

Abstract
Sequence-defined polymers show promise for biomimetics, self-assembly, catalysis, and information storage, wherein the primary structure begets complex chemical processes. Here we report the solution-phase and the high-yielding solid-phase syntheses of discrete oligourethanes and methods for their self-immolative sequencing, resulting in rapid and robust characterization of this class of oligomers and polymers, without the use of MS/MS. Crucial to the sequencing is the inherent reactivity of the terminal alcohol to “unzip” the oligomers, in a controlled and iterative fashion, releasing each monomer as a 2-oxazolidinone. By monitoring the self-immolation reaction via LC/MS, an applied algorithm rapidly produces the sequence of the oligourethane. Not only does this process provide characterization of structurally complex molecules, it works as a reader of molecular information.
Sequence-defined polymers, e.g., β-peptides, γ-peptides, peptoids, polyureas, and polycarbamates,1 have garnered significant interest over recent decades, to the point that their size and structural complexity are nearing those of biopolymers.2 This complexity renders sequence elucidation difficult, at times - impossible. When successful, analysis has relied upon an assortment of 1 and 2D NMR spectroscopy together with sophisticated mass spectrometry techniques.3 Molecular sequencing techniques such as Edman degradation for peptides and Sanger sequencing for DNA are among the most significant chemical achievements of the 20th century. Modern proteomic studies rely on comparisons to databases for protein identification,4 wherein many of the protein sequences were elucidated via Edman degradations. Notably, very few techniques analogous to Edman or Sanger sequencing exist for synthetic macromolecules, likely due to the fact that only recently has synthetic methodology been capable of creating monodisperse macromolecules as structurally complex as biopolymers.1 Peptoids are one exception, with Zuckerman realizing their stepwise chemical degradation on resin.5 As more examples of sequence-defined polymers with controlled primary1,6,7 and secondary2,8 structures emerge, new advanced characterization techniques are necessary.
Controlled chemical degradations,9,10 such as the classic Edman degradation, prevailed in the early days of polymer science but have fallen out of favor with the widespread adoption of NMR11 and mass spectrometry.12,13 However, if the sequence-defined polymer is designed with degradative-sequencing in mind, controlled chain-end depolymerization still offers a powerful approach to primary structure determination. A limit to this approach is that copolymers seldom depolymerize in a controlled manner14,15 as some homopolymers do.16 Triggered and complete depolymerizations, termed self-immolative polymers, are one solution to this problem.17,18 In general, removal of a triggering group from the chain end results in a spontaneous cascading elimination or cyclization event that releases all monomers, the kinetics for which depend upon the structure of the polymer backbone and its breakdown mechanism. The first example of such a system utilized a polyurethane backbone, where upon revealing the terminal amine caused a spontaneous 1,6-elimination and subsequent decarboxylation, revealing the next amine.19 Typically, 1,6- and 1,4-quinone-methide eliminations20 are much faster than alternating cyclization–eliminations21 or intramolecular cyclization mechanisms.22 We recognized if the self-immolation could be induced predictably, producing observable intermediates, it could act as a sequencing routine.
Reported here is the first example of a kinetically slow depolymerization designed for the characterization of the primary structure of a sequence-defined polymer. For the cyclization event that iteratively deconstructs the polymer, we utilized a urethane backbone, wherein a terminal β-alcohol facilitates a favorable 5-exo-trig cyclization that releases a 2-oxazolidinone and a new O-terminus, available for the next cyclization event (Scheme 1). Importantly, as described below, the sequence can be deciphered by a single LC/MS run. This work adds polyurethanes to the very short list of chemically sequenceable abiotic polymers.
Scheme 1.

Self-Immolative Mechanism of Urethanes
As precedent for our concept, random copoly(4-hydroxybutyrate) esters self-immolate via intramolecular cyclization to generate γ-lactones, as monitored by NMR spectroscopy.22 We postulated that urethanes should be capable of a similar intramolecular cyclization. We reasoned that the use of β-amino alcohols as the monomers would allow us to exploit the high effective molarity (EM) of the terminal alcohol, promoting the sequencing in aqueous-alcohol media without hydrolysis or alcoholysis of the urethane linkages.23 Furthermore, β-amino alcohols are an inexpensive and diverse pool of chiral monomers.
We began by synthesizing a trimer as a model with which to study the self-immolation (Figure 1, Scheme S1). We chose 2,4-dimethoxybenzyl amine to act as a functional handle that mimics the Rink-amide resin linkage. The amine was activated with 1,1′-carbonyldiimidazole (CDI), followed by chemoselective addition of the amine of l-phenylalaninol to form a stable urea linkage 1. The alcohol terminus was then activated with CDI, followed once again by chemoselective addition of the amine of l-alaninol, forming the first carbamate 2. This was repeated with l-leucinol to provide the desired trimer 3 (Figure 1a).
Figure 1.

(a) Sequencing of trimer 3 in 2.5:1 MeOH/H2O with K3PO4. Reaction was heated to 70 °C in a microwave and (b) sampled at the denoted time intervals by LC/MS.
For self-immolation, a sufficiently basic media and appropriate temperature to promote the intramolecular cyclization via the terminal alcohol was developed. We chose potassium phosphate (K3PO4), due to its low cost, solubility in aqueous/organic mixtures, and minimal footprint in spectroscopic techniques. Further, the pKa of the conjugate acid is 12.3,24 making it sufficiently basic for reversible deprotonation of the terminal alcohol, while lacking the basicity to cause an uncontrollable and rapid depolymerization.
To precisely control the temperature for reproducible self-immolation, microwave irradiation was utilized over traditional convection heating. Trimer 3 was microwaved in the presence of K3PO4 in a MeOH/H2O mixture at 70 °C and sampled for LC/MS at 20 min intervals (Figure 1). Each molecule, 3, 2, and 1, could be cleanly observed and quantified by absorbance at 280 nm (near the absorbance maximum of 2,4-dimethoxy benzyl amine) in the chromatogram. All three were defined by their distinctive retention time and absorbance and characterized by their m/z. The gradual disappearance of trimer 3, appearance and subsequent disappearance of dimer 2, and final existence of only monomer 1 are indicative of a stepwise sequencing. Upon deprotonation of the terminal alcohol, 3 cyclizes intramolecularly with the proximal urethane to form 2-oxazolidinone 4. The newly released terminal alcohol of dimer 2 then cyclizes forming 5. Both 4 and 5 were observed by their characteristic m/z, but they did not have measurable absorbances due to their lack of a chromophore at the diode array detector (DAD) wavelengths (210–800 nm). As anticipated, the urea linkage in 1 was stable to hydrolysis and cyclization by the terminal alcohol.
Having achieved preliminary success, we studied the importance of various conditions on the reaction. Trimer 3 was subjected to the same basic conditions at 23 °C and monitored by LC/MS. A small amount of initial self-immolation can be seen upon addition of the base (~10% of 3 is consumed over the first hour), but no further reaction is seen through 14 days (Figure S1). As another control, trimer 3 was subjected to the 70 °C immolation conditions, sans K3PO4, and monitored by LC/MS. No immolation, appreciable degradation, or hydrolysis was observed after 90 min (Figure S2). Various solvents and inorganic bases were explored, with varying effects on the sequencing rates and efficiencies (Figures S3-S7). The combination of DMSO with Cs2CO3 was found to be very effective at sequencing and more amenable to 1H NMR spectroscopy. By immolating trimer 3 in d6-DMSO with Cs2CO3, we were able to confirm the formation of 4 and 5 (Figure S8) by 1H NMR spectroscopy, and peak integrals showed that the transformation of 3 into 1, 4, and 5 is quantitative with no observable byproducts.
Urethanes are known to degrade either via addition/elimination mechanisms or via β-eliminations that generate isocyanates.25,26 To confirm addition/elimination via the anticipated 5-exo-trig cyclization, a methoxy terminated trimer, analogous to 3, was synthesized and subjected to the same sequencing conditions (Scheme S2, Figure S9). After microwaving 3a for 1 h with K3PO4 at 70 °C followed by 1 h at 90 °C, no immolation was observed. Likewise, 1H NMR spectroscopy showed no decomposition when the sample was heated at 70 °C in d6-DMSO with Cs2CO3 (Figure S10).
To challenge the sequencing capabilities, a side-chain diverse pentamer was synthesized according to the procedure described above (Scheme S1). Pentamer 6 was subjected to the self-immolation conditions used with 3 and monitored by LC/MS (Figure 2a). The immolation proved slower with the longer oligomer, halting after 2 h of irradiation (Figure S11). However, each iteration of the truncated oligomer (7, 3, 2, and immolation to completion, we used 30 equivalents of K3PO4. The stacked chromatograms (Figure 2b) show that each truncated iteration of the oligomer grows in and then disappears. The lone chromophoric 2-oxazolidinone 8 appears immediately as the first terminal monomer cyclizes, remaining relatively consistent throughout the full immolation. Figure 2c quantifies the progress of the reaction as a function of time by calculating the overall percent composition (from integrated absorbances at 280 nm) of each oligomer in solution.
Figure 2.

Sequencing of pentamer 6 and analysis by LC/MS. (a) Sequencing of pentamer 6 in 2.5:1 MeOH/H2O with K3PO4. Reaction was heated to 70 °C in a microwave reactor and (b) sampled at the delineated time intervals by LC/MS. (c) Progress of the reaction plotted as percent composition of each oligomer vs time.
To access a pool of functionally diverse oligomers and polymers, we turned to solid-phase synthesis (SPS). The synthesis of the oligourethanes on an insoluble matrix was optimized from a method described by Schultz.7 N-Fmoc protected monomers were synthesized by reduction of commercial Fmoc-protected amino acids to the corresponding alcohols, with retention of optical purity,27 in 80–95% yield (Scheme S3). The Fmoc-1,2-amino alcohols were activated with 4-nitrophenyl chloroformate and isolated in >70% yield as the bench stable 4-nitrophenylcarbonate. Stepwise oligomerization was achieved by reacting the activated monomers with resin-bound amines in the presence of DIPEA and HOBt (Scheme S4). Fmoc-deprotection of resin-bound moieties was monitored by UV–vis spectrometry,28 thus determining coupling efficiencies to be 99%, which is comparable to solid-phase peptide synthesis (SPPS).29 Cross-reactive side-chain functionalities were protected with acid-labile protecting groups that would be removed during resin cleavage (Scheme S5), analogous to SPPS.
We chose to use a commercial 2-chlorotrityl polystyrene resin loaded with phenylalaninol. Growth of the oligomer therefore proceeds in an O-to-N manner, and more importantly, cleavage of the oligourethanes from the resin 1) and the corresponding 2-oxazolidinones (8, 9, 4, and 5) were observable in the LC/MS traces. To drive the self-generates the free O-termini that are necessary for sequencing and leaves the amine termini free for coupling or labeling. We postulated that this terminal amine could also either cyclize and rearrange to a urea, or itself could form a 2-oxazolidinone during immolation, potentially creating a two-headed immolative polymer. To prevent both possibilities and to incorporate a long-wavelength chromophore with which to monitor the immolation, the terminal amine was “capped” with 4-fluoro-7-nitrobenzofurazan (NBD-F) prior to resin cleavage. Following this design, heptamer 10 (Figure 3) was isolated in 84% overall yield after six coupling steps, one NBD labeling step, and cleavage from the resin (Scheme S4).
Figure 3.

Self-immolative sequencing of heptamer 10.
Pure heptamer 10 was immolated and easily sequenced in DMSO at 70 °C with Cs2CO3 (Figure 3, Scheme S5). Each iteration (11, 12, 13, 14, 15, and 16, Scheme S5) was observed and characterized by LC/MS. With sights toward using this technology in a combinatorial capacity, we tested whether the oligomers could be accurately sequenced without prior purification. Heptamer 10 was cleaved from the resin, and the crude product was sequenced (Figure S12). The resulting sequencing plot is comparable to the purified heptamer and easily interpreted.
Finally, we looked to further enhance side chain diversity. However, some monomers were problematic. Notably, glycine-derived monomers stalled immolation, which we postulate is due to a lack of any steric compression, akin to the Thorpe–Ingold effect.30 In addition, a glutamate-derived monomer also stalled, resulting in unidentified side products. Taking care to avoid these complications, heptamer 17 (Figure 4) with five unique side chains was synthesized and successfully sequenced (Schemes S6-S7, Figure S13). High-resolution LC/MS of the immolation distinguished each of the 12 components (seven oligomers, five oxazolidinones, Figure S14), confirming clean, iterative, and repetitive terminal 5-exo-trig cyclizations.
Figure 4.
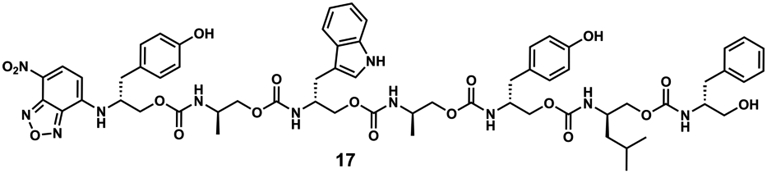
Structure of heptamer 17.
In order to test the simplicity, robustness, and accuracy of the methodology, a blind study was performed. Less than 1 μmol of an octamer, never sequenced prior, was given to a colleague. The colleague was provided with a monomer pool, the knowledge that the unknown is an oligourethane, and an experimental procedure for self-immolation. In less than 4 h, they were able to immolate, collect and analyze the LC/MS data, and decipher the exact structure of the octamer with a single LC/MS trace, as exemplified in Figure 5. This study is detailed in the Supporting Information (SI).
Figure 5.

Sequencing of an “unknown” octamer. The LC trace at 470 nm shows the location of each oligomer’s characteristic molecular ion (M+). The mass difference between each peak corresponds to the mass of each monomer as it is removed from the sequence. The sequence and structure of the oligomer was then easily deciphered.
With a robust and predictable cyclization, we developed an algorithm that automates the read-out process. Taking the data from Figure 5, the algorithm correlates the mass differences to their respective oxazolidinone, which is correlated to their SMILES code. When executed, the algorithm provides the monomers (interpreted from their corresponding oxazolidinones) in the order with which they appear in the oligomer. An explanation of the algorithm can be found in the SI, and the algorithm itself is available on GitHub.
In summary, N-to-O repeating polyurethanes have been identified as a new class of chemically sequenceable, sequence-defined polymers—joining only peptides, peptoids, and nucleic acids in this regard. The ability to sequence with a single LC/MS trace provides a simple and economical approach to sequencing, forsaking MS/MS and databases. This system has potential for combinatorial receptor/catalyst creation, as well as for information storage in digital polymers. Potentially more important than introducing this new sequenceable polymer family to the community, we hope to inspire others to design sequenceability into the linkages of sequence-defined polymers.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support for this work from the Army Research Office (W911NF-17-1-0522), the Howard Hughes Medical Institute (GT10481), and the Welch Reagents Chair to E.V.A. (F-0045). We would like to acknowledge the UT Mass Spectrometry Facility for their instrumental help and the UT NMR facilities for Bruker AVANCE III 500: NIH Grant Number 1 S10 OD021508-01.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b12818.
Detailed experimental procedures, sequencing experiments, sequencing algorithm, supplementary data, and spectral data for all compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Solleder SC; Schneider RV; Wetzel KS; Boukis AC; Meier MAR Recent Progress in the Design of Monodisperse, Sequence-Defined Macromolecules. Macromol. Rapid Commun 2017, 38 (9), 1600711. [DOI] [PubMed] [Google Scholar]
- (2).Martinek TA; Fülöp F Peptidic foldamers: ramping up diversity. Chem. Soc. Rev 2012, 41 (2), 687–702. [DOI] [PubMed] [Google Scholar]
- (3).Mutlu H; Lutz J-F Reading Polymers: Sequencing of Natural and Synthetic Macromolecules. Angew. Chem., Int. Ed 2014, 53 (48), 13010–13019. [DOI] [PubMed] [Google Scholar]
- (4).Cottrell JS Protein identification using MS/MS data. J. Proteomics 2011, 74 (10), 1842–1851. [DOI] [PubMed] [Google Scholar]
- (5).Proulx C; Noë F; Yoo S; Connolly MD; Zuckermann RN On-resin N-terminal peptoid degradation: Toward mild sequencing conditions. Biopolymers 2016, 106 (5), 726–736. [DOI] [PubMed] [Google Scholar]
- (6).Zuckermann RN; Kerr JM; Kent SBH; Moos WH Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc 1992, 114 (26), 10646–10647. [Google Scholar]
- (7).Cho C; Moran E; Cherry; Stephans J; Fodor S; Adams C; Sundaram A; Jacobs J; Schultz P An unnatural biopolymer. Science 1993, 261 (5126), 1303–1305. [DOI] [PubMed] [Google Scholar]
- (8).Hill DJ; Mio MJ; Prince RB; Hughes TS; Moore JS A Field Guide to Foldamers. Chem. Rev 2001, 101 (12), 3893–4012. [DOI] [PubMed] [Google Scholar]
- (9).Harwood HJ Sequence Distribution in Copolymers. Angew. Chem., Int. Ed. Engl 1965, 4 (12), 1051–1060. [Google Scholar]
- (10).Compton RG; Bamford CH; Tipper CFH Degradation of Polymers; Elsevier Science: 1975; Vol. 14. [Google Scholar]
- (11).1 - Polymer Structure and Carbon-13 NMR. In Polymer Sequence Determination, Randall JC, Ed.; Academic Press: 1977; pp 1–27. [Google Scholar]
- (12).Montaudo G; Samperi F; Montaudo MS Characterization of synthetic polymers by MALDI-MS. Prog. Polym. Sci 2006, 31 (3), 277–357. [Google Scholar]
- (13).Gruendling T; Weidner S; Falkenhagen J; Barner-Kowollik C Mass spectrometry in polymer chemistry: a state-of-the-art up-date. Polym. Chem 2010, 1 (5), 599–617. [Google Scholar]
- (14).Smith DA; Youren JW Pyrolysis of polyolefin elastomers. Br. Polym. J 1976, 8 (4), 101–117. [Google Scholar]
- (15).Pan G; Li H; Cao Y 1H-NMR investigation of the thermooxidation degradation of poly(oxymethylene) copolymers. J. Appl. Polym. Sci 2004, 93 (2), 577–583. [Google Scholar]
- (16).McCormick H Quantitative aspects of pyrolysis/gas-liquid chromatography of some vinyl polymers. Journal of Chromatography A 1969, 40, 1–15. [Google Scholar]
- (17).Peterson GI; Larsen MB; Boydston AJ Controlled Depolymerization: Stimuli-Responsive Self-Immolative Polymers. Macromolecules 2012, 45 (18), 7317–7328. [Google Scholar]
- (18).Kim H; Mohapatra H; Phillips ST Rapid, On-Command Debonding of Stimuli-Responsive Cross-Linked Adhesives by Continuous, Sequential Quinone Methide Elimination Reactions. Angew. Chem., Int. Ed 2015, 54 (44), 13063–13067. [DOI] [PubMed] [Google Scholar]
- (19).Sagi A; Weinstain R; Karton N; Shabat D Self-Immolative Polymers. J. Am. Chem. Soc 2008, 130 (16), 5434–5435. [DOI] [PubMed] [Google Scholar]
- (20).Erez R; Shabat D The azaquinone-methide elimination: comparison study of 1,6- and 1,4-eliminations under physiological conditions. Org. Biomol. Chem 2008, 6 (15), 2669–2672. [DOI] [PubMed] [Google Scholar]
- (21).McBride RA; Gillies ER Kinetics of Self-Immolative Degradation in a Linear Polymeric System: Demonstrating the Effect of Chain Length. Macromolecules 2013, 46 (13), 5157–5166. [Google Scholar]
- (22).Zhang L-J; Deng X-X; Du F-S; Li Z-C Chemical Synthesis of Functional Poly(4-hydroxybutyrate) with Controlled Degradation via Intramolecular Cyclization. Macromolecules 2013, 46 (24), 9554–9562. [Google Scholar]
- (23).Kirby AJ, Effective Molarities for Intramolecular Reactions. In Advances in Physical Organic Chemistry; Gold V, Bethell D, Eds.; Academic Press: 1980; Vol. 17, pp 183–278. [Google Scholar]
- (24).Beitia JLL Tripotassium Phosphate: From Buffers to Organic Synthesis. Synlett 2011, 2011 (01), 139–140. [Google Scholar]
- (25).Montaudo G; Puglisi C; Scamporrino E; Vitalini D Mechanism of thermal degradation of polyurethanes. Effect of ammonium polyphosphate. Macromolecules 1984, 17 (8), 1605–1614. [Google Scholar]
- (26).Chapman TM Models for polyurethane hydrolysis under moderately acidic conditions: A comparative study of hydrolysis rates of urethanes, ureas, and amides. J. Polym. Sci., Part A: Polym. Chem 1989, 27 (6), 1993–2005. [Google Scholar]
- (27).Hwang S-H; Blaskovich MA; Kim H-O A Convenient Reduction of alpha-Amino Acids to 1,2-Amino Alcohols With Retention of Optical Purity. Open Org. Chem. J 2008, 2, 107–109. [Google Scholar]
- (28).Eissler S; Kley M; Bächle D; Loidl G; Meier T; Samson D Substitution determination of Fmoc-substituted resins at different wavelengths. J. Pept Sci 2017, 23 (10), 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Collins JM; Porter KA; Singh SK; Vanier GS High-Efficiency Solid Phase Peptide Synthesis (HE-SPPS). Org. Lett 2014, 16 (3), 940–943. [DOI] [PubMed] [Google Scholar]
- (30).Foo SW; Takada Y; Yamazaki Y; Saito S Dehydrative synthesis of chiral oxazolidinones catalyzed by alkali metal carbonates under low pressure of CO2. Tetrahedron Lett. 2013, 54 (35), 4717–4720. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.