

Abstract
Chemoresistance and inadequate therapeutics transport across the blood brain barrier (BBB) remain the major barriers to treating medulloblastoma (MB). Hedgehog (Hh) and IGF/PI3K pathways regulate tumor cell proliferation and resistance in MB. Current Hh inhibitors are effective initially to treat SHH-MB but acquire resistance. Herein, we showed that Hh inhibitor MDB5 and BRD4/PI3K dual inhibitor SF2523 synergistically inhibited the proliferation of DAOY and HD-MB03 cells when used in combination. Treatment of these MB cells with the combination of MDB5 and SF2523 significantly decreased colony formation and expression of MYCN, p-AKT, and cyclin D1 but significantly increased in Bax expression, compared to individual drugs. We used our previously reported copolymer mPEG-b-PCC-g-DC copolymer, which showed 8.7 ± 1.0 and 6.5 ± 0.1% loading for MDB5 and SF2523 when formulated into nanoparticles (NPs). There was sustained drug release from NPs, wherein 100% of MDB5 was released in 50 h, but only 60% of SF2523 was released in 80 h. Targeted NPs prepared by mixing 30:70 ratio of COG-133-PEG-b-PBC and mPEG-b-PCC-g-DC copolymer delivered a significantly higher drug concentration in the cerebellum at 6 and 24h after intravenous injection into orthotopic SHH-MB tumor-bearing NSG mice. Moreover, systemic administration of COG-133-NPs loaded with MDB5 and SF2523 resulted in decreased tumor burden compared to non-targeted drug-loaded NPs, without any hepatic toxicity. In conclusion, our nanomedicine of MDB5 and SF2523 offers a novel therapeutic strategy to treat chemoresistant MB.
Keywords: medulloblastoma, hedgehog inhibitor, blood-brain barrier, mimetic peptide, Bromodomain-containing protein 4, MYC proto-oncogene
Introduction
Medulloblastoma (MB) is an aggressive primary brain tumor in children. MB is classified into SHH, WNT, Group 3, and Group 4 by WHO where, each group shows different origins, pathogenesis, and therapeutic targets [1]. A combination of intensive adjuvant chemotherapy and high-dose radiation remains the standard treatment of MB. However, this treatment has only a 60% cure rate and usually results in growth impairment, endocrine disorders, and neurocognitive deficits. MB treatment with chemotherapy is considered challenging due to diverse genetic make-up, development of resistance, inefficient transfer of drugs through the blood-brain barrier (BBB), and neurotoxicity [2].
SHH-MB and Group 3 MB subgroups represent about 30% each of total MB cases with variable prognoses. The SHH-MB subgroup is characterized by activation of the Hh pathway, whereas Group 3 MB has upregulated MYC expression [3]. While SHH inhibitors are being developed, the absence of an exact ligand-binding domain on MYC has presented an obstacle toward its direct inhibition. However, targeting MYC activating pathways is an indirect way to suppress MYC oncogenic activity. Bromodomain (BRD)-containing complexes bind to the promoter regions of MYC, and by inhibiting these complexes, MYC activity could be curbed. Vismodegib was the first Hh inhibitor approved for the treatment of Basal Cell Carcinoma (BCC). However, in SHH-MB patients, vismodegib had short-term effects, and patients eventually developed drug resistance. Besides, SHH-MB patients with mutations of genes downstream of SMO or mutated PI3K/Akt pathway develop resistance to vismodegib.
There are several mechanisms of resistance specific to the Hh pathway in MB. A mutation in SMO (D473H) disrupts the binding of vismodegib and sonidegib, resulting in resistance. Downstream of SMO germline loss of SUFU gene, which encodes a GLI inhibitor, is the primary cause of resistance to vismodegib [4]. Compounds directly binding to GLI, such as GANT61, ATO, and GlaB could be used as an alternative to SMO inhibitors [5,6]. However, the clinical development of these molecules is hindered by toxicity and lack of specificity.
Moreover, the non-canonically Hh pathway is activated by EGFR, RAS, mTOR, WNT, and NOTCH, thus minimizing the benefits of SMO inhibitors [7]. Therefore, inhibiting GLI activating pathways is a viable strategy to overcome resistance. Notably, the amplification of GLI2, MYCN, and CCND1 help MB switching its oncogenic signaling pathways, thus losing their addiction to the Hh pathway [8]. Therefore, new treatments are urgently needed that would be effective in overcoming resistance and treating SHH-MB. We hypothesize that simultaneously inhibiting both Hh and MYC pathways would have a clinically measurable beneficial effect. To address this question, we started to find drugs targeting the two pathways.
In our previous work, we synthesized a potent SMO inhibitor 2-chloro-N1-[4-chloro-3-(2-pyridinyl) phenyl]-N4, N4-bis (2-pyridinyl methyl)-1, 4-benzene-dicarboxamide (MDB5) [9]. We observed that MDB5 binds to SMO and inhibited Hh components, cancer stem cell (CSC) markers, cell migration, and tumorigenic potential of pancreatic cancer cells better than vismodegib. Further, recently a BRD4/PI3K dual inhibitor SF2523 was synthesized, which inhibited MYC activities [10]. Treatment of different cancer cells such as prostate, renal cell carcinoma, and chondrosarcoma with SF2523 inhibited their growth. BRD4 binds at the promoter of several receptor tyrosine kinases (RTKs), and BET inhibitors downregulate MYC expression. However, only BET inhibitors are ineffective in treating cancer because PI3K and MYC cooperate as part of a single mitogenic signaling pathway to form cancers. Thus, simultaneous inhibition of BET and PI3K show tumor regression and overcomes resistance in tumor cells [11]. As a PI3K/BRD4 inhibitor, SF2523 is highly promising in MB treatment because it edges over several other BET or only PI3K inhibitors being tested in clinics.
Further, the intact blood-brain barrier (BBB) hinders the transport of most drugs into the brain. Reports also suggest that the integrity of the BBB is compromised in certain brain tumors called blood-tumor barrier (BTB); however, it is highly heterogeneous, and permeability is non-uniformly affected by active efflux of molecules by tumor cells [12]. A thorough understanding of the BBB/BTB structure and function will help enhance drug transport to brain tumors. One of the viable approaches is the receptor-mediated BBB/BTB transcytosis through proteins expressed on endothelial cells lining the BBB/BTB. In this case, a targeting ligand (antibody or peptide) binds to its receptor present on the abluminal surface and induces receptor-mediated endocytosis. The targeting ligand could be conjugated to the drug molecule itself or decorated on nanocarriers. Various receptors which have been utilized for drug transport to the brain include transferrin (TfR), insulin receptor (IR) and insulin-like growth factor 1 receptors (IGFR-1), and lipoprotein-related protein-1/2. However, many of these receptors are present in other tissues, which leads to undesired biodistribution and potential toxicity. Since Apolipoprotein E (ApoE) receptor is overexpressed in the brain endothelia, ApoE-targeting peptide COG-133 conjugated nanocarrier was used for enhanced drug delivery to the brain [13].
In this study, we developed an optimized Hh inhibitor MDB5 and SF2523 in COG-133 decorated polymeric NPs formulations. Combining MDB5 and SF2523 could efficiently downregulate SHH and MYCN expression, lowered AKT phosphorylation, and increased G1 arrest and apoptosis of MB cells. Systemic injection of COG-133 conjugated NPs loaded with MDB5 and SF2523 resulted in decreased tumor burden compared to non-targeted NP formulations.
Materials and Methods
Materials
We synthesized MDB5 as reported previously. SF2523 was purchased from Adooq Bioscience (Irvine, CA). ApoE peptide COG-133 was synthesized by the Biomatik Corporation (Ontario, Canada) to a purity of 95% and reconstituted in sterile isotonic phosphate-buffered saline (PBS). DAOY and HD-MB03 cells were purchased from ATCC and Leibniz Institute (Germany), respectively. COG-133 was acetylated at its amino terminus, and the carboxyl terminus was blocked with an amide moiety (Ac-LRVRLASHLRKLRKRLL-amide). mPEG-b-PCC-g-DC and COG-133-PEG-b-PBC copolymer were synthesized as described previously and characterized by 1H NMR and GPC. All other reagents were purchased from Sigma-Aldrich or Fisher Scientific USA and used without any purification/modification.
Synthesis of COG-133-PEG-b-PCC-g-DC and preparation of Nanoparticles (NPs)
Methoxy poly(ethylene glycol)-b-poly(carbonate)-g-dodecanol (PEG-b-PCC-g-DC) and COOH-PEG-b-PBC were synthesized as described previously. For conjugation of COG-133 peptide, COOH-PEG-b-PBC (200 mg) was added under stirring to 1.5 mL (Ac-LRVRLASHLRKLRKRLL-amide, 21 mg) solution in N, N-dimethylformamide (DMF). The reaction was carried out for 24 h at RT, followed by dialysis (MWCO 3500) against 200 mL DMF (× 3) at RT with solvent replacement each hour. COG-133-PEG-b-PBC was purified by precipitation in excess of cold diethyl ether, filtration, and vacuum drying. The copolymer was characterized by 1H NMR and BCA assay to determine the conjugation efficiency of COG-133.
NPs were prepared by film hydration as described before (14). Briefly, PEG-b-PCC-g-DC (30 mg) and drug (MDB5 or SF2523, 1.5 mg) were dissolved in dichloromethane (DCM, 1 mL) in a 20 mL scintillation vial. The solvent was removed using a rotary evaporator, and the film was dried overnight in a desiccator. Next, 1mL PBS was added and bath sonicated (Branson 3800) for 10 min, and the mixture was centrifuged at 10000 RPM for 5 min. The supernatant was collected and subjected to further characterization. Non-incorporated drug was separated by filtration of NP suspension through a 0.2 μm filter [14]. The hydrodynamic diameter and surface charge of these NPs were measured using a Malvern Zetasizer. Fifteen runs were performed for both the NP diameter and zeta potential measurements. An aliquot of the collected NP fraction was lysed with acetonitrile: water (50:50) mixture, centrifuged, and the drug content was measured using an HPLC system (Waters Corp.). The mobile phase was composed of a mixture of acetonitrile: water (60:40) at the flow rate of 1 ml/min. For in vitro drug release, NPs, with the equivalent of 500 μg drug content, were enclosed into a dialysis bag (MWCO −3.5 kDa) and suspended into 50 mL PBS containing + 1% v/v Tween 80 solution for the sink conditions. Samples (1 mL) were drawn at different time points (0–150 h), and the media was replaced with fresh media. Samples were analyzed for drug content using HPLC.
Cell viability assay
DAOY (ATCC) and HD-MB03 cells (Leibniz Institute, Germany) were cultured in EMEM, and DMEM media, respectively, supplemented with 10% fetal bovine serum (FBS), penicillin (50 U/mL), and streptomycin (50 μg/mL). For cell viability assay, DAOY and HD-MB03 cells were seeded in a 96-wells plate at a density of 3 × 103/well containing 200 μL growth medium and then treated with MDB5 or SF2523 designated doses for 24–48 h. The cytotoxic effect of drugs was determined by MTT assay according to the method reported [15]. The Half-maximal inhibitory concentrations (IC50) values were measured using GraphPad Prism software (version 7.0). For combination index (CI), each drug was tested alone and at fixed ratios with 12 two-fold serial dilutions and evaluated using the MTT assay conditions described above. The IC50 was calculated for each drug alone and their respective fixed concentration ratios using software COMPUSYN.
Cell cycle analysis and apoptosis assay
The cell cycle was carried out using FxCycle™ propidium iodide (PI)/RNase staining solution kit (Invitrogen, Waltham, MA). Briefly, samples were incubated with drugs for 24–48 h. Next, cells were collected and fixed with 70% ethanol for 3 h. Samples each containing ~ 1 × 106 cells in suspension were incubated with 0.5 mL of FxCycle™ PI/RNase staining solution for 30 min in the dark. Cells were subjected to flow cytometry (ARIA-II, BD Biosciences) using 488 nm, 532 nm, or similar excitation and collect emission using a 585/42 bandpass filter or equivalent. For apoptosis assay, cells were seeded onto 24-well plates and treated with DMSO (control), MDB5, SF2523, and their combination and incubated for 24 h. The cells were collected and suspended for 10 min with a solution of 5 μL annexin V antibody diluted in a 200 μL binding buffer. Subsequently, the cells were then incubated with 10 μL propidium iodide (PI), and the samples were analyzed using flow cytometry.
Colony formation assay
DAOY and HD-MB03 (500 cells/well) were cultured overnight. The next day, cells were treated with MDB5, SF2523, or their combination at their IC50 for 48 h, washed twice with PBS and added fresh media. After 10–14 days, colonies were fixed in formalin and stained with 0.5% crystal violet, and stained colonies were dissolved in 10% acetic acid solution to measure the optical density.
Western blot analysis
Protein was extracted from the cells with RIPA lysis buffer (Millipore Sigma) and centrifuged at 14,000 rpm for 20 min at 4 °C. Denatured proteins (50 μg/lane) were separated using SDS-PAGE gels (7–15%) and electro-transferred to a polyvinylidene difluoride (PVDF) membrane (Invitrogen). After incubating with blocking buffer (LI-COR) for 1 h, the membrane was probed with antibodies (1:1000) of interest overnight at 4 °C, followed by secondary IR dye-conjugated antibodies (1:10000, LI-COR) for 1 h at RT. all the membranes were probed with β-actin for loading control. Blots were visualized and quantified using iBright FL1000 (Thermo Fisher Scientific, Inc.)
Cell uptake studies
DAOY cells at a density of 50,000/well were seeded into 24 well plates overnight, washed with PBS, and incubated with media containing MDB5 or SF2523 loaded COG-133-NPs or non-targeted NPs. After a designated time, the cells were washed with PBS, counted to the same number, homogenized in 1 mL HPLC grade water, and spiked with MDB57 or GDC-0449 as an internal standard (IS) for MDB5 and SF2523, respectively. Subsequently, proteins were precipitated by adding cold acetonitrile (1.0 mL) followed by vortexing and high-speed centrifugation (15000 rpm) for 15 min. The supernatant was evaporated and reconstituted with 250 μL of water: acetonitrile: (40:60, v/v). The mass spectrometer conditions were for MDB5 (m/z 568.2→341.2) and MDB7 (m/z 604.2→198.3), SF2523 (m/z 372.0→316.1) and GDC-0449 (m/z 421.1→139.0). The separation was done using Shimadzu LC-20 AT equipped with XTerra Shield RP18 Column, 125Å, 5 μm, 4.6 mm × 250 mm (Waters Corp). The elution solvent was a mixture of water (containing 1% formic acid): acetonitrile (40:60, v/v), and a flow rate of 1.0 mL/min was used.
Animal studies
The Institutional Animal Care and Use Committee (IACUC) of the University of Nebraska Medical Center approved all the animal-related experiments and met all the federal guidelines. DAOY cells were transduced with a green fluorescent protein (GFP) and luciferase-expressing lentiviral vector. Stable GFP expression was used for cell sorting to obtain >95 % luciferase-expressing cell population. The orthotopic MB tumor was established by injecting the luciferase-expressing MB cells stereotaxically, 2 mm deep into the cerebellum, as described earlier [13]. Briefly, under deep anesthesia, the skull of NSG mice (male or female, 6–8-week-old) was exposed, and the animal was mounted on a stereotaxic frame. After finding cerebellum location in reference to bregma, a small hole (2 mm) was carefully drilled in the skull avoiding penetration into meningeal membranes. The anatomical landmarks on the skull were located 1 mm posterior of the lambdoid suture of the scalp over the cerebellum and 1 mm lateral of the sagittal suture. Cells (1×105) suspended in 10 μL PBS was injected using a Hamilton syringe slowly at the rate of 50 μL/h. Tumor growth was monitored using IVIS imaging.
Biodistribution of NPs in the MB tumor
Biodistribution of MDB5 and SF2523 loaded in COG-133 peptide decorated and non-targeted NPs was determined in the tumor and other major organs in orthotopic MB-bearing mice. NPs were injected intravenously (i.v.) with drugs at the dose of 20 mg/kg each, and at 6 and 24 h after injection, blood was withdrawn via cardiac puncture, and the brain and other major organs were collected. Approximately 50 mg tissue samples were homogenized in 1 mL water pre spiked with MDB7 or GDC-0449 as an internal standard (IS) for MDB5 and SF2523. Samples were analyzed with LC-MS/MS as described above.
In vivo antitumor effect
After establishing an orthotopic MB tumor, mice were randomly divided into the following seven groups (n = 6): i) mice intravenously injected with PBS; ii) mice treated i.v., with MDB5 and SF2523 solution (propylene glycol (20 % v/v) + Cremophore EL (30% v/v) + 5% dextrose solution (50% v/v), iii) MDB5 loaded NPs; iv) SF2523 loaded NPs; v) NPs loaded with MDB5 and SF2523; vi) MDB5 loaded COG-133-NPs; vii) SF2523 loaded COG-133-NPs, and viii) COG-133 NPs loaded with MDB5 and SF2523 at the dose of 20 mg/kg every third day. The treatment was performed seven times. During treatment, the body weights of the mice were measured before every injection. The tumor growth was monitored by IVIS imaging. For histological examination, the major organs of representative mice from each group of animals were dissected out and stained with H&E. Deidentified frozen MB patient tumors, and five normal cerebellums were obtained from UNMC Tissue Procurement Shared Resource. Tissues after fixing with 10% paraformaldehyde solution for 24 h, embedded in 5 μm thick sections of paraffin were stained for different proteins. Stained slides were imaged at 40 × magnification using an iScan HT Slide Scanner (Ventana Medical Systems, Inc, AZ).
Statistical analysis
Comparisons between two different groups were analyzed by Student’s t-test, and one-way ANOVA was performed for three or more groups. The data obtained were shown as the mean ± S.D., and p < 0.05 was considered statistically significant.
Results
MB patient and orthotopic tumor tissues upregulate PI3K/AKT and Hh signaling pathways
Since p-AKT and p-PI3K are aberrantly expressed in human MB tissues, we compared their expression levels using patient adjacent normal tissue samples. We observed minimal p-AKT and p-PI3K staining in the normal cerebellum (n = 5), whereas their staining was prominent in the MB tissues (Figure S1). We found that GLI1 and MYCN protein expression was also upregulated in these tissue samples but not in normal cerebral tissues (Figure S1A). Overexpression of Hh ligands, PTCH-1, SMO, and GLI1 was much higher in SHH-MB cell line DAOY than Group 3 MB cell line HD-MB03 normal human brain cells SVG p12 (CRL-8621, ATCC) had very low expression of these Hh ligands (Figure S1B).
MDB5 and SF2523 synergistically inhibit MB cell proliferation and colony formation
MDB5 and SF2523 inhibited DAOY and HD-MB03 cell proliferation concentration-dependent manner, as determined by MTT assay (Figure 1A&B). We found SF2523 was more cytotoxic to MB cells with IC50 of 12.6 and 5.8 μM in DAOY and HD-MB03 cells than MDB5 with IC50 of 63.2 and 95.3 μM in these cells, respectively. We also determined the synergistic effect of MDB5 and SF2523 combinations on cell viability by incubating DAOY and HD-MB03 cells with these two drugs at different concentrations (Figure 1C&D). In DAOY cells, the IC50 of combination was achieved at 30.0 μM MDB5 and 6.0 μM SF2523, whereas in HD-MB03 cells, IC50 of combination was achieved at 25.0 μM MDB5 and 3.0 μM SF2523 concentrations. To determine the synergistic effect of combination treatment on colony formation, we treated DAOY and HD-MB03 cells with IC50 of MDB5 and SF2523. Both SF2523 and MDB5 significantly inhibited the colony formation of DAOY and HD-MB03 cells than the control, and the inhibition was significantly higher for their combination therapy (Figures 1E&F and S2).
Figure 1. MDB5 and SF2523 synergistically inhibit HD-MB03 and DAOY cell proliferation and colony formation.
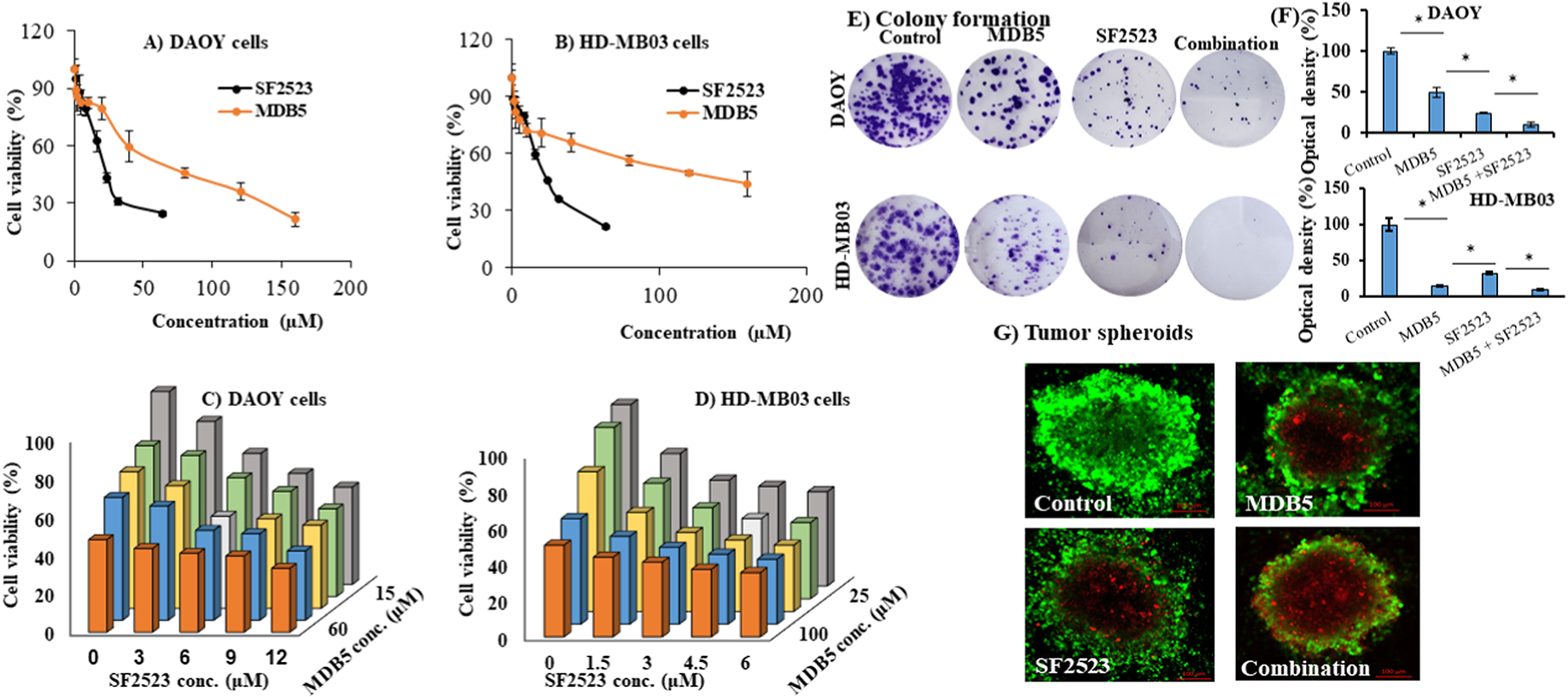
(A-B) MDB5 and SF2523 inhibit DAOY and HD-MB03 proliferation in a dose-dependent manner. (C-D) Synergistic effect of MDB5 and SF2523 combination on the viability of DAOY and HD-MB03 cells. (E-F) The colony formation capacity of DAOY and HD-MB03 cells were synergistically inhibited by MDB5 and SF2523 even when incubated at their IC50 values (63.2 μM MDB5 and 12.6 μM SF2523 for DAOY cells and 95.1 μM MDB5 and 5.8 μM SF2523 for HD-MB03 cells). Quantification of colony formation by measuring the optical density of (O.D.) of crystal violet of the stained cell colonies with 10% acetic acid solution at 590 nm. (G) A representative figure showing higher cell apoptosis in tumor spheroids generated using DAOY cells by MDB5 and SF2523 combination than individual drugs. Confocal laser scanning microscopy (CLSM) images were captured at a depth of 15 μm.
We treated DAOY cell tumor spheroids with MDB5, SF2523 and their combination to mimic human MB physiology. The cells were visualized under a confocal microscope to determine the % of FITC labeled CalceinAM positive live cells, and propidium iodide (PI) stained dead cells. Treatment of tumor spheroids with MDB5 and SF2523 combination for 10 days exhibited the highest percentage of apoptotic cell death (Figure 1G).
MDB5 and SF2523 kill MB cells in SMO dependent and independent manner, respectively
GLI1/2 and PTCH1 mRNA expression was significantly inhibited by MDB5 and SF2523 as determined by real-time RT-PCR after 48h incubation in both DAOY and HD-MB03 cells (Figure 2A&B). SF2523 did not affect the SMO mRNA levels alone or in combination with MDB5. Further, both these drugs showed no change. Like other BRD4 inhibitors, including JQ1, SF2523 inhibits BRD4 binding to acetylated histones and inhibits growth-promoting transcription factor MYC without affecting BRD4 expression (Figure 2A&B). CD15+ cells have been shown to recapitulate the heterogeneity of Ptch+/− MB, thus can regenerate all the tumor cell types (16). Therefore, we isolated CD15+ cells from DAOY cells using flow cytometry and treated them with MDB5, SF2523, and their combination for 48h. We observed MDB5, SF2523, and their combination downregulated CD133, SOX2, and OCT4 expression at mRNA levels (Figure 2C).
Figure 2. MDB5 and SF2523 kill MB cells in SMO dependent and independent manner, respectively.
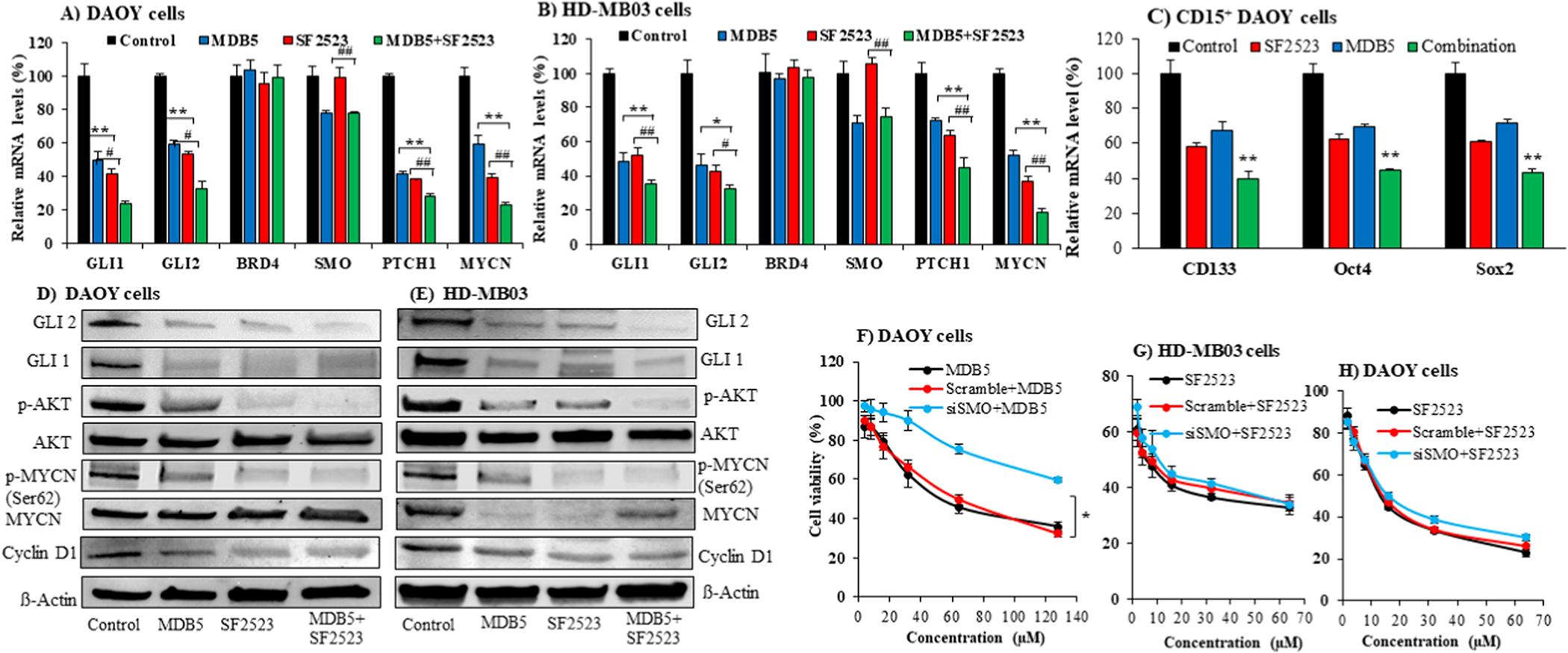
(A-B) Inhibition of GLI1/2, PTCH1, and SMO mRNA expression after incubation of DAOY and HD-MB03 cells with MDB5 (30 μM for DAOY cells and 50 μM for HD-MB03 cells) and SF2523 (12 μM for DAOY cells and 6 μM for HD-MB03 cells) alone and their combination for 48 h as determined by real-time RT-PCR (*, p < 0.05, **, p < 0.01, compared to MDB5, #, p < 0.05, ##, p < 0.01, compared to SF2523). (C) MDB5, SF2523, and their combination also inhibit cancer stem cell markers such as CD133, OCT4, and SOX2 expression after incubation of CD15+ DAOY cells (**, p < 0.01, compared to MDB5, ##, p < 0.0 compared to SF2523). (D-E) GLI1, GLI2, total AKT, p-AKT, MYCN, p-MYCN, Cyclin D1 protein expression in DAOY cells and HD-MB03 cells after treated with MDB5, SF2523, and combination for 48 h. (F) SMO gene silencing with siSMO transfection of DAOY cells inhibits the cell-killing ability of SMO targeting MDB5. (G) SMO gene silencing in HD-MB03 cells had little effect on the cell-killing ability of SF2523, which targets oncogenes MYCN and PI3K/AKT, but not SMO. H) SMO gene silencing in DAOY cells had an insignificant effect on the cell-killing ability of SF2523, which targets oncogenes MYCN and PI3K/AKT, but not SMO.
As expected, MDB5 treatment decreased GLI1 and GLI2 expression at protein levels in DAOY cells as determined by Western blot analysis (Figure 2D, Figure S3A&B). Treatment of DAOY and HD-MB03 cells with SF2523 alone or with MDB5 showed a significant decrease in p-AKT and p-MYCN without affecting the total proteins of both, whereas treatment of DAOY cells with MDB5 alone did not affect these proteins (Figure 2D&E). We probed against phospho MYCN Ser62 antibody in our Western blot assay, thus observed a decrease in p-MYCN (Figure 2D, Figure S3C). Furthermore, their combination showed an additive effect in inhibiting GLI and GLI2 proteins, reconfirming the RT-PCR results.
To confirm that MDB5 shows its effect through SMO inhibition, we first determined the activity of siSMO on SMO expression by incubating DAOY and HD-MB03 cells with siSMO or scrambled siRNA after complex formation with lipofectamine 2000®. As expected, siSMO but not scrambled siRNA inhibited SMO in a dose-dependent manner (Figure S4). Then, we transfected DAOY cells with siSMO (20 nM) using lipofectamine 2000®, before treatment with MDB5. SMO silencing in these cells led to a significant decrease in MDB5 cell killing efficiency compared to scrambled siRNA transfected cells (Figure 2F). Of note, because the silencing effect siSMO was partial, we did not observe complete abrogation of MDB5 activity. However, SMO silencing in DAOY and HD-MB03 cells had no effect on the cell-killing ability of SF2523, which targets MYCN and PI3K/AKT, but not SMO (Figure 2 G&H).
SF2523 and MDB5 arrest MB cells in G0/G1 phase and promote apoptosis
To determine whether the inhibition of cell proliferation by MDB5 and SF2523 is due to cell cycle arrest, we performed cell cycle analysis after their treatment in DAOY cells (Figures S5). Both MDB5 and SF2523 increased the percentage of cells in the G0/G1 phase (75.01% for MDB5 and 74.77% for SF2523) significantly compared to DMSO treated control cells (52.23%) (Figure 3A). Similar effects exhibited on HD-MB03 cells after treatment with MDB5 and SF2523, with 48.88% and 60.84% cells, respectively, were found in G0/G1 phase compared to DMSO treated cells (26.47% cells) (Figure 3B). The combination treatment further increased cell population in G0/G1 phase to 84.10% for DAOY cells and 71.20% for HD-MB03 cells (Figure 3A&B and Figure S5). There was a significant decrease in the percentage of cells in the G2/M phase from 30.79% in control cells to 13.10% in MDB5 treated cells and 12.04% in SF2523 treated cells 9.33% in their combination. The same trend was reserved in HD-MB03 cells with a decrease in the G2/M phase from 34.04% to 19.44% in MDB5 treated cells, 13.11% in SF2523 treated cells, and 12.03% in their combination-treated cells.
Figure 3. MDB5 and SF2523 synergistically arrest cells in G0/G1 phase and promote apoptosis in MB cells.
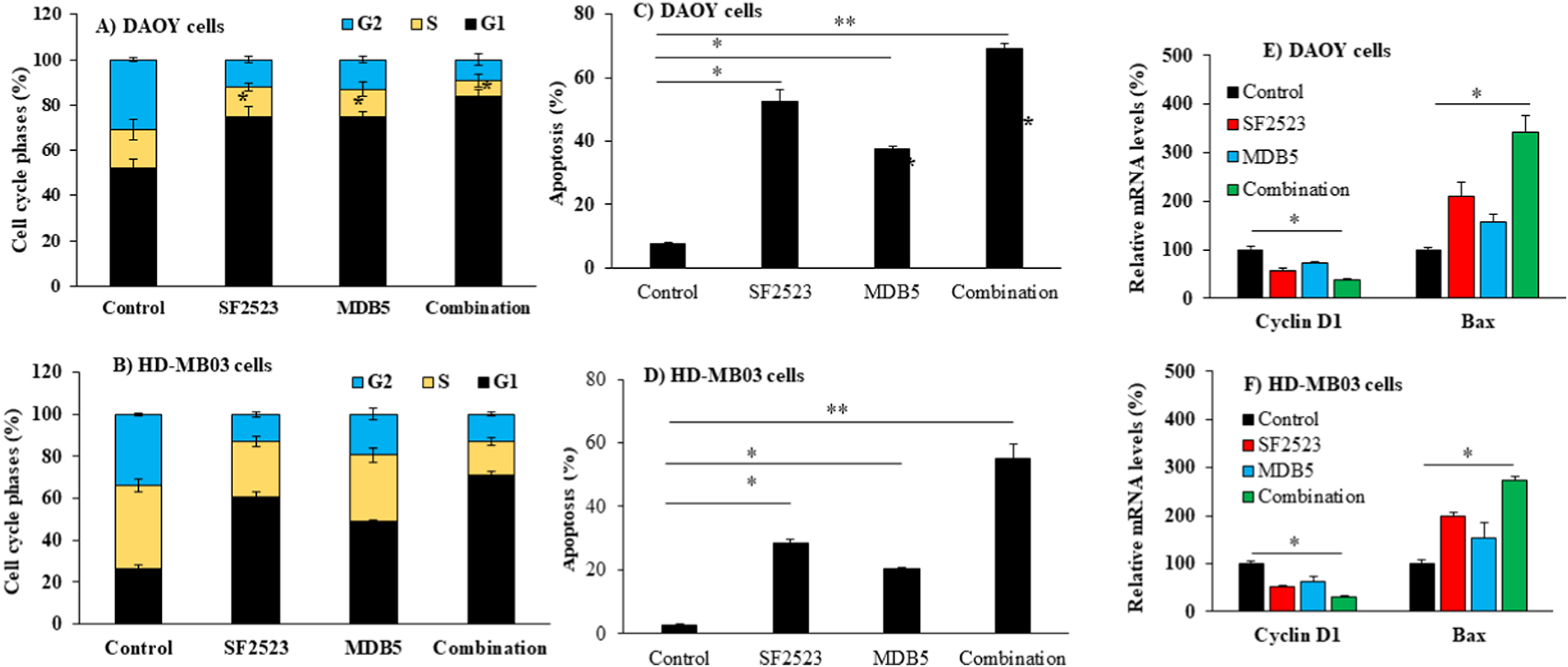
(A-B) Cell cycle analysis after treatment with SF2523 and MDB5 to DAOY and HD-MB03 cells. Induction of apoptosis by SF2523 and MDB5 in (C) DAOY cells and (D) HD-MB03 cells after 48 h of incubation with these two drugs. (E-F) Combination of MDB5 and SF2523 synergistically upregulated Bax and downregulated Cyclin D1 as determined by real-time RT-PCR (n=3, *, p < 0.05, **, p < 0.01, ns, p > 0.05).
The percentage of apoptotic DAOY and HD-MB03 cells after treatment with MDB5, SF2523 and their combination (Figure S6). Figures 3C&3D illustrate that both MDB5 and SF2523 could effectively induce apoptosis in DAOY cells to 37.49%, and 52.35%, respectively, then the control cells (7.45%). Combination treatment resulted in a significantly higher percentage of apoptotic cells (69.04%), as shown in Figure 3C. Similarly, in HD-MB03 cells the percentage of apoptotic in control cells was only 2.27% compared to 20.34% in MDB5 treated, 28.57% in SF2523 treated, and 55.27% in the combination treated cells (Figure 3D).
Next, changes in the expression of cell cycle and apoptosis related gene was determined. MDB5 and SF2523 in combination markedly reduced cyclin D1 expression, while the Bax level was found to be enhanced, which were significant change from the treatment with either MDB5 or SF2523 alone (Figure 3E&F). Taken together, these findings suggest both MDB5 and SF2523 induced G0/G1 cell cycle arrest through modulating the critical signals of DNA damage.
MDB5 and SF2523 are efficiently encapsulated into nanoparticles and release slowly
Since both MDB5 and SF2523 are lipophilic compounds (Table S1), they are excellent candidates for encapsulation into NPs. We used our copolymer mPEG-b-PCC-g-DC for nanoparticle formation as described before [16]. These drugs were encapsulated individually into the NPs by film hydration. NPs exhibited the mean particle size of 80 ± 10 nm with low polydispersity index (PDI) of 0.218 (Figure 4A). The ζ potential of NPs was +0.9 ± 0.1 mV. Previously, we have shown that the critical micelle concentration (CMC) of PEG-PCC-PCD polymer was around 10−8 M indicating high stability of NPs upon dilution [17]. Further, adequate thermodynamic stability of drug loaded micelles in PBS as a function of time was also reported [15]. Drug-loaded NPs were well-dispersed and had a spherical shape with the mean size of 60 ± 10 nm, as determined by transmission electron microscopy (TEM) images (Figure 4B). As determined by HPLC, the encapsulation efficiency (EE) and drug loading capacity of MDB5 in NPs were 85.9 ± 0.3% and 8.7 ± 1.0%, whereas those of SF2523 were 70.3 ± 0.5% and 6.5 ± 1.2% indicative of an effective loading and the favorable structural stability. Therefore, the method used here showed a relative high efficiency and good dispersion quality. There was sustained release of these drugs from NPs in PBS containing 1% Tween 80, wherein 100% of MDB5 was released in 50 h, but only 60% of SF2523 was released in 80 h, respectively (Figure 4C).
Figure 4. Characterization of nanoparticles loaded with MDB5 or SF2523.

A) Particle size distribution by DLS. B) NPs morphology by TEM. C) The cumulative drug release profile of MDB5 and SF2523 in PBS (1% Tween 80) (Mean ±S.D., n=3. D) Cytotoxicity of MDB5 and E) SF2523 NPs in DAOY cells. MB cells viability was determined using the MTT assay. MDB5 and SF2523 both showed a dose-dependent loss of cell viability (mean ± S.D., n = 5).
Effect of MDB5 and SF2523 loaded NPs on cell proliferation was determined after incubating these drugs with DAOY and HD-MB03 cells for 48h. As shown in Figures 4D and 4E, all tested groups exhibited concentration dependent cytotoxicity. The higher IC50 values for drug-loaded NPs was likely because NPs first need to be internalized via endocytosis before releasing their drug payload. This trend is usually reversed in vivo when long circulation and residence times in the tumor become major factors, and NPs are more effective than free drugs.
COG-133 decorated NPs achieve higher cellular uptake and biodistribution to orthotopic MB tumor site
Since free drugs do not cross the BBB effectively, we synthesized COG-133 peptide conjugated PEG-b-PBC copolymer and characterized with 1H NMR as described previously (Figure S7A) [13]. We prepared targeted NPs by taking optimized ratios (20% w/w) of COG-133-PEG-b-PBC and mPEG-b-PCC-g-DC copolymers and evaluated their size, drug loading, and stability. These parameters were non significant between targeted and non-targeted NPs. Similar drug release profiles could be attributed to the same hydrophobic segments of NPs and the same release mechanism. The effect of COG-133 targeting on the cellular uptake of NPs was determined by incubating them with DAOY cells followed by measuring drug concentration by LC-MS/MS. COG-133-NPs displayed significantly higher uptake of MDB5 and SF2523 than those incubated with non-targeted NPs (Figure 5A&5B). Further, recently we reported that pre incubation with free COG-133 peptide reduced the cellular uptake of targeted COG-133-NPs indicating the uptake is via receptor-mediated endocytosis.
Figure 5. COG-133 peptide decorated NPs significantly enhance the cellular uptake in vitro and delivery to MB tumors after systemic administration.
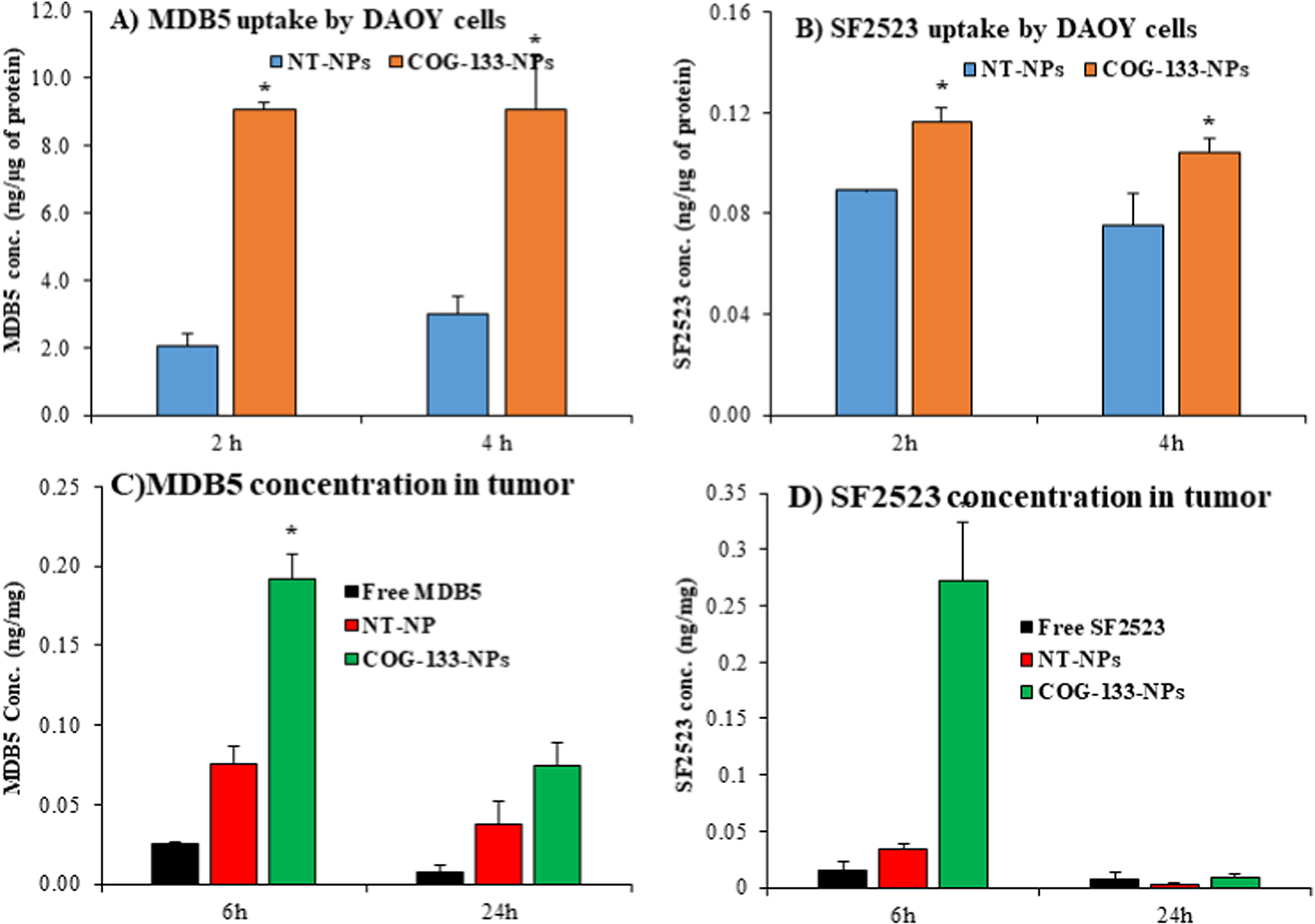
A-B) cellular uptake of MDB5 and SF2523 in DAOY cells at 2 and 4 h post-incubation of drug-loaded COG-133-NPs. C-D) MDB5 and SF2523 concentrations in the brain tumor at 6 and 24 h after systemic administration of drug-loaded COG-133-NPs into orthotopic MB tumor-bearing NSG mice at the dose of 10 mg/kg (mean ± S.D., n= 4, p<0.05).
COG-133 peptide decorated nanoparticles effectively deliver MDB5 and SF2523 to MB tumor in an orthotopic mouse model
We established an orthotopic MB tumor by injecting luciferase expressing DAOY cells stereotaxically into the cerebella of NSG mice, which were imaged by IVIS to monitor tumor growth. DAOY cells generated orthotopic MB tumor showed upregulation of SHH and GLI2 compared to adjacent normal cerebellum tissue (Figure S1C).
For biodistribution, when the bioluminescence reached 1×106 p/sec/cm2/sr mice were injected with free SF2523, free MDB5, non-targeted, and COG-133-NPs encapsulating SF2523 and MDB5. Free SF2523 was not very efficient in crossing the BBB, and low concentration was detected at both 6 h (0.015 ± 0.005 ng/mg) and 24 h (0.0007 ± 0.0001 ng/mg) time points. The decoration of COG-133 to NPs significantly increased SF2523 cerebellar concentrations at 6 (0.272 ± 0.007 ng/mg) and 24 h (0.08 ± 0.001 ng/mg) (n=4) post systemic administration (Figure 5C). Non-targeted NPs exhibited a low concentration of SF2523 in the cerebellum after 6 h (0.033 ± 0.001 ng/mg) and 24 h (0.003 ± 0.0005 ng/mg), respectively. Similarly, MDB5 concentration was significantly enhanced at 6 h (0.19 ± 0.02 ng/mg) and 24 h (0.075 ± 0.001 ng/mg) by COG-133-NPs (Figure 5D). Non-targeted NPs exhibited a low concentration of MDB5 in the cerebellum after 6 h (0.08 ± 0.002 ng/mg) and 24 h (0.035 ± 0.001 ng/mg), respectively (Figure 5D). As expected, the free MDB5 concentration was 0.03 ± 0.001 ng/mg at 6 h and 0.01 ± 0.001 ng/mg at 24 h. These results suggest that targeted NPs resulted in significant increased MDB5 and SF2523 delivery to the brain.
Combination therapy of MDB5 and SF2523 encapsulated into COG-133-NPs exerts superior efficacy against orthotopic MB tumor growth
When orthotopic SHH-MB bearing NSG mice generated with stably luciferase expressing DAOY cells were treated with free solution of MDB5 and SF2523 or with COG-133-NPs or non-targeted NPs loaded with MDB5, SF2523 and their combination at the dose of 20 mg/kg for 3 weeks after every 3 days. Treatment with COG-133-NPs exhibited inhibition in tumor growth, as evidenced by decreased bioluminescence signals compared to the mice treated with their non-targeted NPs and non-treated mice (Figures 6A and Figure S8). Notably, SF2523 loaded NPs was not effective with and without targeting peptide. The most significant antitumor efficacy was observed with combination therapy with COG-133-NPs, which suggests that COG-133-NPs could be used to treat tumors effectively (Figure 6B). An analysis of body weight variations generally defines the safety of different therapy regimens. Figure 6C indicates that COG-133-NPs showed a very low systemic toxicity.
Figure 6. Antitumor efficacy of COG-133-NPs loaded with MDB5 and SF2523 after systemic administration into orthotopic MB bearing NSG mice at the dose of 20 mg/kg.
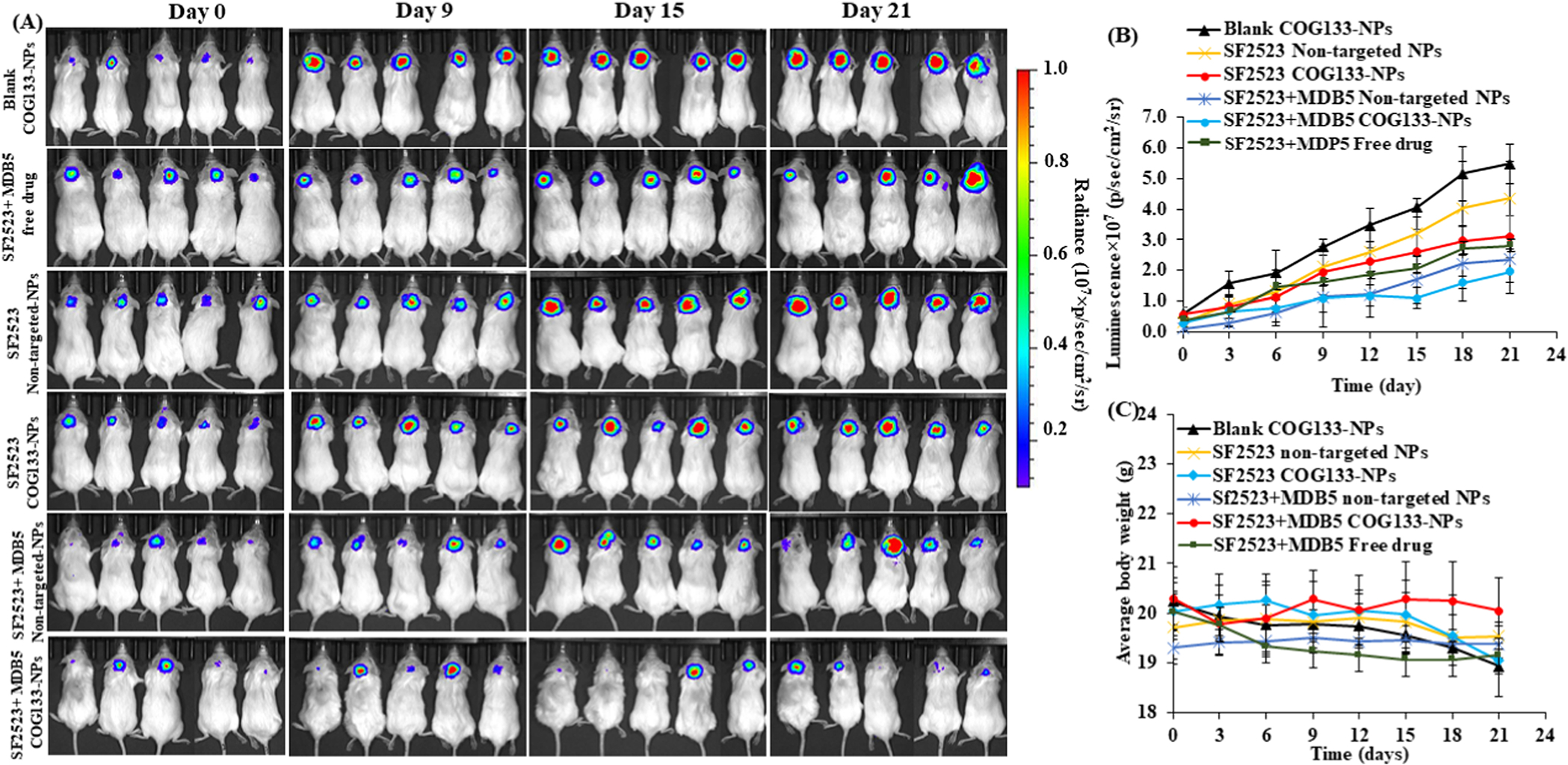
A) Measurement of bioluminescence images of tumors at different times (n=5), B) IVIS signal intensity (photons/s/cm2/sr) over time after systemic administration. C) Changes in mouse body weight during treatment (mean ± S.D., n=5).
Hematoxylin and eosin (H & E) staining of brain tissue sections confirmed the extent of MB tumor growth, which was significantly decreased by the combination treatment group (Figure 7). GLI1 expression was high in untreated, and blank NPs treated samples compared to COG-133 NPs treated samples. The MB tumor treated by MDB5 and SF2523 loaded COG-133-NPs significantly decreased p-MYCN expression compared to the control, free drug, and non-targeted NP groups. MDB5 and SF2523 loaded COG-133-NPs treated tumors showed a significant decrease in proliferation marker Ki-67 while a significant increase in apoptotic marker cleaved caspase 3 in the tumor tissue compared to other treatment groups (Figure 7). These results suggest COG-133-NPs efficiently deliver MDB5 and SF2523 to MB tumors, resulting in maximum tumor reduction without systemic toxicity.
Figure 7.

Hematoxylin and eosin (H&E) and immunohistochemical staining of MB tissues for GLI1, MYCN, Ki-67, and cleaved caspase-3 after systemic administration of SF2523 and MDB5 in solution and in COG-133-NPs and non-targeted NPs into orthotopic MB tumor-bearing NSG mice. Scale bar 100 μM, inset image 20X.
Discussion
About 18% to 20% of childhood brain tumors are MB, making it the most common brain tumor in children (https://medulloblastoma.org/medulloblastoma-statistics/). MB is known for very few mutations and genetic changes compared to adult tumors. During normal cerebellar development, the SHH pathway plays a vital role in granule neuron precursors (GNPs); however, its inappropriate activation leads to MB formation. IGF-II activates PI3K signaling and thus synergizes with SHH to enhance MB progression. We observed higher p-PI3K and p-AKT in MB patient samples than adjacent normal cerebellum (Figure S1). In some cases, treatment of MB-bearing mice with SHH inhibitors is sufficient to cause tumor regression, but their prolonged use develops chemoresistance.
Recently, we synthesized a potent SHH inhibitor, MDB5, which showed improved therapeutic efficacy than its parent compound vismodegib (GDC-0449) [9]. Due to an additional 2-pyridylmethyl group in MDB5 showed additional binding with SMO protein than vismodegib [9]. Further, targeting BET proteins was previously shown to inhibit MYCN-amplified tumors; therefore, we selected a BRD4/PI3K dual inhibitor SF2523 to inhibit MYCN expression in MB. Sustained BRD4 levels were found in both the cell lines, suggesting that MYCN inhibition by SF2523 or MDB5 do not affect the overall BRD4 level (Figure 2A&B). This might be because small-molecule BET inhibitors prevent engagement of the BET protein with chromatin by competitively binding to acetylated lysine residues in the BET bromodomain. In two independent reports, Wen et al. [18] and Andrews et al. [19] have also reported similar observations.
Treatment of DAOY cells and HD-MB03 cells with MDB5 and SF2523 exhibited a significant decrease in GLI1, GLI2, and p-MYCN protein expression (Figures 2D&E). These two drugs showed different levels of cell killing ability, possibly due to different levels of SHH and MYCN expression by these two cell lines. DAOY cells treated with siSMO showed resistance to MDB5 treatment, indicating its exclusive mode of action is through SMO binding. Importantly, DAOY cells and HD-MB03 cells showed no resistance to SF2523 after co-incubation with siSMO (Figure 2G). Interestingly, SF2523 was equipotent in decreasing GLI1 and GLI2 proteins (Figure 2D). These findings are because these cells are less dependent on Hh signaling, and SF2523 inhibited GLI activity by SMO-independent pathways. Conversely, the combination of these could be less prone to SMO resistance. The higher expression levels of apoptosis-associated protein BAX exhibited following drug treatment were also reflected in the apoptosis assay (Figures 3C–F). GLI1 overexpression leads to BCL-2 upregulation and BAX downregulation in tumor cells [20]. Therefore, MDB5 mediated GLI1 downregulation resulted in decreased BCL-2 expression and increased Bax expression [21].
SHH signaling exerts a proliferative effect in MB by amplifying MYCN; the latter activates cyclins while repressing cyclin-dependent kinase inhibitors [22]. Recently, a PI3K inhibitor BEZ235 has been shown to significantly downregulate the phosphorylated levels after treatment of both SHH- and MYC-driven MB cells with PI3K-mTOR pathway molecules [23]. Further, MYC stability is regulated by posttranslational phosphorylation at Ser62 and Thr58. Phosphorylation at Ser62 by ERK stabilizes MYC, while its phosphorylation by BRD4 at Thr58 leads to subsequent ubiquitin-mediated proteolysis [24].
MYCN is directly regulated by GLI proteins in SHH-MB, while BRD4 occupies the GLI promoter and inhibits its expression. Further, activation of the PI3K/AKT/mTOR axis pathway could increase MYCN [25,26]. SHH pathway also cooperates with PI3K signaling and promotes proliferation [27]. Deregulated Hh signaling in cerebellar cells upregulates Cyclins D1 and D2, and thus regulate the proliferation of GNPs and start preneoplastic lesions to MB [28]. Further, MYCN protein also promotes cell cycle progression by upregulating cyclin D1. Consistent with this notion, inhibition of the SHH pathway by MDB5 or inhibition of MYCN activity by SF2523 resulted in Cyclin D1 expression in both DAOY and HD-MB03 cell lines (Figure 3E&F).
While NPs have shown promise for drug delivery to the tumor via passive targeting, several challenges remain to be overcome for controlling their biodistribution to the brain. In general, NPs predominantly accumulate in the cells of the reticuloendothelial system (RES), such as in the liver and spleen, as well as in the tumor and inflammation site. NPs coated with poly(ethylene glycol) (PEG) accumulate more efficiently in the brain with a compromised BBB compared to uncoated NPs, partly due to the greatly improved blood circulation time and the “stealth” properties (avoidance of internalization by RES cells) of PEG-coated NPs [29]. Apart from enhancing drug transport across the BBB, PEG-coated NPs can also improve the penetration of NPs to overcome the barrier created by the extracellular matrix (ECM) to reach the target cells [30,31]. Further, to make these drugs available to the tumor efficiently in vivo, we investigated whether COG-133-NPs loaded with MDB5 and SF2523 can effectively treat orthotopic SHH-MB-bearing mice. ApoE is a lipoprotein that is a significant component of cholesterol transporting carriers in the brain. COG-133-NPs are likely to be endocytosed into the BBB endothelium via low-density lipoprotein (LDL) receptors and transcytoses to the brain. In mPEG-b-PCC-g-DC copolymer, DC conjugated to a polycarbonate backbone with an ester bond makes this lipopolymer biodegradable with CO2 and alcohol as end products, hence no side effects. However, the selective internalization of NPs can be enhanced by functionalization with receptor binding ligands such as COG-133 here. Although we have not determined APOE expression in DAOY cells, but previous studies show the presence of ApoE receptors on several intracranial tumors including MB [32,33]. We have observed similar results in our recent publication [13]. Our results show that COG-133 targeted NPs loaded with MDB5 or SF2523 effectively deliver payloads to MB cell lines in vitro and in orthotopic MB in animals more efficiently than non-targeted drug-loaded NPs (Figure 5).
Combination therapy of two or more drugs is used to lower individual drug doses achieve better therapeutic outcomes with minimal systemic toxicity [34]. Since MDB5 inhibits Hh signaling and SF2523 is a dual inhibitor of PI3K and BRD4, we decreased the dose of MDB5 and SF2523 from 20 mg/kg to 10 mg/kg for their combination therapy to determine whether there is synergism in their therapeutic efficacy. Our data suggests that the free drug combination was not quite effective and rather showed progressive disease during the treatment (Figure 6A&B). As expected, MDB5 and SF2523 loaded COG-133-NPs exhibited the highest synergy in tumor regression after systemic administration (Figure 6A&B), with no loss of mouse weight (Figure 6C) after the treatment. Targeted NPs significantly decreased MDB5 target protein GLI1, SF2523 target protein p-MYCN, and proliferation marker Ki-67 expression. However, they increased cleaved caspase-3 expressions in the combination treatment group (Figure 7), suggesting higher concentrations of these drugs at the tumor site (Figure 5C&D).
In conclusion, the key hurdles to MB therapy are the BBB, resistance, and safety. Systemic administration of MDB5 and SF2523 loaded COG-133-NPs effectively delivered these drugs to MB tumor sites and significantly inhibited MB progression compared to non-targeted NP formulations.
Supplementary Material
Acknowledgments
The NIH (1R01NS116037), pediatric Cancer Research Group of the University of Nebraska Medical Center and Children’s Hospital (LB805), Team Jack Foundation, Nebraska DHHS LB506, Nebraska Research Initiative (NRI) Program, and the Faculty Start-up fund to Ram Mahato are duly acknowledged for providing financial support for this work.
Footnotes
Declaration of competing interest
The authors are declaring no financial interests/personal relationships which may be considered as potential competing interests:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability
The authors declare that all the data related to this study are available within the paper and its supplementary information.
References
- 1.Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012;488:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar V, Kumar V, McGuire T, Coulter DW, Sharp JG, Mahato RI. Challenges and Recent Advances in Medulloblastoma Therapy. Trends Pharmacol Sci 2017;38:1061–1084. [DOI] [PubMed] [Google Scholar]
- 3.Coni S, Mancuso AB, Di Magno L, Sdruscia G, Manni S, Serrao SM, et al. Corrigendum: Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci Rep 2017;7:46645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014;25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A 2007;104:8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A 2010;107:13432–13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res 2015;21:505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015;27:327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar V, Chaudhary AK, Dong Y, Zhong HA, Mondal G, Lin F, et al. Design, Synthesis and Biological Evaluation of novel Hedgehog Inhibitors for treating Pancreatic Cancer. Sci Rep 2017;7:1665–017-01942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morales GA, Garlich JR, Su J, Peng X, Newblom J, Weber K, et al. Synthesis and cancer stem cell-based activity of substituted 5-morpholino-7H-thieno[3,2-b]pyran-7-ones designed as next generation PI3K inhibitors. J Med Chem 2013;56:1922–1939. [DOI] [PubMed] [Google Scholar]
- 11.Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, et al. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015;27:837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arvanitis CD, Ferraro GB, Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer 2020;20:26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Kumar V, Lin F, Sethi B, Coulter DW, McGuire TR, et al. ApoE mimetic peptide targeted nanoparticles carrying a BRD4 inhibitor for treating Medulloblastoma in mice. J Control Release 2020;323:463–474. [DOI] [PubMed] [Google Scholar]
- 14.Sezgin Z, Yuksel N, Baykara T. Preparation and characterization of polymeric micelles for solubilization of poorly soluble anticancer drugs. Eur J Pharm Biopharm 2006;64:261–268. [DOI] [PubMed] [Google Scholar]
- 15.Kumar V, Mondal G, Dutta R, Mahato RI. Co-delivery of small molecule hedgehog inhibitor and miRNA for treating liver fibrosis. Biomaterials 2016;76:144–156. [DOI] [PubMed] [Google Scholar]
- 16.Karaca M, Dutta R, Ozsoy Y, Mahato RI. Micelle Mixtures for Coadministration of Gemcitabine and GDC-0449 To Treat Pancreatic Cancer. Mol Pharm 2016;13:1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li F, Danquah M, Mahato RI. Synthesis and characterization of amphiphilic lipopolymers for micellar drug delivery. Biomacromolecules 2010;11:2610–2620. [DOI] [PubMed] [Google Scholar]
- 18.Wen N, Guo B, Zheng H, Xu L, Liang H, Wang Q, et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int J Oncol 2019;55:879–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews FH, Singh AR, Joshi S, Smith CA, Morales GA, Garlich JR, et al. Dual-activity PI3K-BRD4 inhibitor for the orthogonal inhibition of MYC to block tumor growth and metastasis. Proc Natl Acad Sci U S A 2017;114:E1072–E1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gai X, Tu K, Li C, Lu Z, Roberts LR, Zheng X. Histone acetyltransferase PCAF accelerates apoptosis by repressing a GLI1/BCL2/BAX axis in hepatocellular carcinoma. Cell Death Dis 2015;6:e1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Gao S, Wang W, Xia Y, Liang J. Downregulation of NMyc inhibits neuroblastoma cell growth via the Wnt/betacatenin signaling pathway. Mol Med Rep 2018;18:377–384. [DOI] [PubMed] [Google Scholar]
- 22.Enguita-German M, Schiapparelli P, Rey JA, Castresana JS. CD133+ cells from medulloblastoma and PNET cell lines are more resistant to cyclopamine inhibition of the sonic hedgehog signaling pathway than CD133- cells. Tumour Biol 2010;31:381–390. [DOI] [PubMed] [Google Scholar]
- 23.Chaturvedi NK, Kling MJ, Coulter DW, McGuire TR, Ray S, Kesherwani V, et al. Improved therapy for medulloblastoma: targeting hedgehog and PI3K-mTOR signaling pathways in combination with chemotherapy. Oncotarget 2018;9:16619–16633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas WD, Chen J, Gao YR, Cheung B, Koach J, Sekyere E, et al. Patched1 deletion increases N-Myc protein stability as a mechanism of medulloblastoma initiation and progression. Oncogene 2009;28:1605–1615. [DOI] [PubMed] [Google Scholar]
- 25.Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, et al. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res 2006;66:8139–8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cascon A, Robledo M. MAX and MYC: a heritable breakup. Cancer Res 2012;72:3119–3124. [DOI] [PubMed] [Google Scholar]
- 27.Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 2004;131:217–228. [DOI] [PubMed] [Google Scholar]
- 28.Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development 2006;133:3929–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev 2016;99:28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G, et al. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med 2012;4:149ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nance E, Zhang C, Shih TY, Xu Q, Schuster BS, Hanes J. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano 2014;8:10655–10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murakami M, Ushio Y, Morino Y, Ohta T, Matsukado Y. Immunohistochemical localization of apolipoprotein E in human glial neoplasms. J Clin Invest 1988;82:177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu C, Yokota N, Gao Y, Yamamoto J, Tokuyama T, Namba H. Gene expression of growth signaling pathways is up-regulated in CD133-positive medulloblastoma cells. Oncol Lett 2011;2:357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination therapy in combating cancer. Oncotarget 2017;8:38022–38043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all the data related to this study are available within the paper and its supplementary information.