indicate threshold for positive IL-1RA reactivity (3 SD above the mean optical density for DC and HC groups for the discovery and validation cohorts, respectively). Data represent means ± SD of europium counts (A, C) or optical densities (B, D) of ≥2 independent experiments. *P < 0.05, **P < 0.01, ***P<0.001 by Kruskal-Wallis (A), Mann-Whitney U-tests (B - D).
Elevation in levels of circulating IgG4 antibodies is observed in 90% of IgG4-RD patients.15 Thus, we evaluated the IgG subclasses of circulating anti-IL-1RA autoantibodies of the IgG4-RD patients in our discovery cohort. IL-1RA reactivities were detected via ELISA for each IgG subclass (IgG1–4) in plasma from patients with IgG4-RD (Figure 2, C). We observed significantly higher IgG4 reactivity against IL-1RA in IgG4-RD patients (n = 14, additional patient recruited to discovery cohort at the time of this experiment) as compared to healthy controls (n = 10) (Fig. 2, C). Interestingly, 14.2% of the IgG4-RD patient samples from our validation cohort were positive for IgG4 anti-IL-1RA autoantibodies, representing >90% of the patients that were also positive for total IgG anti-IL-1RA (Fig. 2, D). IgG1- and IgG3-specific reactivities to IL-1RA were also significantly higher in IgG4-RD patients as compared to healthy controls. These results indicate that IgG4 is the predominant subclass in the autoantibody response towards human IL-1RA, although IL-1RA-specific IgG1, IgG2, and IgG3 antibodies are also present in a subset of IgG4-RD patients.
Anti-IL-1RA antibodies promote IL-1-mediated expression and production of inflammatory and fibrotic mediators
We investigated whether anti-IL-1RA autoantibodies mediate inflammatory and fibrotic processes in IgG4-RD by antagonizing IL-1RA using the HEK-Blue IL-1β reporter line in which IL-1 receptor activity is measured through the detection of secreted embryonic alkaline phosphatase (SEAP) (S. Fig 4). Incubating IL-1RA/IL-1β-stimulated reporter cells with the patient-derived mAb G4–21 resulted in a significant increase in SEAP production as compared to control antibodies (Fig. 3, A). Significant increases in SEAP production were also observed in a dose-dependent manner, when reporter cells were incubated with increasing concentrations of a commercial anti-IL-1RA polyclonal antibody (pAb), 200–01RA as compared to a pAb isotype control (Fig. 3, B). Moreover, reporter cells produced significantly more SEAP when incubated with plasma from IgG4-RD patients (n = 13), with 54% positive for neutralizing anti-IL-1RA autoantibodies, as compared plasma from healthy controls (HC; n = 15) (Fig. 3, C). Together, these findings indicate that both recombinantly expressed antibodies, and antibodies present in plasma from patients with IgG4-RD, can antagonize the function of IL-1RA.
FIG 3.
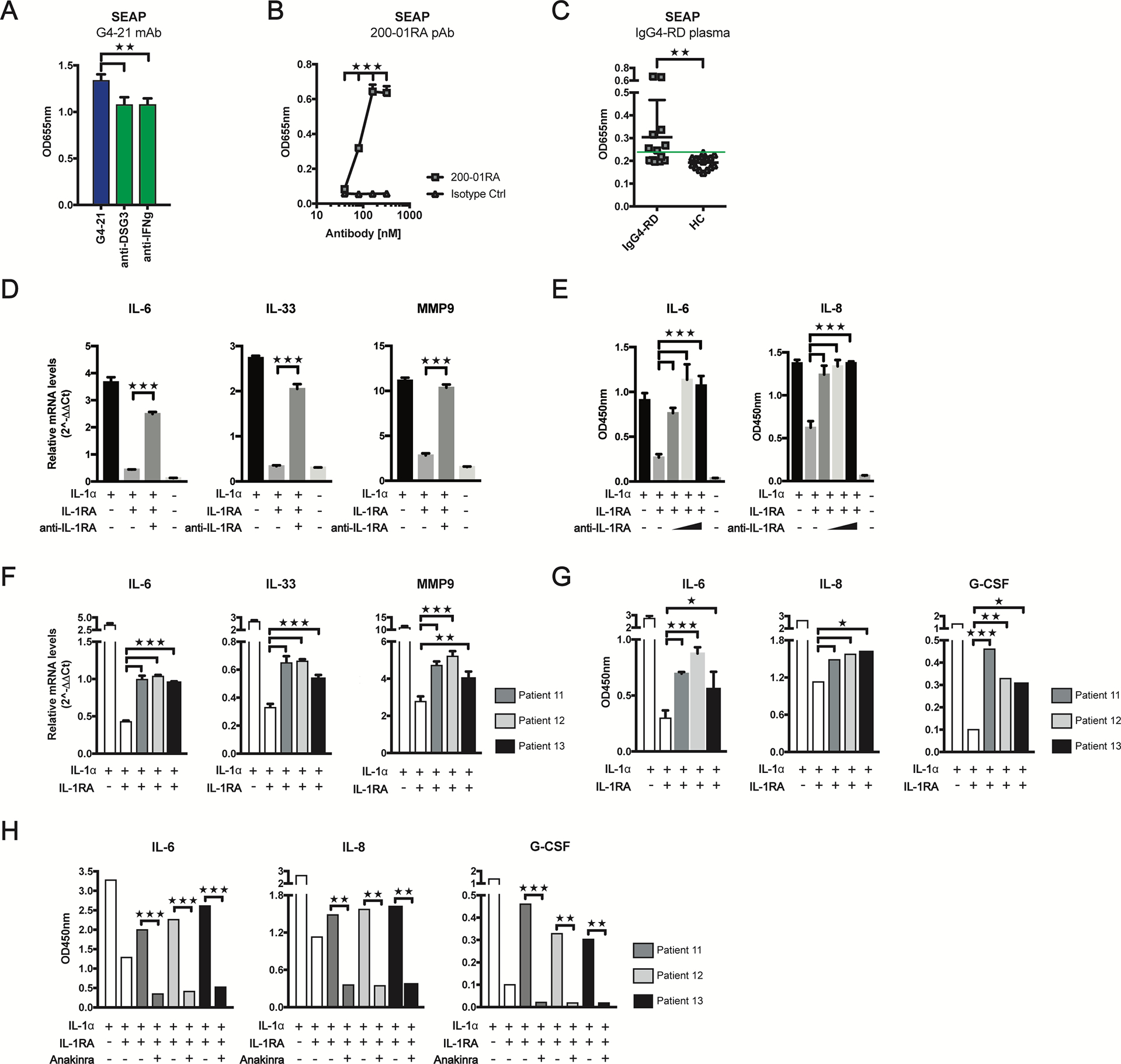
Anti-IL-1RA antibodies exacerbate IL-1-induced expression of inflammatory and fibrotic mediators and are inhibited by anakinra. A - C, Quantification of secreted embryonic alkaline phosphatase (SEAP) produced by HEK-Blue IL-1β reporter cells incubated with human IL-1β and IL-1RA in the presence of mAb G4–21 or control antibodies (A), a commercial anti-IL-1RA pAb at 40, 80, 160 and 320nM, (B), or plasma from IgG4-RD patients (IgG4-RD, n=13) or healthy controls (HC, n=15) (C). Threshold for IL-1RA neutralization (3 SD above mean optical density for HCl group) is indicated by a green line (C). D - G, Quantification of mRNA (D, F) and protein levels (E, G) of inflammation- and fibrosis-associated genes in IL-1α-stimulated A549 epithelial cells incubated with anti-IL-1RA pAb at 100nM (D) and at 50, 100 and 200nM (E) or plasma from three IgG4-RD patients positive for anti-IL-1RA autoantibodies (F, G) in the presence of IL-1RA. H, ELISA quantification of IL-6, IL-8, and G-CSF supernatant levels in IL-1α-stimulated A549 cells incubated with IgG4-RD patient plasma with IL-1RA and anakinra. Data represent mean ± SD of ≥2 wells and represent ≥2 independent experiments. *P<0.05, **P<0.01, ***P<0.001 by Kruskal-Wallis test (A), Mann-Whitney U-test (B, C) or two-tailed unpaired Student’s t tests (D–H).
We investigated whether anti-IL-1RA antibodies could abrogate the regulatory activity of IL-1RA in epithelial and fibroblast cell lines stimulated with IL-1α and promote inflammatory and fibrotic responses. We measured expression of genes encoding multiple inflammatory cytokines (IL-1α, IL-6, IL-8, IL-33, TNFα, G-CSF), growth factors (PDGF-α, PDGF-b, PDGF-c, CTGF), extracellular matrix proteins (MMP-3, MMP-7, MMP-9, MMP-12, MMP-13, TIMP1) and collagen synthesis proteins (COL1A and COL4A) in A549 epithelial cells and MRC5 fibroblasts stimulated with IL-1α and IL-1RA in the presence or absence of pAb 200–01RA. Treatment of IL-1α-stimulated A549 and MRC5 cells with IL-1RA resulted in significant changes in mRNA levels of these genes, compared to IL-1α-stimulated cells alone (Fig. 3, D; S. Figs. 5, A – D and 6, A). Addition of pAb 200–01RA to IL-1α/IL-1RA-stimulated A549 and MRC5 cells also significantly altered expression of these genes, resembling conditions with IL-1α stimulation alone. (Fig. 3, D; S. Fig 5, 6, A). Further, IL-6 and IL-8 protein levels were significantly elevated in supernatants of both IL-1α/IL-1RA-stimulated A549 and MRC5 cells upon treatment with pAb 200–01RA as compared to IL-1α/IL-1RA alone (Fig. 3, E; S. Fig. 6, B).
We investigated whether anti-IL-1RA autoantibodies in the plasma of patients with IgG4-RD could promote expression of these pro-inflammatory and -fibrotic mediators in vitro. We treated IL-1α/IL-1RA-stimulated A549 or MRC5 cells with plasma from three IgG4-RD patients in our discovery cohort that demonstrated reactivity to IL-1RA. Plasma from each patient partially restored IL-6, IL-33, and MMP-9 (for A549 cells) and IL-6, IL-8 and G-CSF (for MRC5 cells) mRNA levels to those observed in the IL-1α-stimulated cells (Fig. 3, F; S. Fig. 6, C). Additionally, IL-6, IL-8, and G-CSF protein levels were significantly elevated in the supernatants of A549 and MRC5 cells treated with IgG4-RD patient plasma for 24 hours (Fig. 3, G; S. Fig. 6, D). These increases were not attributed to endogenous IL-1 present in the plasma samples (S. Fig. 6, E). Together, these findings indicate that both recombinant and IgG4-RD plasma-derived anti-IL-1RA autoantibodies can directly modulate the expression of key mediators involved in inflammation, tissue remodeling and fibrosis through the antagonism of IL-1RA in vitro.
Anti-IL-1RA autoantibodies are linked to the number of affected organs in IgG4-RD and IL-1RA is expressed in lesional tissues
We explored the relationship between organ involvement and anti-IL-1RA autoantibodies by quantifying the number of organs involved in patients with IgG4-RD with or without IgG4 anti-IL-1RA autoantibodies (Fig. 4, A). On average, patients with anti-IL-1RA autoantibodies had greater numbers of organs involved as compared to those negative for anti-IL-1RA autoantibodies (Fig. 4, A), suggesting that anti-IL-1RA antibodies correlate to multi-organ manifestations of IgG4-RD.
FIG 4.
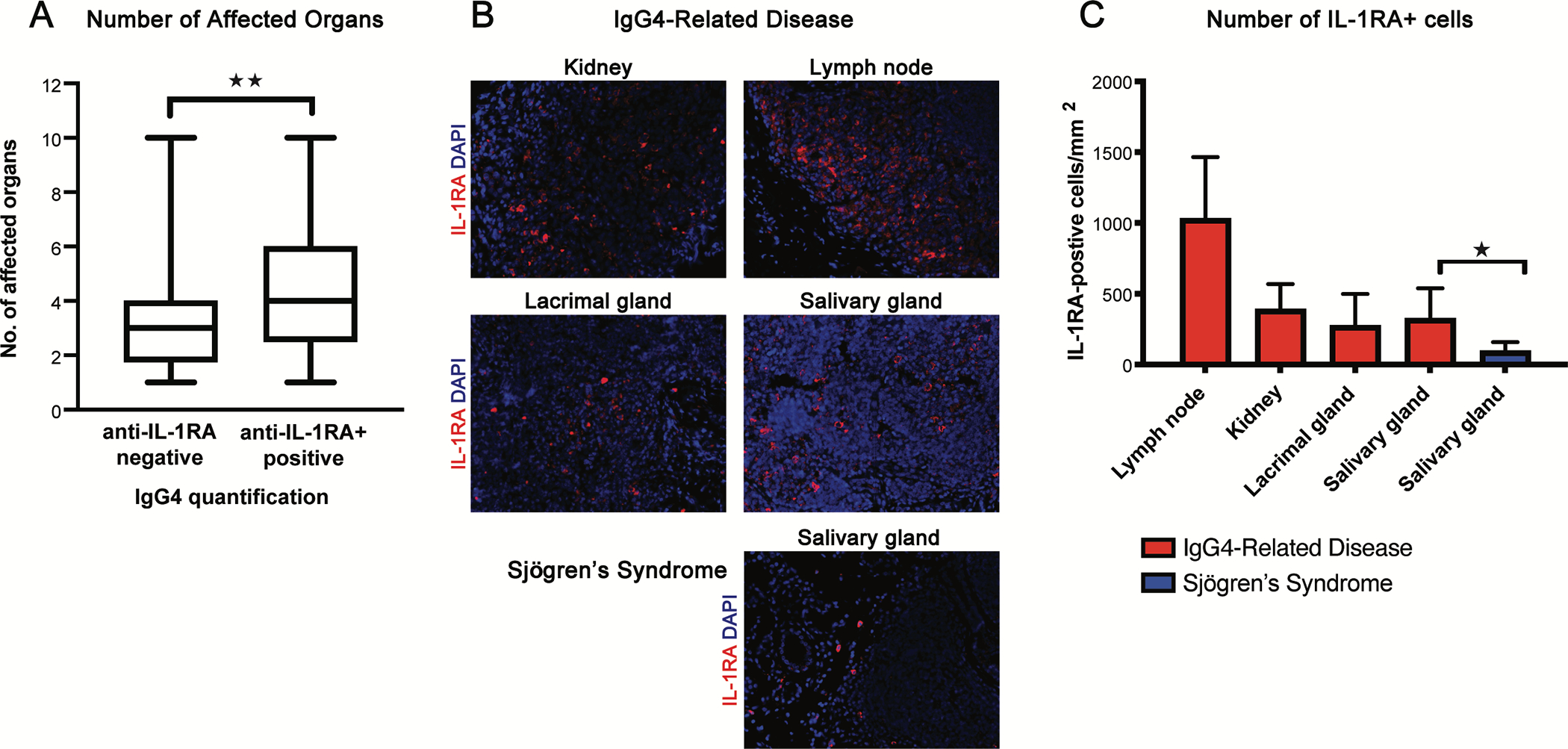
IL-1RA reactivity correlates with multiorgan involvement of IgG4-RD and is expressed in tissue lesions of IgG4-RD patients. A, Number of affected organs in IgG4-RD patients either positive or negative for IgG4 anti-IL-1RA autoantibodies from the validation cohort. B and C, IL-1RA immunofluorescence in tissue-biopsy samples from four patients with IgG4-RD and one patient with Sjögren’s syndrome. IL-1RA (red) and DAPI (blue) staining in biopsied kidney, lymph node, lacrimal gland, and salivary gland tissues from four patients with IgG4-RD and salivary gland tissue from one patient with Sjögren’s syndrome (B). Quantification of IL-1RA staining in tissue biopsies from the patients described (C). Data represent percentage of subjects with and without anti-IL-1RA autoantibodies of ≥2 independent experiments (A) or means ± SD of the cell counts positive for IL-1RA per mm^2 of five fields per tissue sample (C). *P < 0.05, **P < 0.01 by Mann-Whitney U-tests.
Over-expression of self-proteins in the tumefactive lesions of IgG4-RD patients is associated with breaches of humoral immune responses, including the presence of autoantibodies.7 We examined expression of IL-1RA in patients with IgG4-RD, by performing multi-color immunofluorescence in the lesional tissues from four patients with IgG4-RD. IL-1RA was present in the kidney, lacrimal gland, lymph node, and salivary gland tissues from these patients (Fig. 4, B). Quantification indicated that there were significantly more IL-1RA-positive cells in salivary gland tissues affected by IgG4-RD compared to a control sample from a patient with Sjögren’s syndrome (Fig. 4, C). Although our tissue sample size was small, these findings demonstrate that IL-1RA, the target of anti-IL-1RA autoantibodies, is present at sites of tissue damage in IgG4-RD.
Peptide epitope mapping of anti-IL-1RA antibodies.
Given that multiple anti-IL-1RA antibodies were able to inhibit IL-1RA function, we mapped the binding sites of these antibodies on IL-1RA. We tested the binding of multiple anti-IL-1RA antibodies to sixteen overlapping 19-mer peptides spanning the IL-1RA protein by ELISA (S. Fig. 7). mAb G4–21 exhibited increased binding to peptide RA-05 as compared to the human IgG isotype control mAb (Fig. 5, A). The commercial anti-human/murine IL-1RA mAb, MAB4801, demonstrated significantly increased binding to RA-12, with >15X fold change binding reactivity over a rat IgG1 isotype control mAb (Fig. 5, B). In addition, pAb 200–01RA showed significantly increased binding to all IL-1RA peptides, with the highest reactivities (>50X fold change) to peptides RA-04, −05, −06 (cluster 1), RA-08, −09, −10 (cluster 2), RA-12 (cluster 3), −14, −15, and −16 (cluster 4), compared to a pAb isotype control (Fig. 5, C). Comparison of the peptide reactivities across all three anti-IL-1RA antibodies revealed two shared peptide epitopes in clusters 1 and 3, RA-05 and RA-12, that bound each IL-1RA-specific mAb and pAb, 200–01RA, and two unique peptide epitopes representative of clusters 2 and 4, RA-09 and RA-16, uniquely targeted by 200–01RA.
FIG 5.
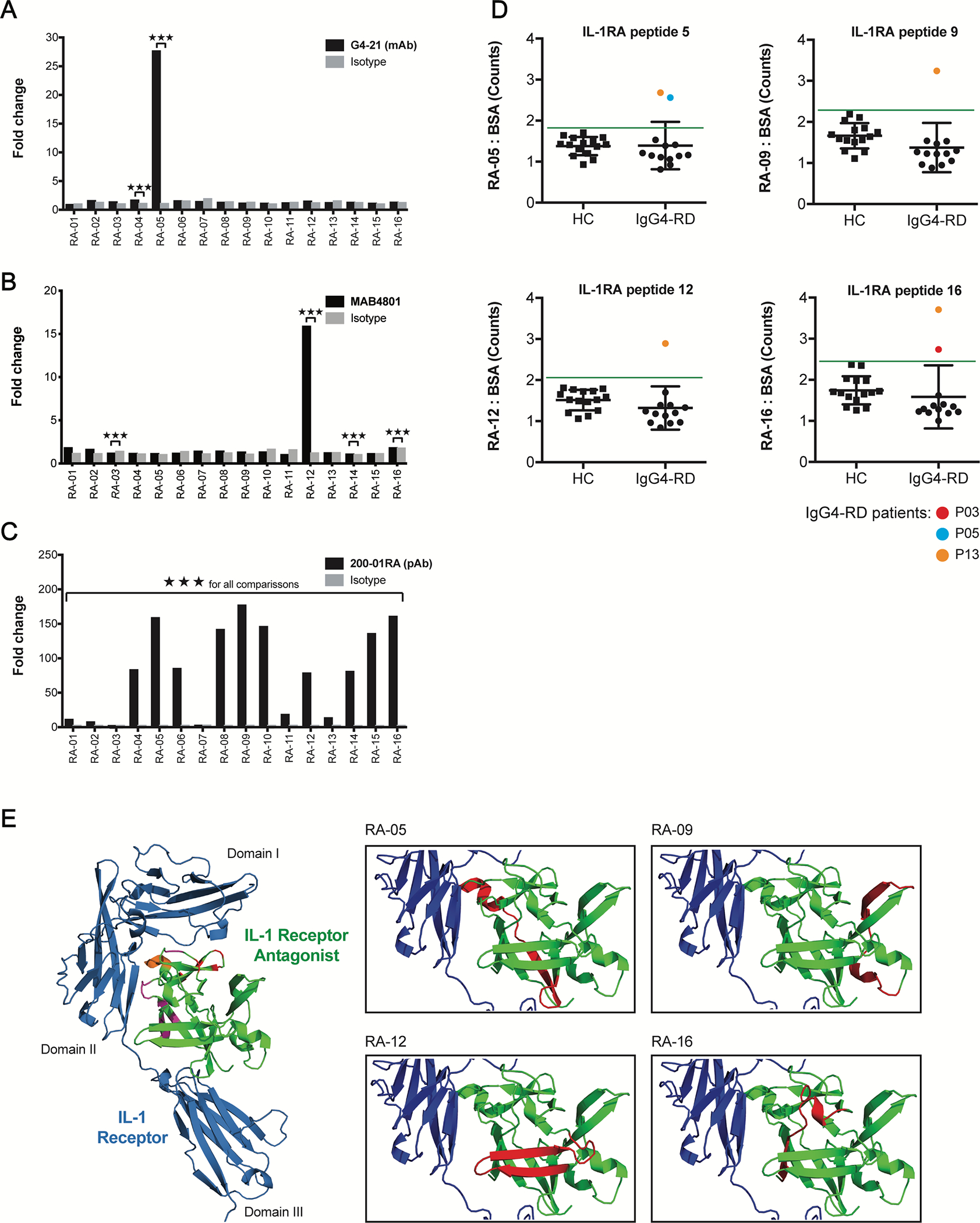
Anti-IL-1RA antibodies target IL-1RA peptides near the IL-1 Receptor interface. A – C, ELISA quantification of the IgG4-RD patient-derived mAb, G4–21 (A), commercial anti-IL-1RA mAb, MAB4801 (B), and commercial anti-IL-1RA pAb, 200–01RA (C), binding to sixteen 19-mer overlapping human IL-1RA peptides. Fold change represents europium counts of each peptide over the minimum peptide reactivity (across all peptides) per antibody. D, ELISA quantification of IgG binding to four immunogenic IL-1RA peptides identified in A – C in the plasma of patients with IgG4-RD (IgG4-RD; n = 13) and healthy controls (HC; n 15). Data represent mean ± SD of europium counts of triplicate wells. Green line indicates threshold for IL-1RA peptide binding (3 SD above the mean europium count from the healthy control group). Colored data points indicate IgG4-RD patient samples positive for autoantibodies for a given peptide. E, 3D structure of human IL-1 Receptor (blue) in complex with human IL-1RA (green; PDB#1IRA). Left: IL-1RA residues implicated in binding IL-1 Receptor at domains I and II and at their interface are highlighted in red, purple, and orange, respectively. Right: Peptides 05, −09, −12, and −16 targeted by anti-IL-1RA autoantibodies are highlighted in red. ***P<0.001 by two-tailed unpaired Student’s t tests.
We evaluated the prevalence of reactivities against these four specific non-overlapping IL-1RA peptides, RA-05, RA-09, RA-12 and RA-16, in plasma from our discovery cohort of IgG4-RD patients (n = 13). Three IgG4-RD patients had positive reactivity against ≥1 peptide epitope, demonstrating that multiple regions of IL-1RA may be targeted by circulating IgG autoantibodies in IgG4-RD patients (Fig. 5, D).
Mapping of these peptides onto a previously solved crystal structure of the IL-1RA/IL-1R complex (PDB:1IRA)16 demonstrated that these anti-IL-1RA antibodies target epitopes at or proximal to (RA-05, RA-12) and distal to (RA-09, RA-16) the IL-1RA/IL-1R binding interface. Identification of these peptides as targets of neutralizing anti-IL-1RA antibodies highlights unique structural and/or allosteric interactions by which anti-IL-1RA antibodies could abrogate IL-1RA-mediated inhibition of IL-1 activity (Fig.5, E).
Anti-IL-1RA autoantibodies exhibit cross reactivity to other IL-1 family members
Based on high degrees of homology, we tested whether anti-IL1-RA autoantibodies cross-react with IL-36RA or IL-38. We observed significantly higher levels of reactivity to IL-36RA and IL-38 with mAb G4–21 and pAb 200–01RA compared to TGFα and BSA controls (Fig. 6, A, B). We did not detect reactivity to IL-36RA or IL-38 in mAbs derived from unrelated plasmablast clonal families (G4–05, G4–25), or the isotype control mAb (Fig. 6, A). Alignment of the sequences of the IL-1RA peptides RA-05, RA-09, RA-12 and RA-16 with the corresponding regions on IL-36RA and IL-38 revealed high sequence homology and topology across the three receptor antagonist cytokines (Fig. 6, C). These findings highlight potential peptide epitopes by which IL-1RA-targeting autoantibodies cross-bind IL-36RA and IL-38.
FIG 6.
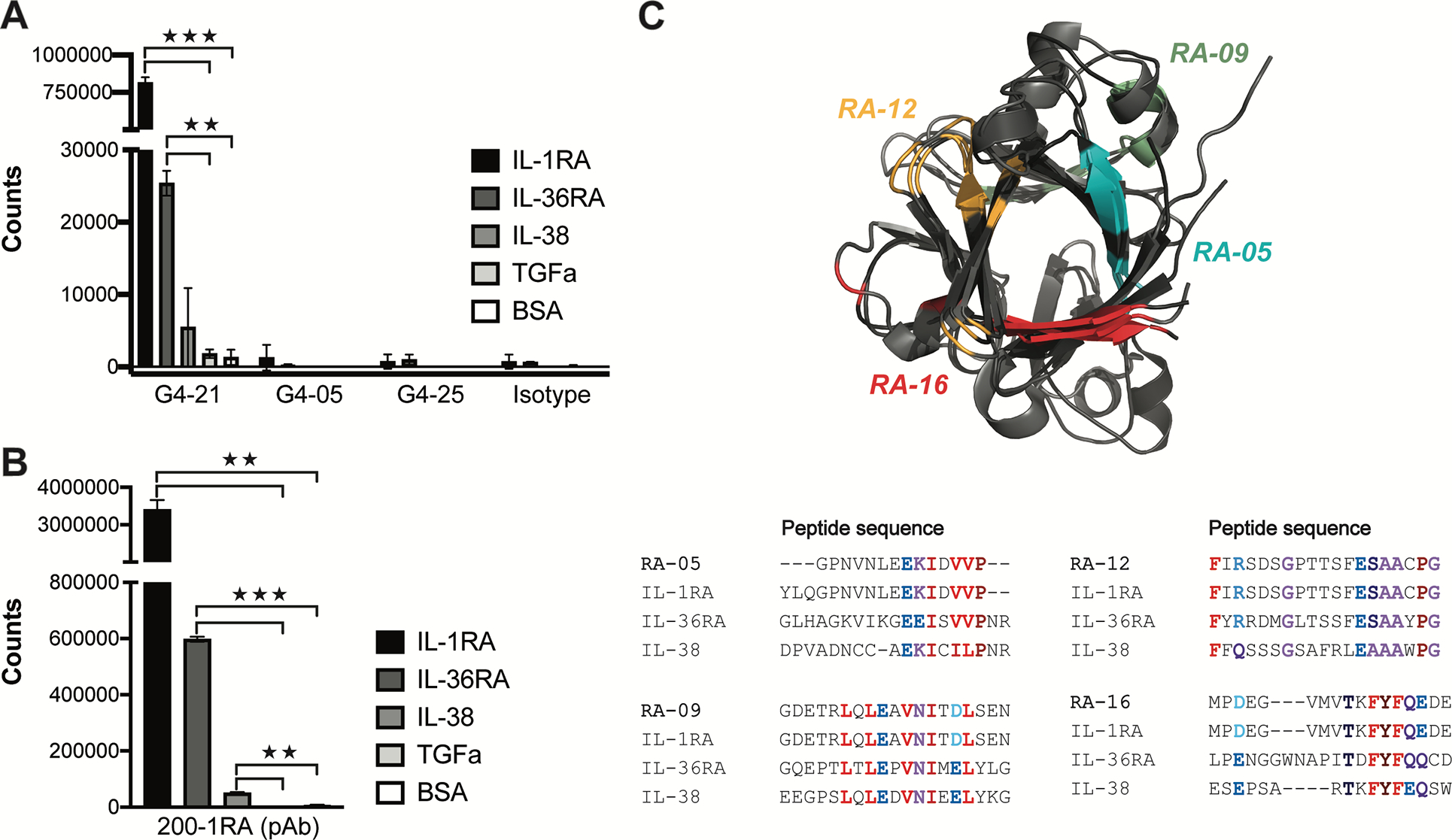
Anti-IL-1RA autoantibodies exhibit cross reactivity to IL-1 family receptor antagonist cytokines. A and B, ELISA quantification of binding reactivities against IL-1RA, IL-36RA and IL-38 by IgG4-RD patient mAbs G4–21, G4–05, and G4–25, and an isotype control (A) and a commercial anti-IL-1RA pAb, 200–01RA (B). TGFα and BSA alone served as controls. C, Top: 3D structure overlay of IL-1RA (PDB#1IRA), IL-36RA (PDB#4P0J), and IL-38 (PDB#5B0W). Bottom: amino acid sequence alignments of peptide epitopes IL-1RA 37–56 (RA-05), IL-1RA 73–92 (RA-09), IL-1RA 100–119 (RA-12), and IL-1RA 136–152 (RA-16) of human IL-1RA, IL-36RA, and IL-38. Bolded colors in sequences indicate residues with similar chemical properties. Corresponding regions are colored in the IL-1RA-IL-36RA-IL-38 3D structure overlay. **P < 0.01, ***P<0.001 by two-tailed unpaired Student’s t tests.
Anti-IL-1RA autoantibodies detected in patients with lupus and rheumatoid arthritis
Patients with systemic lupus erythematosus (SLE) have elevated levels of IgG reactivity to cytokines compared to healthy subjects that correlate with disease severity.14 Therefore, we investigated the presence of anti-IL-1RA autoantibodies in serum samples from a cohort of SLE patients. Serum from SLE (n = 49) contained significantly higher levels of reactivity against IL-1RA compared to heathy controls (n = 25), with 16 of 49 SLE patients (33%) positive for anti-IL-1RA autoantibodies (Fig. 7, A). We also analyzed IL-1RA reactivity in the serum of patients with either multiple sclerosis (MS; n = 36) or rheumatoid arthritis (RA; n = 55). Similar to SLE, levels or IL-1RA reactivity were significantly elevated in 25% of RA patients as compared to healthy controls (n = 50), but not in MS (Fig. 7, B). These results indicate that anti-IL-1RA autoantibodies are detectable in subsets of patients with SLE and RA and that antagonism of IL-1RA-mediated responses could contribute to inflammation in these autoimmune diseases and IgG4-RD.
FIG 7.
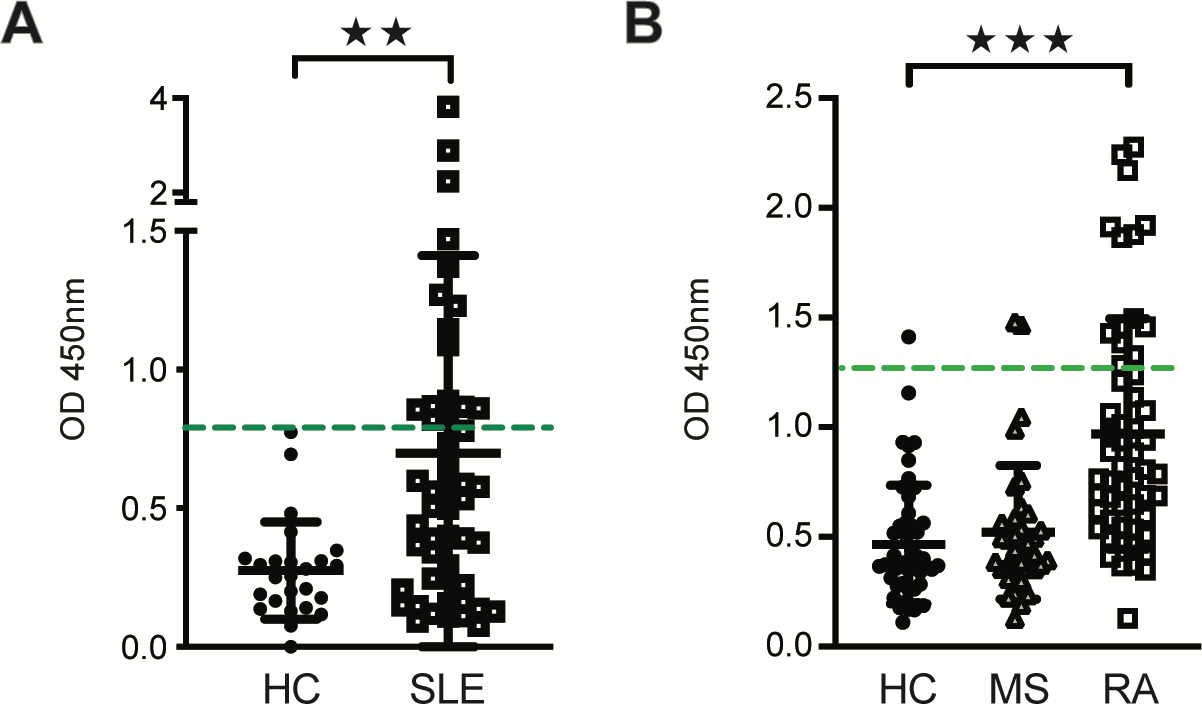
Anti-IL-1RA autoantibodies are elevated in patients with systemic lupus erythematosus and rheumatoid arthritis. A and B, ELISA quantification of IgG reactivities to recombinant human IL-1RA in the serum of patients with systemic lupus erythematosus (SLE; n = 49) and healthy controls (HC; n = 25) (A) and in patients with multiple sclerosis (MS; n = 36), rheumatoid arthritis (RA; n = 55) and healthy controls (HC; n = 50) (B). Threshold for anti-IL-1RA antibody positivity (3 SD above the mean optical density for the healthy control groups) is indicated by a dashed horizontal line in green. Data represent means ± SD of optical densities of triplicate wells. **P < 0.01, ***P<0.001 by Mann Whitney U test.
DISCUSSION
Autoantibodies targeting cytokines are present in several autoimmune and inflammatory diseases,17, 18 and can be pathogenic by promoting pro-inflammatory responses.19–21 Although multiple putative antigen targets of autoreactive IgG antibodies have been identified in IgG4-RD, it remains unclear as to if and how these and other antibodies contribute to the fibroinflammatory nature and diverse organ manifestations observed in this disease. Here we identified neutralizing anti-IL-1RA autoantibodies in IgG4-RD and characterize their role in promoting the expression of pro-inflammatory and -fibrotic mediators, via antagonism of IL-1RA function. We also show that similar antibodies are present in the sera of patients with SLE and RA. Together our findings uncover a potential common mechanism for the promotion of inflammatory and fibrotic pathways that could contribute to pathophysiology in these diseases.
We demonstrated that plasma autoantibodies from IgG4-RD patients can promote expression of pro-inflammatory and pro-fibrotic factors, including MMP-9 and IL-33 in epithelial and fibroblast cell lines stimulated with IL-1α and IL-1RA. These cytokines, enzymes, and growth factors coordinate protective immune responses, including inflammatory responses triggered by infection and wound healing.22 However, dysregulation of these factors can promote pathogenic inflammation and fibrosis causing irreversible tissue damage and dysfunction.23 Activation of TGFβ1 and MMP9 via overexpression of IL-13 can promote airway fibrosis in mice, similar to that observed in chronic kidney disease.24, 25 IL-33, an IL-1 family inflammatory cytokine, can also promote lung fibrosis in bleomycin-treated mice through regulation of MMP-9, TGFβ1and other fibrotic mediators.26–28 Together, these findings support the possibility that anti-IL-1RA autoantibodies can drive inflammation and fibrosis in IgG4-RD and other autoimmune diseases, by promoting expression of these factors, as a result of blocking IL-1RA activity.
We identified four IL-1RA immunogenic peptide epitope targets of monoclonal and polyclonal anti-IL-1RA antibodies and autoantibodies from patients with IgG4-RD. Anti-IL-1RA antibodies targeting peptide epitopes at the IL-1RA/IL-1R interface may sterically block this interaction while anti-IL-1RA antibodies that target peptide epitopes outside of this interaction could induce conformational changes that weaken the affinity of IL-1RA for IL-1R. Neutralizing anti-IL-1RA antibodies that target these epitopes and disrupt the IL-1RA/IL-1R interaction, may thereby allow IL-1R to bind IL-1 and promote signaling. Furthermore, in vitro cross-reactivity between anti-IL-1RA antibodies and functionally related cytokines, IL-36RA and IL-38, that inhibit IL-36 signaling, suggests that the mechanism through which anti-IL-1RA antibodies promote inflammatory and fibrotic mediator production may be more widespread than through targeting of IL-1RA alone. Future studies will need to investigate the role of IL-36 in the pathogenesis of IgG4-RD, and whether abrogation of IL-36RA and IL-38 function by autoantibodies plays a significant role in this disease.
IgG4 antibodies were the predominant subclass of anti-IL-1RA reactivity we identified, and are a diagnostic feature of IgG4-RD.15 IgG4 molecules typically exhibit anti-inflammatory properties, however, they have been shown to promote disease symptoms in Fc-independent manners in pemphigus vulgaris and myasthenia gravis, through binding to skin- and muscle-specific autoantigens.29–31 As we identified epitopes that suggest that anti-IL-1RA antibodies disrupt the IL-1RA/IL-1R interaction, it is possible that a subset of antibodies in IgG4-RD promote pathogenesis independently of Fc signaling or complement activation. We also identified reactivities against IL-1RA in all IgG subclasses in patients with IgG4-RD, suggesting that antibodies of other IgG subclasses (IgG1–3) may contribute to disease pathogenesis via engagement of their Fc regions.
Given the rationale for contributions of all IgG subclasses to disease, autoantibody-driven pathogenesis in IgG4-RD may occur through multiple mechanisms. We recently demonstrated that tissue infiltrating B cells in IgG4-RD autoimmune pancreatitis produce profibrotic mediators.32 As we detected IL-1RA in the tissues of affected organs in IgG4-RD, activated B cells in these tissues may, in part, target IL-1RA, and promote tissue inflammation and fibrosis via overexpression and secretion of inflammatory and fibrotic mediators.
This study has several limitations. First, although large for a rare disease such as IgG4-RD, our patient cohort was small, and depth of sequencing may have limited our ability to identify additional key autoantibodies. Nevertheless, we also identified an additional autoantigen target, macrophage inflammatory protein 3 (MIP3a, CCL20), in our cytokine microarray analysis of plasmablast-derived mAbs and plasma samples from IgG4-RD. Further studies are needed to elucidate its role in IgG4-RD. Second, although we identified peptide epitopes for anti-IL-1RA autoantibodies that structurally implicate disruption of the IL-1RA/IL-1R interaction, further studies are needed to discern the mechanisms by which anti-IL-1RA antibodies inhibit IL-1RA function and whether these autoantibodies mediate pathogenesis in Fc-dependent or -independent manners. Finally, additional studies are needed to characterize the roles of anti-IL-1RA autoantibodies in SLE and RA, and to determine whether these autoantibodies represent a common pathogenic mechanism.
In conclusion, we describe the identification of anti-IL-1RA autoantibodies in patients with IgG4-RD that antagonize the inhibitory effects of IL-1RA, thereby promoting the production of pro-inflammatory and pro-fibrotic mediators implicated in disease pathogenesis. Identification of IL-1RA in IgG4-RD tissue lesions as well as association of multi-organ involvement in individuals with anti-IL-1RA autoantibodies of the IgG4 subclass suggest an underlying molecular mechanism for the varied organ manifestations in this disease. The crosstalk between anti-IL-1RA antibodies and other IL-1 receptor-antagonist family members and the presence of these autoantibodies in SLE and RA suggest a broader function for autoantibodies targeting IL-1 receptor-antagonist family members and an underappreciated mechanism for the pathogenesis of multiple autoimmune diseases. Future studies will be critical for evaluating the diagnostic, prognostic and pathogenic potential of anti-IL-1RA autoantibodies in IgG4-RD, and for addressing whether inhibition of IL-1 may be an advantageous route for the treatment of anti-IL-1RA driven disease.
Supplementary Material
KEY MESSAGES.
Anti-IL-1RA autoantibodies are present in a subset of patients with IgG4-RD and correlate with the number of affected organs.
Anti-IL-1RA antibodies promote the production of pro-inflammatory and pro-fibrotic meditators by abrogating the activity of IL-1RA.
Anti-IL-1RA antibodies bind to IL-1RA at sites proximal and distal to the IL-1RA-IL-1R interface, likely modulating interactions between IL-1RA and IL-1 receptor.
ACKNOWLEDGEMENTS
The authors thank Julia Fukuyama, PhD, for statistical advice, Lisa Blum, PhD, for assistance in sequencing plasmablast antibody repertoires, and Rong Mao, PhD, and Qian Wang, MD, PhD, for critical review of the manuscript. We thank the patients and volunteers who provided blood and tissue samples for these studies.
Funding:
Supported by grants from the Stanford Graduate Fellowship (to J. Jarrell), the Rheumatology Research Foundation (to Dr. Baker and Dr. Perugino), and the National Institutes of Health (T32AI07290, to J. Jarrell, 2T32AR050942-11, to Dr. Baker, T32AR007258, to Dr. Perugino, UM1 AI1144295 to Dr. Stone, U19 AI110495 to Dr. Pillai, and R01AR06367604 and U19AI11049103, to Dr. Robinson).
ABBREVIATIONS:
- IL1-RA
IL-1 receptor-antagonist
- IgG4-RD
IgG4-RD
- SLE
systemic lupus erythematosus
- RA
rheumatoid arthritis
- MS
multiple sclerosis
- Ig
immunoglobulin
Footnotes
Conflict of interest: William Robinson is a Founder, member of the Board of Directors, and consultant to Atreca, Inc. No other potential conflicts of interest relevant to this article were reported.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012; 366:539–51. [DOI] [PubMed] [Google Scholar]
- 2.Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet 2015; 385:1460–71. [DOI] [PubMed] [Google Scholar]
- 3.Frulloni L, Lunardi C, Simone R, Dolcino M, Scattolini C, Falconi M, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–42. [DOI] [PubMed] [Google Scholar]
- 4.Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005; 54:274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubers LM, Vos H, Schuurman AR, Erken R, Oude Elferink RP, Burgering B, et al. Annexin A11 is targeted by IgG4 and IgG1 autoantibodies in IgG4-related disease. Gut 2018; 67:728–35. [DOI] [PubMed] [Google Scholar]
- 6.Endo T, Takizawa S, Tanaka S, Takahashi M, Fujii H, Kamisawa T, et al. Amylase alpha-2A autoantibodies: novel marker of autoimmune pancreatitis and fulminant type 1 diabetes. Diabetes 2009; 58:732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perugino CA, AlSalem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G, et al. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol 2019; 143:736–45 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiokawa M, Kodama Y, Kuriyama K, Yoshimura K, Tomono T, Morita T, et al. Pathogenicity of IgG in patients with IgG4-related disease. Gut 2016; 65:1322–32. [DOI] [PubMed] [Google Scholar]
- 9.Gupta S, Tatouli IP, Rosen LB, Hasni S, Alevizos I, Manna ZG, et al. Distinct Functions of Autoantibodies Against Interferon in Systemic Lupus Erythematosus: A Comprehensive Analysis of Anticytokine Autoantibodies in Common Rheumatic Diseases. Arthritis Rheumatol 2016; 68:1677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Umehara H, Okazaki K, Nakamura T, Satoh-Nakamura T, Nakajima A, Kawano M, et al. Current approach to the diagnosis of IgG4-related disease - Combination of comprehensive diagnostic and organ-specific criteria. Mod Rheumatol 2017; 27:381–91. [DOI] [PubMed] [Google Scholar]
- 11.Tan YC, Kongpachith S, Blum LK, Ju CH, Lahey LJ, Lu DR, et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol 2014; 66:2706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott SE, Kongpachith S, Lingampalli N, Adamska JZ, Cannon BJ, Mao R, et al. Affinity Maturation Drives Epitope Spreading and Generation of Proinflammatory Anti-Citrullinated Protein Antibodies in Rheumatoid Arthritis. Arthritis Rheumatol 2018; 70:1946–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson WH, Steinman L, Utz PJ. Protein and peptide array analysis of autoimmune disease. Biotechniques 2002; Suppl:66–9. [PubMed] [Google Scholar]
- 14.Price JV, Haddon DJ, Kemmer D, Delepine G, Mandelbaum G, Jarrell JA, et al. Protein microarray analysis reveals BAFF-binding autoantibodies in systemic lupus erythematosus. J Clin Invest 2013; 123:5135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Annu Rev Pathol 2014; 9:315–47. [DOI] [PubMed] [Google Scholar]
- 16.Schreuder H, Tardif C, Trump-Kallmeyer S, Soffientini A, Sarubbi E, Akeson A, et al. A new cytokine-receptor binding mode revealed by the crystal structure of the IL-1 receptor with an antagonist. Nature 1997; 386:194–200. [DOI] [PubMed] [Google Scholar]
- 17.Cappellano G, Orilieri E, Woldetsadik AD, Boggio E, Soluri MF, Comi C, et al. Anti-cytokine autoantibodies in autoimmune diseases. Am J Clin Exp Immunol 2012; 1:136–46. [PMC free article] [PubMed] [Google Scholar]
- 18.Lin CH, Chi CY, Shih HP, Ding JY, Lo CC, Wang SY, et al. Identification of a major epitope by anti-interferon-gamma autoantibodies in patients with mycobacterial disease. Nat Med 2016; 22:994–1001. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki H, Ayabe T, Kamimura J, Kashiwagi H. Anti-IL-1 alpha autoantibodies in patients with rheumatic diseases and in healthy subjects. Clin Exp Immunol 1991; 85:407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sjowall C, Ernerudh J, Bengtsson AA, Sturfelt G, Skogh T. Reduced anti-TNFalpha autoantibody levels coincide with flare in systemic lupus erythematosus. J Autoimmun 2004; 22:315–23. [DOI] [PubMed] [Google Scholar]
- 21.Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 2010; 207:291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelso A. Cytokines: principles and prospects. Immunol Cell Biol 1998; 76:300–17. [DOI] [PubMed] [Google Scholar]
- 23.Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta 2013; 1832:1049–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 2001; 194:809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan TK, Zheng G, Hsu TT, Wang Y, Lee VW, Tian X, et al. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am J Pathol 2010; 176:1256–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 2012; 18:1028–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol 2014; 134:1422–32 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu L, Luo Z, Zheng J, Yao P, Yuan Z, Lv X, et al. IL-33 Can Promote the Process of Pulmonary Fibrosis by Inducing the Imbalance Between MMP-9 and TIMP-1. Inflammation 2018; 41:878–85. [DOI] [PubMed] [Google Scholar]
- 29.Ayatollahi M, Joubeh S, Mortazavi H, Jefferis R, Ghaderi A. IgG4 as the predominant autoantibody in sera from patients with active state of pemphigus vulgaris. J Eur Acad Dermatol Venereol 2004; 18:241–2. [DOI] [PubMed] [Google Scholar]
- 30.Koneczny I. A New Classification System for IgG4 Autoantibodies. Front Immunol 2018; 9:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Futei Y, Amagai M, Ishii K, Kuroda-Kinoshita K, Ohya K, Nishikawa T. Predominant IgG4 subclass in autoantibodies of pemphigus vulgaris and foliaceus. J Dermatol Sci 2001; 26:55–61. [DOI] [PubMed] [Google Scholar]
- 32.Della-Torre E, Rigamonti E, Perugino C, Baghai-Sain S, Sun N, Kaneko N, et al. B lymphocytes directly contribute to tissue fibrosis in patients with IgG4-related disease. J Allergy Clin Immunol 2020; 145:968–81 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.