

Abstract
Src family kinases (SFK) are a group of non-receptor tyrosine kinases which play a pivotal role in cellular responses and oncogenesis. Accumulating evidence suggest that SFK also act as a key component in signalling pathways of the central nervous system (CNS) in both physiological and pathological conditions. Despite the crucial role of SFK in signal transduction of the CNS, the relationship between SFK and molecules implicated in pain has been relatively unexplored. This article briefly reviews the recent advances uncovering the interplay of SFK with diverse membrane proteins and intracellular proteins in the CNS and the importance of SFK in the pathophysiology of migraine and neuropathic pain. Mechanisms underlying the role of SFK in these conditions and potential clinical applications of SFK inhibitors in neurological diseases are also summarised. We propose that SFK are the convergent point of signalling pathways in migraine and neuropathic pain and may constitute a promising therapeutic target for these diseases.
Keywords: Src family kinase, NMDA receptors, P2X receptors, neuroinflammation, migraine, neuropathic pain
1. INTRODUCTION
1.1. Overview of SFK Characteristics
Src family kinases (SFK) are a group of non-receptor tyrosine kinases which play a pivotal role in cellular responses. Src were the first identified member of the family. In 1911, a tumor-causing virus called Rous sarcoma virus from the chicken was discovered by Peyton Rous [1] and the oncogene in the virus is called v-Src [2]. The cellular homologue of v-Src was later identified and known as c-Src or Src, which was the first identified proto-oncogene [2]. After the discovery of Src, eight members of SFK (Blk, Fyn, Lyn, Lck,Yes, Hck, Fgr and Yrk) have been found and their expression levels differ according to tissues types [3]. SFK share a conserved structure which is composed of four domains: a unique domain varying between family members (SH4), Src Homology 3 (SH3), Src Homology 2 (SH2), kinase domain (SH1) as well as a C-terminal regulatory segment [4]. From the N-terminus, the second amino acid is co- translationally myristoylated, which is functioned by targeting membrane and transducing signals. The third amino acid is generally post-translationally palmitoylated, which is used for subcellular trafficking of SFK [4], with the exception of Src and Blk that are non-palmitoylated SFK [5]. Thus, different SFK members may serve specific functions as palmitoylation of SFK is critical for SFK localization and trafficking, which implicates distinct trafficking pathways. The activation loop of SFK is located on the kinase domain with a keyphosphorylation site, tyrosine 416, and another important phosphorylation site is tyrosine 527 located at the C-terminus, which are numbered according to chicken Src numbering [6]. Phosphorylation at either of the two sites confers distinct functions for SFK. Full catalytic activity of SFK requires autophosphorylation at tyrosine 416, in which way SFK can achieve a more active conformation with higher accessibility to the substrate-binding site and more proper position of catalytic residues for phosphate transfer [7]; whilst phosphorylation at tyrosine 527 by kinases such as C-terminal Src kinase (Csk) inhibits SFK activity [8] via SH2 domain docking onto the phosphorylated tyrosine 527 and forming the assembled state with SH3 [7]. Therefore, phosphorylation status of the tyrosine 416 and tyrosine 527 sites becomes a key indicator of the participation of SFK in various conditions.
SFK are located downstream of a variety of membrane receptors, gap junctions and cation channels to modulate intracellular signals driving cell development via phosphorylating tyrosine residues of other proteins. These kinases have been extensively studied in the field of oncology, and they were originally recognised as products of proto-oncogenes, whose overexpression, mutation and dysregulation in cells can promote the hallmarks of oncogenesis [9]. In the central nervous system (CNS), six members of SFK (Src, Fyn, Lyn, Lck, Yes and Yrk) have been found [10-14] and they are present in differentiated neurons [11] and glia including astrocytes [15], microglial cells [16], and oligodendrocytes [17]. SFK are involved in regulating the development and activity of both neuron and glia, neuroplasticity and signal transduction [18, 19], suggesting a comprehensive regulatory role of SFK in the CNS. Nevertheless, more information on SFK expression in the neurons and glia of specific brain regions such as the amygdala have been rarely reported. Further work on answering these fundamental questions will pave the way for studies of SFK activity in various diseases. Despite this, increasing evidence support SFK acting as a key component in signalling pathways of the CNS in both physiological and pathological conditions, including neurodegenerative diseases, pain, stroke, epilepsy and psychiatric disorders. For a full review of SFK-related pathogenesis in neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), the readers are referred to previous publications [20, 21]. Pathogenesis of certain psychiatric disorders is also associated with SFK-mediated pathways, a typical example of which is schizophrenia [22]. In this article, we review the recent findings concerning the crosstalk between SFK and multiple proteins in the CNS and the role of SFK in the pathophysiology of migraine and neuropathic pain. The current progress on potential clinical applications of SFK inhibitors in neurological disorders is also discussed.
1.2. SFK Regulate Multiple Proteins in the CNS
SFK are well-known for their ability to interact, either directly or indirectly, with a large range of membrane proteins and intracellular proteins (Table 1), indicating their active roles in regulating these proteins and transmitting signalling from them.
Table 1. SFK play an important role in signal transduction of the CNS via regulating multiple membrane proteins and intracellular proteins.
| Protein | Modification of the Protein | Outcome after SFK Activation | Key References |
|---|---|---|---|
| NMDAR | Phosphorylation | Upregulation of NMDAR activity and membrane translocation, affecting synaptic transmission | [23] |
| AMPAR | Phosphorylation | Regulation of AMPAR membrane trafficking and synaptic transmission | [24] |
| PTPa | N/A | Upregulation of NMDAR activity | [25] |
| PSD-95 | Phosphorylation | Activation of NMDAR | [26] |
| PSD-93 | N/A | Increase in NR2B phosphorylation | [27] |
| GABAAR | Phosphorylation | Upregulation of GABAAR activity and synaptic inhibition | [28] |
| Panx1 | Phosphorylation | Panx1 channel activation | [29] |
| P2X7R | N/A | Upregulation of P2X7R activity | [30] |
| 5-HT1AR | N/A | Inhibition of 5-HT1A R activity | [31] |
| 5-HT2AR | N/A | Increase in 5-HT2A R-mediated glutamate responses induced by 5-HT | [32] |
| Na+,K+-ATPase | Phosphorylation | Increase in phosphorylation on Y260 site of α1 subunits of Na+,K+-ATPase in LLC-PK1 and SYF cells | [33] |
| N/A | Increase in Na+,K+-ATPase-mediated glutamatergic neurotransmission | [34] | |
| N/A | Increase in Na+,K+-ATPase-mediated neuroinflammatory responses | [35] | |
| PKCδ | Phosphorylation | Increase in PKCδ-mediated neuroinflammatory responses | [36] |
| TLR4 | Phosphorylation | Increase in TLR4-mediated neuroinflammatory responses | [37] |
| S100B | N/A | Increase in S100B-mediated proliferation, migration and undifferentiation of astrocytes | [38] |
| P2X4R | N/A | Upregulation of P2X4R expression | [39] |
Abbreviations: NMDAR, N-methyl-D-aspartate receptor; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; PTPα, a receptor-type protein tyrosine phosphatase; PSD-95, postsynaptic density protein 95; PSD-93, postsynaptic density protein 93; GABAAR, γ-aminobutyric acid type-A receptor; Panx1, pannexin-1; P2X7R, purinergic receptors P2X7; 5-HT1AR, 5-hydroxytryptamine receptor subtype 1A; 5-HT2AR, 5-hydroxytryptamine receptor subtype 2A; Na+,K+-ATPase, sodium–potassium adenosine triphosphatase; PKCδ, protein kinase C δ; TLR4, toll-like receptor 4; S100B, S100 calcium-binding protein B; P2X4R, purinergic receptors P2X4. N/A, not applicable.
2. SFK REGULATE EXCITATORY GLUTAMATE RECEPTORS
Among all the molecules which SFK are able to interact with in the brain, the well-studied proteins are ionotropic glumate receptors, including N-methyl-D-aspartate (NMDA) receptors that are consisted of two mandatory NR1 subunits and two modulatory subunits of NR2A-D and/or NR3A-B, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors that are composed of four subunits GluA1-4 [40]. These receptors permit the flow of cation ions, including K+, Na+ and Ca2+, through their channels and are highly associated with synaptic plasticity, cognition and behaviour [40].
Robust evidence show that SFK are able to interact with and mediate both NR2A- and NR2B-containing NMDA receptor function and signalling [41, 42]. SFK are found to link NMDA receptors to pannexin-1 (Panx1) hemi-gap junction protein during anoxia [43], excitotoxicity [29] and migraine [44]. NMDA receptors couple SFK to Panx1 to form a signalling complex by which SFK phosphorylate Panx1 at tyrosine 308 site to mediate the Panx1 channel activity, leading to constant neuronal depolarizations and excitotoxicity [29]. Our recent study also shows that the coupling of SFK and Panx1 as a complex is regulated by NR2A-containing receptors in a rat migraine aura model, suggesting that SFK play an important role in transmitting NR2A signalling in migraine aura pathogenesis [44]. Besides NR2A, SFK also regulate NR2B activity. In Fyn-knockout mice, the tyrosine phosphorylation level of NR2B is lower than that of wild- type mice [41] and PP2, a specific SFK inhibitor, reduces endogenous phosphorylation of NR2B [45]. Fyn can be scaffolded to NR2B by the scaffolding protein, receptor for activated C kinase 1 (RACK1), under normal conditions, while upon acute exposure to ethanol, RACK1 is released from Fyn-NR2B complex, resulting in phosphorylation of NR2B mediated by Fyn and thus the activity of NMDA receptors is amplified [46]. Besides Fyn, Src also constitutes a part of the NMDA receptor complex to regulate NMDA receptor activity via NADPH dehydrogenase subunit 2 (ND2) in the postsynaptic density of the brain [47]. Uncoupling Src from the NMDA receptor complex by a peptide Src40-58 inhibits Src-upregulated NMDA receptor activity in cultured hippocampal neurons [47]. In the amygdala, the inhibitory peptide Tat-Src40-58 prevents NR2B phosphorylation and NR2B surface expression, regulating NMDA receptor trafficking and synaptic plasticity [48]. Furthermore, a component of the NMDA receptor complex, PTPα, which is a receptor- type protein tyrosine phosphatase (PTP), can activate SFK by dephosphorylating the inhibitory phosphorylated tyrosine 527 residues of SFK [23]. Intracellular application of PTPα potentiates miniature excitatory postsynaptic currents mediated by NMDA receptors in rat hippocampal neurons, whilst deactivation of SFK by PP2 inhibits this potentiation, suggesting that PTPα-induced enhancement of NMDA receptor function is mediated by SFK [25]. Therefore, SFK are proposed to be a hub for the regulation of NMDA receptors [23], implicating a crucial role of SFK in NMDA receptor-related activities and diseases.
Another type of excitatory glutamate receptors, AMPA receptors, have also been found to be regulated by SFK. In neurons, the tyrosine 876 site on the C terminus of GluA2 subunits is phosphorylated by SFK, including Src, Fyn and Lyn. This SFK-mediated phosphorylation on the tyrosine 876 site of the GluA2 subunits is tightly associated with AMPA receptor membrane trafficking and synaptic plasticity [24]. On the one hand, SFK-mediated phosphorylation on the tyrosine 876 site contributes to agonist-dependent internalisation of AMPA receptors; On the other hand, it favors the retention of AMPA receptors in synapses in basal states, suppressing long-term depression (LTD) and promoting synaptic upscaling [24, 49-51]. Recently, SFK are identified also to phosphorylate the tyrosine 881 site of GluA3 subunits of AMPA receptors in cortical neurons, but its function remains to be studied [51]. Besides, tyrosine phosphorylation of the C terminus of GluA1 subunits of AMPA receptors by Fyn, but not Src, protects the subunits from calpain-mediated truncation, a mechanism in the regulation of synaptic plasticity, which contributes to the preservation of the integrity of AMPA receptors [52]. Furthermore, Fyn modulates AMPA receptor expression in a non-transcriptionally manner in rodent neocortical neurons [53]. In this study, an SFK-selective protein tyrosine kinase (PTK) inhibitor reduces neurotrophin brain-derived neurotrophic factor (BDNF)-enhan-
ced AMPA receptor level, and the decreased AMPA receptor protein level after exposure to BDNF is also found in the neocortex of Fyn-knockout mice. Since BDNF is able to activate Fyn in an in vitro kinase assay, the authors conclude that BDNF-activated Fyn signalling plays an important role in upregulating AMPA receptor expression [53]. Fyn also transmits intracellular signalling from AMPA receptors by physically interacting with AMPA receptors and being activated upon stimulation of the receptors, leading to activation of the mitogen-activated protein kinase (MAPK) pathway and increased expression of BDNF [54]. The activation of SFK by AMPA receptors is modulated by nitric oxide (NO) pathway, which is critical for excitatory retinal nerve cell death [55]. Taken together, the ability of SFK, mainly Src, Fyn and Lyn, in regulating AMPA receptor activities, supports an important role of SFK in synaptic transmission and plasticity.
SFK are known to also mediate the scaffolding proteins - postsynaptic density protein 95 (PSD-95) and postsynaptic density protein 93 (PSD-93) within the glutamate receptor complex. PSD-95 and PSD-93 are members of the membrane-associated guanylate kinases that mediate the clustering of receptors and ion channels, including NMDA receptors, AMPA receptors, voltage-gated ion channels, and other associated signalling proteins by forming a multimeric scaffold at postsynaptic sites [56]. An increase in the phosphorylation level of PSD-95 by SFK is detected after brain ischemia and reperfusion in rat hippocampus, which contributes to the overexcitation of NMDA receptors [26]. In addition, after transient brain ischemia, interactions among Src/Fyn, α1C subunits of L-type voltage-gated calcium channels and PSD-95 rapidly increase, forming an α1C-PSD-95-Src/Fyn complex [57]. This complex facilitates SFK- mediated tyrosine phosphorylation on α1C, upregulating the activity of the calcium channels in the post-ischemic hippocampus [57]. PSD-93, on the other hand, is shown to upregulate the interaction between Fyn and NR2B subunits of NMDA receptors as well as Fyn-mediated phosphorylation of NR2B; This is evidenced by that knockout of PSD-93 downregulates the Fyn-mediated NR2B phosphorylation, which contributes to the neuroprotective ability against ischemic brain injury in mice [27]. Taken together, SFK play a crucial role in synaptic transmission and plasticity via regulating glutamate receptor complex; despite this, the downstream signalling of this SFK-mediated glutamate receptor functions and responses still await to be understood in order to manifest possible mechanisms for glutamate receptor-related diseases.
2.1. SFK Regulate Inhibitory GABAA Receptors
γ-aminobutyric acid type-A (GABAA) receptors are Cl--selective heteropentameric ionotropic receptors whose activation causes fast synaptic inhibition [58]. These receptors are highly associated with cognition and behaviour, and their functions are tightly regulated mainly via phosphorylation [58]. In neurons, they can be phosphorylated by not only a number of serine or threonine protein kinases but also Src [28]. The primary phosphorylation sites of GABAA receptors by Src are tyrosine 365 and tyrosine 367 within the intracellular domain of γ2 subunits of GABAA receptors, and this phosphorylation raises the possibility of the channel opening [28], thus enhancing GABAA receptor function. It is reported that Src, but not Fyn and Yes, binds directly to the intracellular domains of β and γ2 subunits of GABAA receptors, supporting the Src-mediated mechanism for regulation of GABAA receptor function [59]. However, later evidence shows that Fyn also binds to GABAA receptors, but in a phosphorylation-dependent manner as Fyn contributes to tyrosine phosphorylation of their γ2 subunits [60]. In addition to inhibitory signal transmission, phosphorylation at tyrosine 365 and tyrosine 367 of γ2 subunits of GABAA receptors by SFK also contributes to impairment of spatial memory formation [61]. These evidence demonstrate the participation of SFK in mediating γ2 subunits of GABAA receptors in synaptic inhibition, which yet needs further elucidation in terms of how this SFK-mediated GABAA receptor function may contribute to the receptor-related diseases.
2.2. SFK Regulate P2X7 Receptor-Panx1 Complex
Purinergic receptors P2X are ATP-gated cation channel families. One of the most studied subtypes is the P2X7 receptor that is able to form large molecule-permeable pores on cell membranes upon activation [62]. P2X7 receptors are special among their family members because they can form a large pore with Panx1 by physical association for permeation of larger molecules in addition to the typical formation of an ion channel [63]. The P2X7 receptor-Panx1 complex is crucial for ATP signalling, apoptic cell death and inflammation [63, 64]; however, how this complex functions is largely unknown. A recent study shows that this complex is involved in the regulation of cortical susceptibility to cortical spreading depression (CSD), a causative event of migraine aura, and CSD-associated neuroinflammation in rodents [65]. In this study, albeit lack of direct evidence, it is speculated that SFK may be the intermediate protein linking P2X7 receptors to Panx1 as a complex to regulate the above processes. The involvement of SFK in the P2X7 receptor-Panx1 complex signalling is also supported by that Panx1 currents and membrane permeabilization induced by activation of P2X7 receptors can be decreased by both an SFK inhibitor and an exogenous peptide TAT-P2X7 that targets the SFK-related COOH terminus of P2X7 receptors to reduce activated SFK bound to P2X7 receptors [66]. These authors also find that activation of P2X7 receptors induces SFK activation, which again can be attenuated by interfering with the P2X7 receptor-SFK interaction via the peptide TAT-P2X7. This study delineates that SFK are implicated in the signal transduction between P2X7 receptors and Panx1, which determines the Panx1 activity and the membrane pore permeabilization, despite the fact that the identity of the SFK substrate is unknown. Further investigation on whether SFK may directly modify the P2X7 receptor-Panx1 complex is needed.
2.3. SFK Regulate 5-HT Receptors
5-hydroxytryptamine (5-HT, also known as serotonin) receptors are known to mediate various neural activities and vascular tone. Specific subtypes of the receptors are the target of multiple drugs treating neurological disorders, including migraine, PD, anxiety and depression [67]. 5-HT receptors are a group of G-protein-coupled receptors (GPCRs), the latter of which are known to actively interact with SFK [68]. Direct association of SFK with GPCRs activates SFK, whereas SFK activity is essential in mediating GPCRs trafficking, which affects cell proliferation, cell cytoskeletal rearrangement and neurotransmission [68]. Given that SFK are also involved in diverse signalling pathways, the interplay between SFK and 5-HT receptors can take place in a number of more complex contexts.
SFK are involved in the regulation of serotonergic inhibition via specific subtypes of 5-HT receptors. The activity of 5-HT1A receptors resulting in inhibitory current can be amplified in the pyramidal neurons of mice prefrontal cortex treated with the SFK inhibitor PP2, indicating an inhibitory effect of SFK on 5-HT1A receptor response [31]. SFK activation is also known to provide a pivotal mechanistic explanation for 5-HT-mediated vasoconstriction via 5-HT2A receptors in rat aorta [69] and rat mesenteric artery [70]. Upon exposure to 5-HT, SFK are activated and inhibition of SFK almost completely abolishes 5-HT-induced contraction in rat aorta, the process of which is mediated by the interplay between SFK and 5-HT2A receptors [69]. In the same study, Src, but not other subtypes, is confirmed to play a role in mediating this 5-HT2A receptor-mediated contractile response based on the pharmacological evidence from the SFK inhibitor PP2 that suggest only the half-maximal inhibitory concentration (IC50) of PP2 for Src affects the contraction [69]. The authors validate the existence of an interaction between Src and 5-HT2A receptors by the findings that 5-HT2A receptors co-localize with Src and associate with Src as a signalling complex in HEK 293T cells. A later study reinforces the SFK-mediated mechanism of 5-HT-dependent vasoconstriction [71]. Moreover, a recent study discovers that the interplay between SFK and 5-HT2A receptors modulates 5-HT-enhanced glutamate responses and NR2A phosphorylation in the trigeminal motor nucleus of rat brainstem slices [32]. Additionally, in a study of AD, 5-HT4 receptor activation upregulates the activity of α-secretase that results in less formation of pathogenic amyloid-β (Aβ) peptide and this is regulated by G protein and SFK-dependent pathway [72]. These evidence reveal the mode of SFK activity in 5-HT receptor- mediated signalling, yet how SFK interact with these 5-HT receptors and the role of such interaction in certain neurological diseases remain largely unknown.
2.4. SFK Regulate Na+, K+-ATPase
Sodium–potassium adenosine triphosphatase (Na+,K+-ATPase) is an ion pump that actively transports three Na+ and two K+ across the membrane via consuming ATP [73]. This molecular machine maintains resting membrane potential and cellular volume by regulating ionic concentrations, including Na+ and K+, and provides a Na+ gradient, which is the driving force for transport of glucose, amino acids and other nutrients into the cell via secondary transporters [73]. In addition to pumping ions, Na+,K+-ATPase is also engaged in signal transduction via its dynamic interaction with multiple proteins. Na+,K+-ATPase and its associated proteins are assembled to be a signalosome that integrates signals important for cell physiological and pathological processes [74]. An important partner for Na+,K+-ATPase to conduct signal transduction is SFK.
A large body of research has contributed to the discovery that the α1 subunit of Na+,K+-ATPase/SFK complex constitutes a functional receptor for its ligands such as cardiotonic steroids (CTS) to activate downstream protein kinase cascades that can regulate reactive oxygen species (ROS) production and Ca2+ oscillation, which ultimately affects cell differentiation and growth [75]. This complex enables Na+,K+-ATPase, to regulate the function of SFK directly. SFK activity is inhibited by Na+,K+-ATPase in this complex whereas ouabain, a type of CTS as well as a Na+,K+-ATPase inhibitor, activates the associated SFK by freeing the kinase domain of SFK from Na+,K+-ATPase and recruits more SFK to bind to the complex, initiating signalling cascades [76]. SFK also regulate Na+,K+-ATPase activity via the direct interaction. A SFK inhibitor PP1 inhibits Na+,K+-ATPase current in cortical neurons, which is further shown to be mediated by specific SFK subtypes, Lyn and Yes, but not Fyn [77]. In this study, Lyn is identified to directly associate with the α3 subunit of Na+,K+-ATPase and possibly elevates its phosphorylation [77]. Moreover, tyrosine 260 of the α1 subunit of Na+,K+-ATPase is a SFK-specific binding and phosphorylation site, which is required for Na+,K+-ATPase/SFK signalling [33], but whether and how this SFK-mediated phosphorylation of Na+,K+-ATPase affects activities in the CNS remains unknown.
In the CNS, the Na+,K+-ATPase/SFK complex exerts multiple functions. In rat brain, this complex is essential in regulating glutamate transporter function and glutamatergic neurotransmission by coupling to glutamate transporters and forming a macromolecular signalosome [34]. Besides, Na+,K+-ATPase has a close relationship with NMDA receptors, which is mediated by SFK. This is supported by that inhibition of Na+,K+-ATPase potentiates NMDA-evoked currents in rat hippocampal CA1 pyramidal neurons and SFK modulate this process [78]. Additionally, the Na+,K+-ATPase inhibitor ouabain induces NFκB activation and upregulation of proinflammatory cytokine mRNA levels via NMDA receptor-SFK-MAPK pathways in rat cerebellar cells [35]. The Na+,K+-ATPase/SFK signalling also plays a role in developing neuronal injury [79], neuropathic pain [80] and ammonia-induced astrocyte swelling [81]. Therefore, targeting this signalling pathway by relevant compounds may provide therapeutic potential for treating these neurological abnormalities. Future efforts should be made to better understand the interaction between specific Na+,K+-ATPase isoform and SFK and the downstream signalling events in the context of related neurological diseases.
3. SFK REGULATE GLIA-ASSOCIATED NEUROINFLAMMATION
Neuroinflammation is an emergent property of many neurological disorders, including AD, PD, migraine and chronic pain [82]. The primary players in neuroinflammation are activated glial cells, including microglia and astrocytes, which release inflammatory soup containing IL-1β, IL-6 and TNF-α [83]. Phosphorylation and dephosphorylation of SFK by enzymes such as protein tyrosine phosphatase 1B [84] have profound effects on many downstream signalling pathways, leading to the activation of microglia and astrocytes, which in turn releases inflammatory cytokines, exacerbating damage to the surrounding cells.
Microglia activation is heavily dependent on SFK phosphorylation. In primary microglial cultures, Src is found to be both necessary and sufficient for microglia activation [85]. In another study, using murine microglial cells and a AD transgenic mouse model, blocking SFK by dasatinib reverses the effect of Aβ-induced activation of microglia and TNF-α release both in vitro and in the hippocampus and temporal cortex in vivo, suggesting Aβ-induced microgliosis is SFK-dependent [86]. Src is also recognised to be important in neuroinflammation in a PD model as one potential anti-inflammatory medication extracted from a Chinese herb with the code name, FLZ, is found to suppress Src activity and the downstream inflammatory signalling cascade in mouse microglial BV2 cells, thereby preventing neurodegeneration [87]. Moreover, Fyn can phosphorylate protein kinase C δ (PKCδ) and drive NFκB pathway to facilitate the production of IL-6, IL-12 and TNF-α in microglial cells [36]. Interestingly, this study identifies upregulated transcription and expression of Fyn during chronic inflammation and that Fyn (-/-) mice exhibit attenuated neuroinflammatory responses, uncovering the role of Fyn as a critical upstream mediator in microglial neuroinflammation [36].
Astrocyte activation is another characteristic of the inflamed brain. By using FynT constitutively active and FynT kinase-dead mutants, Lee et al. report that FynT isoform activity enhances proinflammatory cytokines and chemokines levels induced by TNF-α in astrocytes via phosphorylating protein kinase C δ (PKCδ) and activating NFκB [88]. Furthermore, astrocytic inflammatory responses led by alcohol intake is mediated by SFK [37]. More specifically, chronic ethanol induces the phosphorylation of Toll-like receptor 4 (TLR4) and SFK and their physical interaction in astrocytes, leading to activation of cyclooxygenase (COX-2) and secretion of prostaglandin E2 (PGE2) [37]. In addition to astrocytic inflammation, SFK also play a role in S100 calcium binding protein B (S100B)-mediated morphology and migration of astrocytes [38]. SFK display higher activity in the presence of S100B and directly interact with S100B in astrocytoma cell line GL15, which instigates downstream P13K pathways, thereafter modulating astrocytic phenotype [38]. Taken together, the ascertained pivotal activity of SFK in mediating glia-associated inflammatory responses makes these kinases a potential therapeutic target to ameliorate the inflamed brain, which might hamper neuroinflammation-related neurological disorders.
In summary, the participation of SFK in regulating the functions and activities of various proteins in the CNS indicate that SFK are a key point for the convergence of diverse signalling pathways, which sheds light on SFK-centered modulation of CNS function under physiological and pathological conditions. Further studies of the crosstalk between SFK and their identified or new interactive targets are still required in order to obtain a more comprehensive picture of SFK signalling in the brain.
4. SFK IN MIGRAINE
Migraine is a disabling neurovascular disorder characterized by recurrent unilateral headache, which is often accompanied by other neurological symptoms. It has been widely understood that the pathophysiology of migraine contains a neuroimmune interaction between the trigeminovascular pathway and neuroinflammatory responses, leading to peripheral and central sensitization, yet the molecular mechanism of migraine still remains largely unknown [89].
Albeit there is no direct evidence linking SFK with migraine, the emergent role of SFK in experimental models of migraine and the association of SFK with targets implicated in migraine pathogenesis strongly suggest that SFK play an important role in migraine pathophysiology. Most notably, SFK activity is required for propagation of CSD [90], a transient propagating excitation wave followed by depression of neurons and glia that can result in activation of trigeminovascular system and migraine with aura [91]. In this study, SFK phosphorylation at tyrosine 416 site is elevated after induction of a single CSD in rats, whereas inhibition of SFK activity by perfusing PP2 into the rat contralateral cerebroventricle decreases the SFK phosphorylation level induced by CSD and also cortical susceptibility to CSD [90]. Consistent with this, another study by Chen et al. also shows that CSD frequency is decreased by intraperitoneal administration of PP2 [65]. These data support the importance of SFK activity in the mechanism of migraine aura. The critical role of SFK in migraine pathophysiology is further evidenced by a recent study using a chronic migraine model induced by repeated perfusion of inflammatory soup to the dura of rats [92]. This study shows that deactivation of SFK after intracerebroventricular injection of PP2 alleviates mechanical allodynia, and reduces NR2B phosphorylation and PSD-95 expression, which subsequently affect synaptic plasticity and central sensitization that is essential for maintenance of migraine headache [92]. Intesteringly, in a mice model of familial hemiplegic migraine type 2 (FHM2) with mutated Na+,K+-ATPase α2 isoform, SFK are largely activated in the middle cerebral arteries, which initiates downstream signalling cascade, leading to increased sensitization of the arteries to intracellular Ca2+ and arterial contraction [93]. This Na+,K+-ATPase/SFK signalling is the causative mechanism for the brain perfusion abnormalities of FHM2 characterised by initial vasospasm and hypoperfusion followed by vasodilation and hyperperfusion [94], supporting SFK-modulated signal transduction in migraine pathophysiology. Collectively, the involvement of SFK activity in regulating cortical susceptibility to CSD, mechanical allodynia and trigeminovascular activation support that SFK activity plays a key role in migraine pathogenesis via both central and peripheral actions.
Although SFK signalling pathways in migraines still remain elusive, increasing evidence has suggested that SFK activity plays a part in mediating the release and expression of calcitonin gene-related proteins (CGRP) that is a well- known target for migraine therapy [95] and prevention [96]. Pretreatment of PP2, but not its negative analog PP3, blocks the nerve growth factor (NGF)-promoted CGRP release evoked by capsaicin in sensory neuron culture from rat dorsal root ganglia [97]. The facts that NGF mediates SFK activation-dependent activities and SFK contribute to sensitizing sensory neurons [98] reinforces the role of SFK in the pathway of NGF-promoted CGRP release. Notably, in the trigeminal nucleus caudalis of the aforementioned rat chronic migraine model, SFK inhibition by PP2 reduces CGRP expression, indicating that SFK are involved in regulating CGRP level and central sensitization [92]. However, the mechanism underlying SFK-regulated CGRP release and expression in migraine conditions remain unknown. Future studies on the pathways of SFK-mediated CGRP level in the central and peripheral systems may improve our understanding of the pathogenesis of migraine. Besides the ability of SFK to mediate CGRP release and expression, our recent study shows that SFK can mediate CSD-induced Panx1 activation [44]. SFK-Panx1 coupling is promoted after a single CSD, and SFK inhibition by PP2 attenuates the CSD-induced neuronal Panx1 activation in rat ipsilateral cortices; conversely, disrupting SFK-Panx1 interaction by an interfering peptide TAT-Panx308 reduces cortical susceptibility to CSD in mouse brain slices [44]. Given that Panx1 megachannel opening induced by CSD promotes proinflammatory responses which triggers trigeminovascular activation and migraine-like behaviour in mice [99], it is reasonable to propose a key role of SFK-dependent Panx1 activation in migraine aura pathogenesis, by which the CSD-induced neuroinflammatory responses and trigeminovascular activation are initiated [44]. Furthermore, increasing evidence suggest that a crosstalk may exist between SFK and NMDA receptors upstream of Panx1 in migraine aura. Inhibition of NR2A-containg NMDA receptors by the antagonist, NVP-AAM077 (NVP) decreases SFK phosphorylation level induced by CSD in rats and SFK activation by the activator pYEEI is found to reverse the reduced susceptibility to CSD caused by NR2A inhibition in mouse brain slices [90]. Consistently, NR2A inhibition reduces the elevation of ipsilateral cortical SFK-Panx1 interaction, Panx1 activation induced by CSD and cortical susceptibility to CSD in rats [44]. It is well-known that NMDA receptors and their major subunits, NR2A and NR2B, play crucial roles in regulating cortical susceptibility to CSD [100-102] and, as discussed above, SFK activation is not only required for CSD propagation [90] but also for promoting Panx1 activation leading to neuroinflammatory responses [99]. Therefore, we have proposed that NR2A/SFK/Panx1 signalling may play an important role in migraine pathogenesis, indicating that drugs that target this pathway might constitute an effective strategy for migraine prophylaxis [44]. It is likely that SFK facilitate neuroinflammation and nociception via coupling to Panx1, but this possibility remains to be further clarified.
As mentioned in the above sections, SFK also interact directly or indirectly with several other proteins especially GABAA receptors, P2X7 receptors and 5-HT receptors, all of which proteins are involved in migraine pathophysiology. Of these, 5-HT receptors, especially 5-HT1B/1D/1F receptors, are the known targets for current migraine-specific treatments [103, 104], whilst GABAA receptors and P2X7 receptors are promising targets for migraine prevention [65, 105, 106]. However, how SFK mediate their signalling pathways in migraine are lacking, which requires further investigation.
Collectively, we propose that SFK work as a convergent point of multiple signalling pathways, especially NMDA receptor, P2X7 receptor and Na+,K+-ATPase signallings, in migraine pathophysiology (Fig. 1). Future investigations are required to consolidate this proposal.
Fig. (1).
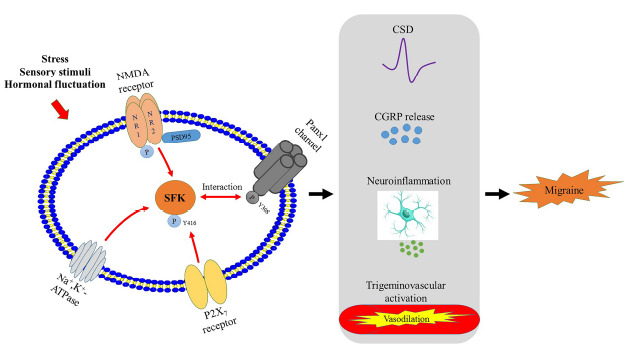
Schemic representation of the proposed SFK-converged signalling pathways in migraine pathogenesis, including NMDA receptor, P2X7 receptor and Na+,K+-ATPase signallings that have identified relationships with SFK in migraine models (red arrows). The SFK-transmitted signallings lead to changes in cortical susceptibility to CSD, CGRP release, neuroinflammation and trigeminovascular activation (black arrows). (A higher resolution / colour version of this figure is available in the electronic copy of the article).
5. SFK IN NEUROPATHIC PAIN
Neuropathic pain is a type of chronic pain and is caused by lesions of neurons or diseases affecting the somatosensory nervous system [107]. The pathophysiology of this pain state encompasses sensitization of peripheral and central nervous system, intense neuroinflammation produced by neuroimmune-glia interactions across the pain pathway, and substantial molecular changes in ion channels, neurotransmitters and their receptors [107].
A vast amount of research corroborates that SFK-mediated signallings are implicated in the neuropathic pain process. As mentioned above, activation of non-neuronal cells, including microglia [85] and astrocytes [88], is heavily dependent on SFK activity, which facilitates expression and secretion of pro-inflammatory factors. Similarly, activation of SFK in spinal microglia contributes to mechanical hypersensitivity after nerve injury [108] and formalin-induced persistent pain [109]. In the rat spinal dorsal horn, SFK are activated in hyperactive microglia after nerve injury rather than in neurons or astrocytes, and their activation promotes mechanical hypersensitivity through SFK/ERK signalling cascade [108], whereas SFK/p38-MAPK signalling pathway is involved in late-stage mechanical hypersensitivity post-formalin-induced pain [109]. Specific SFK subtypes have been identified to play a role in neuropathic pain. Lyn predominantly expresses in spinal microglial cells and is crucial for nerve injury-induced neuropathic pain [39]. This is demonstrated in Lyn knockout mice, which exhibit disabled upregulation of ionotropic ATP receptor subtype, purinergic P2X4 receptors and striking decrease in tactile allodynia, a hallmark of neuropathic pain, after nerve injury [39]. Additionally, nectin-1/Src signalling is also involved in neuropathic pain process [110]. Nectin-1 level is enhanced in rats after chronic constrictive injury and glial cell-derived neurotrophic factor (GDNF) downregulates the phosphorylation level of Src, a downstream target of nectin-1, which displays an analgesic effect on the neuropathic pain resulted from the nerve injury [110]. Furthermore, SFK have been proposed to have a determinant role in the pain-associated hyperalgesia, as evidenced by that intradermal injection of the SFK activator peptide pYEEI decreases nociceptive threshold and induces mechanical hyperalgesia in rats [111]. Consistently, inhibition of SFK activity attenuates hyperalgesia in rat pain models induced by complete Freund adjuvant (CFA) [112], EphrinB2-Fc [113], GDNF [114] and formalin [109].
The crosstalk between SFK and NMDA receptors in neuropathic pain has been well characterised. Tyrosine phosphorylation of the NR2B subunits of NMDA receptors at tyrosine 1472 by SFK in spinal dorsal horn contributes to neuropathic pain, as shown by that inhibition of SFK activity by PTP or its inhibitor PP2 reduces SFK-enhanced NR2B phosphorylation as well as NMDA-increased NR2B synaptic concentration by promoting NR2B endocytosis, eventually ameliorating pain hypersensitivity and behaviour in rodents [115, 116]. As aforementioned, among the 6 subtypes of SFK in the CNS, mainly Src and Fyn regulate NMDA receptor activity and function, which also applies to pain models. Knockout of Fyn in mice with neuropathic pain maintains the paw withdrawal threshold similar to that of wild-type mice [117]. In the spinal dorsal horn of these Fyn-/- mice, NR2B phosphorylation at the tyrosine 1472 site in the postsynaptic density is abolished, and neuronal nitric oxide synthase (nNOS) activity is largely decreased, suggesting the crucial role of Fyn in the pathogenesis of neuropathic pain [117]. In terms of Fyn activity in pain sensitization, a constitutively active Fyn mutant, Fyn(Y528F), expressed in the spinal dorsal horn of intact mice, but not a catalytically inactive Fyn mutant, Fyn(K296M), evokes mechanical allodynia and thermal hyperalgesia, and raises the contents of NR2B- containing NMDA receptors and GluA1-containing AMPA receptors at synaptosomal membrane fraction [118]. Furthermore, prior expression of the Fyn inactive mutant Fyn(K296M) in mice exhibits an analgesic action by attenuating CFA-induced pain, as well as NR2B and GluA1 levels [118]. Fyn-dependent potentiation of NMDA receptor currents can be gated by BDNF, which disinhibits synaptic transmission within lamina I spinal neurons and facilitates the progression of spinal neuropathic pain [119]. In the previous section, we summarised that SFK regulate NMDA receptor function via direct interaction. A recent interesting study shows that disrupting the binding of Src to the NMDA receptor complex by a peptide Src40-49Tat suppresses neuropathic and inflammatory pain behaviours in rodents [120]. This finding might indicate a possibly efficacious treatment of pain with better safety profile than using NMDA receptor blockers, as the latter can bring in deleterious side effects by suppressing the physiological function of NMDA receptors [121, 122]. Taken together, it can be concluded that SFK, especially Src, Fyn and Lyn, play an essential role in the mechanism and progression of neuropathic pain in animal models. We propose that the deactivation of SFK might be a promising strategy in alleviating neuropathic pain through interfering SFK-mediated transduction of NMDA receptor and P2X4 receptor signallings (Fig. 2). The developed SFK inhibitors targeting different subtypes might be considered for pre-clinical study in neuropathic pain models.
Fig. (2).
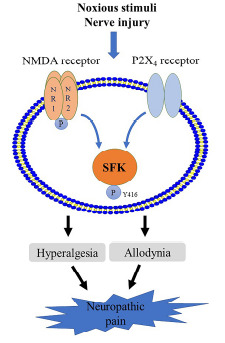
Schemic representation of the proposed SFK-converged signalling pathways in neuropathic pain including NMDA receptor and P2X4 receptor signallings that are known to be mediated by SFK in neuropathic pain models (blue arrows). The SFK-transmitted signallings lead to hyperalgesia and allodynia (black arrows). (A higher resolution / colour version of this figure is available in the electronic copy of the article).
6. SFK INHIBITORS IN CLINICAL APPLICATIONS FOR NEUROLOGICAL DISEASES
SFK have long been targeted in treating cancer with a number of developed SFK inhibitors for the ability of SFK to promote tumour development [123]. Four potent SFK/multikinase inhibitors - dasatinib, bosutinib, ponatinib and vandetanib have been approved by the Food and Drug Administration (FDA) and applied clinically in cancer treatment [124]. Interestingly, studies of SFK in the context of neurological diseases have also been transformed from bench to bedside, which acknowledges the pivotal role of SFK in the abnormalities of the brain. From the clinical studies of SFK inhibitors, several advantages of the drug can be clearly recognised. SFK inhibitors are generally safe without adverse effects. Among all the developed SFK inhibitors, saracatinib, an inhibitor of SFK and Abl family of kinases [125], has received raised attention in its potential application in the treatment of the neurodegenerative diseases [21, 126]. In the previous clinical studies of saracatinib in cancer treatment, it exhibited well-tolerance in humans but was discontinued for this application due to its unsatisfactory efficacy in Phase II trials [127-131]. Subsequently, this drug was investigated as a potential medication for AD since Fyn has been targeted as a therapeutic intervention in AD, and saracatinib effectively blocks Fyn activity [132]. A Phase Ib multiple ascending dose study of saracatinib confirmed the safety and well tolerability of saracatinib in AD patients [133]. However, it was again discontinued for AD treatment as a larger Phase IIa trial suggested that it had no statistically significant effect on observed AD-associated pathological changes [134]. Saracatinib is currently undergoing a Phase I clinical study (NCT03661125) for PD psychosis. Another tyrosine kinase inhibitor masitinib that targets a variety of tyrosine kinases, including Src, Fyn and Lyn shows satisfactory safety profile and efficacy in patients with amyotrophic lateral sclerosis (ALS) when used in combination with an FDA-approved ALS medication called Rilutek in a Phase II trials [135]. The same results are seen with masitinib alone in patients with multiple sclerosis (MS) [136] and AD [137]. Phase III trials of masitinib for the above indications (NCT01433497, NCT03127267, NCT01872598) are therefore being carried on in order to support masitinib to be an innovative treatment in these diseases. SFK inhibitors are generally delivered by the oral route of administration. This makes them convenient and economical for the patients during the course of treatment. Notably, SFK inhibitors show great potential to penetrate CNS via oral delivery. Saracatinib exhibits substantial CNS penetration with oral dose at 100-125 mg in the above-mentioned Phase Ib study [133]. The earliest FDA-approved SFK inhibitor dasatinib is also able to cross the blood-brain barrier to the CNS in mice and patients, the level of which in the cerebrospinal fluid (CSF) of patients corresponds to the antitumour activity observed in the CNS [138]. The ability of CNS penetration of the drug can be essential in its efficacy for the treatment of some neurological disorders if its intended targets are located in the brain.
These features of SFK inhibitors support SFK to be a promising therapeutic target. Nevertheless, the potential caveats of SFK inhibitors should be considered. SFK family members might have distinct functions in various neurological abnormalities. Since current SFK inhibitors target several subtypes, it is important to understand the roles of these targeted SFK members in a certain disorder in order to apply specific SFK inhibitors as a feasible treatment. Additionally, the ubiquitous expression of SFK in diverse tissues and the multi-kinase inhibitory property of some SFK inhibitors (e.g., Masitinib) might increase the risk for off-target effects. These might cause undesirable side effects and reduce the efficacy of the drug. Development of highly selective SFK inhibitors is thus necessary to address these issues. In fact, another direction for clinical applications of SFK inhibitors is combination therapy with other medications. As mentioned above, the combination therapy using masitinib and Rilutek for ALS has been developed [135] and continued for the Phase III trial. Interestingly, dasatinib attenuates Aβ-associated microgliosis in mice model of AD [86], and a combination treatment using dasatinib and quercetin for AD is currently investigated in a clinical trial (NCT04063124). The combination of an approved drug for a certain disorder with an SFK inhibitor might produce optimal effects and be a useful strategy in the management of the diseases. Furthermore, peptides targeting SFK-interacting sites of membrane receptors have been developed and tested in pre-clinical studies. These peptides are Src40-49Tat and TAT-Panx308, which are introduced in the previous sections of this review. In rodents, both peptides show neuroprotective effects: Src40-49Tat reduces pain behaviour [120], while TAT- Panx308 alleviates brain damage caused by stroke [29]. Potential clinical applications of these peptides should be considered as they might be served as an improved therapeutic strategy by targeting SFK-mediated receptor function and signalling, compared to receptor antagonists that often inhibit important basal functions and cause adverse events.
CONCLUSION
Evidences at present are sufficient to establish the important role of SFK in the development of migraine and neuropathic pain through transmitting signalling of multiple membrane proteins and intracellular proteins, including NMDA receptors and P2X receptors that are crucial for these diseases. The safety profile and CNS-penetrating ability of SFK inhibitors make it promising for their clinical applications in neurodegenerative diseases and possibly in more neurological diseases, including migraine and neuropathic pain. Future pre-clinical and clinical studies might be considered for applying SFK inhibitors in alleviating these pain states. Novel therapeutic strategies relevant to SFK, including combination therapy with other medications and targeting SFK signalling might provide new insights into the clinical potential of SFK inhibitors.
ACKNOWLEDGEMENTS
Declared none.
AUTHORS' CONTRIBUTIONS
L. Nie, M.Wang designed the concept. L. Nie, M.Wang and W.R. Ye drafted the paper. S. Chen, and D. Chirchiglia and M. Wang edited the paper. All authors read and approved the manuscript.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors have no conflicts of interest, financial or otherwise.
References
- 1.Rous P. A SARCOMA OF THE FOWL TRANSMISSIBLE BY AN AGENT SEPARABLE FROM THE TUMOR CELLS. J. Exp. Med. 1911;13(4):397–411. doi: 10.1084/jem.13.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stehelin D., Fujita D.J., Padgett T., Varmus H.E., Bishop J.M. Detection and enumeration of transformation-defective strains of avian sarcoma virus with molecular hybridization. Virology. 1977;76(2):675–684. doi: 10.1016/0042-6822(77)90250-1. [DOI] [PubMed] [Google Scholar]
- 3.Parsons S.J., Parsons J.T. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23(48):7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 4.Xiao X., Yang Y., Mao B., Cheng C.Y., Ni Y. Emerging role for SRC family kinases in junction dynamics during spermatogenesis. Reproduction. 2019;157(3):R85–R94. doi: 10.1530/REP-18-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasahara K., Nakayama Y., Kihara A., Matsuda D., Ikeda K., Kuga T., Fukumoto Y., Igarashi Y., Yamaguchi N. Rapid trafficking of c-Src, a non-palmitoylated Src-family kinase, between the plasma membrane and late endosomes/lysosomes. Exp. Cell Res. 2007;313(12):2651–2666. doi: 10.1016/j.yexcr.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Boggon T.J., Eck M.J. Structure and regulation of Src family kinases. Oncogene. 2004;23(48):7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 7.Nagar B., Hantschel O., Young M.A., Scheffzek K., Veach D., Bornmann W., Clarkson B., Superti-Furga G., Kuriyan J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112(6):859–871. doi: 10.1016/S0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 8.Nada S., Okada M., MacAuley A., Cooper J.A., Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351(6321):69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- 9.Guarino M. Src signaling in cancer invasion. J. Cell. Physiol. 2010;223(1):14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
- 10.Cotton P.C., Brugge J.S. Neural tissues express high levels of the cellular src gene product pp60c-src. Mol. Cell. Biol. 1983;3(6):1157–1162. doi: 10.1128/MCB.3.6.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Umemori H., Wanaka A., Kato H., Takeuchi M., Tohyama M., Yamamoto T. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Brain Res. Mol. Brain Res. 1992;16(3-4):303–310. doi: 10.1016/0169-328X(92)90239-8. [DOI] [PubMed] [Google Scholar]
- 12.Omri B., Crisanti P., Marty M.C., Alliot F., Fagard R., Molina T., Pessac B. The Lck tyrosine kinase is expressed in brain neurons. J. Neurochem. 1996;67(4):1360–1364. doi: 10.1046/j.1471-4159.1996.67041360.x. [DOI] [PubMed] [Google Scholar]
- 13.Sudol M., Hanafusa H. Cellular proteins homologous to the viral yes gene product. Mol. Cell. Biol. 1986;6(8):2839–2846. doi: 10.1128/MCB.6.8.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sudol M., Greulich H., Newman L., Sarkar A., Sukegawa J., Yamamoto T. A novel Yes-related kinase, Yrk, is expressed at elevated levels in neural and hematopoietic tissues. Oncogene. 1993;8(4):823–831. [PubMed] [Google Scholar]
- 15.Moia L.J., Matsui H., Nishio H., Tsuchida T., Tokuda M., Itano T., Nagao S., Hatase O. Identification of a src family protein specifically expressed in rat astrocytes by immunohistochemistry and double immunofluorescent study. Brain Res. 1992;585(1-2):283–286. doi: 10.1016/0006-8993(92)91219-5. [DOI] [PubMed] [Google Scholar]
- 16.Krady J.K., Basu A., Levison S.W., Milner R.J. Differential expression of protein tyrosine kinase genes during microglial activation. Glia. 2002;40(1):11–24. doi: 10.1002/glia.10101. [DOI] [PubMed] [Google Scholar]
- 17.Colognato H., Ramachandrappa S., Olsen I.M., ffrench-Constant C. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J. Cell Biol. 2004;167(2):365–375. doi: 10.1083/jcb.200404076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ingraham C.A., Cox M.E., Ward D.C., Fults D.W., Maness P.F. c-src and other proto-oncogenes implicated in neuronal differentiation. Mol. Chem. Neuropathol. 1989;10(1):1–14. doi: 10.1007/BF02969481. [DOI] [PubMed] [Google Scholar]
- 19.Kalia L.V., Gingrich J.R., Salter M.W. Src in synaptic transmission and plasticity. Oncogene. 2004;23(48):8007–8016. doi: 10.1038/sj.onc.1208158. [DOI] [PubMed] [Google Scholar]
- 20.Schenone S., Brullo C., Musumeci F., Biava M., Falchi F., Botta M. Fyn kinase in brain diseases and cancer: the search for inhibitors. Curr. Med. Chem. 2011;18(19):2921–2942. doi: 10.2174/092986711796150531. [DOI] [PubMed] [Google Scholar]
- 21.Nygaard H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry. 2018;83(4):369–376. doi: 10.1016/j.biopsych.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerjee A., Wang H.Y., Borgmann-Winter K.E., MacDonald M.L., Kaprielian H., Stucky A., Kvasic J., Egbujo C., Ray R., Talbot K., Hemby S.E., Siegel S.J., Arnold S.E., Sleiman P., Chang X., Hakonarson H., Gur R.E., Hahn C.G. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol. Psychiatry. 2015;20(9):1091–1100. doi: 10.1038/mp.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salter M.W., Kalia L.V. Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 2004;5(4):317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi T., Huganir R.L. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J. Neurosci. 2004;24(27):6152–6160. doi: 10.1523/JNEUROSCI.0799-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le H.T., Maksumova L., Wang J., Pallen C.J. Reduced NMDA receptor tyrosine phosphorylation in PTPalpha-deficient mouse synaptosomes is accompanied by inhibition of four src family kinases and Pyk2: an upstream role for PTPalpha in NMDA receptor regulation. J. Neurochem. 2006;98(6):1798–1809. doi: 10.1111/j.1471-4159.2006.04075.x. [DOI] [PubMed] [Google Scholar]
- 26.Du C.P., Gao J., Tai J.M., Liu Y., Qi J., Wang W., Hou X.Y. Increased tyrosine phosphorylation of PSD-95 by Src family kinases after brain ischaemia. Biochem. J. 2009;417(1):277–285. doi: 10.1042/BJ20080004. [DOI] [PubMed] [Google Scholar]
- 27.Zhang M., Li Q., Chen L., Li J., Zhang X., Chen X., Zhang Q., Shao Y., Xu Y. PSD-93 deletion inhibits Fyn-mediated phosphorylation of NR2B and protects against focal cerebral ischemia. Neurobiol. Dis. 2014;68:104–111. doi: 10.1016/j.nbd.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Moss S.J., Gorrie G.H., Amato A., Smart T.G. Modulation of GABAA receptors by tyrosine phosphorylation. Nature. 1995;377(6547):344–348. doi: 10.1038/377344a0. [DOI] [PubMed] [Google Scholar]
- 29.Weilinger N.L., Lohman A.W., Rakai B.D., Ma E.M., Bialecki J., Maslieieva V., Rilea T., Bandet M.V., Ikuta N.T., Scott L., Colicos M.A., Teskey G.C., Winship I.R., Thompson R.J. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat. Neurosci. 2016;19(3):432–442. doi: 10.1038/nn.4236. [DOI] [PubMed] [Google Scholar]
- 30.Leduc-Pessah H., Weilinger N.L., Fan C.Y., Burma N.E., Thompson R.J., Trang T. Site-Specific Regulation of P2X7 Receptor Function in Microglia Gates Morphine Analgesic Tolerance. J. Neurosci. 2017;37(42):10154–10172. doi: 10.1523/JNEUROSCI.0852-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodfellow N.M., Sargin D., Ansorge M.S., Gingrich J.A., Lambe E.K. Mice with compromised 5-HTT function lack phosphotyrosine-mediated inhibitory control over prefrontal 5-HT responses. J. Neurosci. 2014;34(17):6107–6111. doi: 10.1523/JNEUROSCI.3762-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dantsuji M., Nakamura S., Nakayama K., Mochizuki A., Park S.K., Bae Y.C., Ozeki M., Inoue T. 5-HT2A receptor activation enhances NMDA receptor-mediated glutamate responses through Src kinase in the dendrites of rat jaw-closing motoneurons. J. Physiol. 2019;597(9):2565–2589. doi: 10.1113/JP275440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banerjee M., Cui X., Li Z., Yu H., Cai L., Jia X., He D., Wang C., Gao T., Xie Z. Na/K-ATPase Y260 Phosphorylation-mediated Src Regulation in Control of Aerobic Glycolysis and Tumor Growth. Sci. Rep. 2018;8(1):12322–12322. doi: 10.1038/s41598-018-29995-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose E.M., Koo J.C.P., Antflick J.E., Ahmed S.M., Angers S., Hampson D.R. Glutamate transporter coupling to Na,K-ATPase. J. Neurosci. 2009;29(25):8143–8155. doi: 10.1523/JNEUROSCI.1081-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Sá Lima L., Kawamoto E.M., Munhoz C.D., Kinoshita P.F., Orellana A.M.M., Curi R., Rossoni L.V., Avellar M.C.W., Scavone C. Ouabain activates NFκB through an NMDA signaling pathway in cultured cerebellar cells. Neuropharmacology. 2013;73:327–336. doi: 10.1016/j.neuropharm.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Panicker N., Saminathan H., Jin H., Neal M., Harischandra D.S., Gordon R., Kanthasamy K., Lawana V., Sarkar S., Luo J., Anantharam V., Kanthasamy A.G., Kanthasamy A. Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson’s Disease. J. Neurosci. 2015;35(27):10058–10077. doi: 10.1523/JNEUROSCI.0302-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Floreani N.A., Rump T.J., Abdul Muneer P.M., Alikunju S., Morsey B.M., Brodie M.R., Persidsky Y., Haorah J. Alcohol-induced interactive phosphorylation of Src and toll-like receptor regulates the secretion of inflammatory mediators by human astrocytes. J. Neuroimmune Pharmacol. 2010;5(4):533–545. doi: 10.1007/s11481-010-9213-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brozzi F., Arcuri C., Giambanco I., Donato R. S100B Protein Regulates Astrocyte Shape and Migration via Interaction with Src Kinase: IMPLICATIONS FOR ASTROCYTE DEVELOPMENT, ACTIVATION, AND TUMOR GROWTH. J. Biol. Chem. 2009;284(13):8797–8811. doi: 10.1074/jbc.M805897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuda M., Tozaki-Saitoh H., Masuda T., Toyomitsu E., Tezuka T., Yamamoto T., Inoue K. Lyn tyrosine kinase is required for P2X(4) receptor upregulation and neuropathic pain after peripheral nerve injury. Glia. 2008;56(1):50–58. doi: 10.1002/glia.20591. [DOI] [PubMed] [Google Scholar]
- 40.Traynelis S.F., Wollmuth L.P., McBain C.J., Menniti F.S., Vance K.M., Ogden K.K., Hansen K.B., Yuan H., Myers S.J., Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010;62(3):405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakazawa T., Komai S., Tezuka T., Hisatsune C., Umemori H., Semba K., Mishina M., Manabe T., Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR ε 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 2001;276(1):693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- 42.Yang M., Leonard J.P. Identification of mouse NMDA receptor subunit NR2A C-terminal tyrosine sites phosphorylated by coexpression with v-Src. J. Neurochem. 2001;77(2):580–588. doi: 10.1046/j.1471-4159.2001.00255.x. [DOI] [PubMed] [Google Scholar]
- 43.Weilinger N.L., Tang P.L., Thompson R.J. Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 2012;32(36):12579–12588. doi: 10.1523/JNEUROSCI.1267-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bu F., Nie L., Quinn J.P., Wang M. Sarcoma Family Kinase-Dependent Pannexin-1 Activation after Cortical Spreading Depression is Mediated by NR2A-Containing Receptors. Int. J. Mol. Sci. 2020;21(4):1–14. doi: 10.3390/ijms21041269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheung H.H., Gurd J.W. Tyrosine phosphorylation of the N-methyl-D-aspartate receptor by exogenous and postsynaptic density-associated Src-family kinases. J. Neurochem. 2001;78(3):524–534. doi: 10.1046/j.1471-4159.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 46.Yaka R., Phamluong K., Ron D. Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J. Neurosci. 2003;23(9):3623–3632. doi: 10.1523/JNEUROSCI.23-09-03623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gingrich J.R., Pelkey K.A., Fam S.R., Huang Y., Petralia R.S., Wenthold R.J., Salter M.W. Unique domain anchoring of Src to synaptic NMDA receptors via the mitochondrial protein NADH dehydrogenase subunit 2. Proc. Natl. Acad. Sci. USA. 2004;101(16):6237–6242. doi: 10.1073/pnas.0401413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sinai L., Duffy S., Roder J.C. Src inhibition reduces NR2B surface expression and synaptic plasticity in the amygdala. Learn. Mem. 2010;17(8):364–371. doi: 10.1101/lm.1765710. [DOI] [PubMed] [Google Scholar]
- 49.Kohda K., Kakegawa W., Matsuda S., Yamamoto T., Hirano H., Yuzaki M. The δ2 glutamate receptor gates long-term depression by coordinating interactions between two AMPA receptor phosphorylation sites. Proc. Natl. Acad. Sci. USA. 2013;110(10):E948–E957. doi: 10.1073/pnas.1218380110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scholz R., Berberich S., Rathgeber L., Kolleker A., Köhr G., Kornau H.C. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron. 2010;66(5):768–780. doi: 10.1016/j.neuron.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 51.Yong A.J.H., Tan H.L., Zhu Q., Bygrave A.M., Johnson R.C., Huganir R.L. Tyrosine phosphorylation of the AMPA receptor subunit GluA2 gates homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA. 2020;117(9):4948–4958. doi: 10.1073/pnas.1918436117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rong Y., Lu X., Bernard A., Khrestchatisky M., Baudry M. Tyrosine phosphorylation of ionotropic glutamate receptors by Fyn or Src differentially modulates their susceptibility to calpain and enhances their binding to spectrin and PSD-95. J. Neurochem. 2001;79(2):382–390. doi: 10.1046/j.1471-4159.2001.00565.x. [DOI] [PubMed] [Google Scholar]
- 53.Narisawa-Saito M., Silva A.J., Yamaguchi T., Hayashi T., Yamamoto T., Nawa H. Growth factor-mediated Fyn signaling regulates alpha-amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor expression in rodent neocortical neurons. Proc. Natl. Acad. Sci. USA. 1999;96(5):2461–2466. doi: 10.1073/pnas.96.5.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayashi T., Umemori H., Mishina M., Yamamoto T. The AMPA receptor interacts with and signals through the protein tyrosine kinase Lyn. Nature. 1999;397(6714):72–76. doi: 10.1038/16269. [DOI] [PubMed] [Google Scholar]
- 55.Socodato R., Santiago F.N., Portugal C.C., Domingues A.F., Santiago A.R., Relvas J.B., Ambrósio A.F., Paes-de-Carvalho R. Calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors trigger neuronal nitric-oxide synthase activation to promote nerve cell death in an Src kinase-dependent fashion. J. Biol. Chem. 2012;287(46):38680–38694. doi: 10.1074/jbc.M112.353961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stathakis D.G., Hoover K.B., You Z., Bryant P.J. Human postsynaptic density-95 (PSD95): location of the gene (DLG4) and possible function in nonneural as well as in neural tissues. Genomics. 1997;44(1):71–82. doi: 10.1006/geno.1997.4848. [DOI] [PubMed] [Google Scholar]
- 57.Hou X-Y., Zhang G-Y., Yan J-Z., Liu Y. Increased tyrosine phosphorylation of α(1C) subunits of L-type voltage-gated calcium channels and interactions among Src/Fyn, PSD-95 and α(1C) in rat hippocampus after transient brain ischemia. Brain Res. 2003;979(1-2):43–50. doi: 10.1016/S0006-8993(03)02845-2. [DOI] [PubMed] [Google Scholar]
- 58.Sieghart W., Sperk G. Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr. Top. Med. Chem. 2002;2(8):795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- 59.Brandon N.J., Delmas P., Hill J., Smart T.G., Moss S.J. Constitutive tyrosine phosphorylation of the GABA(A) receptor γ 2 subunit in rat brain. Neuropharmacology. 2001;41(6):745–752. doi: 10.1016/S0028-3908(01)00121-6. [DOI] [PubMed] [Google Scholar]
- 60.Jurd R., Tretter V., Walker J., Brandon N.J., Moss S.J. Fyn kinase contributes to tyrosine phosphorylation of the GABA(A) receptor γ2 subunit. Mol. Cell. Neurosci. 2010;44(2):129–134. doi: 10.1016/j.mcn.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tretter V., Revilla-Sanchez R., Houston C., Terunuma M., Havekes R., Florian C., Jurd R., Vithlani M., Michels G., Couve A., Sieghart W., Brandon N., Abel T., Smart T.G., Moss S.J. Deficits in spatial memory correlate with modified gamma-aminobutyric acid type A receptor tyrosine phosphorylation in the hippocampus. Proc. Natl. Acad. Sci. USA. 2009;106(47):20039–20044. doi: 10.1073/pnas.0908840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adinolfi E., Callegari M.G., Ferrari D., Bolognesi C., Minelli M., Wieckowski M.R., Pinton P., Rizzuto R., Di Virgilio F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol. Biol. Cell. 2005;16(7):3260–3272. doi: 10.1091/mbc.e04-11-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Locovei S., Scemes E., Qiu F., Spray D.C., Dahl G. Pannexin1 is part of the pore forming unit of the P2X(7) receptor death complex. FEBS Lett. 2007;581(3):483–488. doi: 10.1016/j.febslet.2006.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pelegrin P., Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25(21):5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen S.P., Qin T., Seidel J.L., Zheng Y., Eikermann M., Ferrari M.D., van den Maagdenberg A.M.J.M., Moskowitz M.A., Ayata C., Eikermann-Haerter K. Inhibition of the P2X7-PANX1 complex suppresses spreading depolarization and neuroinflammation. Brain. 2017;140(6):1643–1656. doi: 10.1093/brain/awx085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iglesias R., Locovei S., Roque A., Alberto A.P., Dahl G., Spray D.C., Scemes E. P2X7 receptor-Pannexin1 complex: pharmacology and signaling. Am. J. Physiol. Cell Physiol. 2008;295(3):C752–C760. doi: 10.1152/ajpcell.00228.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pithadia A.B., Jain S.M. 5-Hydroxytryptamine Receptor Subtypes and their Modulators with Therapeutic Potentials. J. Clin. Med. Res. 2009;1(2):72–80. doi: 10.4021/jocmr2009.05.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luttrell D.K., Luttrell L.M. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23(48):7969–7978. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- 69.Lu R., Alioua A., Kumar Y., Kundu P., Eghbali M., Weisstaub N.V., Gingrich J.A., Stefani E., Toro L. c-Src tyrosine kinase, a critical component for 5-HT2A receptor-mediated contraction in rat aorta. J. Physiol. 2008;586(16):3855–3869. doi: 10.1113/jphysiol.2008.153593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sung D.J., Noh H.J., Kim J.G., Park S.W., Kim B., Cho H., Bae Y.M. Serotonin contracts the rat mesenteric artery by inhibiting 4-aminopyridine-sensitive Kv channels via the 5-HT2A receptor and Src tyrosine kinase. Exp. Mol. Med. 2013;45:e67. doi: 10.1038/emm.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim J.G., Leem Y-E., Kwon I., Kang J-S., Bae Y.M., Cho H. Estrogen modulates serotonin effects on vasoconstriction through Src inhibition. Exp. Mol. Med. 2018;50(12):1–9. doi: 10.1038/s12276-018-0193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pimenova A.A., Thathiah A., De Strooper B., Tesseur I. Regulation of amyloid precursor protein processing by serotonin signaling. PLoS One. 2014;9(1):e87014. doi: 10.1371/journal.pone.0087014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aperia A., Akkuratov E.E., Fontana J.M., Brismar H. Na+-K+-ATPase, a new class of plasma membrane receptors. Am. J. Physiol. Cell Physiol. 2016;310(7):C491–C495. doi: 10.1152/ajpcell.00359.2015. [DOI] [PubMed] [Google Scholar]
- 74.Xie Z., Cai T. Na+-K+--ATPase-mediated signal transduction: from protein interaction to cellular function. Mol. Interv. 2003;3(3):157–168. doi: 10.1124/mi.3.3.157. [DOI] [PubMed] [Google Scholar]
- 75.Cui X., Xie Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules. 2017;22(6):990. doi: 10.3390/molecules22060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tian J., Cai T., Yuan Z., Wang H., Liu L., Haas M., Maksimova E., Huang X-Y., Xie Z-J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell. 2006;17(1):317–326. doi: 10.1091/mbc.e05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang X.Q., Yu S.P. Novel regulation of Na, K-ATPase by Src tyrosine kinases in cortical neurons. J. Neurochem. 2005;93(6):1515–1523. doi: 10.1111/j.1471-4159.2005.03147.x. [DOI] [PubMed] [Google Scholar]
- 78.Zhang L., Guo F., Su S., Guo H., Xiong C., Yin J., Li W., Wang Y. Na(+)/K(+)-ATPase inhibition upregulates NMDA-evoked currents in rat hippocampal CA1 pyramidal neurons. Fundam. Clin. Pharmacol. 2012;26(4):503–512. doi: 10.1111/j.1472-8206.2011.00947.x. [DOI] [PubMed] [Google Scholar]
- 79.Rhee Y-H., Moon J.H., Jung J.Y., Oh C., Ahn J-C., Chung P-S. Effect of photobiomodulation therapy on neuronal injuries by ouabain: the regulation of Na, K-ATPase; Src; and mitogen-activated protein kinase signaling pathway. BMC Neurosci. 2019;20(1):19–19. doi: 10.1186/s12868-019-0499-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Penniyaynen V.A., Plakhova V.B., Rogachevskii I.V., Terekhin S.G., Podzorova S.A., Krylov B.V. Molecular mechanisms and signaling by comenic acid in nociceptive neurons influence the pathophysiology of neuropathic pain. Pathophysiology. 2019;26(3-4):245–252. doi: 10.1016/j.pathophys.2019.06.003. [DOI] [PubMed] [Google Scholar]
- 81.Dai H., Song D., Xu J., Li B., Hertz L., Peng L. Ammonia-induced Na,K-ATPase/ouabain-mediated EGF receptor transactivation, MAPK/ERK and PI3K/AKT signaling and ROS formation cause astrocyte swelling. Neurochem. Int. 2013;63(6):610–625. doi: 10.1016/j.neuint.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 82.DiSabato D.J., Quan N., Godbout J.P. Neuroinflammation: the devil is in the details. J. Neurochem. 2016;139(Suppl. 2):136–153. doi: 10.1111/jnc.13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Milligan E.D., Watkins L.R. Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 2009;10(1):23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Song G.J., Jung M., Kim J.H., Park H., Rahman M.H., Zhang S., Zhang Z.Y., Park D.H., Kook H., Lee I.K., Suk K. A novel role for protein tyrosine phosphatase 1B as a positive regulator of neuroinflammation. J. Neuroinflammation. 2016;13(1):86. doi: 10.1186/s12974-016-0545-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Socodato R., Portugal C.C., Domith I., Oliveira N.A., Coreixas V.S., Loiola E.C., Martins T., Santiago A.R., Paes-de-Carvalho R., Ambrósio A.F., Relvas J.B. c-Src function is necessary and sufficient for triggering microglial cell activation. Glia. 2015;63(3):497–511. doi: 10.1002/glia.22767. [DOI] [PubMed] [Google Scholar]
- 86.Dhawan G., Combs C.K. Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer’s disease. J. Neuroinflammation. 2012;9:117. doi: 10.1186/1742-2094-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tai W., Ye X., Bao X., Zhao B., Wang X., Zhang D. Inhibition of Src tyrosine kinase activity by squamosamide derivative FLZ attenuates neuroinflammation in both in vivo and in vitro Parkinson’s disease models. Neuropharmacology. 2013;75:201–212. doi: 10.1016/j.neuropharm.2013.07.020. [DOI] [PubMed] [Google Scholar]
- 88.Lee C., Low C.Y., Wong S.Y., Lai M.K., Tan M.G. Selective induction of alternatively spliced FynT isoform by TNF facilitates persistent inflammatory responses in astrocytes. Sci. Rep. 2017;7:43651. doi: 10.1038/srep43651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Noseda R., Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain. 2013;154(Suppl. 1):S44–S53. doi: 10.1016/j.pain.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 90.Bu F., Wang Y., Jiang L., Ma D., Quinn J.P., Wang M. Sarcoma family kinase activity is required for cortical spreading depression. Cephalalgia. 2018;38(11):1748–1758. doi: 10.1177/0333102417748572. [DOI] [PubMed] [Google Scholar]
- 91.Ayata C. Cortical spreading depression triggers migraine attack: pro. Headache. 2010;50(4):725–730. doi: 10.1111/j.1526-4610.2010.01647.x. [DOI] [PubMed] [Google Scholar]
- 92.Wang X.Y., Zhou H.R., Wang S., Liu C.Y., Qin G.C., Fu Q.Q., Zhou J.Y., Chen L.X. NR2B-Tyr phosphorylation regulates synaptic plasticity in central sensitization in a chronic migraine rat model. J. Headache Pain. 2018;19(1):102. doi: 10.1186/s10194-018-0935-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Staehr C., Hangaard L., Bouzinova E.V., Kim S., Rajanathan R., Boegh Jessen P., Luque N., Xie Z., Lykke-Hartmann K., Sandow S.L., Aalkjaer C., Matchkov V.V. Smooth muscle Ca2+ sensitization causes hypercontractility of middle cerebral arteries in mice bearing the familial hemiplegic migraine type 2 associated mutation. J. Cereb. Blood Flow Metab. 2019;39(8):1570–1587. doi: 10.1177/0271678X18761712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Staehr C., Rajanathan R., Matchkov V.V. Involvement of the Na+, K+ -ATPase isoforms in control of cerebral perfusion. Exp. Physiol. 2019;104(7):1023–1028. doi: 10.1113/EP087519. [DOI] [PubMed] [Google Scholar]
- 95.Durham P.L. Calcitonin gene-related peptide (CGRP) and migraine. Headache. 2006;46(Suppl. 1):S3–S8. doi: 10.1111/j.1526-4610.2006.00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dodick D.W. CGRP ligand and receptor monoclonal antibodies for migraine prevention: Evidence review and clinical implications. Cephalalgia. 2019;39(3):445–458. doi: 10.1177/0333102418821662. [DOI] [PubMed] [Google Scholar]
- 97.Park K.A., Fehrenbacher J.C., Thompson E.L., Duarte D.B., Hingtgen C.M., Vasko M.R. Signaling pathways that mediate nerve growth factor-induced increase in expression and release of calcitonin gene-related peptide from sensory neurons. Neuroscience. 2010;171(3):910–923. doi: 10.1016/j.neuroscience.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jin X., Morsy N., Winston J., Pasricha P.J., Garrett K., Akbarali H.I. Modulation of TRPV1 by nonreceptor tyrosine kinase, c-Src kinase. Am. J. Physiol. Cell Physiol. 2004;287(2):C558–C563. doi: 10.1152/ajpcell.00113.2004. [DOI] [PubMed] [Google Scholar]
- 99.Karatas H., Erdener S.E., Gursoy-Ozdemir Y., Lule S., Eren-Koçak E., Sen Z.D., Dalkara T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science. 2013;339(6123):1092–1095. doi: 10.1126/science.1231897. [DOI] [PubMed] [Google Scholar]
- 100.Bu F., Du R., Li Y., Quinn J.P., Wang M. NR2A contributes to genesis and propagation of cortical spreading depression in rats. Sci. Rep. 2016;6(23679):23576. doi: 10.1038/srep23576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marrannes R., Willems R., De Prins E., Wauquier A. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res. 1988;457(2):226–240. doi: 10.1016/0006-8993(88)90690-7. [DOI] [PubMed] [Google Scholar]
- 102.Peeters M., Gunthorpe M.J., Strijbos P.J., Goldsmith P., Upton N., James M.F. Effects of pan- and subtype-selective N-methyl-D-aspartate receptor antagonists on cortical spreading depression in the rat: therapeutic potential for migraine. J. Pharmacol. Exp. Ther. 2007;321(2):564–572. doi: 10.1124/jpet.106.117101. [DOI] [PubMed] [Google Scholar]
- 103.Moreno-Ajona D., Chan C., Villar-Martínez M.D., Goadsby P.J. Targeting CGRP and 5-HT1F Receptors for the Acute Therapy of Migraine: A Literature Review. Headache. 2019;59(Suppl. 2):3–19. doi: 10.1111/head.13582. [DOI] [PubMed] [Google Scholar]
- 104.Lambru G., Andreou A.P., Guglielmetti M., Martelletti P. Emerging drugs for migraine treatment: an update. Expert Opin. Emerg. Drugs. 2018;23(4):301–318. doi: 10.1080/14728214.2018.1552939. [DOI] [PubMed] [Google Scholar]
- 105.Gölöncsér F., Sperlágh B. Effect of genetic deletion and pharmacological antagonism of P2X7 receptors in a mouse animal model of migraine. J. Headache Pain. 2014;15:24. doi: 10.1186/1129-2377-15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fan P.C., Lai T.H., Hor C.C., Lee M.T., Huang P., Sieghart W., Ernst M., Knutson D.E., Cook J., Chiou L.C. The α6 subunit-containing GABAA receptor: A novel drug target for inhibition of trigeminal activation. Neuropharmacology. 2018;140:1–13. doi: 10.1016/j.neuropharm.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cohen S.P., Mao J. Neuropathic pain: mechanisms and their clinical implications. BMJ. 2014;348:f7656. doi: 10.1136/bmj.f7656. [DOI] [PubMed] [Google Scholar]
- 108.Katsura H., Obata K., Mizushima T., Sakurai J., Kobayashi K., Yamanaka H., Dai Y., Fukuoka T., Sakagami M., Noguchi K. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J. Neurosci. 2006;26(34):8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tan Y-H., Li K., Chen X-Y., Cao Y., Light A.R., Fu K-Y. Activation of Src family kinases in spinal microglia contributes to formalin-induced persistent pain state through p38 pathway. J. Pain. 2012;13(10):1008–1015. doi: 10.1016/j.jpain.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 110.Gao Y.Y., Hong X.Y., Wang H.J. Role of Nectin-1/c-Src Signaling in the Analgesic Effect of GDNF on a Rat Model of Chronic Constrictive Injury. J. Mol. Neurosci. 2016;60(2):258–266. doi: 10.1007/s12031-016-0792-x. [DOI] [PubMed] [Google Scholar]
- 111.Alessandri-Haber N., Dina O.A., Joseph E.K., Reichling D.B., Levine J.D. Interaction of transient receptor potential vanilloid 4, integrin, and SRC tyrosine kinase in mechanical hyperalgesia. J. Neurosci. 2008;28(5):1046–1057. doi: 10.1523/JNEUROSCI.4497-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lai C.Y., Lin T.B., Hsieh M.C., Chen G.D., Peng H.Y. SIRPα1-SHP2 Interaction Regulates Complete Freund Adjuvant-Induced Inflammatory Pain via Src-Dependent GluN2B Phosphorylation in Rats. Anesth. Analg. 2016;122(3):871–881. doi: 10.1213/ANE.0000000000001116. [DOI] [PubMed] [Google Scholar]
- 113.Slack S., Battaglia A., Cibert-Goton V., Gavazzi I. EphrinB2 induces tyrosine phosphorylation of NR2B via Src-family kinases during inflammatory hyperalgesia. Neuroscience. 2008;156(1):175–183. doi: 10.1016/j.neuroscience.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bogen O., Joseph E.K., Chen X., Levine J.D. GDNF hyperalgesia is mediated by PLCgamma, MAPK/ERK, PI3K, CDK5 and Src family kinase signaling and dependent on the IB4-binding protein versican. Eur. J. Neurosci. 2008;28(1):12–19. doi: 10.1111/j.1460-9568.2008.06308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Suo Z.W., Yang X., Li L., Liu Y.N., Shi L., Hu X.D. Inhibition of protein tyrosine phosphatases in spinal dorsal horn attenuated inflammatory pain by repressing Src signaling. Neuropharmacology. 2013;70:122–130. doi: 10.1016/j.neuropharm.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 116.Li S., Cao J., Yang X., Suo Z.W., Shi L., Liu Y.N., Yang H.B., Hu X.D. NR2B phosphorylation at tyrosine 1472 in spinal dorsal horn contributed to N-methyl-D-aspartate-induced pain hypersensitivity in mice. J. Neurosci. Res. 2011;89(11):1869–1876. doi: 10.1002/jnr.22719. [DOI] [PubMed] [Google Scholar]
- 117.Abe T., Matsumura S., Katano T., Mabuchi T., Takagi K., Xu L., Yamamoto A., Hattori K., Yagi T., Watanabe M., Nakazawa T., Yamamoto T., Mishina M., Nakai Y., Ito S. Fyn kinase-mediated phosphorylation of NMDA receptor NR2B subunit at Tyr1472 is essential for maintenance of neuropathic pain. Eur. J. Neurosci. 2005;22(6):1445–1454. doi: 10.1111/j.1460-9568.2005.04340.x. [DOI] [PubMed] [Google Scholar]
- 118.Liu Y.N., Yang X., Suo Z.W., Xu Y.M., Hu X.D. Fyn kinase-regulated NMDA receptor- and AMPA receptor-dependent pain sensitization in spinal dorsal horn of mice. Eur. J. Pain. 2014;18(8):1120–1128. doi: 10.1002/j.1532-2149.2014.00455.x. [DOI] [PubMed] [Google Scholar]
- 119.Hildebrand M.E., Xu J., Dedek A., Li Y., Sengar A.S., Beggs S., Lombroso P.J., Salter M.W. Potentiation of synaptic GluN2B NMDAR currents by fyn kinase is gated through BDNF-mediated disinhibition in spinal pain processing. Cell Rep. 2016;17(10):2753–2765. doi: 10.1016/j.celrep.2016.11.024. [DOI] [PubMed] [Google Scholar]
- 120.Liu X.J., Gingrich J.R., Vargas-Caballero M., Dong Y.N., Sengar A., Beggs S., Wang S.H., Ding H.K., Frankland P.W., Salter M.W. Treatment of inflammatory and neuropathic pain by uncoupling Src from the NMDA receptor complex. Nat. Med. 2008;14(12):1325–1332. doi: 10.1038/nm.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Muir K.W. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr. Opin. Pharmacol. 2006;6(1):53–60. doi: 10.1016/j.coph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 122.Smith P.F. Therapeutic N-methyl-D-aspartate receptor antagonists: will reality meet expectation? Curr. Opin. Investig. Drugs. 2003;4(7):826–832. [PubMed] [Google Scholar]
- 123.Kim L.C., Song L., Haura E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009;6(10):587–595. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 124.Roskoski R., Jr Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015;94:9–25. doi: 10.1016/j.phrs.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 125.Hennequin L.F., Allen J., Breed J., Curwen J., Fennell M., Green T.P., Lambert-van der Brempt C., Morgentin R., Norman R.A., Olivier A., Otterbein L., Plé P.A., Warin N., Costello G. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J. Med. Chem. 2006;49(22):6465–6488. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 126.Sanz-Blasco S., Bordone M.P., Damianich A., Gomez G., Bernardi M.A., Isaja L., Taravini I.R., Hanger D.P., Avale M.E., Gershanik O.S., Ferrario J.E. The Kinase Fyn As a Novel Intermediate in L-DOPA-Induced Dyskinesia in Parkinson’s Disease. Mol. Neurobiol. 2018;55(6):5125–5136. doi: 10.1007/s12035-017-0748-3. [DOI] [PubMed] [Google Scholar]
- 127.Reddy S.M., Kopetz S., Morris J., Parikh N., Qiao W., Overman M.J., Fogelman D., Shureiqi I., Jacobs C., Malik Z., Jimenez C.A., Wolff R.A., Abbruzzese J.L., Gallick G., Eng C. Phase II study of saracatinib (AZD0530) in patients with previously treated metastatic colorectal cancer. Invest. New Drugs. 2015;33(4):977–984. doi: 10.1007/s10637-015-0257-z. [DOI] [PubMed] [Google Scholar]
- 128.Molina J.R., Foster N.R., Reungwetwattana T., Nelson G.D., Grainger A.V., Steen P.D., Stella P.J., Marks R., Wright J., Adjei A.A. A phase II trial of the Src-kinase inhibitor saracatinib after four cycles of chemotherapy for patients with extensive stage small cell lung cancer: NCCTG trial N-0621. Lung Cancer. 2014;85(2):245–250. doi: 10.1016/j.lungcan.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gucalp A., Sparano J.A., Caravelli J., Santamauro J., Patil S., Abbruzzi A., Pellegrino C., Bromberg J., Dang C., Theodoulou M., Massague J., Norton L., Hudis C., Traina T.A. Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin. Breast Cancer. 2011;11(5):306–311. doi: 10.1016/j.clbc.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mackay H.J., Au H.J., McWhirter E., Alcindor T., Jarvi A., MacAlpine K., Wang L., Wright J.J., Oza A.M. A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: a trial of the PMH phase II consortium. Invest. New Drugs. 2012;30(3):1158–1163. doi: 10.1007/s10637-011-9650-4. [DOI] [PubMed] [Google Scholar]
- 131.Gubens M.A., Burns M., Perkins S.M., Pedro-Salcedo M.S., Althouse S.K., Loehrer P.J., Wakelee H.A. A phase II study of saracatinib (AZD0530), a Src inhibitor, administered orally daily to patients with advanced thymic malignancies. Lung Cancer. 2015;89(1):57–60. doi: 10.1016/j.lungcan.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 132.Nygaard H.B., van Dyck C.H., Strittmatter S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimers Res. Ther. 2014;6(1):8. doi: 10.1186/alzrt238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nygaard H.B., Wagner A.F., Bowen G.S., Good S.P., MacAvoy M.G., Strittmatter K.A., Kaufman A.C., Rosenberg B.J., Sekine-Konno T., Varma P., Chen K., Koleske A.J., Reiman E.M., Strittmatter S.M., van Dyck C.H. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res. Ther. 2015;7(1):35. doi: 10.1186/s13195-015-0119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.van Dyck C.H., Nygaard H.B., Chen K., Donohue M.C., Raman R., Rissman R.A., Brewer J.B., Koeppe R.A., Chow T.W., Rafii M.S., Gessert D., Choi J., Turner R.S., Kaye J.A., Gale S.A., Reiman E.M., Aisen P.S., Strittmatter S.M. Effect of AZD0530 on Cerebral Metabolic Decline in Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2019;76(10):1219–1229. doi: 10.1001/jamaneurol.2019.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mora J.S., Genge A., Chio A., Estol C.J., Chaverri D., Hernández M., Marín S., Mascias J., Rodriguez G.E., Povedano M., Paipa A., Dominguez R., Gamez J., Salvado M., Lunetta C., Ballario C., Riva N., Mandrioli J., Moussy A., Kinet J.P., Auclair C., Dubreuil P., Arnold V., Mansfield C.D., Hermine O. AB10015 STUDY GROUP. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph. Lateral Scler. Frontotemporal Degener. 2020;21(1-2):5–14. doi: 10.1080/21678421.2019.1632346. [DOI] [PubMed] [Google Scholar]
- 136.Vermersch P., Benrabah R., Schmidt N., Zéphir H., Clavelou P., Vongsouthi C., Dubreuil P., Moussy A., Hermine O. Masitinib treatment in patients with progressive multiple sclerosis: a randomized pilot study. BMC Neurol. 2012;12:36. doi: 10.1186/1471-2377-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Piette F., Belmin J., Vincent H., Schmidt N., Pariel S., Verny M., Marquis C., Mely J., Hugonot-Diener L., Kinet J.P., Dubreuil P., Moussy A., Hermine O. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: a randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 2011;3(2):16. doi: 10.1186/alzrt75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Porkka K., Koskenvesa P., Lundán T., Rimpiläinen J., Mustjoki S., Smykla R., Wild R., Luo R., Arnan M., Brethon B., Eccersley L., Hjorth-Hansen H., Höglund M., Klamova H., Knutsen H., Parikh S., Raffoux E., Gruber F., Brito-Babapulle F., Dombret H., Duarte R.F., Elonen E., Paquette R., Zwaan C.M., Lee F.Y. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008;112(4):1005–1012. doi: 10.1182/blood-2008-02-140665. [DOI] [PubMed] [Google Scholar]