

Abstract
Alzheimer’s disease (AD) is a complex neurodegenerative disease that leads to insidious deterioration of brain functions and is considered the sixth leading cause of death in the world. Alzheimer’s patients suffer from memory loss, cognitive deficit and behavioral changes; thus, they eventually follow a low-quality life. AD is considered as a multifactorial disorder involving different neuropathological mechanisms. Recent research has identified more than 20 pathological factors that are promoting disease progression. Three significant hypotheses are said to be the root cause of disease pathology, which include acetylcholine deficit, the formation of amyloid-beta senile plaques and tau protein hyperphosphorylation. Apart from these crucial factors, pathological factors such as apolipoprotein E (APOE), glycogen synthase kinase 3β, notch signaling pathway, Wnt signaling pathway, etc., are considered to play a role in the advancement of AD and therefore could be used as targets for drug discovery and development. As of today, there is no complete cure or effective disease altering therapies for AD. The current therapy is assuring only symptomatic relief from the disease, and progressive loss of efficacy for these symptomatic treatments warrants the discovery of newer drugs by exploring these novel drug targets. A comprehensive understanding of these therapeutic targets and their neuropathological role in AD is necessary to identify novel molecules for the treatment of AD rationally.
Keywords: Alzheimer’s disease, neurodegenerative pathways, novel drug targets, acetylcholinesterase, glycogen synthase kinase, notch signaling, apolipoprotein E
1. INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease with no cure. It is the most common cause of dementia. There are about 40 million people with AD; most of them are above 65 years of age [1]. People affected with AD show disorientation, cognitive impairment, memory loss, behavioral changes leading to poor-quality life. The pathophysiology behind this disease is complex and not fully figured out. Leading causes behind the disease are found to be the increased activity of acetylcholinesterase, which eventually leads to a decrease in the levels of acetylcholine in the brain, hyperphosphorylation of tau proteins, the formation of senile plaques of Aβ and inflammation of neuronal cells in the brain [2]. Besides this, there are some other factors, which modulate the progression of AD, and they include genetic factors, apoptosis, mitochondria-mediated, atrophy, etc. These factors were figured out recently, and the recent studies
are rolling on these factors, how they are involved in the advancement of the disease, and their role in developing effective drugs for AD. Currently, there are only four drugs in the market that could be used for the treatment of AD, and they are donepezil, rivastigmine, galantamine and memantine. One more drug, tacrine, which was an acetylcholinesterase inhibitor, was withdrawn from the market due to its toxicity. These drugs can provide only symptomatic relief from the disease [3, 4]. One of the widely accepted mechanisms for AD development is considered to be low levels of acetylcholine in the brain due to increased acetylcholinesterase activity, eventually leading to progressive cognitive decline. The use of acetylcholinesterase inhibitors has been proved to be beneficial in improving the disease symptoms and also could reduce amyloid protein plaques.
In this context, extensive research has been carried out in search of promising acetylcholinesterase inhibitors that could be used in the treatment of AD. Recent advances in molecular modelling software enable the medicinal chemist to design novel molecules based on the three-dimensional structure of acetylcholinesterase. In silico docking studies trace out the drug-receptor interaction, which could be synthesized and evaluated for anti-Alzheimer’s activity. Recent research in the field has made remarkable progress by identifying novel small molecules such as selenium-containing hydrochloride derivatives [5], triazole ring-containing compounds [6], chalcones [7], and sulfur-containing motifs [8] exhibiting remarkable acetylcholinesterase inhibitory activity. Fig. (1) shows significant changes in the brain during the progression of AD.
Fig. (1).
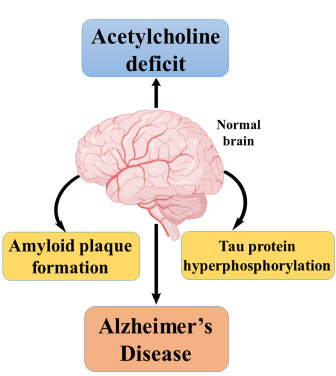
Major hypotheses behind AD.
2. TYPES OF ALZHEIMER’S DISEASE
There are two primary forms of AD i) early-onset AD, ii) late-onset AD [9]. They are subdivided as sporadic as well as familial type. In most of the cases, AD happens after the age of 65. However, rarely AD occurs at an early age and known as early-onset AD or autosomal dominant AD (ADAD). It is a rare type of AD and happens at the age of 30 to 50 years, and here comes the relationship between genetics and the occurrence of AD. Familial Alzheimer’s disease (FAD) is another type of AD, which inherited through generations. It is also associated with genes and their mutations. Mutations to genes encoding presenilins and Amyloid Precursor Proteins (PSEN1, PSEN2 and APP) are responsible for the familial as well as early onset of AD. Presenilins (PES) is a significant component of γ secretase enzyme. Any change in the structure of the enzyme will impair the normal functioning of the same, which will promote the development of AD. Sporadic AD is a prevalent type, which does not pass through generations [10]. In this review, we will be discussing the various factors and pathophysiological markers behind AD.
3. NEUROLOGICAL PATHWAYS INVOLVED IN AD
3.1. Acetylcholine Esterase Inhibition
Earliest, the pathological factor behind AD is found to be acetylcholinesterase (AChE). There are two classes of cholinesterases, acetylcholinesterase and butyrylcholinesterase, in which acetylcholinesterase is located in the brain and neuronal synapses. Acetylcholine (ACh) is a neurotransmitter and plays an essential role in the neuronal transmission of impulses as well as learning and memory [11, 12]. According to the cholinergic hypothesis, an Alzheimer’s brain shows a low level of acetylcholine, a neurotransmitter, happens when there is increased hydrolysis of ACh into acetate and choline in the presence of AChE [13]. According to the cholinergic hypothesis, AD brain shows low levels of ACh as well as it promotes the aggregation of Aβ, thereby the formation of senile plaques. Consequently, the inhibition of AChE could be considered as a promising approach for the treatment of AD [14]. Below Fig. (2) illustrates a pictorial representation of the relationship between AChE and AD [15].
Fig. (2).
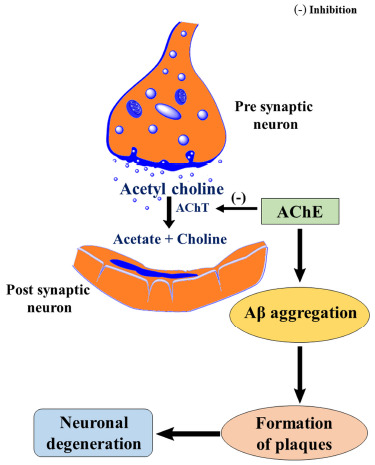
Cholinergic hypothesis of neurodegeneration.
3.2. Amyloid Beta (Aβ) and the Role of APP in Alzheimer
Amyloid-beta deposits typically seen in the brain as senile plaques due to dysregulation of Aβ lead to the formation of insoluble complexes, which get deposited in different parts of the brain [16]. The deposition of Aβ is a significant risk factor in the inception and development of AD [17]. In healthy individuals, there will be the production of Aβ at a healthy level. However, in a diseased condition, there will be an overproduction of Aβ. It is usually produced from amyloid precursor proteins (APP) [18]. APP is a transmembrane protein, which is present in the brain at high levels and cleaved by different enzymes such as α, β, and γ secretases. In those enzymes, cleavage through β and γ secretases will lead to the formation of Aβ fragments [19]. Aβ formed will oligomerize and form plaques or complex structures. In a healthy individual, there will be a balance between the production and cleavage of Aβ in the brain. Impairment of this homeostasis will lead to the deposition of Aβ and plaques; ultimately, the dysfunction of the healthy brain [20, 21]. Fig. (3) shows the connection of APP with the formation of amyloid plaques [22].
Fig. (3).
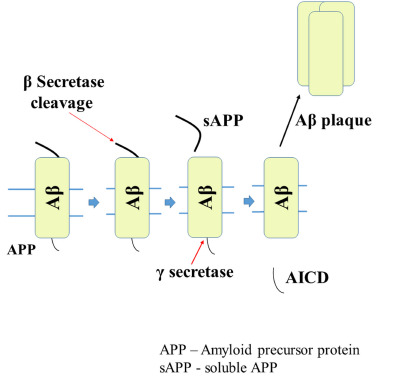
Amyloidogenic pathway of APP.
3.3. Tau Proteins
The neuronal cytoskeleton has several components in which microtubules have a vital role in the formation of normal morphology of neurons, and they provide structural support for neurons [23]. Microtubules are generally made up of tubulin and tau proteins and are responsible for the production of microtubules from tubulin. Tau proteins belong to the category of Microtubular Associated Proteins (MAPs) [24].
Fig. (4) represents the role of tau protein and microtubules in disease progression. Usually, tau proteins bind to tubulin and are regulated by phosphorylation through kinases and phosphatases. But in pathological conditions like AD, tau proteins are abnormally phosphorylated, and lead to a decrease in binding with tubulin and thereby microtubule disorganization occurs, resulting in morphological changes in neuronal structure. Generally, the tau proteins self-polymerize and aggregate to form neurofibrillary tangles (NFTs) [25, 26].
Fig. (4).
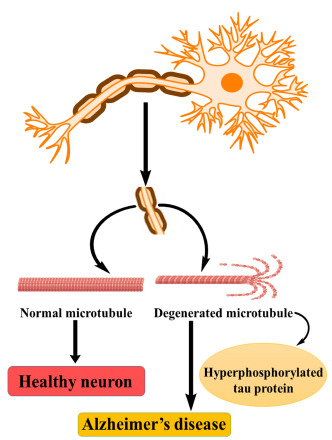
Tau proteins and AD.
3.4. N-methyl-D-aspartate (NMDA)
Glutamate is the major excitatory neurotransmitter present in the mammalian brain, involved in the excitatory postsynaptic transmission of impulses via several receptors. In those, NMDA receptors are abundantly seen throughout the CNS [27]. NMDA receptors play a significant role in synaptic plasticity and cellular processes that form the basis for learning and memory. Therefore, the blockade of NMDA included in the potential disease-modifying treatment for AD. Various studies also point out the role of the N-methyl-D-aspartate receptor (NMDAR) in AD [28]. Fig. (5) demonstrates NMDAR and neuronal death are connected and lead to AD.
Fig. (5).
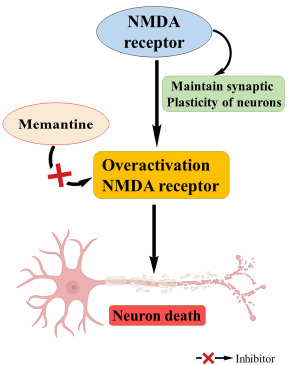
Role of NMDA in AD.
Normal essential functions of the central nervous system like neuronal development, synaptic plasticity, learning, memory formation etc., are mediated by glutamatergic neurotransmission through NMDAR, which is an ionotropic glutamate receptor. It is found that overactivation of NMDAR leads to pathological excitotoxicity in neurological disorders such as AD [29].
Memantine, the only FDA approved NMDA receptor antagonist is active only at high concentrations of glutamate associated excitotoxicity of NMDARs, and they are ineffective at lower levels of the same. At standard conditions, channels for NMDA are open for several milliseconds, and memantine cannot act in this condition. However, during prolonged activation of NMDAR, it will work and accumulate in the channels and blocks the pathway [30].
3.5. Apolipoprotein (ApoE)
An arginine-rich protein seen in human plasma is associated with cholesteryl rich VLDLs. CNS ApoE is synthesized locally in the brain by astrocytes, microglial cells and neurons, and acts as a major transporter of cholesterol [31]. It is discoidal in shape and is synthesized in pathological as well as in physiological conditions. In Fig. (6), the relationship between ApoE and the formation of Aβ is depicted [32]. It is involved mainly in the regulation of the metabolism of lipoproteins [33]. ApoE encoded by a gene positioned on human chromosome 19 and variations in the gene sequence result in three alleles, namely ε2, ε3, and ε4. AD patients bear ApoE4 allele [34]. ApoE modulates morphology as well as homeostasis of the brain and thus holds a major role in ageing. ApoE may also regulate intracellular calcium levels [35].
Fig. (6).
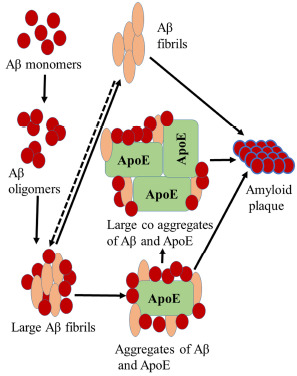
ApoE and Aβ plaque formation.
3.6. Wnt Signaling Pathway
In AD brains, neurons undergo rapid apoptotic death. Wnt signaling pathway promotes the proliferation of progenitor cells typically during brain development [36]. β-amyloid proteins (βAP) induce changes to the Wnt signaling pathway and cause downregulation of the same, and this may lead to the formation of neurofibrillary tangles (NFT). As amyloid plaques and NFT are the histopathological hallmarks of AD, the formation of NFT through impaired Wnt signaling pathway may promote the development of AD [37]. Below is the comparison between the roles of Wnt signaling in both regular and Alzheimer’s condition (Fig. 7).
Fig. (7).
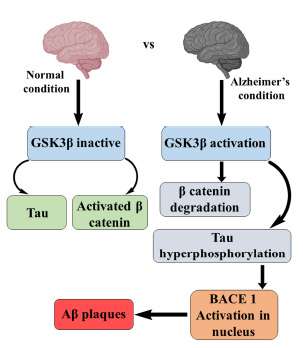
Relation of Wnt signaling pathway with AD.
Wnt signaling pathway includes a variety of secreted glycoproteins like presenilins that involve in the proliferation and differentiation of neurons. Presenilin complexed with β- catenin, and GSK3β leads to the deposition of β-amyloid (βA). They act as transducers in the Wnt signaling pathway. β-amyloid protein (βAP) induces the formation of NFT through the hyperphosphorylation of tau proteins with the help of enzymes like CDK5 and GSK3β. GSK3β acts as a vital component of the Wnt signaling pathway [38].
Besides, increased expression of p53 is seen in neurons impaired with βAP, and this may trigger apoptotic death of neurons and accordingly increase the expression of p53 in the BAX gene. Moreover, the Wnt pathway is negatively modulated by dickkopf-1 (DKK-1). The gene that encodes DKK-1 also targets p53. DKK-1 induces the downregulation of the Wnt signaling pathway [39].
3.7. Sirtuins and AD
Sirtuins are NAD+ dependent enzymes, having an essential role in age-related disorders. They come under the category of histone deacetylase (HDAC). Sirtuin 1 is the regulator of proteins and genes involved in different biological activities including antioxidant responses and anti-apoptotic responses, DNA repair and also it acts as the leading agent in maintaining synaptic plasticity and learning and memory [6]. The relation between sirtuins and the progression of AD is demonstrated by Fig. (8).
Fig. (8).
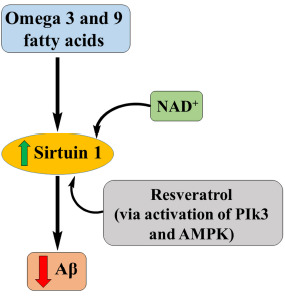
Role of sirtuins in AD.
Calorie restriction (CR) is a regimen that increases the life span of animals in different ways, including reducing the usage of carbohydrates, reducing the production of ROS and thereby reducing the cell damage. CR needs some regulatory proteins such as sirtuins, a group of antiaging enzymes. Also, they are involved in different disease pathology, including diabetes, cancer, and neurodegenerative disorders like AD, Parkinson’s and Huntington disease [40].
By activating sirtuins, we can attain either healthspan extension or a lifespan extension, which is highly associated with AD. Activated levels of sirtuins will reduce the formation of oligomerized Aβ. Activation of sirtuins can be done by two methods, either by using sirtuin-activating compounds such as resveratrol or by providing cellular NAD+ [41]. The most effective and general way involves the usage of direct NAD+ precursors like nicotinamide mononucleotide (NMN) or nicotinamide ribose to supply cellular NAD+. Resveratrol studies show better clearance of Aβ as well as the reduced formation of plaques. Resveratrol acts either directly or indirectly by activating AMPK and PIK3.
DNA impairment may cause the diminution of NAD+. Sirtuins and other DNA repairing enzymes use NAD+ as cofactor so that there will be depletion of NAD+ and loss of sirtuin function and leads to DNA damage [42].
3.8. mTOR
Mammalian target of rapamycin (mTOR) is a serine/threonine-protein kinase involved in the major biological processes such as protein translation and cell growth in response to different signals like growth factors, nutrient state and energy level [43].
At the cellular level, it is essential to keep the homeostasis in the body. Diseases like cancer and diabetes are characterized by impairment of mTOR signaling. It is also associated with neuronal activity, synaptic plasticity, and axon regeneration that is vital in the development of AD [44].
The mammalian target of rapamycin (mTOR) is a conserved protein kinase that plays a significant role in governing a balance between protein synthesis and degradation. It controls protein translation through phosphorylation reaction. It is directly linked to learning and memory [45].
mTOR is associated with different proteins and forms two complexes, mTORC1 and mTORC2. mTOR integrates these signals and controls ribosome biogenesis, transcription, translation and macroautophagy. Reducing mTOR signaling may have positive effects on age-related disorders, including neurodegenerative diseases. The level of mTOR increases the level of Aβ oligomers.
Complete blockage, as well as hyperactive mTOR signaling, worsens AD and results in impaired synaptic plasticity and learning and memory. Reduction of hyperactivity with specific agents like rapamycin may lead to improved synaptic plasticity, learning, and memory [46].
From the studies, it is evident that there is an optimum level of mTOR signaling, which is ideal for learning and memory. Any alterations in this will lead to a detrimental effect on learning and memory.
Hyperactive mTOR signaling may lead to the progression of tau pathology and thereby accumulation of the same through an increased translation of tau protein (Fig. 9). Aβ or tau accumulation produces mTOR hyperactivity, which may arbitrate neuronal death and Aβ-induced cognitive deficit [47].
Fig. (9).
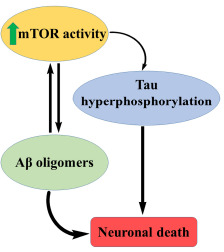
mTOR in AD.
3.9. Caspases
Apoptosis shows a prime role in the proper functioning and appearance of the brain. However, inapt activation of this pathway in an ageing brain leads to the degeneration of neurons. In AD, caspases produce neurodegeneration as well as other underlying pathologies of the disease [48]. Fig. (10) exhibits the involvement of caspases in the progression of AD.
Fig. (10).
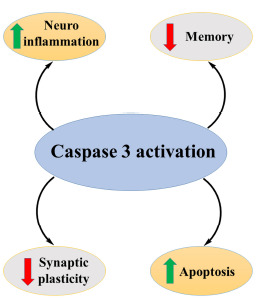
Activity of caspases in AD.
Caspases are protease enzymes that facilitate programmed cell death or apoptosis. Activation of the caspase pathway facilitates a cascade of events that will promote the demolition of cell components and, ultimately, cell death. Dysregulation of caspases underlies diseases like cancer and inflammatory diseases, including AD [49].
Initiator caspases (caspases 8 and 9) eventually activate executioner caspases (caspases 3, 6 and 7) followed by coordination of their activities which lead to demolition of different proteins of cell components and finally cell death through DNA fragmentation. Most of the apoptotic programmes are executed via an intrinsic pathway or extrinsic pathway [50].
In AD, there is NFT formation and neuronal cell death. Amyloid precursor proteins (APP) are cleaved by the action of β and γ secretases to form Aβ, and this Aβ oligomerizes and leads to the production of reactive oxygen species and oxidative stress and finally cell death. The formation of Aβ may lead to activation of caspases, which will lead to the cleavage of different cellular proteins, mainly tau and result in the breakdown of the cytoskeleton and hyperphosphorylation of tau. As tau proteins are essential in the structural stability of neurons, hyperphosphorylation of tau leads to additional instability of the cytoskeleton. Due to hyperphosphorylation, there is an interruption of the microtubular network, which may lead to damaged axonal transport and finally cell death of neurons. Cleavage of APP produces further production of Aβ [51, 52].
3.10. Ubiquitin and Autophagy
Proteins in the body may undergo mutations and misfold into another product. This mutation may lead to abnormal activities. This is probably prevented by controlling the system through different mechanisms such as ubiquitin, biological chaperons and autophagy [53]. This controlling mechanism will prevent the accretion of toxic levels of corrupted proteins by breaking them down into polypeptide chains. If this system is disturbed, it may accumulate in different parts of the body and may cause the development of neurodegenerative diseases like AD and Parkinson’s disease [54].
In AD, memory loss and other behavioral changes are caused by the synthesis of defective proteins, i.e. APP is cleaved into Aβ, and later it oligomerizes and accumulates in the brain to cause neuronal death [55]. Oligomerized Aβ promotes the hyperphosphorylation of tau proteins. Hyperphosphorylation of tau produces NFT, and this impairs the microtubular skeleton of the axon and leads to the damaged axonal transmission of impulses [56].
Degradation of intracellular proteins is mediated by ubiquitin proteasomal system (UPS), and the extracellular proteins by lysosomes. In AD brain, there occurs impairment of UPS, which leads to accumulation of oligomerized Aβ and tau, eventually causing synaptic dysfunction in different neurological disorders like AD, Huntington’s disease and Parkinson’s disease [57].
Autophagy or self-eating is the process through which the lysosome removes cell debris for total degradation of damaged cell components. There are 3 types of autophagy, micro, macro and chaperone-mediated autophagy in which macroautophagy is the most prevalent one [58]. Autophagy is associated with an array of normal physiological processes as well as pathological conditions, such as development, cell death, ageing, antigen presentation, bacterial degradation, and tumor suppression [59]. Fig. (11) shows that the impairment of autophagy is associated with AD.
Fig. (11).
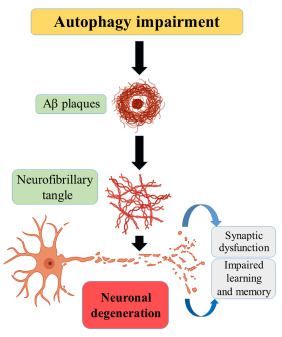
Role of autophagy in Alzheimer’s.
Endosomal and lysosomal pathways are involved in the regulation of levels of APP. Abnormalities in this pathway may lead to the accumulation and deposition of APP [60]. Early endosomes cleave APP into Aβ from healthy cells. An upregulation of the endosomal lysosomal path results in an increased number of lysosomes so that an increased formation of Aβ occurs. As the levels of Aβ increase, oligomerization of Aβ takes place and finally, the accumulation and formation of senile plaques [61, 62].
3.11. Polyunsaturated Fatty Acids (PUFA)
Polyunsaturated fatty acids (PUFA) have a crucial part in neuronal function, and the modification of these compounds in the brain could influence neurodegenerative diseases like AD [63]. Arachidonic acid is the second most PUFA in the brain, and it is converted into different components, which facilitate inflammation and synaptic transmission.
Studies show that various PUFA, ω-3 fatty acids like docosahexaenoic acid (DHA), eicosatetraenoic acid (EPA), and α-linolenic acid (ALA), prevent oligomerization and self-aggregation of Aβ [64]. They interact with aggregation-prone phe19- ala21 and inhibit aggregation. Nevertheless, other ω-6 PUFAs like arachidonic acid (ARA) and linoleic acid (LNA) act as proinflammatory agents and are involved in the pathology of AD. Also, ARA can boost up the production of Aβ peptides [65]. ω-9 fatty acids such as monounsaturated fatty acids (MUFA) and oleic acid (OA) may reduce the levels of Aβ42.
The interconnection between PUFA and AD is shown in Fig. (12). However, in recent findings, it is proven that all the PUFAs, including ARA and LNA, can prevent the aggregation of Aβ and so that they reduce the progression of AD [66].
Fig. (12).
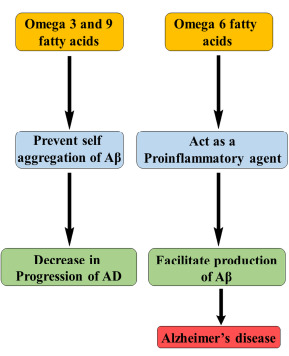
PUFA and AD.
3.12. Calcium Signaling Pathway
Calcium ion (Ca2+) is the significant component that modifies the function and structure of nerve cells in the brain, mainly in the neuronal growth, synaptic and cognitive function. From various studies, it is evident that Ca2+ homeostasis has a vital role in the progression of AD and its associated conditions, such as memory loss and cognitive impairment [67]. Whenever there is an upregulation of calcium release from the endoplasmic reticulum (ER), through ryanodine receptors (RYR), the activity of β and γ secretase increases and thereby oligomerization of Aβ leads to a progression of AD.
Fig. (13) shows the calcium homeostasis in the formation of amyloid plaques and changes in synaptic plasticity. Calcium is essential for the synaptic transmission of impulses as well as synaptic plasticity, which is responsible for learning and memory [68]. The high concentration of Ca2+ may lead to long-term potentiation, and small elevation may cause durable depression. In another way, Ca2+ can either form or remove memories. Any changes in the calcium homeostasis lead to the upsurge of apoptosis, leading to neuronal death and, thereby, dementia [69].
Fig. (13).
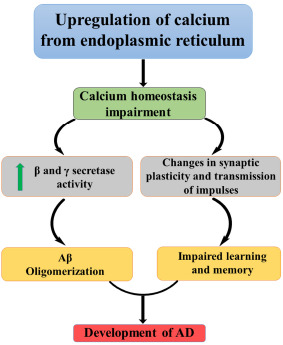
Calcium in AD.
3.13. Ceramide
Ceramide is a principal component of sphingolipids and plays an essential role in vital cellular progressions like growth, cell cycle arrest, survival, and apoptosis. It is composed of a sphingosine lipid and fatty acid, which is connected by an amide bond [70]. Ceramides are suggested to be the crucial players in the pathogenesis of AD. Raised basal serum levels of ceramide species are linked with a higher risk for developing AD [71].
Recently, histopathological studies showed that AD brains have a higher number of ceramides when compared to healthy control brains. Free radicals generated by the ceramide are the principal source of neurodegeneration. With an increased level of ceramides, there may be upregulation of production of reactive oxygen species (ROS) which leads to neuronal damage via Aβ oligomerization and tau protein hyperphosphorylation. The exact role of ceramides is still uncertain [72, 73]. Safeguarding of ceramide homeostasis is essential for normal brain functioning. Studies show that dysregulation of ceramide metabolism is associated with quite a lot of pathological characteristics of AD such as Aβ oligomerization, tau protein hyperphosphorylation, generation of ROS, impairment of different signaling pathways, apoptosis and finally neuronal death [74, 75]. Activities of ceramides in Alzheimer’s brain are portrayed in Fig. (14).
Fig. (14).
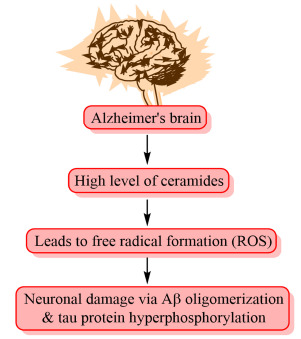
Activity of ceramides in Alzheimer’s brain.
3.14. Notch Signaling Pathway
Presenilin-1 (PES-1) is one of the four core proteins in the γ secretase complex, which is considered crucial in the generation of amyloid-beta (Aβ) from amyloid precursor protein (APP) by γ secretase. Mutation to PES-1 is a significant source of early onset of FAD [76].
A notch signaling pathway is involved in the differentiation and development of various systems, including neuronal development. It is involved in maintaining synaptic plasticity, long-term learning and memory and neurogenesis.
The notch is a transmembrane protein and substrate of γ secretase and or PES-1 considered necessary in the progression of AD. Notch proteins interact with amyloid precursor protein (APP) as well as presenilins. Presenilins facilitate proteolysis of APP and help notch to release the Notch intracellular domain (NICD), which is essential for the notch-signaling pathway. NICD shifts into the nucleus, and this can modify transcription via binding with CSL, a group of DNA binding proteins [77]. Fig. (15) illustrates the role of notch signaling in AD.
Fig. (15).
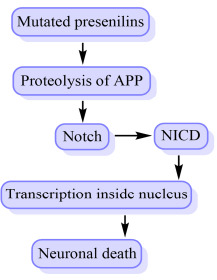
Notch signaling in the progression of AD.
Studies have shown that in a notch mutant mouse, when a loss of notch signaling function occurs, it consequently results in a long-term memory deficit. The permanent gap in notch signaling may show learning and memory impairment. From this, it is evident that notch signaling linked transcription is essential for spatial learning [78]. Notch pathway is also involved in the neuronal death, and γ secretase-facilitated notch signaling deteriorates brain damage. Because of this brain damage, ischemia and stroke, there will be an advancement in AD and vascular dementia [79].
3.15. Neurotrophins in AD
Neurotrophins are a family of proteins, which include nerve growth factor (NGF), Neurotrophin 3 (NT-3), brain-derived neurotrophic factor (BDNF) and Neurotrophin 4 (NT 4). They all bind to specific tyrosine kinase receptors (Trks), NGF to Trk A, NT 4 and BDNF to Trk B and NT 3 binds to Trk C [80, 81].
Pathophysiology of AD primarily focuses on BDNF and NGF. Trk A in forebrain responds to NGF, and it maintains synaptic communication with the hippocampus and cortex neurons [82]. BDNF present in the hippocampus is responsible for the synaptic plasticity of neurons and memory establishment. BDNF will bind to Trk B and is the critical component of memory formation. Therefore, any changes in the expression of NGF or BDNF or levels of subsequent receptors may result in memory deficit (Fig. 16) [83].
Fig. (16).
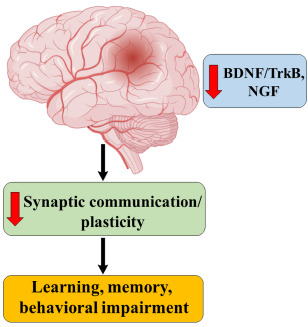
Neurotrophins in AD.
3.16. BACE-1 (β APP Cleaving Enzyme-1 or β Secretase)
BACE-1 is the enzyme involved in the cleavage of APP, which is the significant step in the formation of Aβ [84, 85]. It is a transmembrane protease enzyme, which cleaves APP at its β site. After cleavage,the production of amyloid-beta takes place and oligomerization of the same produces plaques and leads to neuronal damage and AD [86]. The inhibition of BACE 1 shows a reduced level of Aβ and effectively a better synaptic activity [87, 88], as it is related to the formation of amyloid-beta, well described in Fig. (3).
3.17. JNK and AD
c-Jun N terminal kinase (JNK) is vital in the maintenance of neuronal plasticity, cell degeneration, and regeneration. It is activated after the induction of stress factors like oxidative stress, cytokines as well as Aβ. Activated JNK is accountable for the regulation of apoptosis so that it is critical in neurodegenerative diseases like AD [89]. JNK is associated with neuronal death, amyloid-beta production and production of neurofibrillary tangles. JNK 3 is the most prominent one which responds to stress stimuli and is more expressed in the AD brain [90]. Fig. 17 highlights the relation between JNK and AD.
Fig. (17).
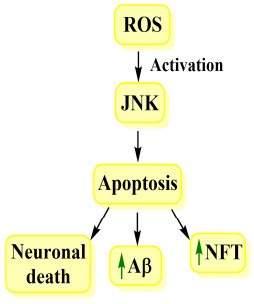
JNK and AD.
From studies, it is evident that there is an overexpression of phosphorylated JNK in AD brains along with Aβ plaques. Also, it promotes the formation of NFT and hyperphosphorylated tau [91, 92].
3.18. Glycogen Synthase Kinase 3β (GSK 3β)
Glycogen synthase kinase is a serine-threonine kinase involved in many vital activities of the body. There are two isoforms of GSK3, namely, GSK3α and GSK3β in which GSK3β is abundantly seen and the level of the same increases with age. From various studies, GSK3β is hyperactive in AD patients. Insulin and the Wnt signaling pathway modulate the activity of GSK3β [93].
Insulin signaling facilitates PI3 kinase activation, which will eventually lead to the phosphorylation of GSK3β and GSK3α serine at positions 9 and 21 by AKT. GSK3 could also be regulated by auto phosphorylating tyrosine residue at positions 279/216, and it augments its activity [94]. Wnt signaling modulates GSK3 by dislocating different binding partners that are bound to GSK3 to stimulate action and prevent phosphorylation and degradation of β catenin.
Deregulation of GSK3 leads to many pathological conditions, including neurological disorders like AD. It is involved in the hyperphosphorylation of tau proteins, inflammation and production of senile plaques by regulating APP and BACE1. GSK3 decreases the production of acetylcholine, leading to the cholinergic deficit and is involved in apoptosis by which there will be neuronal loss [95].
Studies depict the relationship between GSK3 and presenilin 1 (PS-1), and it is demonstrated in Fig. (18). Hyperactivation of GSK3 leads to a functional change in PS1. PS 1 is involved in the pathology of FAD. PS 1 is associated with the formation of a complex of β catenin and serine residues of GSK3. When there is overactivation of GSK3, it downregulates the complex and leads to changes in synaptic and neural functions and hence AD.
Fig. (18).
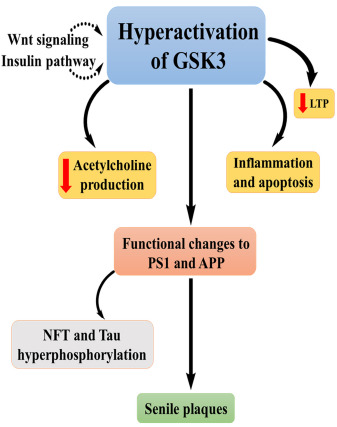
Role of GSK3β in the progression of AD.
Long-term potentiation (LTP) is associated with Wnt signaling pathway, the suppression of which declines LTP. Wnt signaling and insulin negatively regulate GSK3 and so that hyperactivation of GSK3 leads to impairment of LTP. Novel drug development, which targets GSK3, is considered to be a potential drug for AD, mood disorders, and diabetes [96]. In a recent study of a chemical moiety, benzisoxazoles were found to be a promising target as a GSK3 inhibitor [97].
4. FUTURE PERSPECTIVES
AD is the most common type of neurodegenerative disease with no cure. Accumulation of toxic proteins, including alpha-synuclein (Lewy bodies), beta-amyloid plaques, Tau tangles, and TDP-43 is considered as pathophysiology involved in neurodegeneration and these are often used as probable molecular biomarkers to predict disease progression. Significantly altered neurotransmission of dopamine and acetylcholine can also lead to neuronal degeneration.
Currently, available drugs for the treatment of AD belong to the category of either acetylcholinesterase inhibitors or NMDA glutamate receptor antagonist. These drugs are only useful during the first year of treatment for patients with mild to moderate AD and provide some degree of improvement in cognitive functions. The efficacy of these drugs declines wholly and subsequently vanishes after 2 to 3 years of treatment. The progressive loss of efficacy for these symptomatic treatments warrants the necessity of identifying newer targets for novel small molecule drug interventions.
The major difficulty in treating AD is due to the activation of several harmful processes in the brain. To effectively prevent the progress of these destructive processes in the brain, it is necessary to address them simultaneously. In case of such complex disease pathologies, single-target drugs are inadequate to achieve the desired therapeutic effect. Therefore newer multi-target drugs (MTDs) should be designed to simultaneously target several aspects of disease pathology leading to neuronal damage rather than just the symptoms currently addressed by single-target drugs. Further, it is well proven that MTDs have a better safety profile when compared to single-targeted ones.
CONCLUSION
AD is the sixth leading cause of death worldwide. It is a complex neurological disorder with learning and memory impairment and other behavioral changes. There is no such cure for the disease, and only four drugs are available in the market for symptomatic relief of this illness. Major hypotheses include the deficit of acetylcholine, the formation of Aβ plaques, neurofibrillary tangle formation (NFT) and hyperphosphorylation of tau proteins. Other than these hypotheses, there underlie many other pathophysiologies that modify the causes of AD.
In various studies, it is evident that there are more than 20 pathophysiological factors involved in altering this disease progression and development. Therefore, inhibition or activation of these factors could modify the advancement of AD, and those agents used for the same could be another choice of drug for the condition. These factors are all interconnected in a way or the other and imbalance of any of these may facilitate disease progression. Every one of them ends up to the same cardinal hypotheses such as the formation of senile Aβ plaques, tau protein hyperphosphorylation or formation of neurofibrillary tangles (NFTs).
In this review, we discuss the main pathways that underlie neurodegeneration in AD, and this could provide a thorough understanding of different neurodegenerative mechanisms that help in the design and development of novel target-based small molecules for the treatment of AD.
ACKNOWLEDGEMENTS
The author Anu Kunnath Ramachandran is thankful to AICTE, New Delhi for financial assistance in the form of National Doctoral Fellowship (NDF) and another author Subham Das to Manipal Academy of Higher Education for providing Dr.T.M.A. Pai Fellowship. The authors also acknowledge with thanks, to the Manipal College of Pharmaceutical Sciences, for providing facilities for this work.
LIST OF ABBREVIATIONS
- AD
Alzheimer’s Disease
- ADAD
Autosomal Dominant Alzheimer’s Disease
- AChE
Acetylcholinesterase
- Aβ
Amyloid-beta
- APP
Amyloid Precursor Proteins
- ApoE
Apolipoprotein
- AMPK
Adenosine Monophosphate-activated Protein Kinase
- ALA
α-linolenic Acid
- ARA
Arachidonic Acid
- AKT
Protein Kinase B
- βAP
β-amyloid Proteins
- BDNF
Brain-derived Neurotrophic Factor
- BACE-1
β APP Cleaving Enzyme 1
- CNS
Central Nervous System
- CR
Calorie Restriction
- Ca+
Calcium ion
- DKK-1
Dickkopf-1
- DKK-1
Dickkopf-1
- DNA
Deoxyribonucleic Acid
- DHA
Docosahexaenoic acid
- EPA
Eicosatetraenoic Acid
- ER
Endoplasmic Reticulum
- FAD
Familial Alzheimer’s Disease
- GSK 3β
Glycogen Synthase Kinase 3β
- HDAC
Histone D Acetylase
- JNK
c-Jun N terminal Kinase
- LTP
Long-term Potentiation
- LNA
Linoleic Acid
- MAPs
Microtubular Associated Proteins
- mTOR
Mammalian Target of Rapamycin
- MUFA
Monounsaturated Fatty Acids
- NFTs
Neurofibrillary Tangles
- NMDA
N Methyl D Aspartate
- NMDAR
N-methyl-D-aspartate Receptor
- NAD+
Nicotinamide Dinucleotide
- NMN
Nicotinamide Mononucleotide
- NICD
Notch Intracellular Domain
- NGF
Nerve Growth Factor
- NT-3
Neurotrophin 3
- NT-4
Neurotrophin 4
- OA
Oleic Acid
- PES
Presenilins
- PIK3
Phosphoinositide 3 Kinase
- PUFA
Polyunsaturated Fatty Acids
- PI3
Phosphoinositide 3
- PS-1
Presenilin 1
- RYR
Ryanodine Receptors
- ROS
Reactive Oxygen Species
- Trks
Tyrosine kinase receptors
- UPS
Ubiquitin Proteasomal System
- VLDL
Very Low-density Lipoprotein
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors have no conflicts of interest, financial or otherwise.
REFERENCES
- 1.Querfurth H.W., LaFerla F.M. Alzheimer’s disease. N. Engl. J. Med. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.Bornstein J.J., Eckroat T.J., Houghton J.L., Jones C.K., Green K.D., Garneau-Tsodikova S. Tacrine-Mefenamic Acid Hybrids for Inhibition of Acetylcholinesterase. MedChemComm. 2011;2(5):406–412. doi: 10.1039/c0md00256a. [DOI] [Google Scholar]
- 3.Zhu J., Wang L.N., Cai R., Geng S.Q., Dong Y.F., Liu Y.M. Design, synthesis, evaluation and molecular modeling study of 4-N-phenylaminoquinolines for Alzheimer disease treatment. Bioorg. Med. Chem. Lett. 2019;29(11):1325–1329. doi: 10.1016/j.bmcl.2019.03.050. [DOI] [PubMed] [Google Scholar]
- 4.Fedeli C., Filadi R., Rossi A., Mammucari C., Pizzo P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy. 2019;15(12):2044–2062. doi: 10.1080/15548627.2019.1596489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang W.Y., Ravindar L., Rakesh K.P., Manukumar H.M., Shantharam C.S., Alharbi N.S., Qin H.L. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019;173:117–153. doi: 10.1016/j.ejmech.2019.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu M., Peng Y., Zhu L., Wang S., Ji J., Rakesh K.P. Triazole derivatives as inhibitors of Alzheimer’s disease: Current developments and structure-activity relationships. Eur. J. Med. Chem. 2019;180:656–672. doi: 10.1016/j.ejmech.2019.07.059. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X., Rakesh K.P., Bukhari S.N.A., Balakrishna M., Manukumar H.M., Qin H.L. Multi-targetable chalcone analogs to treat deadly Alzheimer’s disease: Current view and upcoming advice. Bioorg. Chem. 2018;80:86–93. doi: 10.1016/j.bioorg.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Zhao C., Rakesh K.P., Ravidar L., Fang W.Y., Qin H.L. Pharmaceutical and medicinal significance of sulfur (SVI)-Containing motifs for drug discovery: A critical review. Eur. J. Med. Chem. 2019;162:679–734. doi: 10.1016/j.ejmech.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itzcovich T., Chrem-Méndez P., Vázquez S., Barbieri-Kennedy M., Niikado M., Martinetto H., Allegri R., Sevlever G., Surace E.I. A novel mutation in PSEN1 (p.T119I) in an Argentine family with early- and late-onset Alzheimer’s disease. Neurobiol. Aging. 2020;85:155.e9–155.e12. doi: 10.1016/j.neurobiolaging.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Wong T.H., Seelaar H., Melhem S., Rozemuller A.J.M., van Swieten J.C. Genetic screening in early-onset Alzheimer’s disease identified three novel presenilin mutations. Neurobiol. Aging. 2020;86:201.e9–201.e14. doi: 10.1016/j.neurobiolaging.2019.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S., Jana K. Revealing the Mechanistic Pathway of Cholinergic Inhibition of Alzheimer ? s Disease by Donepezil : A Metadynamics Simulation Study. 2019:13578–13589. doi: 10.1039/c9cp02613d. [DOI] [PubMed] [Google Scholar]
- 12.Mehta M., Adem A., Sabbagh M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimers Dis. 2012;2012:728983. doi: 10.1155/2012/728983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGleenon B.M., Dynan K.B., Passmore A.P. Acetylcholinesterase inhibitors in Alzheimer’s disease. Br. J. Clin. Pharmacol. 1999;48(4):471–480. doi: 10.1046/j.1365-2125.1999.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allain H., Bentué-Ferrer D., Tribut O., Gauthier S., Michel B.F., Drieu-La Rochelle C. Alzheimer’s disease: the pharmacological pathway. Fundam. Clin. Pharmacol. 2003;17(4):419–428. doi: 10.1046/j.1472-8206.2003.00153.x. [DOI] [PubMed] [Google Scholar]
- 15.Dulawa S.C., Janowsky D.S. Cholinergic regulation of mood: from basic and clinical studies to emerging therapeutics. Mol. Psychiatry. 2019;24(5):694–709. doi: 10.1038/s41380-018-0219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy M.P., LeVine H., III Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010;19(1):311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadigh-Eteghad S., Sabermarouf B., Majdi A., Talebi M., Farhoudi M., Mahmoudi J. Amyloid-beta: a crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015;24(1):1–10. doi: 10.1159/000369101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen G.F., Xu T.H., Yan Y., Zhou Y.R., Jiang Y., Melcher K., Xu H.E. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017;38(9):1205–1235. doi: 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng H., Koo E.H. Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 2011;6(1):27. doi: 10.1186/1750-1326-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Brien R.J., Wong P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yun S.M., Cho S.J., Jo C., Park M.H., Han C., Koh Y.H. Elevation of plasma soluble amyloid precursor protein beta in Alzheimer’s disease. Arch. Gerontol. Geriatr. 2020;87:103995. doi: 10.1016/j.archger.2019.103995. [DOI] [PubMed] [Google Scholar]
- 22.Kolarova M., García-Sierra F., Bartos A., Ricny J., Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012;2012:731526. doi: 10.1155/2012/731526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bachurin S.O., Bovina E.V., Ustyugov A.A. Drugs in Clinical Trials for Alzheimer’s Disease: The Major Trends. Med. Res. Rev. 2017;37(5):1186–1225. doi: 10.1002/med.21434. [DOI] [PubMed] [Google Scholar]
- 24.Cohen T.J., Guo J.L., Hurtado D.E., Kwong L.K., Mills I.P., Trojanowski J.Q., Lee V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011;2(1):252–259. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andorfer C., Acker C.M., Kress Y., Hof P.R., Duff K., Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 2005;25(22):5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iqbal K., Liu F., Gong C.X., Alonso Adel.C., Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118(1):53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R., Reddy P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017;57(4):1041–1048. doi: 10.3233/JAD-160763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olivares D., Deshpande V.K., Shi Y., Lahiri D.K., Greig N.H., Rogers J.T., Huang X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr. Alzheimer Res. 2012;9(6):746–758. doi: 10.2174/156720512801322564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau A., Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460(2):525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- 30.Ray B., Banerjee P.K., Greig N.H., Lahiri D.K. Memantine treatment decreases levels of secreted Alzheimer’s amyloid precursor protein (APP) and amyloid beta (A beta) peptide in the human neuroblastoma cells. Neurosci. Lett. 2010;470(1):1–5. doi: 10.1016/j.neulet.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandon J.A., Farmer B.C., Williams H.C., Johnson L.A. APOE and Alzheimer’s Disease: Neuroimaging of Metabolic and Cerebrovascular Dysfunction. Front. Aging Neurosci. 2018;10:180. doi: 10.3389/fnagi.2018.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanson A.J., Bayer J.L., Baker L.D., Cholerton B., VanFossen B., Trittschuh E., Rissman R.A., Donohue M.C., Moghadam S.H., Plymate S.R., Craft S. Differential Effects of Meal Challenges on Cognition, Metabolism, and Biomarkers for Apolipoprotein E ɛ4 Carriers and Adults with Mild Cognitive Impairment. J. Alzheimers Dis. 2015;48(1):205–218. doi: 10.3233/JAD-150273. [DOI] [PubMed] [Google Scholar]
- 33.Safieh M., Korczyn A.D., Michaelson D.M. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019;17(1):64. doi: 10.1186/s12916-019-1299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uddin M.S., Kabir M.T., Al Mamun A., Abdel-Daim M.M., Barreto G.E., Ashraf G.M. APOE and alzheimer’s disease: evidence mounts that targeting APOE4 may combat alzheimer’s pathogenesis. Mol. Neurobiol. 2019;56(4):2450–2465. doi: 10.1007/s12035-018-1237-z. [DOI] [PubMed] [Google Scholar]
- 35.de Chaves E.P., Narayanaswami V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008;3(5):505–530. doi: 10.2217/17460875.3.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Ferrari G.V., Inestrosa N.C. Wnt signaling function in Alzheimer’s disease. Brain Res. Brain Res. Rev. 2000;33(1):1–12. doi: 10.1016/S0165-0173(00)00021-7. [DOI] [PubMed] [Google Scholar]
- 37.Palomer E., Buechler J., Salinas P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019;13(May):227. doi: 10.3389/fncel.2019.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garner B, Ooi L. Wnt is here! Could Wnt signaling be promoted to protect against Alzheimer disease?: An Editorial for 'Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer's disease in J20- APP transgenic and wild-type mice. J Neurochem. 2018:144–4. 356–359. doi: 10.1111/jnc.14276. [DOI] [PubMed] [Google Scholar]
- 39.Elliott C., Rojo A.I., Ribe E., Broadstock M., Xia W., Morin P., Semenov M., Baillie G., Cuadrado A., Al-Shawi R., Ballard C.G., Simons P., Killick R. A role for APP in Wnt signalling links synapse loss with β-amyloid production. Transl. Psychiatry. 2018;8(1):179. doi: 10.1038/s41398-018-0231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wątroba M., Dudek I., Skoda M., Stangret A., Rzodkiewicz P., Szukiewicz D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017;40:11–19. doi: 10.1016/j.arr.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Braidy N., Jayasena T., Poljak A. Sirtuins in Cognitive Ageing and Alzheimer ? s Disease. 2012;25(3):226–230. doi: 10.1097/YCO.0b013e32835112c1. [DOI] [PubMed] [Google Scholar]
- 42.Guarente L. 2013. Chapter 1 Introduction : Sirtuins in Aging and Diseases; p. 1077. [DOI] [PubMed] [Google Scholar]
- 43.Yin J., Han P., Song M., Nielsen M., Beach T.G., Serrano G.E., Liang W.S., Caselli R.J., Shi J. Amyloid-β increases tau by mediating sirtuin 3 in alzheimer’s disease. Mol. Neurobiol. 2018;55(11):8592–8601. doi: 10.1007/s12035-018-0977-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oddo S. The role of mTOR signaling in Alzheimer disease. Front. Biosci. (Schol. Ed.) 2012;4:941–952. doi: 10.2741/s310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. published correction appears in PLoS One. 2010;6(11) doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maiese K. Targeting molecules to medicine with mTOR, autophagy and neurodegenerative disorders. Br. J. Clin. Pharmacol. 2016;82(5):1245–1266. doi: 10.1111/bcp.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crino P.B. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat. Rev. Neurol. 2016;12(7):379–392. doi: 10.1038/nrneurol.2016.81. [DOI] [PubMed] [Google Scholar]
- 48.Ma T., Hoeffer C.A., Capetillo-Zarate E., Yu F., Wong H., Lin M.T., Tampellini D., Klann E., Blitzer R.D., Gouras G.K. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One. 2010;5(9):1–10. doi: 10.1371/journal.pone.0012845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McIlwain D.R., Berger T., Mak T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015;7(4):a026716. doi: 10.1101/cshperspect.a026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rohn T.T. The role of caspases in Alzheimer’s disease; potential novel therapeutic opportunities. Apoptosis. 2010;15(11):1403–1409. doi: 10.1007/s10495-010-0463-2. [DOI] [PubMed] [Google Scholar]
- 51.Rohn T.T., Head E. Caspases as therapeutic targets in Alzheimer’s disease: is it time to “cut” to the chase? Int. J. Clin. Exp. Pathol. 2009;2(2):108–118. [PMC free article] [PubMed] [Google Scholar]
- 52.LeBlanc A., Liu H., Goodyer C., Bergeron C., Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem. 1999;274(33):23426–23436. doi: 10.1074/jbc.274.33.23426. [DOI] [PubMed] [Google Scholar]
- 53.Upadhya SC, Hegde AN. Role of the ubiquitin proteasome system in Alzheimer's disease. BMC Biochem. 2007;8(1):S12. doi: 10.1186/1471-2091-8-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gadhave K., Bolshette N., Ahire A., Pardeshi R., Thakur K. The Ubiquitin Proteasomal System : A Potential Target for the Management of Alzheimer ? s Disease The Ubiquitin Proteasomal System. 2016;20(7):1392–1407. doi: 10.1111/jcmm.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gong B., Radulovic M., Figueiredo-Pereira M.E., Cardozo C. The Ubiquitin-Proteasome System: Potential Therapeutic Targets for Alzheimer’s Disease and Spinal Cord Injury. Front. Mol. Neurosci. 2016;9:4. doi: 10.3389/fnmol.2016.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sulistio Y.A., Heese K. The Ubiquitin-Proteasome System and Molecular Chaperone Deregulation in Alzheimer’s Disease. Mol. Neurobiol. 2016;53(2):905–931. doi: 10.1007/s12035-014-9063-4. [DOI] [PubMed] [Google Scholar]
- 57.Oddo S., editor. The Ubiquitin-Proteasome System in Alzheimer ? s Disease. 2008;12(2):363–373. doi: 10.1111/j.1582-4934.2008.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Funderburk S.F., Marcellino B.K., Yue Z. Cell “self-eating” (autophagy) mechanism in Alzheimer’s disease. Mt. Sinai J. Med. 2010;77(1):59–68. doi: 10.1002/msj.20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uddin S., Stachowiak A., Mamun A., Al, Tzvetkov N. T., Takeda S., Atanasov A. G., Bergantin L. B. Autophagy and alzheimer?s disease : from molecular mechanisms to therapeutic implications. 2018:1–18. doi: 10.3389/fnagi.2018.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeng Q., Siu W., Li L., Jin Y., Liang S., Cao M., Ma M., Wu Z. Autophagy in Alzheimer’s disease and promising modulatory effects of herbal medicine. Exp. Gerontol. 2019;119(119):100–110. doi: 10.1016/j.exger.2019.01.027. [DOI] [PubMed] [Google Scholar]
- 61.Li Q., Liu Y., Sun M. Autophagy and Alzheimer’s Disease. Cell. Mol. Neurobiol. 2017;37(3):377–388. doi: 10.1007/s10571-016-0386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zare-Shahabadi A., Masliah E., Johnson G.V., Rezaei N. Autophagy in Alzheimer’s disease. Rev. Neurosci. 2015;26(4):385–395. doi: 10.1515/revneuro-2014-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Avallone R., Vitale G., Bertolotti M. Omega-3 Fatty Acids and Neurodegenerative Diseases: New Evidence in Clinical Trials. Int. J. Mol. Sci. 2019;20(17):4256. doi: 10.3390/ijms20174256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.El Shatshat A., Pham A.T., Rao P.P.N. Interactions of polyunsaturated fatty acids with amyloid peptides Aβ40 and Aβ42. Arch. Biochem. Biophys. 2019;663:34–43. doi: 10.1016/j.abb.2018.12.027. [DOI] [PubMed] [Google Scholar]
- 65.Sanchez-Mejia R.O., Mucke L. Phospholipase A2 and arachidonic acid in Alzheimer’s disease. Biochim. Biophys. Acta. 2010;1801(8):784–790. doi: 10.1016/j.bbalip.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cunnane S.C., Plourde M., Pifferi F., Bégin M., Féart C., Barberger-Gateau P. Fish, docosahexaenoic acid and Alzheimer’s disease. Prog. Lipid Res. 2009;48(5):239–256. doi: 10.1016/j.plipres.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 67.Hermes M., Eichhoff G., Garaschuk O. Intracellular Calcium Signaling in Alzheimer?s Disease. 2020;4(1-2):30–41. doi: 10.1111/j.1582-4934.2009.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y., Shi Y., Wei H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J. Alzheimers Dis. Parkinsonism. 2017;7(5):374. doi: 10.4172/2161-0460.1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berridge M.J. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion. 2013;7(1):2–13. doi: 10.4161/pri.21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jazvinšćak Jembrek M., Hof P.R., Šimić G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell. Longev. 2015;2015:346783. doi: 10.1155/2015/346783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Czubowicz K., Jęśko H., Wencel P., Lukiw W.J., Strosznajder R.P. The role of ceramide and sphingosine-1-phosphate in alzheimer’s disease and other neurodegenerative disorders. Mol. Neurobiol. 2019;56(8):5436–5455. doi: 10.1007/s12035-018-1448-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Le Stunff H., Véret J., Kassis N., Denom J., Meneyrol K., Paul J.L., Cruciani-Guglielmacci C., Magnan C., Janel N. Deciphering the link between hyperhomocysteinemia and ceramide metabolism in alzheimer-type neurodegeneration. Front. Neurol. 2019;10(JUL):807. doi: 10.3389/fneur.2019.00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Filippov V., Song M.A., Zhang K., Vinters H.V., Tung S., Kirsch W.M., Yang J., Duerksen-Hughes P.J. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimers Dis. 2012;29(3):537–547. doi: 10.3233/JAD-2011-111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tong M., de la Monte S.M. Mechanisms of ceramide-mediated neurodegeneration. J. Alzheimers Dis. 2009;16(4):705–714. doi: 10.3233/JAD-2009-0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woo H.N., Park J.S., Gwon A.R., Arumugam T.V., Jo D.G. Alzheimer’s disease and Notch signaling. Biochem. Biophys. Res. Commun. 2009;390(4):1093–1097. doi: 10.1016/j.bbrc.2009.10.093. [DOI] [PubMed] [Google Scholar]
- 76.Zhang S., Cai F., Wu Y., Bozorgmehr T., Wang Z., Zhang S., Huang D., Guo J., Shen L., Rankin C., et al. A Presenilin-1 Mutation Causes Alzheimer Disease without Affecting Notch Signaling. Mol. Psychiatry. 2018;3293:1–11. doi: 10.1038/s41380-018-0101-x. [DOI] [PubMed] [Google Scholar]
- 77.Brown T. Alzheimer’s Disease Genetic Mutations: Mini Review. J. Investig. Genomics. 2016;3(1):12–14. doi: 10.15406/jig.2016.03.00039. [DOI] [Google Scholar]
- 78.Kopan R., Goate A. A common enzyme connects notch signaling and Alzheimer’s disease. Genes Dev. 2000;14(22):2799–2806. doi: 10.1101/gad.836900. [DOI] [PubMed] [Google Scholar]
- 79.Allen S.J., Watson J.J., Dawbarn D. The neurotrophins and their role in Alzheimer’s disease. Curr. Neuropharmacol. 2011;9(4):559–573. doi: 10.2174/157015911798376190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sampaio T.B., Savall A.S., Gutierrez M.E.Z., Pinton S. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: implications for pathogenesis and therapy. Neural Regen. Res. 2017;12(4):549–557. doi: 10.4103/1673-5374.205084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iulita M.F., Bistué Millón M.B., Pentz R., Aguilar L.F., Do Carmo S., Allard S., Michalski B., Wilson E.N., Ducatenzeiler A., Bruno M.A., Fahnestock M., Cuello A.C. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol. Dis. 2017;108:307–323. doi: 10.1016/j.nbd.2017.08.019. [DOI] [PubMed] [Google Scholar]
- 82.Puglielli L. Aging of the Brain , Neurotrophin Signaling , and Alzheimer?s Disease : Is IGF1-R the Common Culprit ? 2008;29:795–811. doi: 10.1016/j.neurobiolaging.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Coimbra J. R. M., Marques D. F. F., Baptista S. J. Highlights in BACE1 Inhibitors for Alzheimer ? s Disease Treatment. 2018;6:1–10. doi: 10.3389/fchem.2018.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Das B., Yan R. Role of BACE1 in Alzheimer’s synaptic function. Transl. Neurodegener. 2017;6:23. doi: 10.1186/s40035-017-0093-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mandal M., Zhu Z., Cumming J.N., Liu X., Strickland C., Mazzola R.D., Caldwell J.P., Leach P., Grzelak M., Hyde L., Zhang Q., Terracina G., Zhang L., Chen X., Kuvelkar R., Kennedy M.E., Favreau L., Cox K., Orth P., Buevich A., Voigt J., Wang H., Kazakevich I., McKittrick B.A., Greenlee W., Parker E.M., Stamford A.W. Design and validation of bicyclic iminopyrimidinones as beta amyloid cleaving enzyme-1 (BACE1) inhibitors: conformational constraint to favor a bioactive conformation. J. Med. Chem. 2012;55(21):9331–9345. doi: 10.1021/jm301039c. [DOI] [PubMed] [Google Scholar]
- 86.Mullard A. BACE inhibitor bust in Alzheimer trial. Nat. Rev. Drug Discov. 2017;16(3):155. doi: 10.1038/nrd.2017.43. [DOI] [PubMed] [Google Scholar]
- 87.Yan G., Hao L., Niu Y., Huang W., Wang W., Xu F., Liang L., Wang C., Jin H., Xu P. 2-Substituted-thio-N-(4-substituted-thiazol/1H-imidazol-2-yl)acetamides as BACE1 inhibitors: Synthesis, biological evaluation and docking studies. Eur. J. Med. Chem. 2017;137:462–475. doi: 10.1016/j.ejmech.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 88.Barr R. K., Ramirez M. J. C-Jun N-Terminal Kinase( JNK) Signaling as a Therapeutic Target for Alzheimer ? s Disease. 2016;6:1–12. doi: 10.3389/fphar.2015.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou Q., Wang M., Du Y., Zhang W., Bai M., Zhang Z., Li Z., Miao J. Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 2015;77(4):637–654. doi: 10.1002/ana.24361. [DOI] [PubMed] [Google Scholar]
- 90.Hong Y.K., Lee S., Park S.H., Lee J.H., Han S.Y., Kim S.T., Kim Y.K., Jeon S., Koo B.S., Cho K.S. Inhibition of JNK/dFOXO pathway and caspases rescues neurological impairments in Drosophila Alzheimer’s disease model. Biochem. Biophys. Res. Commun. 2012;419(1):49–53. doi: 10.1016/j.bbrc.2012.01.122. [DOI] [PubMed] [Google Scholar]
- 91.Cao M., Liu F., Ji F., Liang J., Liu L., Wu Q., Wang T. Effect of c-Jun N-terminal kinase (JNK)/p38 mitogen-activated protein kinase (p38 MAPK) in morphine-induced tau protein hyperphosphorylation. Behav. Brain Res. 2013;237:249–255. doi: 10.1016/j.bbr.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 92.Maqbool M., Mobashir M., Hoda N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016;107:63–81. doi: 10.1016/j.ejmech.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 93.Hooper C., Killick R., Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008;104(6):1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hernandez F., Lucas J.J., Avila J. GSK3 and tau: two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 2013;33(Suppl. 1):S141–S144. doi: 10.3233/JAD-2012-129025. [DOI] [PubMed] [Google Scholar]
- 95.Lauretti E., Dincer O., Praticò D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020;1867(5):118664. doi: 10.1016/j.bbamcr.2020.118664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu M., Wang S.L., Zhu L., Wu P.Y., Dai W.B., Rakesh K.P. Structure-Activity Relationship (SAR) Studies of Synthetic Glycogen Synthase Kinase-3 b Inhibitors. Crit. Rev. 2019;164:448–470. doi: 10.1016/j.ejmech.2018.12.073. [DOI] [PubMed] [Google Scholar]
- 97.Rakesh K.P., Shantharam C.S., Sridhara M.B., Manukumar H.M., Qin H.L. Benzisoxazole: a privileged scaffold for medicinal chemistry. MedChemComm. 2017;8(11):2023–2039. doi: 10.1039/C7MD00449D. [DOI] [PMC free article] [PubMed] [Google Scholar]