

Abstract
Background
Congenital myasthenic syndromes (CMSs) are a heterogeneous group of neuromuscular disorders. Mutations of the nicotinic acetylcholine receptor epsilon subunit gene (CHRNE) are the most common causes of these disorders. CMSs are gaining increasing recognition by clinicians. However, pharmacological treatment of CMS with CHRNE mutations has only been discussed in a small number of case reports.
Objective
This study aims to determine how to choose an appropriate pharmacological strategy for CMS with CHRNE mutations.
Methods
A meta-analysis was performed. PubMed, MEDLINE, Web of Science, and Cochrane Library databases were searched for studies published in English prior to June 1, 2020. The extracted data included clinical information, gene mutations, pharmacological treatment, and treatment effects.
Results
A total of 48 studies and 208 CMS patients with CHRNE mutations were included in our meta-analysis. Ten different pharmacological strategies were used in these patients. Our research found that β2-adrenergic receptor agonists had the best treatment effect for CMS patients with CHRNE mutations, especially in patients with primary AChR deficiency. In addition, our analysis found no evidence that age at disease onset influences the treatment results.
Conclusion
This meta-analysis provides evidence that (1) β2-adrenergic receptor agonist therapy could be the first choice of pharmacological strategy for treating CMS with CHRNE mutations; (2) a single-drug-regime, rather than a combination therapy, should be the first choice of treatment; and (3) it is never too late to initiate pharmacological treatment.
Keywords: CHRNE, congenital myasthenic syndrome, β2-adrenergic receptor agonist, fast-channel syndrome, slow-channel syndrome, primary AChR deficiency
1. INTRODUCTION
Congenital myasthenic syndromes (CMSs) are a heterogeneous group of inherited disorders in which the safety margin of neuromuscular transmission is impaired due to mutations of proteins involved in the organization, function, maintenance, and modulation of the neuromuscular junction (NMJ) [1]. The incidence of CMSs was estimated to be 1.8 per million in a total population [2, 3] and 2.3 to 22.2 per million in the pediatric population [4-6], but due to complexity of the procedures that are used to reach an accurate diagnosis, these incidence rates are likely underestimations. Currently, approximately 30 proteins are involved in various types of CMSs; these proteins are either located at the presynaptic, synaptic, or postsynaptic part of the NMJ or they undergo abnormal glycosylation (Fig. 1a). Among them, mutations of the nicotinic acetylcholine receptor epsilon subunit gene (CHRNE) are the most frequent; the incidence of this mutations was estimated to be 3.4 per million children in the UK [4], and it accounted for nearly 49% of all the CMS cases in a large-scale analysis of 680 patients [7].
Fig. (1).
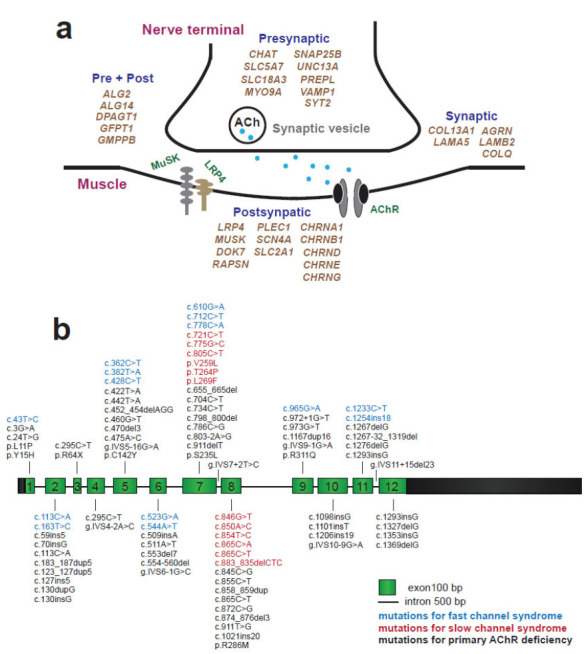
Heterogeneity of genetic defects in CMS. (a) Genes related to CMS are highlighted in khaki. Over 30 genes are involved in varied types of CMS. (b) Schematic diagram of CHRNE showing the exons and introns of different mutations identified within the cohort of patients. Mutations of fast-channel syndrome, slow-channel syndrome, and primary AChR deficiency are in blue, red, and black, respectively. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
The CHRNE gene encodes the ε-subunit of acetylcholine receptor (AChR). Mutations of CHRNE fall into two major groups: kinetic mutations with or without minor AChR deficiency and low-expressor mutations with or without minor kinetic effects, which are also called primary AChR deficiency (Fig. 1b). The kinetic mutations consist of two classes: slow-channel syndromes or fast-channel syndromes [8]. Slow-channel syndrome is named after the abnormally slow decay of synaptic currents caused by prolonged opening events of the AChR channel. In contrast, the fast-channel syndrome is named after the abnormally fast decay of the synaptic response caused by brief channel opening events due to decreased affinity of acetylcholine (ACh), decreased gating efficiency, or impaired fidelity of gating [9]. In fast-channel syndrome and primary AChR deficiency, the NMJ is frequently underdeveloped, providing a relatively explicit basis for muscle weakness. In contrast, in slow-channel syndrome, the opening time of AChR is increased, producing prolonged endplate currents and excess calcium leakage into the endplate region of the muscle fiber [10]. Mutations underlying the fast-channel syndrome or primary AChR deficiency cause a “loss of function” and show recessive inheritance, whereas mutations of the slow-channel syndrome cause a “gain of function” and usually show dominant inheritance. Compared with the other two types of CMS with CHRNE mutations, many patients with the slow-channel syndrome have a prominent involvement of the neck, forearm extensors, wrist and finger extensor muscle [11]. Mutations of not only CHRNE but also other subunits of AChR can lead to kinetic mutations.
The clinical phenotype of CMS with CHRNE mutations is highly heterogeneous. Patients with CMS usually have a neonatal-onset; however, symptoms can also present as late as in the second decade of life. Although the existence of CMS has been characterized for decades, pharmacological treatment is still lacking and unsatisfactory. Establishing optimal therapeutic management of CMS with CHRNE mutations is always a difficult task for clinicians due to (1) the low prevalence, (2) the lack of characteristic manifestation or clinical features, and (3) the absence of consensual criteria for the assessment of pharmacological strategy efficacy among previously published studies. Therefore, the treatment of CMS with CHRNE mutations currently relies mainly upon the clinicians' experience, with no consensus or priority of a specific pharmacological strategy over the others. Acetylcholinesterase inhibitors (AChEIs), a kind of drug that inhibits the hydrolysis of ACh, and 3,4-diaminopyridine (DAP), a drug that increases ACh release from the nerve terminal by blocking voltage-gated potassium channels (VGKCs), were efficient in some CMS cases with CHRNE mutations; however, the improvement was unsatisfying. In some reports, fluoxetine (FLX) and quinidine (QUIN), both of which are long-lived open-channel blockers of AChR, and salbutamol, a selective β2-adrenergic receptor agonist (BA), showed striking amelioration effects. Thus, the pharmacological treatment for CMS with CHRNE mutations remains controversial for many clinicians. This study aims to determine how to choose an appropriate pharmacological strategy for CMS with CHRNE mutations.
2. METHODS
2.1. Search Strategy
The literature search was restricted to articles published in English. Two authors (K.H. and YB.L.) independently searched the PubMed (1966-2020), MEDLINE (1950-2020), Web of Science (1864-2020), and Cochrane Library (2020) databases using the keywords “CHRNE mutations”, “CHRNE” or “congenital myasthenic syndrome, CHRNE”. The most recent search was performed on June 1, 2020. In addition, a manual search was carried out to identify references in the identified studies to find possible other studies. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) [12] chart for the search strategy is shown in (Fig. 2). No randomized clinical trial was identified, and only observational case reports or studies were included in our analysis. The studies were read thoroughly to assess the eligibility to be included in the meta-analysis.
Fig. (2).
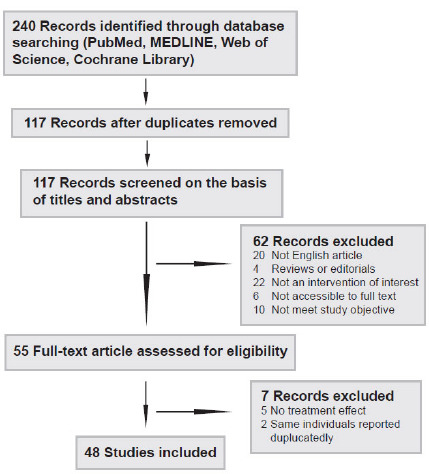
Flow chart presenting the process of the study selection for systematic reviews and meta-analysis.
2.2. Inclusion and Exclusion Criteria for the Literature
Case reports and studies were included if they fulfilled the following criteria: (1) English-language article; (2) patients with CHRNE mutations with no restriction regarding their age, gender, ethnicity and treatment; (3) genetic tests confirmed the CHRNE mutation regardless of the details of the mutations. The exclusion criteria were as follows: (1) articles without pharmacological treatment and (2) articles that do not mention the treatment effect of pharmacologic therapy.
2.3. Data Extraction
The following data were extracted directly from the articles: clinical information, gene mutation, pharmacological treatment, and follow-up data. Clinical data included the age at symptom onset and the age when the case study was reported. If more than one patient had been reported in different studies or by the same group, a comparison of the participants in the studies was made, and data appearing more than once were excluded. Data were independently extracted by two authors (K.H. and YB.L.) with a standardized data extraction form and were inspected by other authors (FF.B. and H.Y.). We examined the published information provided in the original studies. Any contradictory data were discussed until we reached a consensus.
2.4. Quality Assessment of Individual Studies
The quality of individual studies was assessed using the recently published “Tool for evaluating the methodological quality of case reports and case series” proposed by Murad et al. [13] based on the previous criteria from Pierson, Bradford Hills, and Newcastle-Ottawa scale. Each study was independently evaluated according to four domains (selection, ascertainment, causality, and reporting) by two authors (K.H. and YB.L.), leading to an overall assessment for each study (Table 1). The quality assessment included 8 leading exploratory questions with a binary response (yes/no) to determine whether the item suggested the presence of bias.
Table 1. Qualitative assessment of the included studies.
| - | Domains for Evaluating the Methodological Quality of Case Reports and Case Series [13] | |||||||
|---|---|---|---|---|---|---|---|---|
| - | Selection | Ascertainment | Causality | Reporting | ||||
| References | Question 1 | Question 2 | Question 3 | Question 4 | Question 5 | Question 6 | Question 7 | Question 8 |
| Mihaylova et al. [2] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Natera-de Benito et al. [3] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Anja et al. [6] | Yes | Yes | Yes | Yes | No | No | No | No |
| Angelini et al. [14] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Yang et al. [15] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| McMacken et al. [16] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Estephan et al. [17] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Durmus et al. [18] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Ardissone et al. [19] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Verma et al. [20] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Tan et al. [21] | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| Shen et al. [22] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Natera-de Benito et al. [23] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Chang et al. [24] | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| Santos et al. [25] | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| Rodriguez Cruz et al. [26] | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| Azuma et al. [27] | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Webster et al. [28] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Pavone et al. [29] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Finlayson et al. [30] | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes |
| Webster et al. [31] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Shen et al. [32] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Chaouch et al. [33] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Salih et al. [34] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Sadeh et al. [35] | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| Maselli et al. [36] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Brugnoni et al. [37] | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Outteryck et al. [38] | Yes | Yes | Yes | Yes | No | No | No | No |
| Faber et al. [39] | Yes | Yes | Yes | Yes | No | No | No | No |
| Richard et al. [40] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Richard et al. [41] | Yes | Yes | Yes | Yes | No | No | No | No |
| Colomer et al. [42] | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| Soltanzadeh et al. [43] | Yes | Yes | Yes | Yes | No | Yes | No | No |
| Ohno et al. [44] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Muller et al. [45] | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Fidzianska et al. [46] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Ohno et al. [47] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Sine et al. [48] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Ealing et al. [49] | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Ohno et al. [50] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Milone et al. [51] | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Harper et al. [52] | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Ohno et al. [53] | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Engel et al. [54] | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Shen et al. [55] | Yes | Yes | Yes | Yes | No | Yes | No | Yes |
| Karimzadeh et al. [56] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Gul Mert et al. [57] | Yes | Yes | Yes | Yes | No | No | Yes | No |
| Uchitel et al. [58] | Yes | Yes | Yes | Yes | No | No | No | No |
Selection: 1. Does the patient(s) represent(s) the whole experience of the investigator (center) or is the selection method unclear to the extent that other patients with similar presentation may not have been reported? Ascertainment: 2. Was the exposure adequately ascertained? 3. Was the outcome adequately ascertained? Causality:4. Were other alternative causes that may explain the observation ruled out? 5. Was there a challenge/rechallenge phenomenon? 6. Was there a dose–response effect? 7. Was follow-up long enough for outcomes to occur? Reporting: 8. Is the case(s) described with sufficient details to allow other investigators to replicate the research or to allow practitioners make inferences related to their own practice?
2.5. Statistical Analysis
Data analysis was performed using Statistical Package for Social Sciences (SPSS Inc., Chicago, IL, USA) Version 25. Statistical differences were estimated by the Kruskal-Wallis test or one-way ANOVA followed by the Bonferroni post hoc test. A p-value < 0.05 was considered statistically significant.
3. RESULTS
3.1. Search and Selection Results
A total of 48 studies fulfilled the inclusion and exclusion criteria mentioned in the methods’ section. Among the 48 articles selected [2, 3, 6, 14-58], most are case reports, and none are randomized controlled trials. A total of 208 CMS patients with CHRNE mutations were treated with ten different pharmacological strategies in these studies. Since the effect of different pharmacological strategies is described as incomplete, moderated or remarkable, the treatment effect of different pharmacological strategies was categorized into four types: (-) no effect; (+) partial, incomplete, moderate, modest or mild effect; (++) beneficial, positive or clear effect; (+++) remarkable, dramatical or satisfying effect. The treatment details are listed in the Supplementary Table S1 (172.5KB, pdf) -S3 (172.5KB, pdf) .
3.2. Pharmacological Treatment Effects in Patients with the Fast-Channel Syndrome
In the included studies, nine patients were diagnosed with the fast-channel syndrome. The pharmacological strategies used in these patients mainly included AChEIs, prednisone (PDN), AChEIs+BA, AChEIs+PDN, and other treatments. All the patients reported were first prescribed with AChEIs alone and were all responsive. Specifically, due to being misdiagnosed with myasthenia gravis (MG), one patient was prescribed with PDN [58]. Between different pharmacological strategies used in the treatment of fast-channel syndrome, no significant differences were found, partially due to the limited data (p = 0.294) (Fig. 3a).
Fig. (3).
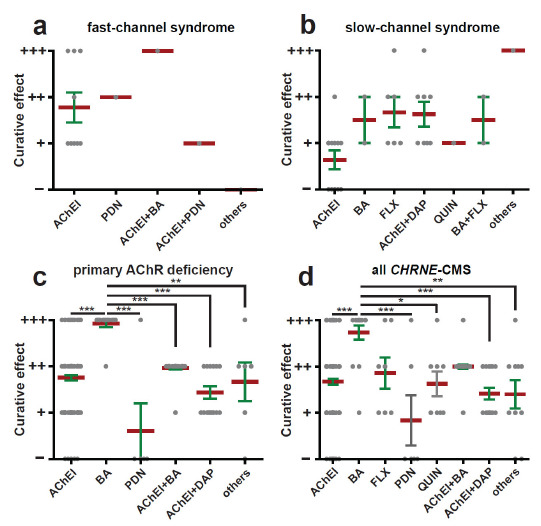
The treatment effect of different pharmacologic strategies based on a review of the literature. Treatment effect was categorised into four grades: (-) no effect; (+) partial, incomplete, moderate, modest or mild effect; (++) beneficial, positive or clear effect; (+++) remarkable, dramatical or satisfying effect. The mean and SEM in each group are indicated. The data were analyzed by the Kruskal-Wallis test. *p < 0.05, **p < 0.01, ***p < 0.001.
3.3. Pharmacological Treatment Effects in Patients with Slow-Channel Syndrome
Twenty patients were diagnosed with the slow-channel syndrome. All the mutations of the slow-channel syndrome in our study were restricted to exon 7 and exon 8. Seven different pharmacological strategies, i.e., AChEIs, BA, FLX, QUIN, AChEIs+DAP, BA+FLX, and other treatments, were prescribed to improve the symptoms; however, no significant differences were found between these strategies (p = 0.057) (Fig. 3b). In contrast to all fast-channel syndrome patients responding to AChEIs to some extent, almost half (5/11) of the patients with slow-channel syndrome showed no response to AChEIs. Moreover, all the slow-channel syndrome patients showed an alleviation in symptoms after other pharmacologic strategies, which suggests that AChEIs should not be the first option for patients with the slow-channel syndrome.
3.4. Pharmacological Treatment Effects in Patients with Primary AChR Deficiency
A total of 179 patients with primary AChR deficiency, which is the main type of CMS with CHRNE mutations, were examined in the included studies. Once the patients were diagnosed with a genetic test, AChEIs were tried in all the patients alone or with other drugs. Intriguingly, BA was used in ten patients with primary AChR deficiency, and all ten patients showed a remarkable clinical improvement. Interestingly, PDN was prescribed to several patients who were initially diagnosed with MG; however, the amelioration was limited. Compared with other pharmacological strategies, BA treatment was the best strategy (p = 0.000, 0.000, 0.000, 0.000, 0.007 compared with AChEIs, PDN, AChEIs+BA, AChEIs+DAP and other treatments, respectively) (Fig. 3c).
3.5. Pharmacological Treatment Effects in CMS Patients with CHRNE Mutations
A total of 208 CMS patients were diagnosed with CHRNE mutations, and ten different pharmacological strategies were prescribed: AChEIs, BA, FLX, PDN, QUIN, AChEIs+BA, AChEIs+DAP, AChEIs+PDN, BA+FLX, and other treatments. Since both AChEIs+PDN and BA+FLX were only used in three patients, we categorized them into the “other treatments” group. Among them, BA had a better treatment effect than AChEIs, PDN, QUIN, AChEIs+DAP and others (p = 0.000, 0.000, 0.021, 0.000 and 0.005, respectively) (Fig. 3d). Moreover, the treatment effect of drugs used alone was not significantly different from the effect of combination therapy, suggesting that it is better to use drugs alone rather than drug combination.
3.6. Influence of Age at Symptom Onset on the Treatment Effect of Pharmacological Treatment
Age at onset varied widely among CMS patients with CHRNE mutations. Information on age at symptom onset was available in 5 of 9 cases, 17 of 20 cases, and 86 of 179 cases of the fast-channel syndrome, slow-channel syndrome, and primary AChR deficiency, respectively (Supplementary Table S1 (172.5KB, pdf) -S3 (172.5KB, pdf) ). The age of symptom onset of the slow-channel syndrome was significantly higher than that of the fast-channel syndrome (p = 0.000 and 0.000, compared with fast-channel syndrome and primary AChR deficiency, respectively) (Fig. 4a). Responses to medicine, however, were not influenced by this variability in the age of symptom onset in each group (p = 0.143 in fast-channel syndrome, p = 0.067 in slow-channel syndrome, p = 0.099 in primary AChR deficiency, and p = 0.361 in all CMS with CHRNE mutations) (Fig. 4b-e). These results suggest that it is never too late to initiate pharmacological treatment in CMS patients with CHRNE mutations.
Fig. (4).
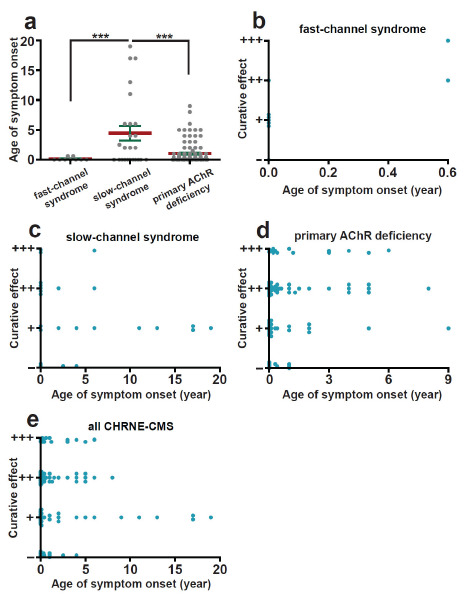
Age at onset of symptoms and treatment effect based on a review of the literature. (a) Age of symptoms onset of each group. The data were analyzed by one-way ANOVA followed by Bonferroni post hoc test. ***p < 0.001. (b-e) The relationship between age of symptoms onset and treatment effect in each group. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
4. DISCUSSION
CHRNE mutations account for up to almost half of the cases of CMS among humans [7]. Our meta-analysis found that primary AChR syndrome is the most common syndrome, as it was found in 86% of cases of CMS with CHRNE mutations. Homozygous or more frequently heterozygous mutations in AChR subunit genes, such as CHRNA1(α), CHRNB1 (β), CHRND (δ) and CHRNE(ε), can cause primary AChR deficiency. However, mutations of ε subunit gene are the most common cause for primary AChR deficiency, which may be partly because the expression of fetal type γ subunit, even at a low level, compensates for the absence of the ε subunit. In contrast, patients harboring low- expression or null mutations in subunits other than the ε subunit might not survive due to the lack of a substituting subunit [54].
Currently, evidence-based medicine primarily considers that randomized clinical trials (RCTs) provide the most robust evidence regarding the efficacy of new treatments in patients with a certain disease. However, RCTs can be nearly impossible to conduct for rare diseases due to the lack of patients available for enrollment [13]. In such cases, a meta-analysis for clinical knowledge regarding the efficacy of pharmacological treatments and other kinds of interventions may be based on observational studies and case reports. An increasing number of meta-analyses of case reports examining rare diseases have been proven to be useful for evidence-based medicine [59, 60]. In our current study, although all the studies reviewed in our meta-analysis are case reports rather than RCTs, the conclusion that β2-adrenergic receptor agonists are an effective first-line treatment for congenital myasthenic syndrome with CHRNE mutations in our analysis is reliable.
Unlike the related autoimmune NMJ disorders such as MG and Lambert-Eaton syndrome, CMS with CHRNE mutations is not caused by an immune response, so theoretically, immunosuppressors are not effective [61, 62]. Most pharmacological strategies for CMS with CHRNE mutations widely depend upon whether it is beneficial to upregulate the amount of available ACh in the synaptic cleft (such as AChEIs and DAP) or to shorten the excessive duration of synaptic current by reducing the channel-open time (such as FLX and QUIN). AChEIs are inhibitors that inhibit ACh from breaking down to choline and acetate, thereby increasing the level and duration of action of the neurotransmitter ACh. DAP is a drug that blocks VGKCs at the presynaptic membrane, prolonging nerve terminal depolarization and increasing ACh release from the nerve terminal into the synaptic cleft [63]. The mechanism of both AChEIs and DAP for the treatment of CMS with CHRNE mutations is a temporary aggregation of ACh, which activates AChR on the postsynaptic muscle membrane. FLX and QUIN, which are both long-lived AChR open-channel blockers, shorten the duration of pathologically long synaptic currents, leading to the prevention of endplate depolarization block and AChR desensitization at the NMJ [64, 65]. FLX and QUIN work in some slow-channel syndromes functionally characterized by prolonged AChR single-channel currents, which may shorten the duration of otherwise prolonged synaptic currents [66]. In the current study, we found that AChEIs, DAP, FLX, and QUIN are effective in some CMS patients with CHRNE mutations but are still not the right choice for all CMS patients with CHRNE mutations.
Specifically, we found that AChEIs had no effect or only a slight effect, while AChEIs+DAP had a beneficial effect on most slow-channel syndromes with CHRNE mutations. In addition, no detrimental effect was found on the slow-channel syndrome with CHRNE mutations, which is interestingly opposite to the opinion that AChEIs or DAP may even worsen slow-channel syndrome. The mechanism underlying this phenomenon is still unclear. The following points may partially account for this phenomenon. First, in slow-channel syndrome with other gene mutations, such as CHRNA1 [33], AChEIs showed overt side effects or even worsened muscle weakness, suggesting that the different gene mutations in slow-channel syndrome may lead to different curative effects of AChEIs and DAP. We hypothesize that the pharmacological strategies are different for slow-channel syndrome according to different mutations. Second, most of the curative effects we extracted from the original literature were descriptive and lacked objective evaluations, such as manual muscle testing (MMT) [67] or quantitative myasthenia gravis (QMG) scores [68], which is also a limitation of our study due to the scarcity of CMS with CHRNE mutations per se, possibly leading to bias. Third, in slow-channel syndrome with CHRNE mutations, the effect of prolonged AChR activation events is to increase the entry of Ca2+ into the postjunctional sarcoplasm through the endplate AChRs, rather than increase the binding of ACh to AChR [69, 70]. The disturbance of the intracellular ionic milieu rather than ACh may also partially explain why AChEIs and DAP were found to have no deleterious effect in slow-channel syndrome with CHRNE mutations.
Compared with the use of AChEIs, where the treatment effect mostly varies from incomplete to moderate, BA demonstrates a robust improvement in the treatment response. BA, such as ephedrine and salbutamol, is also broadly used in CMS patients with CHRNE mutations, which was accidentally discovered to be beneficial for MG first [71]. β2-adrenergic receptors are abundantly expressed in skeletal muscle, and β2-adrenergic receptor stimulation results in various effects on muscle function, including muscle growth, protein regulation, and muscle fiber transitions [72]. Recently, salbutamol, a BA, was found to affect proteins located at the NMJ and exert a stabilizing effect on AChR clusters in normal C2C12 myogenic cells and the C2C12 cell model of CMS with downstream of tyrosine kinase 7 (DOK7) mutation [73]. In zebrafish models of CMS with DOK7 mutation and CMS with muscle-specific receptor tyrosine kinase (MuSK) mutation, β2-adrenergic receptor stimulation was found to improve AChR clustering, motor axon guidance, and the development of prepatterned AChR clusters [74]. Moreover, in a mouse model of another CMS with collagen Q (ColQ) mutation, salbutamol leads to a gradual improvement in muscle strength and significant improvements in several postsynaptic morphological defects of NMJ [75]. The exact mode of action of BA in CMS with CHRNE mutations is still unknown; however, a compensatory mechanism of stabilizing AChR clusters is one of the plausible explanations.
In addition, it has been recently suggested that in skeletal muscle, β2-adrenergic receptor signaling might contribute to exercise training-mediated adaptations [76]. Other researchers found that BA increases skeletal muscle mass by inducing autophagy without any histological abnormalities and enhances muscle force and power output as a result of skeletal muscle hypertrophy [77,78]. Taken together, another possible mechanism of BA on CMS with CHRNE mutations may be that β2-adrenergic receptors can be beneficial by promoting skeletal muscle mass and force.
There are several limitations to this meta-analysis. First, clinical reports have a risk of publication bias, and it is expected that only positive results will be published [79]. Second, we cannot estimate the effect size of an outcome. We found that almost all the clinical cases do not report enough information to aggregate study results using mean, median, or a proportion with a confidence interval. Third, our data may be biased due to incorrect reporting. All studies included are observational studies that lack a QMG score or other objective scores that can be used to help clinicians estimate the treatment effects of different pharmacological strategies. Last, most of the studies do no specify the dosage of the drugs prescribed, and thus, the specific therapeutic response that could not be adequately evaluated.
CONCLUSION
Our meta-analysis advocates for the use of BA, such as salbutamol or ephedrine, as the first-line pharmacological treatment for CMS with CHRNE mutations, especially for patients with primary AChR deficiency, which is the most common type of CMS with CHRNE mutations. Compared with CMS with DOK7 mutation, in which AChEIs worsened symptoms in 40% of the patients [80], AChEIs were never reported to worsen symptoms in CMS with CHRNE mutations, which suggests that AChEIs are safe and may also be an option for CMS with CHRNE mutations. However, more clinical trials are needed to provide solid insight regarding the pharmacological strategy to treat CMS with CHRNE mutations.
ACKNOWLEDGEMENTS
Declared none.
AUTHOR'S CONTRIBUTION
KH, FF-B and HY conceived the project. KH and YB-L extracted data and performed the analysis. KH wrote the manuscript. YB-L polished the manuscript. FF-B and HY critically revised the manuscript for valuable intellectual content. All authors approved the final manuscript.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This research was supported by the National Natural Science Foundation of China (Grant No. 81571256 and 81760238).
CONFLICT OF INTEREST
The authors have no conflicts of interest, financial or otherwise.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.
REFERENCES
- 1.Engel A.G., Shen X.M., Selcen D., Sine S.M. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015;14(4):420–434. doi: 10.1016/S1474-4422(14)70201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mihaylova V., Scola R.H., Gervini B., Lorenzoni P.J., Kay C.K., Werneck L.C., Stucka R., Guergueltcheva V., von der Hagen M., Huebner A., Abicht A., Müller J.S., Lochmüller H. Molecular characterisation of congenital myasthenic syndromes in Southern Brazil. J. Neurol. Neurosurg. Psychiatry. 2010;81(9):973–977. doi: 10.1136/jnnp.2009.177816. [DOI] [PubMed] [Google Scholar]
- 3.Natera-de Benito D., Töpf A., Vilchez J.J., González-Quereda L., Domínguez-Carral J., Díaz-Manera J., Ortez C., Bestué M., Gallano P., Dusl M., Abicht A., Müller J.S., Senderek J., García-Ribes A., Muelas N., Evangelista T., Azuma Y., McMacken G., Paipa Merchan A., Rodríguez Cruz P.M., Camacho A., Jiménez E., Miranda-Herrero M.C., Santana-Artiles A., García-Campos O., Dominguez-Rubio R., Olivé M., Colomer J., Beeson D., Lochmüller H., Nascimento A. Molecular characterization of congenital myasthenic syndromes in Spain. Neuromuscul. Disord. 2017;27(12):1087–1098. doi: 10.1016/j.nmd.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Parr J.R., Andrew M.J., Finnis M., Beeson D., Vincent A., Jayawant S. How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch. Dis. Child. 2014;99(6):539–542. doi: 10.1136/archdischild-2013-304788. [DOI] [PubMed] [Google Scholar]
- 5.Mansukhani S.A., Bothun E.D., Diehl N.N., Mohney B.G. Incidence and Ocular Features of Pediatric Myasthenias. Am. J. Ophthalmol. 2019;200:242–249. doi: 10.1016/j.ajo.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Troha Gergeli A., Neubauer D., Golli T., Butenko T., Loboda T., Maver A., Osredkar D. Prevalence and genetic subtypes of congenital myasthenic syndromes in the pediatric population of Slovenia. Eur. J. Paediatr. Neurol. 2020;26:34–38. doi: 10.1016/j.ejpn.2020.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Abicht A., Dusl M., Gallenmüller C., Guergueltcheva V., Schara U., Della Marina A., Wibbeler E., Almaras S., Mihaylova V., von der Hagen M., Huebner A., Chaouch A., Müller J.S., Lochmüller H. Congenital myasthenic syndromes: achievements and limitations of phenotype-guided gene-after-gene sequencing in diagnostic practice: a study of 680 patients. Hum. Mutat. 2012;33(10):1474–1484. doi: 10.1002/humu.22130. [DOI] [PubMed] [Google Scholar]
- 8.Beeson D., Hantaï D., Lochmüller H., Engel A.G. 126th International Workshop: congenital myasthenic syndromes, 24-26 September 2004, Naarden, the Netherlands. Neuromuscul. Disord. 2005;15(7):498–512. doi: 10.1016/j.nmd.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Engel A.G., Ohno K., Sine S.M. Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nat. Rev. Neurosci. 2003;4(5):339–352. doi: 10.1038/nrn1101. [DOI] [PubMed] [Google Scholar]
- 10.Engel A.G., Lambert E.H., Mulder D.M., Torres C.F., Sahashi K., Bertorini T.E., Whitaker J.N. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann. Neurol. 1982;11(6):553–569. doi: 10.1002/ana.410110603. [DOI] [PubMed] [Google Scholar]
- 11.Otero-Cruz J.D., Báez-Pagán C.A., Dorna-Pérez L., Grajales-Reyes G.E., Ramírez-Ordoñez R.T., Luciano C.A., Gómez C.M., Lasalde-Dominicci J.A. Decoding pathogenesis of slow-channel congenital myasthenic syndromes using recombinant expression and mice models. P. R. Health Sci. J. 2010;29(1):4–17. [PMC free article] [PubMed] [Google Scholar]
- 12.Moher D., Liberati A., Tetzlaff J., Altman D.G., Group P. PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. doi: 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murad M.H., Sultan S., Haffar S., Bazerbachi F. Methodological quality and synthesis of case series and case reports. BMJ Evid Based Med. 2018;23(2):60–63. doi: 10.1136/bmjebm-2017-110853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angelini C., Lispi L., Salvoro C., Mostacciuolo M.L., Vazza G. Clinical and genetic characterization of an Italian family with slow-channel syndrome. Neurol. Sci. 2019;40(3):503–507. doi: 10.1007/s10072-018-3645-2. [DOI] [PubMed] [Google Scholar]
- 15.Yang K., Cheng H., Yuan F., Meng L., Yin R., Zhang Y., Wang S., Wang C., Lu Y., Xi J., Lu Q., Chen Y. CHRNE compound heterozygous mutations in congenital myasthenic syndrome: A case report. Medicine (Baltimore) 2018;97(17):e0347. doi: 10.1097/MD.0000000000010347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McMacken G., Whittaker R.G., Evangelista T., Abicht A., Dusl M., Lochmüller H. Congenital myasthenic syndrome with episodic apnoea: clinical, neurophysiological and genetic features in the long-term follow-up of 19 patients. J. Neurol. 2018;265(1):194–203. doi: 10.1007/s00415-017-8689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Estephan E.P., Sobreira C.F.D.R., Dos Santos A.C.J., Tomaselli P.J., Marques W., Jr, Ortega R.P.M., Costa M.C.M., da Silva A.M.S., Mendonça R.H., Caldas V.M., Zambon A.A., Abath Neto O., Marchiori P.E., Heise C.O., Reed U.C., Azuma Y., Töpf A., Lochmüller H., Zanoteli E. A common CHRNE mutation in Brazilian patients with congenital myasthenic syndrome. J. Neurol. 2018;265(3):708–713. doi: 10.1007/s00415-018-8736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Durmus H., Shen X.M., Serdaroglu-Oflazer P., Kara B., Parman-Gulsen Y., Ozdemir C., Brengman J., Deymeer F., Engel A.G. Congenital myasthenic syndromes in Turkey: Clinical clues and prognosis with long term follow-up. Neuromuscul. Disord. 2018;28(4):315–322. doi: 10.1016/j.nmd.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ardissone A., Moroni I., Bernasconi P., Brugnoni R. Congenital myasthenic syndrome: phenotypic variability in patients harbouring p.T159P mutation in CHRNE gene. Acta Myol. 2017;36(1):28–32. [PMC free article] [PubMed] [Google Scholar]
- 20.Verma S., Mazell S.N., Shah D.A. Amifampridine phosphate in congenital myasthenic syndrome. Muscle Nerve. 2016;54(4):809–810. doi: 10.1002/mus.25230. [DOI] [PubMed] [Google Scholar]
- 21.Tan J.Z., Man Y., Xiao F. A missense mutation in epsilon-subunit of acetylcholine receptor causing autosomal dominant slow-channel congenital myasthenic syndrome in a chinese family. Chin. Med. J. (Engl.) 2016;129(21):2596–2602. doi: 10.4103/0366-6999.192780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen X.M., Okuno T., Milone M., Otsuka K., Takahashi K., Komaki H., Giles E., Ohno K., Engel A.G. Mutations causing slow-channel myasthenia reveal that a valine ring in the channel pore of muscle achr is optimized for stabilizing channel gating. Hum. Mutat. 2016;37(10):1051–1059. doi: 10.1002/humu.23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Natera-de Benito D., Domínguez-Carral J., Muelas N., Nascimento A., Ortez C., Jaijo T., Arteaga R., Colomer J., Vilchez J.J. Phenotypic heterogeneity in two large Roma families with a congenital myasthenic syndrome due to CHRNE 1267delG mutation. A long-term follow-up. Neuromuscul. Disord. 2016;26(11):789–795. doi: 10.1016/j.nmd.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Chang T., Cossins J., Beeson D. A rare c.183_187dupCTCAC mutation of the acetylcholine receptor CHRNE gene in a South Asian female with congenital myasthenic syndrome: a case report. BMC Neurol. 2016;16(1):195. doi: 10.1186/s12883-016-0716-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santos E., Moreira I., Coutinho E., Gonçalves G., Lopes C., Lopes Lima J., Leite M.I. Congenital myasthenic syndrome due to mutation in CHRNE gene with clinical worsening and thymic hyperplasia attributed to association with autoimmune-myasthenia gravis. Neuromuscul. Disord. 2015;25(12):928–931. doi: 10.1016/j.nmd.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Rodríguez Cruz P.M., Palace J., Ramjattan H., Jayawant S., Robb S.A., Beeson D. Salbutamol and ephedrine in the treatment of severe AChR deficiency syndromes. Neurology. 2015;85(12):1043–1047. doi: 10.1212/WNL.0000000000001952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azuma Y., Nakata T., Tanaka M., Shen X.M., Ito M., Iwata S., Okuno T., Nomura Y., Ando N., Ishigaki K., Ohkawara B., Masuda A., Natsume J., Kojima S., Sokabe M., Ohno K. Congenital myasthenic syndrome in Japan: ethnically unique mutations in muscle nicotinic acetylcholine receptor subunits. Neuromuscul. Disord. 2015;25(1):60–69. doi: 10.1016/j.nmd.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 28.Webster R., Liu W.W., Chaouch A., Lochmüller H., Beeson D. Fast-channel congenital myasthenic syndrome with a novel acetylcholine receptor mutation at the α-ε subunit interface. Neuromuscul. Disord. 2014;24(2):143–147. doi: 10.1016/j.nmd.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Pavone P., Polizzi A., Longo M.R., Romano K., Vecchio M., Praticò A.D., Falsaperla R. Congenital myasthenic syndromes: Clinical and molecular report on 7 Sicilian patients. J. Pediatr. Neurosci. 2013;8(1):19–21. doi: 10.4103/1817-1745.111416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finlayson S., Spillane J., Kullmann D.M., Howard R., Webster R., Palace J., Beeson D. Slow channel congenital myasthenic syndrome responsive to a combination of fluoxetine and salbutamol. Muscle Nerve. 2013;47(2):279–282. doi: 10.1002/mus.23534. [DOI] [PubMed] [Google Scholar]
- 31.Webster R., Maxwell S., Spearman H., Tai K., Beckstein O., Sansom M., Beeson D. A novel congenital myasthenic syndrome due to decreased acetylcholine receptor ion-channel conductance. Brain. 2012;135(Pt 4):1070–1080. doi: 10.1093/brain/aws016. [DOI] [PubMed] [Google Scholar]
- 32.Shen X.M., Brengman J.M., Edvardson S., Sine S.M., Engel A.G. Highly fatal fast-channel syndrome caused by AChR ε subunit mutation at the agonist binding site. Neurology. 2012;79(5):449–454. doi: 10.1212/WNL.0b013e31825b5bda. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaouch A., Müller J.S., Guergueltcheva V., Dusl M., Schara U., Rakocević-Stojanović V., Lindberg C., Scola R.H., Werneck L.C., Colomer J., Nascimento A., Vilchez J.J., Muelas N., Argov Z., Abicht A., Lochmüller H. A retrospective clinical study of the treatment of slow-channel congenital myasthenic syndrome. J. Neurol. 2012;259(3):474–481. doi: 10.1007/s00415-011-6204-9. [DOI] [PubMed] [Google Scholar]
- 34.Salih M.A., Oystreck D.T., Al-Faky Y.H., Kabiraj M., Omer M.I., Subahi E.M., Beeson D., Abu-Amero K.K., Bosley T.M. Congenital myasthenic syndrome due to homozygous CHRNE mutations: report of patients in Arabia. J. Neuroophthalmol. 2011;31(1):42–47. doi: 10.1097/WNO.0b013e3181f50bea. [DOI] [PubMed] [Google Scholar]
- 35.Sadeh M., Shen X.M., Engel A.G. Beneficial effect of albuterol in congenital myasthenic syndrome with epsilon-subunit mutations. Muscle Nerve. 2011;44(2):289–291. doi: 10.1002/mus.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maselli R.A., Arredondo J., Cagney O., Mozaffar T., Skinner S., Yousif S., Davis R.R., Gregg J.P., Sivak M., Konia T.H., Thomas K., Wollmann R.L. Congenital myasthenic syndrome associated with epidermolysis bullosa caused by homozygous mutations in PLEC1 and CHRNE. Clin. Genet. 2011;80(5):444–451. doi: 10.1111/j.1399-0004.2010.01602.x. [DOI] [PubMed] [Google Scholar]
- 37.Brugnoni R., Maggi L., Canioni E., Moroni I., Pantaleoni C., D’Arrigo S., Riva D., Cornelio F., Bernasconi P., Mantegazza R. Identification of previously unreported mutations in CHRNA1, CHRNE and RAPSN genes in three unrelated Italian patients with congenital myasthenic syndromes. J. Neurol. 2010;257(7):1119–1123. doi: 10.1007/s00415-010-5472-0. [DOI] [PubMed] [Google Scholar]
- 38.Outteryck O., Richard P., Lacour A., Fournier E., Zéphir H., Gaudon K., Eymard B., Hantaï D., Vermersch P., Stojkovic T. Novel epsilon subunit mutation of the muscle acetylcholine receptor causing a slow-channel congenital myasthenic syndrome. J. Neurol. Neurosurg. Psychiatry. 2009;80(4):450–451. doi: 10.1136/jnnp.2008.148189. [DOI] [PubMed] [Google Scholar]
- 39.Faber C.G., Molenaar P.C., Vles J.S., Bonifati D.M., Verschuuren J.J., van Doorn P.A., Kuks J.B., Wokke J.H., Beeson D., De Baets M. AChR deficiency due to epsilon-subunit mutations: two common mutations in the Netherlands. J. Neurol. 2009;256(10):1719–1723. doi: 10.1007/s00415-009-5190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richard P., Gaudon K., Haddad H., Ammar A.B., Genin E., Bauché S., Paturneau-Jouas M., Müller J.S., Lochmüller H., Grid D., Hamri A., Nouioua S., Tazir M., Mayer M., Desnuelle C., Barois A., Chabrol B., Pouget J., Koenig J., Gouider-Khouja N., Hentati F., Eymard B., Hantaï D. The CHRNE 1293insG founder mutation is a frequent cause of congenital myasthenia in North Africa. Neurology. 2008;71(24):1967–1972. doi: 10.1212/01.wnl.0000336921.51639.0b. [DOI] [PubMed] [Google Scholar]
- 41.Richard P., Gaudon K., Fournier E., Jackson C., Bauché S., Haddad H., Koenig J., Echenne B., Hantaï D., Eymard B. A synonymous CHRNE mutation responsible for an aberrant splicing leading to congenital myasthenic syndrome. Neuromuscul. Disord. 2007;17(5):409–414. doi: 10.1016/j.nmd.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 42.Colomer J., Müller J.S., Vernet A., Nascimento A., Pons M., Gonzalez V., Abicht A., Lochmüller H. Long-term improvement of slow-channel congenital myasthenic syndrome with fluoxetine. Neuromuscul. Disord. 2006;16(5):329–333. doi: 10.1016/j.nmd.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 43.Soltanzadeh P., Müller J.S., Ghorbani A., Abicht A., Lochmüller H., Soltanzadeh A. An Iranian family with congenital myasthenic syndrome caused by a novel acetylcholine receptor mutation (CHRNE K171X). J. Neurol. Neurosurg. Psychiatry. 2005;76(7):1039–1040. doi: 10.1136/jnnp.2004.059436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohno K., Tsujino A., Shen X.M., Milone M., Engel A.G. Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries. J. Med. Genet. 2005;42(8):e53. doi: 10.1136/jmg.2004.026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Müller J.S., Stucka R., Neudecker S., Zierz S., Schmidt C., Huebner A., Lochmüller H., Abicht A. An intronic base alteration of the CHRNE gene leading to a congenital myasthenic syndrome. Neurology. 2005;65(3):463–465. doi: 10.1212/01.wnl.0000172346.26219.fd. [DOI] [PubMed] [Google Scholar]
- 46.Fidzianska A., Ryniewicz B., Shen X.M., Engel A.G. IBM-type inclusions in a patient with slow-channel syndrome caused by a mutation in the AChR epsilon subunit. Neuromuscul. Disord. 2005;15(11):753–759. doi: 10.1016/j.nmd.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 47.Ohno K., Milone M., Shen X.M., Engel A.G. A frameshifting mutation in CHRNE unmasks skipping of the preceding exon. Hum. Mol. Genet. 2003;12(23):3055–3066. doi: 10.1093/hmg/ddg334. [DOI] [PubMed] [Google Scholar]
- 48.Sine S.M., Shen X.M., Wang H.L., Ohno K., Lee W.Y., Tsujino A., Brengmann J., Bren N., Vajsar J., Engel A.G. Naturally occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J. Gen. Physiol. 2002;120(4):483–496. doi: 10.1085/jgp.20028568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ealing J., Webster R., Brownlow S., Abdelgany A., Oosterhuis H., Muntoni F., Vaux D.J., Vincent A., Beeson D. Mutations in congenital myasthenic syndromes reveal an epsilon subunit C-terminal cysteine, C470, crucial for maturation and surface expression of adult AChR. Hum. Mol. Genet. 2002;11(24):3087–3096. doi: 10.1093/hmg/11.24.3087. [DOI] [PubMed] [Google Scholar]
- 50.Ohno K., Anlar B., Ozdirim E., Brengman J.M., DeBleecker J.L., Engel A.G. Myasthenic syndromes in Turkish kinships due to mutations in the acetylcholine receptor. Ann. Neurol. 1998;44(2):234–241. doi: 10.1002/ana.410440214. [DOI] [PubMed] [Google Scholar]
- 51.Milone M., Wang H.L., Ohno K., Prince R., Fukudome T., Shen X.M., Brengman J.M., Griggs R.C., Sine S.M., Engel A.G. Mode switching kinetics produced by a naturally occurring mutation in the cytoplasmic loop of the human acetylcholine receptor epsilon subunit. Neuron. 1998;20(3):575–588. doi: 10.1016/S0896-6273(00)80996-4. [DOI] [PubMed] [Google Scholar]
- 52.Harper C.M., Engel A.G. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Ann. Neurol. 1998;43(4):480–484. doi: 10.1002/ana.410430411. [DOI] [PubMed] [Google Scholar]
- 53.Ohno K., Wang H.L., Milone M., Bren N., Brengman J.M., Nakano S., Quiram P., Pruitt J.N., Sine S.M., Engel A.G. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 1996;17(1):157–170. doi: 10.1016/S0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 54.Engel A.G., Ohno K., Bouzat C., Sine S.M., Griggs R.C. End-plate acetylcholine receptor deficiency due to nonsense mutations in the epsilon subunit. Ann. Neurol. 1996;40(5):810–817. doi: 10.1002/ana.410400521. [DOI] [PubMed] [Google Scholar]
- 55.Shen X.M., Brengman J.M., Shen S., Durmus H., Preethish-Kumar V., Yuceyar N., Vengalil S., Nalini A., Deymeer F., Sine S.M., Engel A.G. Mutations causing congenital myasthenia reveal principal coupling pathway in the acetylcholine receptor ε-subunit. JCI Insight. 2018;3(2):97826. doi: 10.1172/jci.insight.97826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karimzadeh P., Parvizi Omran S., Ghaedi H., Omrani M.D. A Novel c.973G>T Mutation in the ε-subunit of the Acetylcholine Receptor Causing Congenital Myasthenic Syndrome in an Iranian Family. Balkan J. Med. Genet. 2019;22(1):95–98. doi: 10.2478/bjmg-2019-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gül Mert G., Özcan N., Hergüner Ö., Altunbaşak Ş., Incecik F., Bişgin A., Ceylaner S. Congenital myasthenic syndrome in Turkey: clinical and genetic features in the long-term follow-up of patients. Acta Neurol. Belg. 2019 doi: 10.1007/s13760-019-01246-9. [DOI] [PubMed] [Google Scholar]
- 58.Uchitel O., Engel A.G., Walls T.J., Nagel A., Atassi M.Z., Bril V. Congenital myasthenic syndromes: II. Syndrome attributed to abnormal interaction of acetylcholine with its receptor. Muscle Nerve. 1993;16(12):1293–1301. doi: 10.1002/mus.880161205. [DOI] [PubMed] [Google Scholar]
- 59.Sampayo-Cordero M., Miguel-Huguet B., Pardo-Mateos A., Moltó-Abad M., Muñoz-Delgado C., Pérez-López J. Agreement between the results of meta-analyses from case reports and from clinical studies regarding the efficacy of laronidase therapy in patients with mucopolysaccharidosis type I who initiated enzyme replacement therapy in adult age: An example of case reports meta-analyses as an useful tool for evidence-based medicine in rare diseases. Mol. Genet. Metab. 2018;123(2):69–75. doi: 10.1016/j.ymgme.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Jeong W., Keighley C., Wolfe R., Lee W.L., Slavin M.A., Kong D.C.M., Chen S.C. The epidemiology and clinical manifestations of mucormycosis: a systematic review and meta-analysis of case reports. Clin. Microbiol. Infect. 2019;25(1):26–34. doi: 10.1016/j.cmi.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 61.Lee M., Beeson D., Palace J. Therapeutic strategies for congenital myasthenic syndromes. Ann. N. Y. Acad. Sci. 2018;1412(1):129–136. doi: 10.1111/nyas.13538. [DOI] [PubMed] [Google Scholar]
- 62.Huang K., Luo Y.B., Yang H. Autoimmune Channelopathies at Neuromuscular Junction. Front. Neurol. 2019;10:516. doi: 10.3389/fneur.2019.00516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ng F., Lee D.C., Schrumpf L.A., Mazurek M.E., Lee Lo V., Gill S.K., Maselli R.A. Effect of 3,4-diaminopyridine at the murine neuromuscular junction. Muscle Nerve. 2017;55(2):223–231. doi: 10.1002/mus.25208. [DOI] [PubMed] [Google Scholar]
- 64.Sieb J.P., Milone M., Engel A.G. Effects of the quinoline derivatives quinine, quinidine, and chloroquine on neuromuscular transmission. Brain Res. 1996;712(2):179–189. doi: 10.1016/0006-8993(95)01349-0. [DOI] [PubMed] [Google Scholar]
- 65.Zhu H., Grajales-Reyes G.E., Alicea-Vázquez V., Grajales-Reyes J.G., Robinson K., Pytel P., Báez-Pagán C.A., Lasalde-Dominicci J.A., Gomez C.M. Fluoxetine is neuroprotective in slow-channel congenital myasthenic syndrome. Exp. Neurol. 2015;270:88–94. doi: 10.1016/j.expneurol.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thompson R., Bonne G., Missier P., Lochmüller H. Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerg Top Life Sci. 2019;3(1):19–37. doi: 10.1042/ETLS20180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mendell J.R., Florence J. Manual muscle testing. Muscle Nerve. 1990;13:S16–20. doi: 10.1002/mus.880131307. [DOI] [PubMed] [Google Scholar]
- 68.Jaretzki A., III, Barohn R.J., Ernstoff R.M., Kaminski H.J., Keesey J.C., Penn A.S., Sanders D.B. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Myasthenia gravis: recommendations for clinical research standards. Neurology. 2000;55(1):16–23. doi: 10.1212/WNL.55.1.16. [DOI] [PubMed] [Google Scholar]
- 69.Fucile S., Sucapane A., Grassi F., Eusebi F., Engel A.G. The human adult subtype ACh receptor channel has high Ca2+ permeability and predisposes to endplate Ca2+ overloading. J. Physiol. 2006;573(Pt 1):35–43. doi: 10.1113/jphysiol.2006.108092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Castro A., Martinello K., Grassi F., Eusebi F., Engel A.G. Pathogenic point mutations in a transmembrane domain of the epsilon subunit increase the Ca2+ permeability of the human endplate ACh receptor. J. Physiol. 2007;579(Pt 3):671–677. doi: 10.1113/jphysiol.2007.127977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Edgeworth H. A report of progress on the use of ephedrine in a case of myasthenia gravis. J. Am. Med. Assoc. 1930;94:1136–1136. doi: 10.1001/jama.1930.27120410003009c. [DOI] [Google Scholar]
- 72.Lynch G.S., Ryall J.G. Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol. Rev. 2008;88(2):729–767. doi: 10.1152/physrev.00028.2007. [DOI] [PubMed] [Google Scholar]
- 73.Clausen L., Cossins J., Beeson D. Beta-2 Adrenergic Receptor Agonists Enhance AChR Clustering in C2C12 Myotubes: Implications for Therapy of Myasthenic Disorders. J. Neuromuscul. Dis. 2018;5(2):231–240. doi: 10.3233/JND-170293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McMacken G., Cox D., Roos A., Müller J., Whittaker R., Lochmüller H. The beta-adrenergic agonist salbutamol modulates neuromuscular junction formation in zebrafish models of human myasthenic syndromes. Hum. Mol. Genet. 2018;27(9):1556–1564. doi: 10.1093/hmg/ddy062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McMacken G.M., Spendiff S., Whittaker R.G., O’Connor E., Howarth R.M., Boczonadi V., Horvath R., Slater C.R., Lochmüller H. Salbutamol modifies the neuromuscular junction in a mouse model of ColQ myasthenic syndrome. Hum. Mol. Genet. 2019;28(14):2339–2351. doi: 10.1093/hmg/ddz059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brandt N., Nielsen L., Thiellesen Buch B., Gudiksen A., Ringholm S., Hellsten Y., Bangsbo J., Pilegaard H. Impact of β-adrenergic signaling in PGC-1α-mediated adaptations in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2018;314(1):E1–E20. doi: 10.1152/ajpendo.00082.2017. [DOI] [PubMed] [Google Scholar]
- 77.Ito A., Ohnuki Y., Suita K., Ishikawa M., Mototani Y., Shiozawa K., Kawamura N., Yagisawa Y., Nariyama M., Umeki D., Nakamura Y., Okumura S. Role of β-adrenergic signaling in masseter muscle. PLoS One. 2019;14(4):e0215539. doi: 10.1371/journal.pone.0215539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hostrup M., Kalsen A., Onslev J., Jessen S., Haase C., Habib S., Ortenblad N., Backer V., Bangsbo J. Mechanisms underlying enhancements in muscle force and power output during maximal cycle ergometer exercise induced by chronic beta2-adrenergic stimulation in men. J Appl Physiol. 2015;119(5):475–486. doi: 10.1152/japplphysiol.00319.2015. [DOI] [PubMed] [Google Scholar]
- 79.Nissen T., Wynn R. The clinical case report: a review of its merits and limitations. BMC Res. Notes. 2014;7:264. doi: 10.1186/1756-0500-7-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Witting N., Vissing J. Pharmacologic treatment of downstream of tyrosine kinase 7 congenital myasthenic syndrome. JAMA Neurol. 2014;71(3):350–354. doi: 10.1001/jamaneurol.2013.5590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material is available on the publisher’s web site along with the published article.