

Biochemical, structural and functional characterization of Iripin-5, the most abundant serpin from the salivary glands of Ixodes ricinus, together with interface analysis, revealed the possible interactions with proteases that are necessary to explain the mechanism of action of serpins.
Keywords: serpins, serine protease inhibitors, Iripin-5, X-ray structure, Ixodes ricinus, tick saliva
Abstract
Iripin-5 is the main Ixodes ricinus salivary serpin, which acts as a modulator of host defence mechanisms by impairing neutrophil migration, suppressing nitric oxide production by macrophages and altering complement functions. Iripin-5 influences host immunity and shows high expression in the salivary glands. Here, the crystal structure of Iripin-5 in the most thermodynamically stable state of serpins is described. In the reactive-centre loop, the main substrate-recognition site of Iripin-5 is likely to be represented by Arg342, which implies the targeting of trypsin-like proteases. Furthermore, a computational structural analysis of selected Iripin-5–protease complexes together with interface analysis revealed the most probable residues of Iripin-5 involved in complex formation.
1. Introduction
The castor bean tick (Ixodes ricinus) has a wide geographical distribution throughout the Northern Hemisphere of Europe, Asia and Africa that points towards its resistance to various environmental conditions. This has helped this tick to become one of the major factors in the spread of zoonotic diseases, as it serves as a vector for multiple vector-borne pathogens (Tirloni et al., 2014 ▸; Francischetti et al., 2009 ▸). These include tick-borne diseases such as Lyme disease, Helvetica spotted fever, tick-borne meningoencephalitis, babesiosis and tick paralysis (Sprong et al., 2018 ▸). I. ricinus represents a model organism used in the development of new sustainable tick-control approaches such as acaricides and repellents. The saliva of ticks helps them to stay attached to the host until the long-lasting blood-feeding process is finished. This is facilitated by many immunomodulatory, anti-inflammatory and antihemostatic proteins, peptides and nonpeptide molecules in the saliva (Francischetti et al., 2009 ▸; Kotál et al., 2015 ▸).
Serpins (serine protease inhibitors) are the largest superfamily of protease inhibitors and are broadly distributed in nature (Silverman et al., 2001 ▸; Spence et al., 2021 ▸). The vast majority of serpins act as serine protease inhibitors, but during evolution some serpins switched to non-inhibitory functions such as molecular chaperones (for example heat-shock serpin 47; Nagata, 1996 ▸), tumour suppressors (for example maspin; Zou et al., 1994 ▸), storage proteins (for example ovalbumin; Mellet et al., 1996 ▸; Law et al., 2006 ▸) and hormone-binding globulins (for example thyroxine-binding globulin and cortisol-binding globulin; Pemberton et al., 1988 ▸). The typical process of serpin inhibition is irreversible and leads to substrate suicide: inactivation of both the serpin and the target protease. At the beginning of the inhibitory pathway, serpins form a Michaelis complex with the protease (Huntington, 2011 ▸). Subsequently, translocation of the reactive-centre loop (RCL) with the bound protease takes place and leads to the formation of a covalent complex with the trapped protease and the addition of a new strand in β-sheet A (Silverman et al., 2001 ▸). Inhibitory serpins vary in function according to their specificities, and their importance can be illustrated by serpinopathies, diseases caused by serpin dysfunction or deficiency (Belorgey et al., 2007 ▸). Many well known diseases, for example emphysema, cirrhosis, angioedema, hypertension and familial dementia, are caused at least partially by serpin dysfunction (Law et al., 2006 ▸; Huntington, 2011 ▸). This makes serpins interesting candidates for drug design and development, for which a high-resolution structure is necessary. All serpins possess a structurally similar core domain consisting of ∼380 residues. This domain is made up of three β-sheets (A, B and C) and eight or more α-helices (hA–hI; Gettins, 2002 ▸). Another typical characteristic feature of serpins is the presence of an exposed, extended RCL that acts as a bait for the target protease during inhibition. The RCL consists of ∼17 residues and is located between β-sheet A and β-sheet C (Dunstone & Whisstock, 2011 ▸). It was found that serpins show different structural conformations such as native (S, stressed state), cleaved (R, relaxed state), latent (a result of auto-inactivation due to a mutation or self-stabilization) and the δ-conformation (inappropriate partial insertion of the RCL due to a mutation) as well as the possible formation of complexes as a result of the inhibitory mechanism (Dunstone & Whisstock, 2011 ▸). The inhibitory mechanism can result in successful inhibition by covalent complex formation with the target protease or a cleaved conformation. During conformational change of both states, and the incorporation of the RCL into β-sheet A, energy release occurs and a rise in serpin stability is reported as a consequence of this transition. In the case where this process is not sufficiently fast, it results in unsuccessful inhibition of the protease and its release from the acyl-intermediate, followed by the formation of a cleaved conformation of the serpin (Gettins, 2002 ▸; Gettins & Olson, 2016 ▸; Yamasaki et al., 2002 ▸).
Tick salivary serpins play important roles in tick physiology. They are necessary to modulate the immune-system responses of the host and to inhibit various defence mechanisms such as hemostasis, which can result in the facilitated transmission of the aforementioned tick-borne pathogens (Kotál et al., 2015 ▸). High structural conservation of serpins across tick species has been observed (Porter et al., 2015 ▸). The tick I. ricinus expresses over 30 serpins with different specificities, of which only Iris, IRS-2 and Iripin-3 have been characterized in detail functionally, while IRS-2 and Iripin-3 have also been structurally characterized (Prevot et al., 2006 ▸; Chmelař et al., 2011 ▸; Páleníková et al., 2015 ▸; Chlastáková et al., 2021 ▸).
Here, we present the structural, biochemical and functional characterization of the serpin from I. ricinus named Iripin-5 (I. ricinus serpin-5) that is highly expressed in the salivary glands of the tick; its expression is induced by feeding on blood and it displays anti-inflammatory and anticomplement features. Structural analysis revealed that Iripin-5 crystallized in a cleaved conformation and its structure was solved at 1.50 Å resolution. The structure was used for interface and computational analyses of its complexes with chosen proteases.
2. Materials and methods
2.1. Protein cloning, expression and purification
The full-length Iripin-5 sequence was cloned into pET-17b vector and transformed into Escherichia coli strain BL21-pLysS (Novagen, USA). 6 l LB medium (100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol) was inoculated with an overnight culture of BL21-pLysS cells containing the Iripin-5 gene. Protein overexpression was induced by 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) on reaching an OD600 of 0.6 and the cells were harvested 3 h after induction. Inclusion bodies were isolated by sonication in 20 mM Tris-buffered saline (TBS), 150 mM NaCl pH 8.0 with 1%(v/v) Triton X-100 buffer and washed three times with TBS to remove traces of Triton X-100. The inclusion bodies were dissolved in 20 mM TBS, 6 M guanidine–HCl pH 8 and undissolved impurities were removed by centrifugation (12 000g). Refolding was achieved by rapid dilution in a 160-fold excess of 50 mM Tris, 300 mM NaCl, 0.8 mM KCl, 250 mM l-arginine pH 8.5 with 0.25 g wet inclusion bodies per litre of refolding buffer. After filtration, Iripin-5 was purified by ion-exchange and size-exclusion chromatography (Supplementary Figs. S1 and S2). Pure protein was decontaminated from lipopolysaccharide (LPS) by Arvys Proteins (Trumbull, USA) using a detergent-based method. The LPS was removed from the sample because of its proven activation effect on cells, especially the stimulation of cells responsible for immune responses. This would interfere in subsequent experiments. The final concentration of protein was 1.14 mg ml−1 in 20 mM Tris, 150 mM NaCl pH 8.0 buffer and the protein was stored at −80°C.
2.2. Nitric oxide production by IC-21 macrophages
Macrophages of the IC-21 cell line were pre-incubated with various concentrations of Iripin-5 for 4 h. After stimulation with 100 ng ml−1 LPS and 5 ng ml−1 interferon-γ (IFNγ), the cells were incubated for 24 or 48 h. The nitric oxide (NO) concentration was assessed after incubation with a modified Griess reagent (Sigma–Aldrich, Germany).
2.3. Antiprotease selectivity
Assays were performed according to a previous publication (Chmelar et al., 2011 ▸). The enzyme concentrations do not reflect their ratio in the plasma or skin of the tick host. The used concentrations were chosen based on the biochemical properties of particular proteases in order to detect substrate hydrolysis and do not reach saturation of reaction at the same time. Generally, the assay conditions were chosen as half of the V max of each particular protease. Briefly, assays were performed at 30°C and tested in triplicate. The used protein concentration in the reaction was from 400 nM and the serpin was pre-incubated with the target enzyme (listed in Table 1 ▸) for 10 min before adding substrate (250 µM final concentration). For each target enzyme, appropriate buffers at different final concentrations were used. The substrate-hydrolysis rate was determined using an Infinite 200 PRO 96-well plate fluorescence reader (Tecan, Switzerland; excitation at 365 nm, emission at 450 nm).
Table 1. Antiprotease selectivity of Iripin-5.
| Enzyme | Amount of enzyme used (nM) | Remaining enzymatic activity (%) |
|---|---|---|
| Thrombin | 0.01 | 95.2 ± 3.2 |
| Factor Xa | 0.33 | 97.6 ± 4 |
| Kallikrein | 0.04 | 100.9 ± 2.5 |
| Chymase | 0.45 | 81.1 ± 3.3 |
| Trypsin | 0.1 | 55.9 ± 1.5 |
| α-Chymotrypsin | 0.05 | 68.6 ± 1.4 |
| β-Tryptase | 0.01 | 104.2 ± 1.4 |
| Human neutrophil elastase | 0.06 | 13 ± 2.2 |
| Cathepsin G | 8.8 | 80 ± 1.8 |
| u-PA | 0.5 | 101 ± 1.5 |
| Plasmin | 1.2 | 94.1 ± 2.2 |
| Matriptase | 0.03 | 100 ± 1.9 |
| Factor XIa | 0.06 | 98.8 ± 3 |
| Factor XIIa | 0.1 | 98.8 ± 1.2 |
| t-PA | 0.02 | 100.8 ± 3.7 |
| Proteinase 3 | 1.7 | 4.6 ± 0.8 |
2.4. Complement assay
Fresh rabbit erythrocytes were collected in Alsever’s solution from the rabbit marginal ear artery, washed three times in an excess of PBS buffer (1.8 mM KH2PO4, 137 mM NaCl, 10 mM Na2HPO4·7H2O) and finally diluted to a final 2%(v/v) suspension. Fresh human serum was obtained from three healthy individuals. The assay was performed in a 96-well round-bottomed microtiter plate (Nunc, Denmark). In each well, a concentration of 50% human serum in PBS premixed with different concentrations of Iripin-5 (156 nM to 5 µM) was added to a volume of 100 µl. After 10 min of incubation at room temperature, 100 µl of erythrocyte suspension was added. Since the human serum lysed rabbit erythrocytes immediately after their addition to the reaction, we used only 50% concentration (i.e. a 25% final serum concentration after addition of the erythrocyte suspension); the final dilution had been empirically established as optimal. Reaction wells were observed individually under a Olympus SZX7 stereomicroscope with oblique illumination (Olympus KL 1500) using an aluminium pad. The time needed for erythrocyte lysis was measured using a chronometer. When full lysis was achieved, the reaction mixture turned from opaque to transparent. Negative controls did not contain either serpin or human serum. Additional controls were performed with heat-inactivated serum (56°C, 30 min) and the serpin Iripin-3 (156 nM to 10 µM). The assay was evaluated in technical and biological triplicates.
2.5. Neutrophil-migration assay
Neutrophils were obtained from the bone marrow of C57BL/6J mice by magnetic separation using a Neutrophil Isolation Kit (Miltenyi Biotec, Germany). Isolated neutrophils were pre-incubated in RPMI1640 growth medium containing 0.5%(m/v) bovine serum albumin (BSA) in the presence or absence of Iripin-5 (3 µM) for 1 h at 37°C and 5% CO2. The cells were then seeded on the upper inserts of 3.0 µm pore Corning Transwell chambers (24-well format; Sigma–Aldrich, Germany). Chemoattractant solution (1 µM N-formyl-l-methionyl-l-leucyl-phenylalanine-fMLP in RPMI1640 with 0.5% BSA) was placed in the lower compartments. After incubation for 1 h at 37°C and 5% CO2, migration was determined by counting the cells in the lower chamber using a hemocytometer (Meopta, Czech Republic).
2.6. Iripin-5 expression profiles
I. ricinus nymphs were fed on C3H/HeN mice for one day, two days and until full engorgement (3–4 days); I. ricinus females were fed on guinea pigs for one, two, three, four, six and eight days. Adult salivary glands, midguts and ovaries, as well as nymph whole bodies, were dissected under RNAse-free conditions and total RNA was isolated using TriReagent (MRC). cDNA preparations were made from 1 µg total RNA from independent biological triplicates using a Transcriptor First Strand cDNA Synthesis kit (Roche, Czech Republic) according to the manufacturer’s instructions. The cDNA was subsequently used for the analysis of Iripin-5 transcription by qPCR in a RotorGene 6000 cycler (Corbett Research, UK) using Fast Start Universal SYBR Green Master Mix (Roche, Czech Republic), forward primer 5′-CGA GAA CGC AAC CAC TAA GA-3′ and reverse primer 5′-GCT CAA CGT GAC CAA TGT AAT C-3′. Iripin-5 expression profiles were calculated using Livak’s mathematical model (Livak & Schmittgen, 2001 ▸) and normalized to I. ricinus elongation factor 1α (ef1α; GU074829.1; forward primer 5′-CTG GGT GTG AAG CAG ATG AT-3′ and reverse primer 5′-GTA GGC AGA CAC TTC CTT CTG-3′). The amplicon lengths were ef1α, 105 bp; Iripin-5, 251 bp.
2.7. Protein crystallization, X-ray data collection and processing
Crystallization screening using commercial kits (JCSG++ from Jena Bioscience, SG1 and PGA Screen from Molecular Dimensions, and PEGRx and PEG/Ion from Hampton Research, USA) was carried out at room temperature (20°C) and at 4°C by the sitting-drop vapour-diffusion method using an OryxNano crystallization robot (Douglas Instruments). A suitable protein concentration for crystallization screening was determined using the Pre-Crystallization Test (Hampton Research, California, USA) as 1.14 mg ml−1. Drops of protein solution composed of 20 mM Tris, 150 mM NaCl pH 8.0 buffer (1 µl) mixed with reservoir solution (1 or 0.5 µl) were equilibrated against 50 µl reservoir solution and sealed in 96-well Swissci MRC 2-drop crystallization plates (Molecular Dimensions).
For data collection, crystals of Iripin-5 that grew for about one month were flash-cooled in liquid nitrogen with 20%(v/v) glycerol as an additional cryoprotectant. Measurements were carried out on beamline BL14.1 at the BESSY II electron-storage ring operated by Helmholtz-Zentrum Berlin (Mueller et al., 2012 ▸). Collection of diffraction data was performed at 100 K with a 295.165 mm crystal-to-detector (PILATUS 6M) distance. Diffraction intensity data were processed using XDS (Kabsch, 2010 ▸) with the XDSAPP graphical user interface (Sparta et al., 2016 ▸). Data-collection statistics are summarized in Table 2 ▸.
Table 2. X-ray data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray diffraction source | BL14.1, BESSY II, Germany |
| Wavelength (Å) | 0.9184 |
| Detector | PILATUS 6M |
| Crystal-to-detector distance (mm) | 295.165 |
| Rotation range per image (°) | 0.1 |
| Total rotation range (°) | 360 |
| Exposure time per image (s) | 0.1 |
| Resolution range (Å) | 48.09–1.50 (1.59–1.50) |
| Space group | P1211 |
| Molecules in asymmetric unit | 2 |
| a, b, c (Å) | 76.24, 63.78, 81.99 |
| α, β, γ (°) | 90.0, 116.78, 90.0 |
| Mosaicity (°) | 0.199 |
| Total No. of reflections | 752984 (117495) |
| No. of unique reflections | 112133 (17637) |
| Multiplicity | 6.72 |
| Average I/σ(I) | 11.17 (1.41) |
| Completeness (%) | 98.7 (96.5) |
| CC1/2 | 99.8 (61.3) |
| R meas † (%) | 11.0 (124.1) |
| Overall B factor from Wilson plot (Å2) | 24.46 |
| Refinement | |
| Resolution range (Å) | 48.09–1.50 |
| No. of reflections in working set | 110024 (7612) |
| Final R ‡/R free § (%) | 0.153/0.185 |
| Mean B value (Å) | 17.725 |
| No. of non-H atoms in the asymmetric unit | |
| Protein | 6010 |
| Water | 1097 |
| Magnesium | 4 |
| Chlorine | 6 |
| Total | 7117 |
| R.m.s. deviations | |
| Bonds (Å) | 0.012 |
| Angles (°) | 1.672 |
| Average B factor (Å2) | 17.517 |
| Ramachandran plot | |
| Most favoured (%) | 98.64 |
| Allowed (%) | 100.00 |
| PDB code | 7b2t |
R meas = \textstyle \sum_{hkl}\{N(hkl)/[N(hkl)-1]\}^{1/2}\sum_{i}|I_{i}(hkl)- \langle I(hkl)\rangle|/\textstyle \sum_{hkl}\sum_{i}I_{i}(hkl), where the average intensity 〈I(hkl)〉 is taken over all symmetry-equivalent measurements and I i (hkl) is the measured intensity for the ith observation of reflection hkl.
R = \textstyle \sum_{hkl}\big ||F_{\rm obs}|-|F_{\rm calc}|\big |/ \textstyle \sum_{hkl}|F_{\rm obs}|, where F obs and F calc are the observed and calculated structure factors, respectively.
R free is equivalent to the R value but is calculated for 1.87% of the reflections that were chosen at random and omitted from the refinement process.
2.8. Structure determination and refinement
Crystallographic and structural analyses were performed using the CCP4 package (Winn et al., 2011 ▸). The structure of Iripin-5 was solved by the molecular-replacement method using MOLREP (Vagin & Teplyakov, 2010 ▸) with the structure of the serpin IRS-2 (PDB entry 3nda; Chmelar et al., 2011 ▸) as the search model. The structure was refined with REFMAC5 (Murshudov et al., 2011 ▸) and further manually in Coot (Emsley et al., 2010 ▸) from evaluation of the electron-density peaks. The improvement during refinement was monitored by structure validation throughout the refinement process. Water molecules were added to the model using the REFMAC5 interface. Accepted solvent molecules had tolerable hydrogen-bonding geometry contacts of 2.5–3.5 Å with protein atoms or with existing solvent. At this point, residues with two possible conformations were included and their alternative conformations were added for further refinement. In the last steps of refinement, glycerol was built into the appropriate (2F o − F c) and (F o − F c) electron-density maps using coordinates from the ligand data bank in Coot (Emsley et al., 2010 ▸). The MolProbity server (Williams et al., 2018 ▸) and wwPDB validation server (Berman et al., 2003 ▸) were used for final qualitative validation of the model. All figures were prepared using PyMOL (DeLano, 2002 ▸). A summary of the data-collection and refinement statistics is given in Table 2 ▸.
2.9. Structural analysis and molecular dynamics of the modelled Michaelis complexes
The structures of the predominantly inhibited proteases proteinase 3 and human neutrophil elastase were fetched from the PDB as PDB entries 1fuj at 2.20 Å resolution (Fujinaga et al., 1996 ▸) and 3q76 at 1.86 Å resolution (Hansen et al., 2011 ▸), respectively. The cleaved Iripin-5 crystal structure was modelled to match the native conformation of serpins. The inserted RCL from β-sheet A was modelled above the Iripin-5 structure and the missing residues (Leu343, Ile344, Glu345, Val346 and Pro347) were modelled into the structure to complete the native structure. The crystal structures of the chosen proteases were modified by removing alternative conformations of the amino-acid side chains, ligands and ions from the structures as required for further docking calculations using the prepared native Iripin-5 model. The HADDOCK2.2 web server (van Zundert et al., 2016 ▸) was used for Michaelis complex docking and the best results were used to run molecular-dynamics (MD) simulations. MD simulations were performed using the GROMACS simulation suite (Berendsen et al., 1995 ▸) with the CHARMM27 all-atom force field and the SPC/E (extended simple point charge) model for water (Feller & MacKerell, 2000 ▸; Klauda et al., 2005 ▸). Both Michaelis complex models were prepared for simulation by removing the solvent and were then solvated using the SPC/E water model (Berendsen et al., 1987 ▸) in a rhombic dodecahedral box. The protein was centred in the box and the size of the box was such that the protein was at least 1 nm from all edges. Na+ ions were added to the system at a concentration of 150 mM together with an appropriate amount of Cl− ions to neutralize the system. The entire system was minimized using a steepest-descent minimization procedure. The energy-minimized structure was then further equilibrated in two phases for 100 ps each: first under an NVT ensemble (constant number of particles, volume and temperature) followed by an NPT ensemble (constant number of particles, pressure and temperature) to ensure that the system remained stable. Simulations were then performed for 100 ns each, during which time equilibrium of the system was achieved. MD simulations were performed fully in triplicate (i.e. from the minimization to production run) to ensure reproducibility. The results of the molecular simulations were analyzed using VMD (Humphrey et al., 1996 ▸) with the use of the r.m.s.d. trajectory tool. An interface analysis of the resulting structures (the final frames of each simulation) was then performed using the PDBePISA web server (Krissinel & Henrick, 2007 ▸).
2.10. Structural analysis and protein docking of modelled covalent complex conformations
Structures were analyzed and compared with those of the other I. ricinus serpins using PyMOL version 2.0 (DeLano, 2002 ▸; Schrödinger). The HADDOCK2.2 web server (van Zundert et al., 2016 ▸) was used for protein docking to generate covalent complexes. The possible target proteases selected as the best candidates from the antiprotease selectivity assays, namely proteinase 3, human neutrophil elastase, trypsin, α-chymotrypsin, cathepsin G and chymase, were used for analysis. The crystal structures of the human proteases were taken from the Protein Data Bank: PDB entries 1fuj at 2.20 Å resolution (Fujinaga et al., 1996 ▸), 3q76 at 1.86 Å resolution (Hansen et al., 2011 ▸), 1h4w at 1.70 Å resolution (Katona et al., 2002 ▸), 4cha at 1.68 Å resolution (Tsukada & Blow, 1985 ▸), 1au8 at 1.90 Å resolution (F. J. Medrano, W. Bode, A. Banbula & J. Potempa, unpublished work) and 3n7o at 1.80 Å resolution (Kervinen et al., 2010 ▸). Alternative conformations of the amino-acid side chains, ligands and ions were removed from the structures as required. Interface analysis was performed using the PDBePISA (Krissinel & Henrick, 2007 ▸) and COCOMAPS (Vangone et al., 2011 ▸) web servers.
2.11. PDB deposition
The atomic coordinates of Iripin-5 have been deposited in the Protein Data Bank with accession code 7b2t.
2.12. Statistical analyses
All immunological experiments were performed as at least three biological replicates. Data are presented as mean ± standard error of mean (SEM) in all graphs. Student’s t-test or one-way ANOVA were used to calculate statistical differences between two or more groups, respectively. Statistically significant results are marked as follows in the figures: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; n.s., not significant.
3. Results
3.1. Iripin-5 expression profiles
Expression of the Iripin-5 gene was upregulated during tick feeding in all tested tissues. The highest expression was found in semi-engorged nymphs (D2), and expression was also high in fully engorged nymphs (D3) as well as in female salivary glands during finishing of the blood meal (D6 and D8; Fig. 1 ▸). Furthermore, the Iripin-5 transcripts in engorged nymphs and female salivary glands were the most abundant among all tested tick serpins (data not shown). Thus, Iripin-5 is likely to be the most abundant serpin that is secreted from the salivary glands to the host.
Figure 1.

Iripin-5 expression is upregulated in I. ricinus nymphs and adults during feeding. The analysis was performed on tissues of flat, semi-engorged and fully engorged nymphs and female salivary glands, midguts and ovaries. RT-qPCR expression data are normalized against elongation factor 1α (ef1α) and the highest expression was set as 100%. The data represent mean + SEM from three biological replicates. D0–D8: days of feeding.
3.2. Antiprotease selectivity and neutrophil migration
In the protease-selectivity assay, Iripin-5 needed to be in a high excess compared with the target enzyme in order to obtain even a low level of inhibition. The remaining protease inhibition after 10 min incubation with 200 nM Iripin-5 is given in Table 1 ▸. Iripin-5 showed the highest inhibitory specificity against two neutrophil proteases: human neutrophil elastase (87% inhibition) and proteinase 3 (95% inhibition). Statistically significant results are noted in bold. Based on physiologically relevant proteases for tick–host interaction, it was found that only chymase and cathepsin G were inhibited significantly, and only very weakly. Another two inhibited proteases, trypsin and α-chymotrypsin, show importance during digestion.
Since Iripin-5 primarily inhibited neutrophil proteases, the effect on neutrophil functions was also studied. Static migration was tested using a Transwell chamber and purified mouse neutrophils isolated from bone marrow. Pre-incubation with 3 µM Iripin-5 led to a greater than 70% decrease in neutrophil migration, thus showing a significant antineutrophil effect of Iripin-5 (Fig. 2 ▸).
Figure 2.
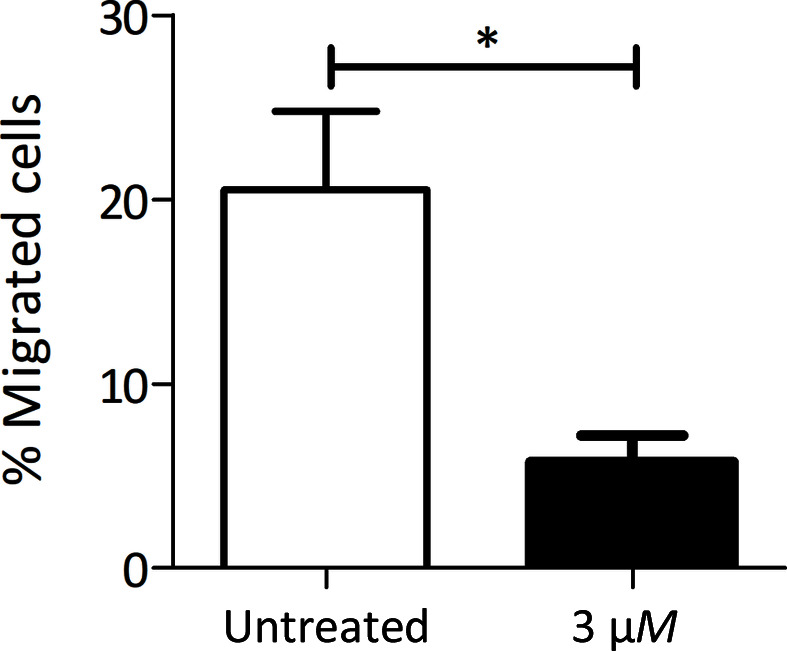
Iripin-5 inhibits neutrophil migration. Mouse bone-marrow neutrophils were pre-incubated with 3 µM Iripin-5 and subjected to migration towards fMLP in a Transwell chamber. The average of three independent experiments (±SEM) is shown. *, p ≤ 0.05
3.3. NO production by IC-21 macrophages
The incubation of macrophages in the presence of Iripin-5 led to a decrease in NO production in a dose-dependent manner. At a concentration of 1 µM, Iripin-5 inhibited NO production slightly, but not significantly, at 24 h, but not at 48 h. At a higher concentration of 5 µM, Iripin-5 significantly decreased the amount of NO at both time points: by 35% and 36%, respectively (Fig. 3 ▸).
Figure 3.

Iripin-5 decreased NO production by activated IC-21 macrophages. Iripin-5 inhibited NO production by IC-21 macrophages when used at high concentration. Macrophages were pre-incubated with 1 and 5 µM Iripin-5, stimulated with LPS and IFN-γ, and the NO concentration was assessed after 24 or 48 h. The mean of three independent experiments (±SEM) is shown. ***, p ≤ 0.001; n.s., not significant.
3.4. Complement assay
Since Iripin-5 affected two major immune-cell types involved in innate immune response, the interference of the tested serpin with another innate immune mechanism involved in antitick immunity of the complement was tested. Iripin-5 inhibited the lysis of erythrocytes by human complement. Human plasma was pre-incubated with different concentrations of Iripin-5 from 156 nM to 5 µM. After the addition of rabbit erythrocytes, their lysis time by complement was measured. A statistically significant reduction in complement-driven lysis activity against erythrocytes when incubating human plasma with Iripin-5 at concentrations of 625 nM and higher was observed. No lysis of any erythrocytes was detected when using 5 µM Iripin-5. The results were compared with those for another serpin, Iripin-3 (Chlastáková et al., 2021 ▸), which had no effect on complement activity, demonstrating the specificity of our assay. The lysis of rabbit erythrocytes in the presence of 25% human serum was achieved within 7 min 57 s ± 0.12 s on average in the control group, which corresponds to the zero value in the graph (Fig. 4 ▸).
Figure 4.

Inhibition of complement by Iripin-5 compared with another I. ricinus salivary serpin, Iripin-3. Human plasma was pre-incubated with an increasing concentration of Iripin-5 (156 nM to 5 µM) or Iripin-3 (312 nM to 10 µM). After the addition of rabbit erythrocytes, their lysis time by complement was measured. For each point in the graph, the mean of three independent experiments (±SEM) is shown. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
3.5. Crystal structure of Iripin-5
In order to obtain a deeper view into the mechanisms of Iripin-5 activity, a detailed structural analysis was performed. To generate a protein structure of Iripin-5, crystallization experiments were performed and the structure of the serpin was solved from the best-diffacting crystals to a resolution of 1.5 Å. Iripin-5 crystals with a monoclinic shape (Fig. 5 ▸) grew after one month at 4°C in a precipitant composed of 0.2 M magnesium chloride hexahydrate pH 8.5, 0.1 M Tris, 30%(w/v) PEG 4000 (condition No. 1-1 of SG1 from Molecular Dimensions). The crystal space group and unit-cell parameters are reported in Table 2 ▸.
Figure 5.

Crystals of Iripin-5 from I. ricinus. (a) Crystals of protein grown in 0.2 M magnesium chloride hexahydrate pH 8.5, 0.1 M Tris, 30%(w/v) PEG 4000. (b) The same crystallization droplet is shown under UV light. (c) Focus on the best-shaped crystals for diffraction measurements. (a) was taken using an Olympus SZX9 microscope and (b) and (c) were taken using a Minstrel Desktop Crystal Imaging System (Rigaku, Japan). The scale bar represents 100 µm.
The structure of Iripin-5 was solved by molecular replacement using the previously published structure of the serpin IRS-2 (PDB entry 3nda) as the model structure, which has a sequence identity of 55.70% (Chmelar et al., 2011 ▸). The crystal structure contains two molecules per asymmetric unit, with a solvent content of 39.97% and a Matthews coefficient of 2.05 Å3 Da−1. The Iripin-5 structure has a typical cleaved serpin secondary-structure fold in both molecules. The structure consists of a mixed αβ secondary structure with an N-terminal helical region and a C-terminal β-sheet fold (Huntington, 2011 ▸; Fig. 6 ▸). The structure is composed of eight α-helices and three β-sheets sequentially arranged in the order α1–β1–α2–α3–β2–α4–β3–α5–β4–β5–β6–β7–β8–α6–α7–β9–β10–α8–β11–β12–β13–β14–β15. Sheet A consists of six β-strands (β2, β3, β4, β10, β11 and β12), sheet B of five β-strands (β1, β7, β8, β14 and β15) and sheet C of four β-strands (β5, β6, β9 and β13) (Fig. 6 ▸).
Figure 6.

Cleaved protein form with colour-distinguished β-sheets: sheet A (blue), sheet B (magenta) and sheet C (violet). The insertion of the RCL between β-strands S3 and S5 (blue) is marked as β-strand S4 (dark pink). The location of protease cleavage is marked with black stars.
The final model of Iripin-5 contains 373 residues in chain A and chain B out of a total of 378, with five missing residues (Leu343–Thr347) in both chains. The missing residues in the crystal structure were detected as an absence of electron density due to the high flexibility of the cleaved regions (Fig. 6 ▸), and thus these regions were not modelled in the final structure. The cleavage is probably a consequence of the presence of protease, most probably during storage. The cleavage of the sample used for crystallization was confirmed by MALDI mass-spectrometric protein analysis (Supplementary Fig. S3), and led to the structural change and thus to the insertion of the cleavage site inside the β-sheet to form the extra β-strand (S4). The cleavage site is homologous to the RCL of other serpin inhibitors and the cleaved state is the most stable, so-called hyperstable or R form, of inhibitory serpins (Huntington, 2011 ▸; Fig. 6 ▸). Moreover, analysis of the protein interfaces by PDBePISA (Krissinel & Henrick, 2007 ▸) did not reveal any specific interactions resulting in the formation of stable quaternary structures. Most probably the structures do not form any complexes in solution (Schlee et al., 2019 ▸).
3.6. Structural analysis and molecular dynamics of the theoretical Michaelis complex
To identify the specific interactions that are potentially responsible for the mechanism of inhibition between the target proteases and Iripin-5, protein docking and subsequent MD simulations of the Michaelis complexes were performed. Three simulations for modelled Michaelis complexes with both neutrophil elastase and proteinase 3, each 100 ns long, were performed.
The stability of the complex was monitored by r.m.s.d. evaluation (Fig. 7 ▸). The results showed that triplicates of both simulated complexes reached equilibrium within the simulation time and the average r.m.s.d.s from the initial starting structure for the Michaelis complexes were 5.3 Å (Fig. 7 ▸ b, orange), 6.7 Å (Fig. 7 ▸ b, grey) and 8.0 Å (Fig. 7 ▸ b, yellow) for the Iripin-5–proteinase 3 complex and 8.4 Å (Fig. 7 ▸ a, blue), 9.6 Å (Fig. 7 ▸ a, yellow) and 5.8 Å (Fig. 7 ▸ a, green) for the Iripin-5–neutrophil elastase complex. The difference between the average r.m.s.d. of the Iripin-5–proteinase 3 complex triplicates was 3.8 Å and that for the Iripin-5–neutrophil elastase complex was 2.7 Å.
Figure 7.

All-atom r.m.s.d. of MD simulations of Michaelis complex models. (a) R.m.s.d. for the Iripin-5–neutrophil elastase Michaelis complex and (b) r.m.s.d. for the Iripin-5–proteinase 3 Michaelis complex, each for 100 ns simulation. Triplicates are distinguished by different colours (corresponding to the visualization of Michaelis complex models in Fig. 8 ▸).
Representations of the Michaelis complexes between Iripin-5 and neutrophil elastase and proteinase 3 are shown in Fig. 8 ▸. Structural alignment performed by PyMOL (DeLano, 2002 ▸) showed that the average r.m.s.d. between the Iripin-5–neutrophil elastase triplicates was 1.709 Å and that between the Iripin-5–proteinase 3 triplicates was 1.958 Å. These results show the flexibility of the Michaelis complex conformation and, more precisely, the flexibility of the Iripin-5 RCL (Fig. 8 ▸).
Figure 8.

Results of MD simulation of the Michaelis complex. The structures are shown at the 100 ns point of simulation for each triplicate of the chosen target protease. The Iripin-5 (magenta) structures are aligned to show the RCL dynamics. Triplicates are distinguished by different colours for the target protease: neutrophil elastase, blue, green and yellow; proteinase 3, grey, orange and yellow. The Iripin-5 RCL is also distinguished in a corresponding colour to the interacting protease. A detailed view of the Michaelis complex interfaces is presented in Supplementary Fig. S6.
The interface analysis of the Michaelis complex triplicates is summarized in Table 3 ▸. The data in bold indicate the importance of the interface for complex formation (PDBePISA; Krissinel & Henrick, 2007 ▸). A more detailed summary of the interacting residues is presented in Supplementary Table S3.
Table 3. Area of the accessible surface interface between Iripin-5 and the tested proteases in the Michaelis complex conformation, the number of hydrogen bonds and the number of salt bridges formed after 100 ns of MD simulation (from PDBePISA; Krissinel & Henrick, 2007 ▸).
| Protease | Surface interface (Å2) | No. of hydrogen bonds | No. of salt bridges |
|---|---|---|---|
| Human neutrophil elastase | 684.9 | 5 | 4 |
| 662.9 | 4 | 5 | |
| 655.7 | 2 | 2 | |
| Proteinase 3 | 864.2 | 4 | 2 |
| 804.2 | 3 | 2 | |
| 827.0 | 2 | 2 |
3.7. Structural analysis of theoretical protein–protease covalent complex conformation
To test the hypothesis of the presence of polar contacts between Iripin-5 and six chosen proteases (proteinase 3, human neutrophil elastase, trypsin, α-chymotrypsin, cathepsin G and chymase), docking calculations of protein–protein interactions were performed using HADDOCK (van Zundert et al., 2016 ▸). The results of the docking studies of interacting residues at the Iripin-5–protease complex interfaces, listed in Table 4 ▸ and shown in Fig. 9 ▸, show different characters for the interactions in complexes.
Table 4. Area of the accessible surface interface between Iripin-5 and the tested proteases, the number of hydrogen bonds and the number of salt bridges formed (from PDBePISA; Krissinel & Henrick, 2007 ▸).
| Protease | Surface interface (Å2) | No. of hydrogen bonds | No. of salt bridges |
|---|---|---|---|
| Proteinase 3 | 892.8 | 13 | 10 |
| Human neutrophil elastase | 733.6 | 8 | 4 |
| Trypsin | 919.5 | 12 | 10 |
| α-Chymotrypsin | 787.5 | 7 | 1 |
| Cathepsin G | 947.1 | 8 | 4 |
| Chymase | 849.8 | 5 | 6 |
Figure 9.

Cartoon representation of the docking results of Iripin-5 (magenta) with chosen proteases: cathepsin G (violet), trypsin (cyan), elastase (hot pink), α-chymotrypsin (blue), chymase (orange) and proteinase 3 (lemon). The residues interacting with the protease catalytic triad are shown in detail in Supplementary Fig. S5.
Only the α-chymotrypsin catalytic triad interacted with Arg342; thus, the potential Iripin-5 P1 site was a candidate for binding the protease. However, the proteinase 3 and chymase side-chain residues of the catalytic triad were not in contact with Arg342 of Iripin-5. The remaining proteases (cathepsin G, elastase and trypsin) interacted with Arg342 of Iripin-5 via side-chain residues other than the catalytic triad (Supplementary Fig. S5). Detailed information about atomic interface analysis is shown in Supplementary Table S2. These results were calculated using PDBePISA (Krissinel & Henrick, 2007 ▸).
4. Discussion
The I. ricinus sialome (transcriptome from the salivary glands) contains four major types of protease inhibitors, cystatins, TIL-domain inhibitors, Kunitz inhibitors and serpins, which are proven or presumed to be modulators of host-defence mechanisms (Chmelař et al., 2017 ▸). Among them, the serpins stand out thanks to their omnipresence across all living organisms and their indispensability for many crucial biochemical pathways, such as coagulation or complement and other fundamental functions (Huntington, 2011 ▸; Law et al., 2006 ▸). Considering the fact that tick serpins usually do not form multigenic families, as are typical for other salivary protease inhibitors such as Kunitz-domain and TIL-domain inhibitors, they seem to be suitable candidates for targeting in tick-control attempts. Moreover, the structural conservation and use of serpins by vertebrates makes them promising candidates for novel drug development combined with the use of protein engineering (Chmelař et al., 2017 ▸). Tick serpins can be utilized as specific regulators of dysregulated processes, such as inflammation, immune-system regulation or hemostasis. Several tick serpins have been shown to interfere with vertebrate immunity (Chmelař et al., 2017 ▸). To date, three of them have been functionally characterized in I. ricinus. It has been shown that the salivary serpin Iris modulates host innate and acquired immunity (Leboulle et al., 2002 ▸). Likewise, IRS-2 and Iripin-3 modulated adaptive immune responses (Chmelar et al., 2011 ▸; Chlastáková et al., 2021 ▸). Moreover, crystal structures were determined for the last two, which are the only two tick serpins with resolved 3D structures to date.
Iripin-5 belongs to the salivary serpins, the role of which is considered to be as modulators of host defence mechanisms. Iripin-5 seems to be one of the main salivary serpins since its mRNA expression is by far the highest compared with other I. ricinus serpins. This serpin is massively induced by the blood meal. Here, several effects supporting immunomodulatory and anti-inflammatory roles of Iripin-5 are reported. The observed inhibition of neutrophil migration suggests anti-inflammatory activity at the very beginning of the immune reaction. Macrophages play an important role in the interaction between ticks, the immune system of the host and transmitted pathogens. Activated macrophages secrete signalling molecules such as cytokines or NO to recruit immune cells to sites of inflammation or towards pathogens (Laroux et al., 2001 ▸). The saliva of different tick species has been shown to suppress the ability of macrophages to produce NO (Kýčková & Kopecký, 2006 ▸). Since Iripin-5 inhibits this very feature of macrophages, Iripin-5 is likely to be at least partially responsible for this activity observed in I. ricinus saliva.
The inhibition of complement described here is interesting, but not surprising, as vertebrate serpins are natural regulators of the complement cascade (Bos et al., 2002 ▸). There are other tick salivary protein families in which the members have been described as complement inhibitors (Daix et al., 2007 ▸; Tyson et al., 2008 ▸), but our case is the first observation of complement inhibition by a tick serpin. This finding confirms the hypotheses about the functional redundancy of tick salivary proteins (Chmelař et al., 2016 ▸).
Structural analysis of Iripin-5 shows the typical serpin fold in the relaxed state that was observed in other known crystal structures of I. ricinus serpins (IRS-2 and Iripin-3; Chmelař et al., 2017 ▸; Chlastáková et al., 2021 ▸). The relaxed cleaved state of Iripin-5 was caused by the presence of contaminating proteases, probably during protein storage, and this cleavage has been observed previously (Kovářová et al., 2010 ▸). The crystal structure of Iripin-5 was compared with those of IRS-2 (PDB entry 3nda; Chmelar et al., 2011 ▸) and Iripin-3 (PDB entry 7ahp; Chlastáková et al., 2021 ▸) both by sequence alignment (Fig. 10 ▸ a) and structural superimposition (Fig. 10 ▸ b). The comparison of I. ricinus serpins with known structures reveals an almost identical fold (Fig. 10 ▸ b) with some divergence in the loop regions. The r.m.s.d. between molecules was calculated by PyMOL (DeLano, 2002 ▸). On alignment of Iripin-5 and Iripin-3 the r.m.s.d. was 0.616 Å, while the r.m.s.d. between Iripin-5 and IRS-2 was 0.804 Å across all atoms. In contrast, sequence alignments of Iripin-3 and IRS-2 with Iripin-5 showed only 53.89% and 55.70% sequence identity, respectively (Fig. 10 ▸ a). Electrostatic surface potentials support complex formation and stability and consequently the inhibition of proteases. This can be achieved by charge–charge repulsion or attraction in accordance with their function as a protease substrate or inhibitor (Marijanovic et al., 2019 ▸). A comparison of surface electrostatics among I. ricinus serpins reveals that Iripin-5 has a more negatively charged surface than the other two aforementioned serpins; Iripin-3 has only a slightly more negatively charged surface than Iripin-5, but shows much greater inhibition (Fig. 10 ▸ c).
Figure 10.

(a) Amino-acid sequence alignment between the serpins IRS-2, Iripin-3 and Iripin-5. Well conserved amino-acid motifs are indicated in red and the P1 site of the RCL is marked as a bold rectangle. This sequence alignment was obtained using Clustal Omega (Madeira et al., 2019 ▸) and ESPript (http://espript.ibcp.fr; Robert & Gouet, 2014 ▸). (b) Superposition of I. ricinus crystal structures, namely Iripin-5 (magenta), IRS-2 (blue) and Iripin-3 (cyan). (c) Comparison of the electrostatic potentials of IRS-2 (PDB entry 3nda), Iripin-5 (PDB entry 7b2t) and Iripin-3 (PDB entry 7ahp). As shown in the figure, blue indicates positive potential and red indicates negative potential.
The amino acids of the RCL, specifically the P1 residue, determine the protease specificity (Marijanovic et al., 2019 ▸). This was confirmed by structural analysis of the I. ricinus salivary serpins IRS-2, Iripin-3 and Iripin-5. Iris, with Met340 at the P1 site, is an inhibitor of leukocyte elastase and elastase-like serine proteases (Prevot et al., 2007 ▸), although its inhibition is managed by several exosites in α-helices A and D (Prevot et al., 2009 ▸). However, IRS-2 has Tyr341 at its P1 site, which signifies the inhibition of chymotrypsin-like proteases (Chmelař et al., 2017 ▸), and Iripin-3 has Arg342 at the P1 site, indicating its trypsin-like protease inhibition (Chlastáková et al., 2021 ▸). Nevertheless, diverse RCL residues can represent potential cleavage sites, but only a few residues (16–17 residues from the C-terminal β-sheet) manage to successfully inhibit the target protease (Gettins, 2002 ▸). For Iripin-5, the last visible residue of the inserted RCL is Arg342 (Supplementary Table S1 and Fig. S4), which is the potential P1 site, suggesting the targeting of trypsin-like proteases preferring Arg or Lys side chains at the P1 site rather than elastase-like (Ala, Gly and Val) or chymotrypsin-like (Tyr, Phe and Trp) proteases (Barrett et al., 2004 ▸). However, Iripin-5 mainly inhibited neutrophil elastase and proteinase 3, which is the previously described behaviour of some serpins that inhibited serine proteases, despite that fact that these serpins have an inappropriate P1 recognition site and should have inhibited different proteases (Gettins, 2002 ▸). The presence of Arg at the P1 site is common for salivary serpins from prostriate ticks (Mulenga et al., 2009 ▸) and led to the proposal of an interaction with blood-coagulation proteases.
Michaelis and covalent complex studies were performed to reveal the possible residues responsible for the inhibition of target proteases. The Michaelis complex is the initial step of protease inhibition; more specifically, it enables the cleavage of the scissile bond and the subsequent acylation step, and therefore represents the most informative structural conformation of serpins (Gettins, 2002 ▸). Apart from the primary recognition site of the serpin, some serpins also employ specific surface regions called exosites that can specify protease inhibition (Gettins & Olson, 2016 ▸). For this reason, MD simulations of Michaelis complexes were performed. No exosites were found to be directly involved in formation of the Michaelis complex (Fig. 8 ▸). In Iripin-5–neutrophil elastase the Michaelis complex was observed to involve engagement of Glu330 in the Iripin-5 RCL to form salt bridges with Arg36 of neutrophil elastase. Similarly, in Iripin-5–proteinase 3 the Michaelis complex was observed to involve the formation of salt bridges between Glu345 in the Iripin-5 RCL and Lys103 of proteinase 3 and between Val340 of the RCL and Glu101 after the MD simulation in triplicate. The two resulting structures of the Iripin-5–neutrophil elastase Michaelis complex and the single structure of the Iripin-5–proteinase 3 Michaelis complex were confirmed to involve interfaces that play important roles in complex formation (PDBePISA; Krissinel & Henrick, 2007 ▸). Previously, it was observed that not only the position of the specific residues in RCL but also the dynamics of the RCL play an important role in protease inhibition by serpins (Marijanovic et al., 2019 ▸). It is probable that these two aspects are responsible for protease inhibition of the I. ricinus serpin Iripin-5.
Docking studies of covalent complexes revealed probable interactions between the chosen proteases and Iripin-5. The docking covalent complexes exhibit quite a large interface area, as observed previously for serpin–trypsin covalent complexes, with around 12 interacting interface residues. In the Iripin-5–trypsin complex more residues were involved in the formation of hydrogen bonds compared with other Iripin-5–protease complexes. These results are similar to the results of interface interaction comparison of antithrombin–trypsin and antithrombin–elastase complexes, in which the complex with trypsin made more hydrogen bonds (Rashid et al., 2015 ▸). This could probably explain the important role of Glu310 in Iripin-5, which forms salt bridges in the complex with protease. Moreover, some residues of Iripin-5 were involved in hydrogen-bond formation more frequently, namely Gln299, Asp301, Glu51, Lys288, Glu294 and the abovementioned Glu310 and Arg342. We propose that these residues should play an important role in the formation of a covalent complex between Iripin-5 and protease.
5. Conclusions
The continuing structural studies of arthropod (ectoparasite) serpins provide an understanding of their specific functions and protease targets. Structural information on complexes with targets and cofactors would help to understand the exact mechanism of action of these functionally diverse serpins. Iripin-5 is the third described crystal structure of a tick serpin, and despite its cleaved form it provides important experimental proof of the specificity of Iripin-5 and its possible interactions with proteases. Iripin-5 appears to be an immunomodulatory and anti-inflammatory protein used by I. ricinus ticks to overcome host defensive mechanisms. The presence of Arg at the P1 site led to the proposal of an interaction with blood-coagulation proteases. MD simulations of the Michaelis complex revealed flexibility of the RCL to be one of the factors responsible for inhibition. A more detailed study of the dynamic behaviour of Iripin-5 during the inhibition mechanism may be beneficial for a better understanding of inhibition. The residues with the most important roles in the formation of a covalent complex between Iripin-5 and proteases were proposed based on docking and MD simulations and it was found that Glu310 should play a crucial role in the interaction between Iripin-5 and proteases, with the exception of α-chymotrypsin.
6. Related literature
The following references are cited in the supporting information for this article: Cox & Mann (2008 ▸), Cox et al. (2011 ▸), Rappsilber et al. (2007 ▸) and Shevchenko et al. (2006 ▸).
Supplementary Material
PDB reference: Iripin-5, 7b2t
Supplementary Methods and Materials, Supplementary Figures and Supplementary Tables. DOI: 10.1107/S2059798321007920/rr5207sup1.pdf
Acknowledgments
The diffraction data were collected on BL14.1 at the BESSY II electron-storage ring operated by Helmholtz-Zentrum Berlin (HZB). We thank HZB for the allocation of synchrotron-radiation beam time. Author contributions were as follows. IKS, JC and MK designed the project. JK performed the cloning, expression and purification and determined the antiprotease selectivity and NO production. LAM performed the complement assay. ZB performed the neutrophil migration assay. HL determined Iripin-5 expression profiles. BK, PH and TP carried out crystallization experiments and performed X-ray diffraction analysis. BK analyzed the crystallographic data, solved the structure and drafted the manuscript. BK, MK and JAC performed the docking and analyzed the docking data.
Funding Statement
This work was funded by European Regional Development Fund, MEYS grant CZ.02.1.01/0.0/0.0/15_003/00004; Grantová Agentura České Republiky grant 19-14704Y; Jihočeská Univerzita v Českých Budějovicích grants 105/2019/P and 04-039/2019/P.
References
- Barrett, A. J., Rawlings, N. D. & Woessner, J. F. (2004). Handbook of Proteolytic Enzymes, 2nd ed, Vol. 1. London: Academic Press.
- Belorgey, D., Hägglöf, P., Karlsson-Li, S. & Lomas, D. A. (2007). Prion, 1, 15–20. [DOI] [PMC free article] [PubMed]
- Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. (1987). J. Phys. Chem. 91, 6269–6271.
- Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. (1995). Comput. Phys. Commun. 91, 43–56.
- Berman, H., Henrick, K. & Nakamura, H. (2003). Nat. Struct. Mol. Biol. 10, 980. [DOI] [PubMed]
- Bos, I. G. A., Hack, C. E. & Abrahams, J. P. (2002). Immunobiology, 205, 518–533. [DOI] [PubMed]
- Chlastáková, A., Kotál, J., Beránková, Z., Kaščáková, B., Martins, L. A., Langhansová, H., Prudnikova, T., Ederová, M., Kutá Smatanová, I., Kotsyfakis, M. & Chmelař, J. (2021). Front. Immunol. 12, 626200. [DOI] [PMC free article] [PubMed]
- Chmelař, J., Kotál, J., Kopecký, J., Pedra, J. H. F. & Kotsyfakis, M. (2016). Trends Parasitol. 32, 368–377. [DOI] [PMC free article] [PubMed]
- Chmelař, J., Kotál, J., Langhansová, H. & Kotsyfakis, M. (2017). Front. Cell. Infect. Microbiol. 7, 216. [DOI] [PMC free article] [PubMed]
- Chmelar, J., Oliveira, C. J., Rezacova, P., Francischetti, I. M. B., Kovarova, Z., Pejler, G., Kopacek, P., Ribeiro, J. M. C., Mares, M., Kopecky, J. & Kotsyfakis, M. (2011). Blood, 117, 736–744. [DOI] [PMC free article] [PubMed]
- Cox, J. & Mann, M. (2008). Nat. Biotechnol. 26, 1367–1372. [DOI] [PubMed]
- Cox, J., Neuhauser, N., Michalski, A., Scheltema, R. A., Olsen, J. V. & Mann, M. (2011). J. Proteome Res. 10, 1794–1805. [DOI] [PubMed]
- Daix, V., Schroeder, H., Praet, N., Georgin, J.-P., Chiappino, I., Gillet, L., de Fays, K., Decrem, Y., Leboulle, G., Godfroid, E., Bollen, A., Pastoret, P.-P., Gern, L., Sharp, P. M. & Vanderplasschen, A. (2007). Insect Mol. Biol. 16, 155–166. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL. http://www.pymol.org.
- Dunstone, M. A. & Whisstock, J. C. (2011). Methods Enzymol. 501, 63–87. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Feller, S. E. & MacKerell, A. D. (2000). J. Phys. Chem. B, 104, 7510–7515.
- Francischetti, I. M. B., Sa-Nunes, A., Mans, B. J., Santos, I. M. & Ribeiro, J. M. C. (2009). Front. Biosci. 14 2051–2088. [DOI] [PMC free article] [PubMed]
- Fujinaga, M., Chernaia, M. M., Halenbeck, R., Koths, K. & James, M. N. G. (1996). J. Mol. Biol. 261, 267–278. [DOI] [PubMed]
- Gettins, P. G. W. (2002). Chem. Rev. 102, 4751–4804. [DOI] [PubMed]
- Gettins, P. G. W. & Olson, S. T. (2016). Biochem. J. 473, 2273–2293. [DOI] [PMC free article] [PubMed]
- Hansen, G., Gielen-Haertwig, H., Reinemer, P., Schomburg, D., Harrenga, A. & Niefind, K. (2011). J. Mol. Biol. 409, 681–691. [DOI] [PubMed]
- Humphrey, W., Dalke, A. & Schulten, K. (1996). J. Mol. Graph. 14, 33–38. [DOI] [PubMed]
- Huntington, J. A. (2011). J. Thromb. Haemost. 9, Suppl. 1, 26–34. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Katona, G., Berglund, G. I., Hajdu, J., Gráf, L. & Szilágyi, L. (2002). J. Mol. Biol. 315, 1209–1218. [DOI] [PubMed]
- Kervinen, J., Crysler, C., Bayoumy, S., Abad, M. C., Spurlino, J., Deckman, I., Greco, M. N., Maryanoff, B. E. & de Garavilla, L. (2010). Biochem. Pharmacol. 80, 1033–1041. [DOI] [PubMed]
- Klauda, J. B., Brooks, B. R., MacKerell, A. D., Venable, R. M. & Pastor, R. W. (2005). J. Phys. Chem. B, 109, 5300–5311. [DOI] [PubMed]
- Kotál, J., Langhansová, H., Lieskovská, J., Andersen, J. F., Francischetti, I. M. B., Chavakis, T., Kopecký, J., Pedra, J. H. F., Kotsyfakis, M. & Chmelař, J. (2015). J. Proteomics, 128, 58–68. [DOI] [PMC free article] [PubMed]
- Kovářová, Z., Chmelař, J., Šanda, M., Brynda, J., Mareš, M. & Řezáčová, P. (2010). Acta Cryst. F66, 1453–1457. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Kýčková, K. & Kopecký, J. (2006). J. Med. Entomol. 43, 1208–1214. [PubMed]
- Laroux, F. S., Pavlick, K. P., Hines, I. N., Kawachi, S., Harada, H., Bharwani, S., Hoffman, J. M. & Grisham, M. B. (2001). Acta Physiol. Scand. 173, 113–118. [DOI] [PubMed]
- Law, R. H. P., Zhang, Q., McGowan, S., Buckle, A. M., Silverman, G. A., Wong, W., Rosado, C. J., Langendorf, C. G., Pike, R. N., Bird, P. I. & Whisstock, J. C. (2006). Genome Biol. 7, 216. [DOI] [PMC free article] [PubMed]
- Leboulle, G., Crippa, M., Decrem, Y., Mejri, N., Brossard, M., Bollen, A. & Godfroid, E. (2002). J. Biol. Chem. 277, 10083–10089. [DOI] [PubMed]
- Livak, K. J. & Schmittgen, T. D. (2001). Methods, 25, 402–408. [DOI] [PubMed]
- Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., Basutkar, P., Tivey, A. R. N., Potter, S. C., Finn, R. D. & Lopez, R. (2019). Nucleic Acids Res. 47, W636–W641. [DOI] [PMC free article] [PubMed]
- Marijanovic, E. M., Fodor, J., Riley, B. T., Porebski, B. T., Costa, M. G. S., Kass, I., Hoke, D. E., McGowan, S. & Buckle, A. M. (2019). Sci. Rep. 9, 3870. [DOI] [PMC free article] [PubMed]
- Mellet, P., Michels, B. & Bieth, J. G. (1996). J. Biol. Chem. 271, 30311–30314. [DOI] [PubMed]
- Mueller, U., Darowski, N., Fuchs, M. R., Förster, R., Hellmig, M., Paithankar, K. S., Pühringer, S., Steffien, M., Zocher, G. & Weiss, M. S. (2012). J. Synchrotron Rad. 19, 442–449. [DOI] [PMC free article] [PubMed]
- Mulenga, A., Khumthong, R. & Chalaire, K. C. (2009). BMC Genomics, 10, 217. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nagata, K. (1996). Trends Biochem. Sci. 21, 23–26. [DOI] [PubMed]
- Páleníková, J., Lieskovská, J., Langhansová, H., Kotsyfakis, M., Chmelař, J. & Kopecký, J. (2015). Infect. Immun. 83, 1949–1956. [DOI] [PMC free article] [PubMed]
- Pemberton, P. A., Stein, P. E., Pepys, M. B., Potter, J. M. & Carrell, R. W. (1988). Nature, 336, 257–258. [DOI] [PubMed]
- Porter, L., Radulović, Ž., Kim, T., Braz, G. R. C., Da Silva Vaz, I. & Mulenga, A. (2015). Ticks Tick Borne Dis. 6, 16–30. [DOI] [PMC free article] [PubMed]
- Prevot, P.-P., Adam, B., Boudjeltia, K. Z., Brossard, M., Lins, L., Cauchie, P., Brasseur, R., Vanhaeverbeek, M., Vanhamme, L. & Godfroid, E. (2006). J. Biol. Chem. 281, 26361–26369. [DOI] [PubMed]
- Prevot, P.-P., Beschin, A., Lins, L., Beaufays, J., Grosjean, A., Bruys, L., Adam, B., Brossard, M., Brasseur, R., Zouaoui Boudjeltia, K., Vanhamme, L. & Godfroid, E. (2009). FEBS J. 276, 3235–3246. [DOI] [PubMed]
- Prevot, P.-P., Couvreur, B., Denis, V., Brossard, M., Vanhamme, L. & Godfroid, E. (2007). Vaccine, 25, 3284–3292. [DOI] [PubMed]
- Rappsilber, J., Mann, M. & Ishihama, Y. (2007). Nat. Protoc. 2, 1896–1906. [DOI] [PubMed]
- Rashid, Q., Kapil, C., Singh, P., Kumari, V. & Jairajpuri, M. A. (2015). J. Biomol. Struct. Dyn. 33, 1352–1362. [DOI] [PubMed]
- Robert, X. & Gouet, P. (2014). Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed]
- Schlee, S., Straub, K., Schwab, T., Kinateder, T., Merkl, R. & Sterner, R. (2019). Proteins, 87, 815–825. [DOI] [PubMed]
- Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V. & Mann, M. (2006). Nat. Protoc. 1, 2856–2860. [DOI] [PubMed]
- Silverman, G. A., Bird, P. I., Carrell, R. W., Church, F. C., Coughlin, P. B., Gettins, P. G., Irving, J. A., Lomas, D. A., Luke, C. J., Moyer, R. W., Pemberton, P. A., Remold-O’Donnell, E., Salvesen, G. S., Travis, J. & Whisstock, J. C. (2001). J. Biol. Chem. 276, 33293–33296. [DOI] [PubMed]
- Sparta, K. M., Krug, M., Heinemann, U., Mueller, U. & Weiss, M. S. (2016). J. Appl. Cryst. 49, 1085–1092.
- Spence, M. A., Mortimer, M. D., Buckle, A. M., Minh, B. Q. & Jackson, C. J. (2021). Mol. Biol. Evol. 38, 2915–2929. [DOI] [PMC free article] [PubMed]
- Sprong, H., Azagi, T., Hoornstra, D., Nijhof, A. M., Knorr, S., Baarsma, M. E. & Hovius, J. W. (2018). Parasites Vectors, 11, 145. [DOI] [PMC free article] [PubMed]
- Tirloni, L., Seixas, A., Mulenga, A., da Silva Vaz, I. Jr & Termignoni, C. (2014). Exp. Parasitol. 137, 25–34. [DOI] [PubMed]
- Tsukada, H. & Blow, D. M. (1985). J. Mol. Biol. 184, 703–711. [DOI] [PubMed]
- Tyson, K. R., Elkins, C. & de Silva, A. M. (2008). J. Immunol. 180, 3964–3968. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vangone, A., Spinelli, R., Scarano, V., Cavallo, L. & Oliva, R. (2011). Bioinformatics, 27, 2915–2916. [DOI] [PubMed]
- Williams, C. J., Headd, J. J., Moriarty, N. W., Prisant, M. G., Videau, L. L., Deis, L. N., Verma, V., Keedy, D. A., Hintze, B. J., Chen, V. B., Jain, S., Lewis, S. M., Arendall, W. B., Snoeyink, J., Adams, P. D., Lovell, S. C., Richardson, J. S. & Richardson, D. C. (2018). Protein Sci. 27, 293–315. [DOI] [PMC free article] [PubMed]
- Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. & Wilson, K. S. (2011). Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed]
- Yamasaki, M., Arii, Y., Mikami, B. & Hirose, M. (2002). J. Mol. Biol. 315, 113–120. [DOI] [PubMed]
- Zou, Z., Anisowicz, A., Hendrix, M. J., Thor, A., Neveu, M., Sheng, S., Rafidi, K., Seftor, E. & Sager, R. (1994). Science, 263, 526–529. [DOI] [PubMed]
- Zundert, G. C. P. van, Rodrigues, J. P. G. L. M., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., Melquiond, A. S. J., van Dijk, M., de Vries, S. J. & Bonvin, A. M. J. J. (2016). J. Mol. Biol. 428, 720–725. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Iripin-5, 7b2t
Supplementary Methods and Materials, Supplementary Figures and Supplementary Tables. DOI: 10.1107/S2059798321007920/rr5207sup1.pdf