

Abstract
Background & Aim:
Besides their physiological role in bile formation and fat digestion, bile acids (BAs) synthesized from cholesterol in hepatocytes act as signaling molecules that modulate hepatocellular carcinoma (HCC). Trafficking of cholesterol to mitochondria through steroidogenic acute regulatory protein 1 (STARD1) is the rate-limiting step in the alternative pathway of BAs generation, whose physiological relevance is not well understood. Moreover, the specific contribution of the STARD1-dependent BA synthesis pathway to HCC has not been explored.
Methods:
STARD1 expression was analyzed in a cohort of human NASH derived HCC specimens. Experimental NASH-driven HCC models included MUP-uPA mice fed high fat high cholesterol diet (HFHC) and diethylnitrosamine (DEN) treatment in wild type (WT) mice fed HFHC. Molecular species of BAs and oxysterols were analyzed by mass spectrometry. Effects of NASH-derived BAs profile were investigated in tumor-initiated stem-like cells (TICs) and primary mouse hepatocytes (PMH).
Results:
We show that patients with NASH-associated HCC exhibit increased hepatic expression of STARD1 and enhanced BAs pool. Using NASH-driven HCC models, STARD1 overexpression in WT mice increased liver tumor multiplicity, whereas hepatocyte-specific STARD1 deletion (Stard1ΔHep) in WT or MUP-uPA mice reduced tumor burden. These findings mirrored the levels of unconjugated primary BAs, β-muricholic acid and cholic acid, and their tauroconjugates in STARD1 overexpressing and Stard1ΔHep mice. Incubation of TICs or PMH with a mix of BAs mimicking this profile stimulated expression of genes involved in pluripotency, stemness and inflammation.
Conclusions:
We show a previously unrecognized role of STARD1 in HCC pathogenesis by promoting the synthesis of primary BAs through the mitochondrial pathway, whose products act in TICs to stimulate self-renewal, stemness and inflammation.
Keywords: Cholesterol, mitochondria, bile acids, hepatocellular carcinoma, oxysterols, STARD1
LAY SUMMARY
The incidence of hepatocellular carcinoma (HCC) in Western countries had tripled in the past 40 years due to the obesity, type-2 diabetes, non-alcoholic fatty liver disease and steatohepatitis (NASH) epidemic. Effective therapy is limited due to the incomplete understanding of HCC pathogenesis and the aggressive nature of the disease. In addition to their physiological role in bile formation and fat digestion, bile acids (BAs) act as signaling molecules and modulate liver tumorigenesis. The contribution of the alternative pathway of BAs synthesis to HCC development is unknown. We uncover a key role of STARD1 in NASH-driven HCC by stimulating the generation of BAs in the mitochondrial acidic pathway, whose products stimulate hepatocytes for pluripotenty, self-renewal and inflammation.
Graphical Abstract
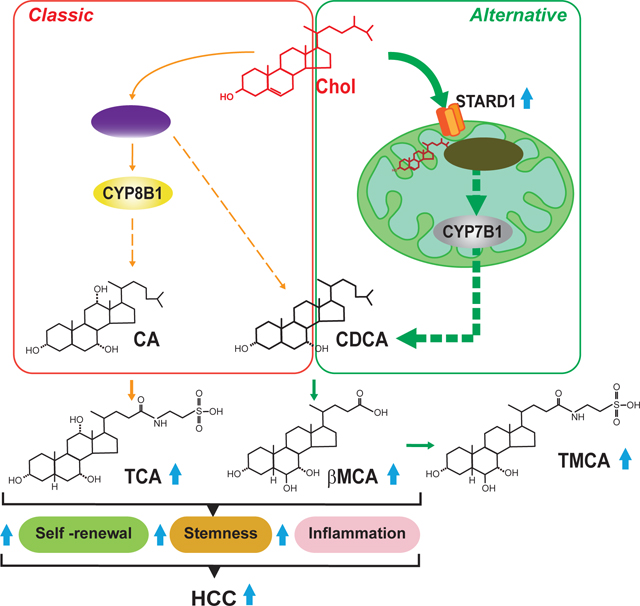
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common type of liver cancer and the end-stage of chronic liver disease caused by different etiologies, including non-alcoholic steatohepatitis (NASH). The incidence of NASH-driven HCC is expected to increase worldwide due to its association with the obesity and type-2 diabetes epidemic. Overweight (body mass index >25) and obesity are known risk factors for cancer development, especially HCC 1, 2. HCC has a poor prognosis with frequent recurrence and metastasis 2, 3. Although important improvements in the management of HCC have been made in the last 2–3 decades, effective treatment options such as local ablative therapies, resection or transplantation are mainly limited to early disease stages 4, 5. Unfortunately, the therapeutic armamentarium for HCC is limited, ineffective and subject to secondary or acquired chemoresistance by poorly understood mechanisms 6. Hence, there is an urgent need to understand HCC pathogenesis and identify new therapeutic targets.
Diet-induced NASH and chronic endoplasmic reticulum (ER) stress have been shown to lead to HCC development 7–9. Cancer cells are under anabolic pressure for the synthesis of membrane lipids to sustain dysregulated cell proliferation, and increased cholesterol and fatty acid synthesis support HCC growth 10. Consistent with a key structural and functional role of cholesterol in membrane bilayers, recent reports indicated that dietary or de novo synthesized cholesterol fosters HCC development, in part, through the generation of bile acids (BAs) 11–14. BAs are synthesized in hepatocytes from cholesterol predominantly through the classical (neutral) pathway, which is regulated by the rate-limiting enzyme 7-α-hydroxylase (encoded by CYP7A1). Besides, sterol 12α-hydroxylation by 12α-hydroxylase (encoded by CYP8B1) is specifically required for cholic acid (CA) synthesis. In addition to their key role in fat digestion and vitamin metabolism, BAs are critical signaling molecules that regulate gene expression by targeting nuclear (e.g. FXR) and membrane (e.g. TGR5) receptors and have been linked to NASH progression and HCC promotion 14–16. Indeed, the severity of human NASH has been associated with specific changes in plasma levels of BAs while mouse models (e.g. FXR−/−, BSEP−/− or MDR2−/− mice) with an increase in total circulating BAs exhibited spontaneous formation of HCC 16–19.
The mitochondrial pool of cholesterol is minor compared to its plasma membrane content and modulates vital mitochondrial functions, such as oxidative phosphorylation, mitochondrial apoptosis, chemotherapy resistance or susceptibility to TNF/Fas-mediated NASH progression 20–26. The mitochondrial cholesterol level is regulated by specific carriers most notably STARD1, which mediates the trafficking of cholesterol to the mitochondrial inner membrane for metabolism 27–29. In the liver, mitochondrial cholesterol is metabolized by 27-hydroxylase (encoded by CYP27A1) to 27-hydroxycholesterol followed by 25-hydroxycholesterol 7-α-hydroxylase (encoded by CYP7B1), which then feeds the alternative mitochondrial pathway of BA synthesis leading mainly to chenodeoxycholic acid (CDCA) generation 15, 30, 31. In mouse liver, CDCA is metabolized to α-muricholic acid (αMCA) and its 7β-epimer β-muricholic acid (βMCA) 32. The mitochondrial acidic pathway of BA synthesis is considered to contribute to a minor extent to the total BA pool and its physiological relevance is not well understood.
Patients with NASH exhibit elevated free cholesterol 33, 34 and enhanced STARD1 expression 33. Since the contribution of the alternative pathway of BAs synthesis to HCC has not been previously addressed, we investigated the role of STARD1 in NASH-driven HCC. The present study shows a previously unrecognized role for STARD1 in HCC by stimulating the generation of BAs from cholesterol via the alternative pathway, whose products act in tumor-initiating stem-like cells (TICs) and hepatocytes to stimulate expression of genes involved in pluripotency, stemness and inflammation.
MATERIALS AND METHODS
Human NASH-derived HCC cohort
Human liver samples were obtained from donors and recipients undergoing liver transplantation at the Liver Transplantation Unit of the Hospital Clinic, Barcelona (Table S1). During the donor sample procurement, an intra-operative assessment of the liver is systematically carried out to rule out fibrosis, cirrhosis, steatosis and other abnormalities before transplantation. A biopsy of the resected liver from the recipient was performed right after the hepatectomy and samples were fixed in formalin for histological evaluation or quickly snap-frozen. Samples from control (donors) with signs of steatosis, fibrosis, inflammation were discarded. Recipients with NASH-derived HCC eligible for liver transplantation complied with the Milan Criteria and were stratified by the BCLC score (single tumor ≤5 cm or 2–3 tumors ≤3 cm each) 35. Samples from individuals with viral hepatitis, alcoholic steatohepatitis or cryptogenic cirrhosis were excluded. The protocol (HCB/2012/8011) was approved by the HCB/UB Ethics Committee of the Hospital Clinic of Barcelona Spain.
Stard1ΔHep and MUP-uPA-Stard1ΔHep mice.
Liver-specific Stard1 knockout (Stard1ΔHep) mice were created by crossing Stard1f/f mice, which were generated by the Cre-lox technology, with Alb-Cre mice and have been recently characterized 36. Stard1ΔHep and Stard1f/f littermates were used in this study. MUP-uPA transgenic mice were generated and previously characterized 37. MUP-uPA transgenic animals were crossed with Stard1ΔHep mice and backcrossed with Stard1f/f to select homozygous Stard1f/f–MUP-uPA tg positives with or without Alb-Cre expression (MUP-uPA-Stard1f/f and MUP-uPA-Stard1ΔHep) and used in the present study.
NASH-driven HCC development and treatment
For the induction of HCC, C57Bl/6j mice were injected i.p. with a single dose of DEN (25 mg/kg) on postnatal day 14 and 4 weeks later were introduced to different diets (Table S2). Animals were fed either with high fat diet (HFD, containing 60% calories from fat) or high fat-high cholesterol diet (HFHC, containing 60% calories from fat and added 0.5% cholesterol) up to 32 weeks. Additionally, a regular diet with added cholesterol was custom-made (Teklad diet 2014 with 2% cholesterol, HC diet). In some cases, DEN-treated mice were fed HFHC with added ezetimibe (EZE) (100 mg Ezetrol/kg of diet, equivalent to 10 mg/Kg/day) and fed for 24 weeks. To determine the effect of EZE treatment on survival, DEN-treated mice were fed HFHC diet for 52 weeks. At time of sacrifice, animals were anesthesized, exanguinated, macroscopic tumors counted and liver was harvested and processed for subsequent analysis.
For the induction of heterotopic tumors induced by TICs, 8-week old atimic nude immunodeficient mice (Charles River) were subcutaneously injected with 1 × 106 TICs in 100 μL at 1:1 PBS matrigel high concentration (Corning #354248) either stably overexpressing Stard1 or green fluorescent protein (Gfp) at the right or left flanks, respectively, and tumors allowed to grow for 3 weeks.
All procedures involving animals and their care were approved by the Ethics Committee of the University of Barcelona following national and European guidelines for maintenance and husbandry of research animals.
TICs isolation and treatment.
TICs (CD133+/CD49f+) were isolated from murine HCC, as described previously 38. Briefly, resected HCC tissues were immediately dissected into small pieces and digested with collagenase. Suspended liver cells were stained with PE-anti-CD133, APC-anti-CD49f, and FITC-anti-CD45 antibodies (BD Biosciences) followed by FACS analysis, as described before 39. TICs and primary mouse hepatocytes (PMH) were treated with the specified concentrations of BAs: CA (Sigma, C1129), βMCA (Sigma, SML2372) or TCA (Sigma, T4009) for 24 or 48 hours and analyzed for expression of pluripotency, stemness and inflammatory genes by qPCR.
Quantification and statistical analysis
All data are presented as mean ± SEM. In each experiment, N defines sample size. The Student’s t-test was used to define differences between two groups. To define differences between more than two groups One-way Analysis of Variance (ANOVA) was used with a Bonferroni multiple comparison post-test, or Kruskall-wallis non-parametric test for data displaying a non-gaussian distribution. The criterion for significance was set at P<0.05. Statistical analyses were performed using GraphPad Prism version 5. Given the variability of the in vivo studies, 6–12 mice were included per group to ensure statistical power.
RESULTS
Patients with NASH-driven HCC exhibit increased expression of STARD1 and a high BA burden.
Although the basal expression of STARD1 in the liver is low, STARD1 is upregulated in patients with NASH but not in subjects with steatosis alone 33. However, the role of STARD1 in NASH-driven HCC had not been explored. We examined the expression of STARD1 in a cohort of patients with NASH-derived HCC (Table S1). Histology analyses revealed alterations in the parenchymal architecture of human HCC samples (Figure S1A), exhibiting fat infiltration, increased liver triglycerides (TG), and fibrosis (Figure S1B–C), reflected by Sirius red staining and increased expression of fibrogenic genes ACTA2 and COL1A1 (Figure S1D). Liver samples from patients with NASH-driven HCC exhibited increased levels of STAR gene transcript and STARD1 protein, compared to samples from control subjects (Figure 1A). Moreover, liver sections from patients with HCC displayed increased immunohistochemical STARD1 staining (Figure 1B). STARD1 was expressed predominantly in hepatocytes as indicated by the colocalization of STARD1 with asialoglycoprotein receptor 1 (ASGPR1) and to a minor extent with Kupffer cells and hepatic stellate cells labeled with F4/80 and α-SMA, respectively (Figure S2). Higher hepatic free cholesterol levels were observed by staining liver sections with GST-perfringolysin (GST-PFO) (Figure 1C), which detects free cholesterol in membranes 40, as well as by filipin staining and HPLC analysis (Figure S1E, F). GST-PFO staining of liver sections from patients with NASH-driven HCC colocalized with cytochrome c immunofluorescence (Figure 1D), indicating the presence of free cholesterol in mitochondria in human HCC, consistent with findings in experimental HCC22, 26. Furthermore, human HCC samples exhibited increased expression of HMGCS, HMGCR and SREBP2, the master transcription factor for cholesterol homeostasis (Figure 1E). Although SREBP2 is negatively regulated by cholesterol, we addressed whether the activation of SREBP2 in association with increased cholesterol was linked to a refractory feedback loop triggered by the TNFR1-Caspase-2-S1P-SREBP2 axis 41. Human HCC samples exhibited increased expression of CASP-2 and MBPTS1 (S1P) compared to control subjects (Figure 1E). Moreover, samples from human HCC displayed increased expression of HIF1A and target genes, such as PDK1, SLC2A1 (Glut 1), SLC2A3 (Glut 3) and SLC25A11 (2-OGC) (Figure 1F), which has been shown to regulate mitochondrial GSH homeostasis in HCC 38.
Figure 1. Increased STARD1 expressed in human NASH-driven HCC.
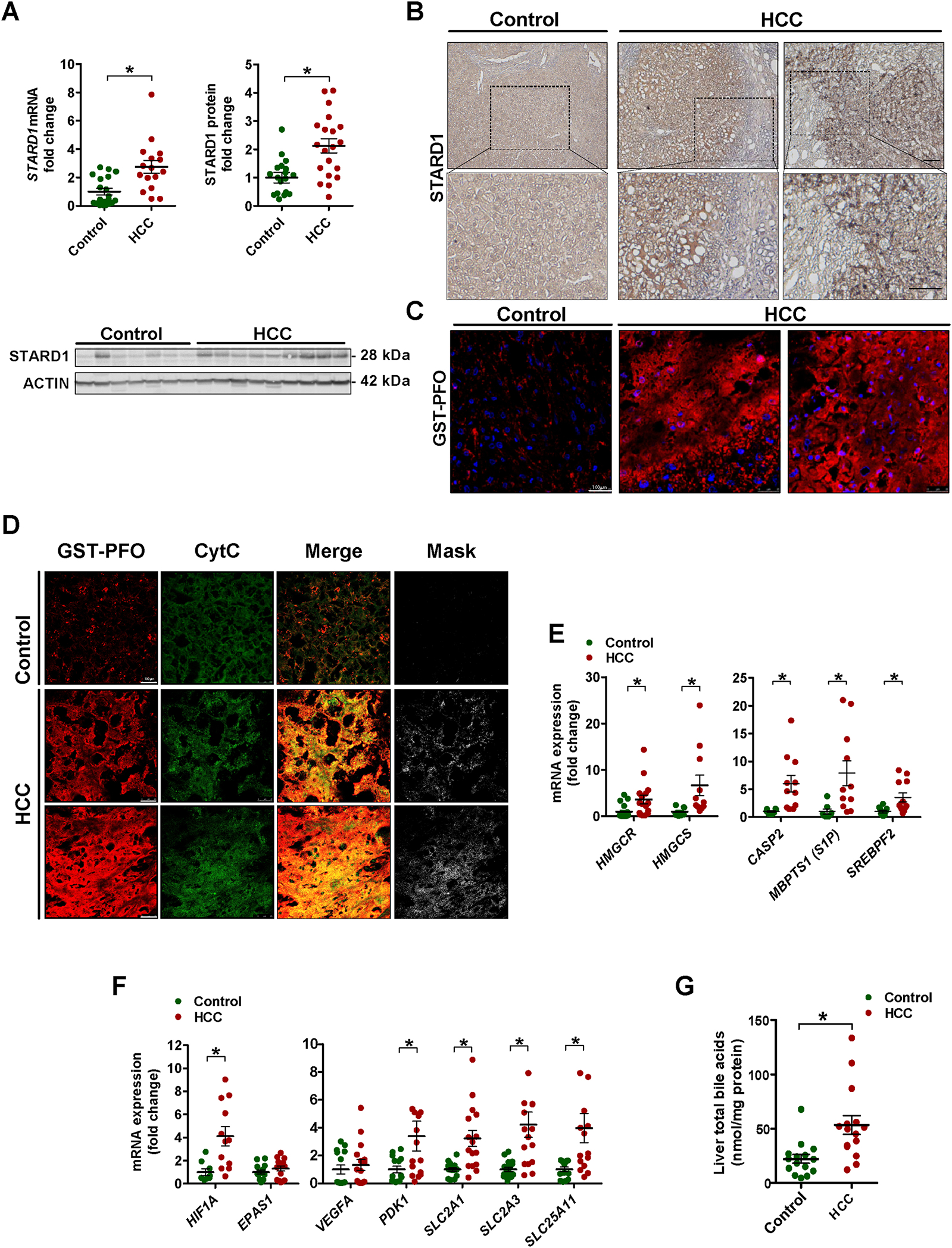
A) Expression of STAR gene mRNA by qPCR and STARD1 protein in liver tissue from control donors and NASH-driven HCC (N of controls =15, N of HCC patients 15–20).
B) Representative immunohistochemical expression of STARD1 from control and NASH-HCC patient liver samples.
C) Staining of liver sections from control and HCC samples with GST-PFO (red) to detect free cholesterol. Nuclei were stained with DAPI.
D) Immunostaining of liver sections from control and HCC samples with GST-PFO and cytochrome c (Cyt C), showing their colocalization as merge and mask. Bar 75 μm.
E) Transcript quantification by qPCR of genes controlling cholesterol biosynthesis, ER stress-driven activation of SREBP2.
F) mRNA levels of HIF1A and HIF2A (EPAS1) and HIF-1α regulated genes. N of control 13–18, N of HC 13–20.
G) Hepatic levels of total BAs in samples from control and human HCC. (N=15 both groups).
All values are mean ± SEM. * indicate statistically significant differences between the indicated groups (p<0.05) in Student’s t test. Magnification bar in histology pictures, 100 μm.
We next examined the levels of hepatic BAs pool in patients with HCC. Compared to control subjects, human HCC samples revealed a two-fold increase in total hepatic BAs levels (Figure 1G) that paralleled the increased expression of CYP7A1, CYP8B1, CYP27A1 and CYP7B1 as well as CYP7A1 and CYP27A1 (Figure S1G, H), suggesting the activation of the classical (neutral) and alternative (acidic) pathways of BAs synthesis.
Cholesterol promotes NASH-driven HCC and induces STARD1 expression in mice.
Next, we addressed the specific contribution of the alternative mitochondrial pathway of BAs generation to HCC. First, we validated the tumor promoter role of cholesterol in HCC because despite accumulating evidence linking cholesterol with HCC development 11–13, there have been studies showing a tumor suppressor effect of cholesterol in HCC 42–46. As high fat diet (HFD) alone does not induce NASH and the diethylnitrosamine (DEN) plus HFD feeding does not completely model NASH-driven HCC 9, we established a dietary NASH-driven HCC approach by feeding DEN-pretreated mice with a HFD diet supplemented with cholesterol (HFHC) (Figure 2A), as this diet has been shown to induce NASH 47, 48. Compared to mice fed DEN+HFD alone, DEN+HFHC-fed mice exhibited higher serum ALT levels (Figure 2B), enhanced liver cholesterol content and decreased Hmgcr expression (Figure 2C). Serum HDL or LDL levels were independent on whether DEN-treated mice were fed HFD or HFHC diet (Figure 2D). Moreover, the degree of macrovesicular steatosis detected by Oil-Red staining and TG levels was similar between DEN+HFD and DEN+HFHC-fed mice (Figure 2C, E), although fibrosis was more severe in DEN+HFHC-fed mice compared to DEN+HFD-fed mice (Figure 2E, F). In addition, GST-PFO staining of liver sections of DEN+HFHC-fed mice revealed increased free cholesterol levels, which colocalized with cytochrome c (Figure 2E). In addition, DEN+HFHC feeding increased liver inflammation revealed by enhanced expression of Tnfα and Ccl2 (Figure 2G). Of significance, while DEN+HFD feeding for 24 weeks had a modest impact in tumor burden relative to DEN alone, DEN+HFHC feeding resulted in a much larger increase in tumor number and maximal area (Figure 2H), which increased even further after 32 weeks of HFHC feeding (Figure 2I). Tumor burden increased the levels of Afp in serum, especially in the DEN+HFHC group and these tumors displayed higher expression of Afp and Yap, two bona fide HCC markers (Figure 2J). Moreover, DEN+HFHC feeding increased liver expression of Stard1 (Figure 2K), which was preferentially expressed in HCC tumors (Figure 2L). Furthermore, DEN+HFHC-fed mice exhibited increased expression of markers involved in tumorigenesis (Gpc3, Ly6d, Golm1), cell adhesion and interactions (Birc5, Cd44, Lyve1) and cellular proliferation (Mki67) with respect to DEN+HFD-fed mice (Figure 2M).
Figure 2. HFHC feeding promotes NASH-driven HCC development in DEN-treated wild type.
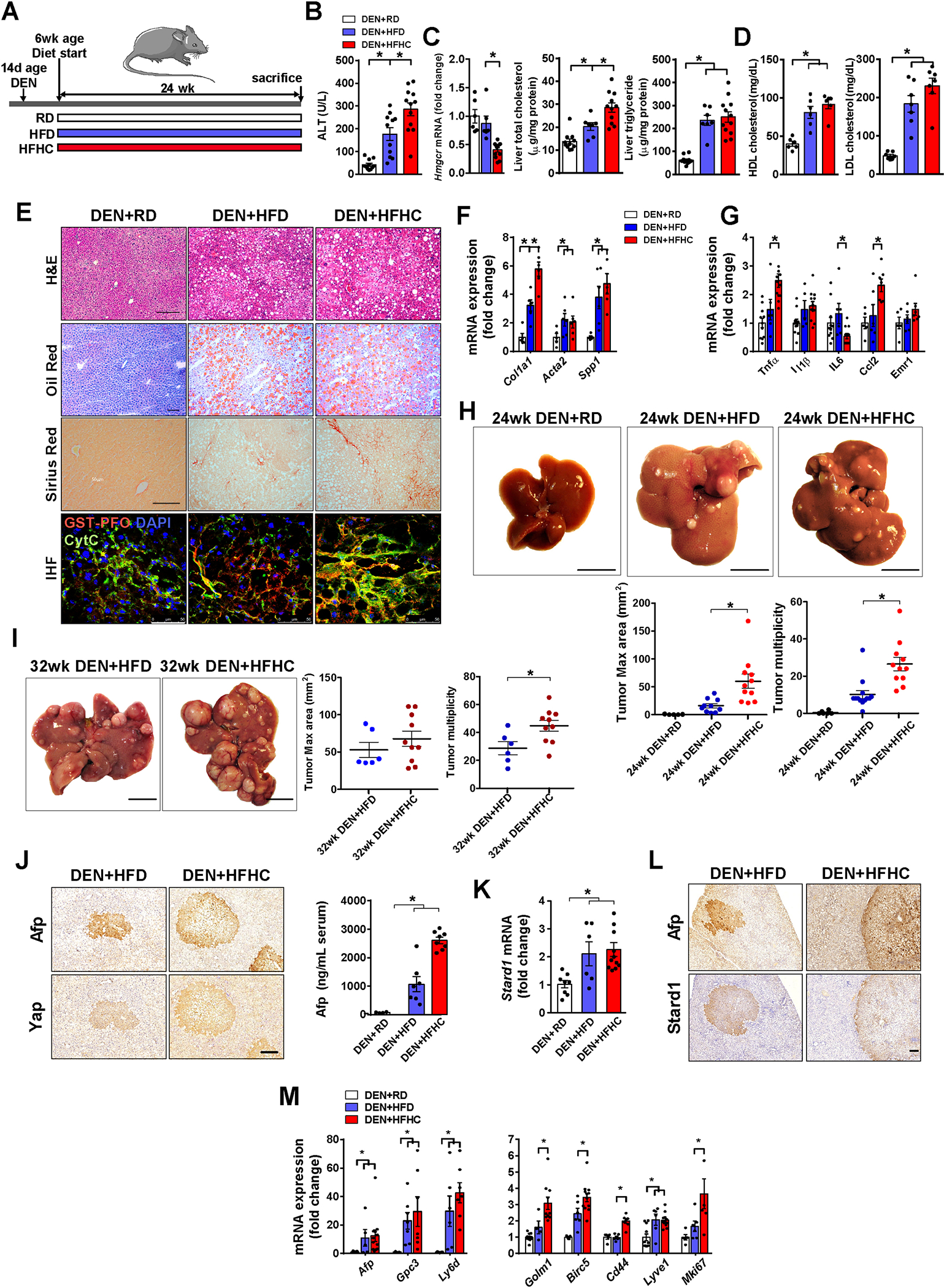
A) Schematic illustration of the experimental design, with induction of tumorogenesis in liver of mice with DEN at 14 days of age, feeding with regular diet (RD), high fat diet (HFD) or cholesterol-supplemented HFD (HFHC) diet for 24 weeks. N per group: RD (11), HFD (12), HFHC (12).
B) Transaminase serum levels (ALT) of mice after the corresponding treatments.
C) Liver Hmgcr transcript, total cholesterol and triglycerides liver composition. N=6 per group.
D) HDL and LDL levels in serum from DEN+HFD or DEN+HFHC fed mice. N=6 per group.
E) Representative histological staining for hematoxylin-eosin (H&E), neutral lipid (oil red o) and collagen fibers (sirius red) of liver sections. Immunohistofluorescence of liver sections stained for free cholesterol with GST-PFO probe (red), mitochondria with anti-cytochrome c (green) and nuclei with Dapi (blue). Size bar 100 μm.
F) mRNA levels of fibrogenesis-associated genes (Col1a1, Acta2, Spp1). All values are corrected by a housekeeping gene (Actb) and relative to values from the animals of DEN- RD diet. N=6–10 per group.
G) mRNA levels of inflammation genes (Tnfa, Il1b, Il6, Ccl2, Emr1).
H) Representative macroscopic images and quantification of tumor multiplicity and maximal area from DEN-treated mice fed HFHC diet for 24 weeks. RD, N=6; HFD, N=10; HFHC, N=11.
I) As in H) except that DEN-treated mice were fed HFHC diet for 32 weeks. HFD, N=6; HFHC, N=10.
J) Immunohistochemical expression of Afp and Yap of liver consecutive sections from DEN-treated mice fed HFC or HFHC diet for 24 weeks.
K) mRNA levels of Stard1 of whole liver tissue from DEN-treated mice fed RD or HFHC diet. N=6–10 per group.
L) Immunohistochemistry of consecutive sections (T, tumor) stained for Afp, or Star. Size bar, 500 μmeter.
M) mRNA levels of tumor markers and inflammatory genes of whole liver tissue from DEN-treated mice fed RD, HFD or HFHC. N=6–10 per group.
All values are mean ± SEM; symbol * indicates statistically significant differences (p<0.05) on a one-way ANOVA test or student’s t test.
Ezetimibe treatment attenuates DEN plus HFHC-driven HCC.
To further determine the role of cholesterol in NASH-driven HCC, we tested the effect of ezetimibe, which prevents the intestinal absorption of cholesterol. Although its role in human NASH is not well established 49, we addressed its impact in DEN+HFHC-driven HCC (Figure S3A). Ezetimibe treatment decreased hepatic cholesterol accumulation in mice pretreated with DEN and fed HFHC diet (Figure S3B). The ability of ezetimibe to decrease liver cholesterol was due to its effect in blocking absorption of dietary cholesterol rather than affecting the novo cholesterol synthesis, consistent with the decreased the expression of Hmgcr and Hmgcs1 following HFHC feeding, indicating that dietary cholesterol exerts its expected feedback inhibition on the de novo synthesis of cholesterol (Figure S3C). Interestingly, the presence of ezetimibe reversed the downregulation of Hmgcr and Hmgcs1 in HFHC fed mice (Figure S3C). In line with this outcome, treatment of HFHC-fed mice with atorvastatin resulted in a modest effect in decreasing liver cholesterol (Figure S4), in agreement with the lower expression of Hmgcr by dietary cholesterol. Of note, ezetimibe did not change the expression of Stard1 in DEN plus HFHC fed mice (Figure S3C). Moreover, ezetimibe ameliorated liver fibrosis in DEN+HFHC-fed mice, as seen by Sirius Red staining and the decreased expression of fibrosis genes (Figure S3D, E). Consistent with findings in PtenΔHep mice fed HFD 50, the number of tumors in DEN+HFHC-fed mice significantly decreased upon ezetimibe administration (Figure S3F), which paralleled the attenuation of serum Afp levels (Figure S3G), the expression of markers of tumorigenesis, cell adhesion/migration and hepatic proliferation (Figure S3H) and the decrease in the levels of Gp73 and cytokeratin 19 (Figure S3I). More importantly, while the median survival of DEN+HFHC-fed mice was 7-months, ezetimibe treatment significantly increased the survival rate with 50% of mice surviving 12 months post DEN+HFHC feeding (Figure S3J). Overall, these findings indicate that dietary cholesterol promotes NASH-driven HCC development.
STARD1 deletion in hepatocytes attenuates NASH-driven HCC
To address the role of STARD1 in NASH-driven HCC, we recently generated Stard1ΔHep mice 36 and examined their susceptibility to NASH-driven HCC using two different approaches. First, we deleted STARD1 in hepatocytes in MUP-uPA mice, a model characterized by an endogenous chronic ER stress due to the expression of urokinase plasminogen activator (uPA), which develop HCC by the synergism between ER stress and overfeeding 8, 9. MUP-uPA mice were crossed with Stard1ΔHep mice to generate MUP-uPA-Stard1ΔHep mice and fed HFHC (Figure 3A). MUP-uPA-Stard1ΔHep mice exhibited a profound depletion of Stard1 expression in liver extracts with respect to MUP-uPA-Stard1f/f mice (Figure 3B). While feeding MUP-uPA-Stard1f/f mice with HFHC diet for 26 weeks led to the development of liver tumors, the number and maximal area of these tumors in MUP-uPA-Stard1ΔHep mice were markedly reduced (Figure 3C, D). This outcome was accompanied by a decrease in serum Afp levels (Figure 3E) and lower expression of genes involved in fibrosis (Col1a1, Acta2) and inflammation (Il6, Il1β), and an attenuation in the levels of tumor markers (Afp, Cd44 and Ly6d) (Figure 3F–H). Interestingly, ablation of Stard1 did not affect the expression of ER stress markers in MUP-uPA-Stard1ΔHep mice (Figure 3I, J), indicating that the inhibitory effect of Stard1 deletion in this model of NASH-driven HCC is not linked to the prevention of ER stress.
Figure 3. Hepatocyte Stard1 deletion in MUP-uPA mice attenuates NASH-driven HCC in mice.
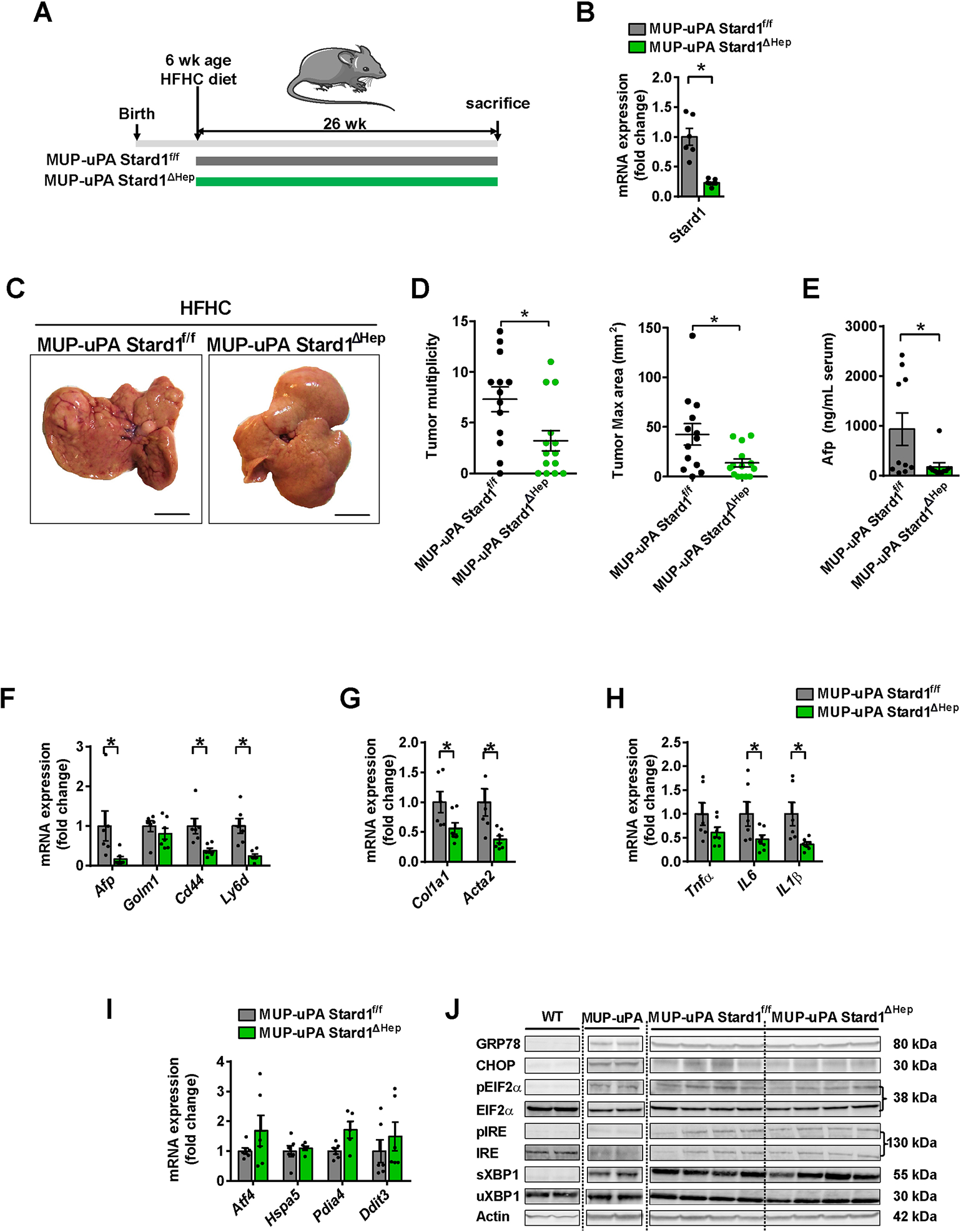
A) Feeding of MUP-uPA Stard1f/f and MUP-uPA Stard1 ΔHep mice with HFHC diet for 26 weeks.
B) mRNA levels of Stard1 in MUP-uPA Stard1f/f (N=6) and MUP-uPA Stard1 ΔHep mice (N=7).
C-D) Macroscopic images of livers from MUP-uPA Stard1f/f (N=13) and MUP-uPA Stard1 ΔHep mice (N=14) fed HFHC diet for 26 weeks, with quantification of tumor multiplicity and maximal area.
E) Serum Afp levels of MUP-uPA Stard1f/f (N=10) and MUP-uPA Stard1 ΔHep mice (N=10) fed HFHC diet.
F-H) mRNA levels tumor markers, fibrosis and inflammation genes of whole liver tissue from MUP-uPA Stard1f/f (N=6) and MUP-uPA Stard1 ΔHep mice (N=7) fed HFHC.
I) mRNA levels of ER stress markers of whole liver tissue from MUP-uPA Stard1f/f (N=6) and MUP-uPA Stard1 ΔHep mice (N=7) fed HFHC.
J) Western blot of ER stress markers as in H). All values are mean ± SEM. *p<0.05, denote statistically significant differences respect to MUP-uPAStard1f/f or Stard1f/f mice in Student’s t test.
In addition to this spontaneous NASH-driven HCC model, Stard1ΔHep mice were treated with DEN and then fed the HFHC diet for 24 weeks. Similar to the MUP-uPA model, DEN-treated Stard1ΔHep mice were relatively resistant to HFHC-mediated HCC development, exhibiting decreased tumor multiplicity and maximal area (Figure 4A, B) and a decrease in the serum Afp levels (Figure 4C). Tumors from Stard1ΔHep mice exhibited decreased Yap and Afp expression (Figure 4D, E) and lower mRNA levels of tumor markers without change in inflammation-related genes (Figure 4F, G). This outcome was accompanied by unchanged expression of ER stress markers in DEN+HFHC-treated Stard1ΔHep mice (Figure 4H, I). Thus, these findings support a critical role of STARD1 in NASH-driven HCC independently of ER stress.
Figure 4. Stard1 ΔHep mice are less sensitive to DEN plus HFHC induced HCC.
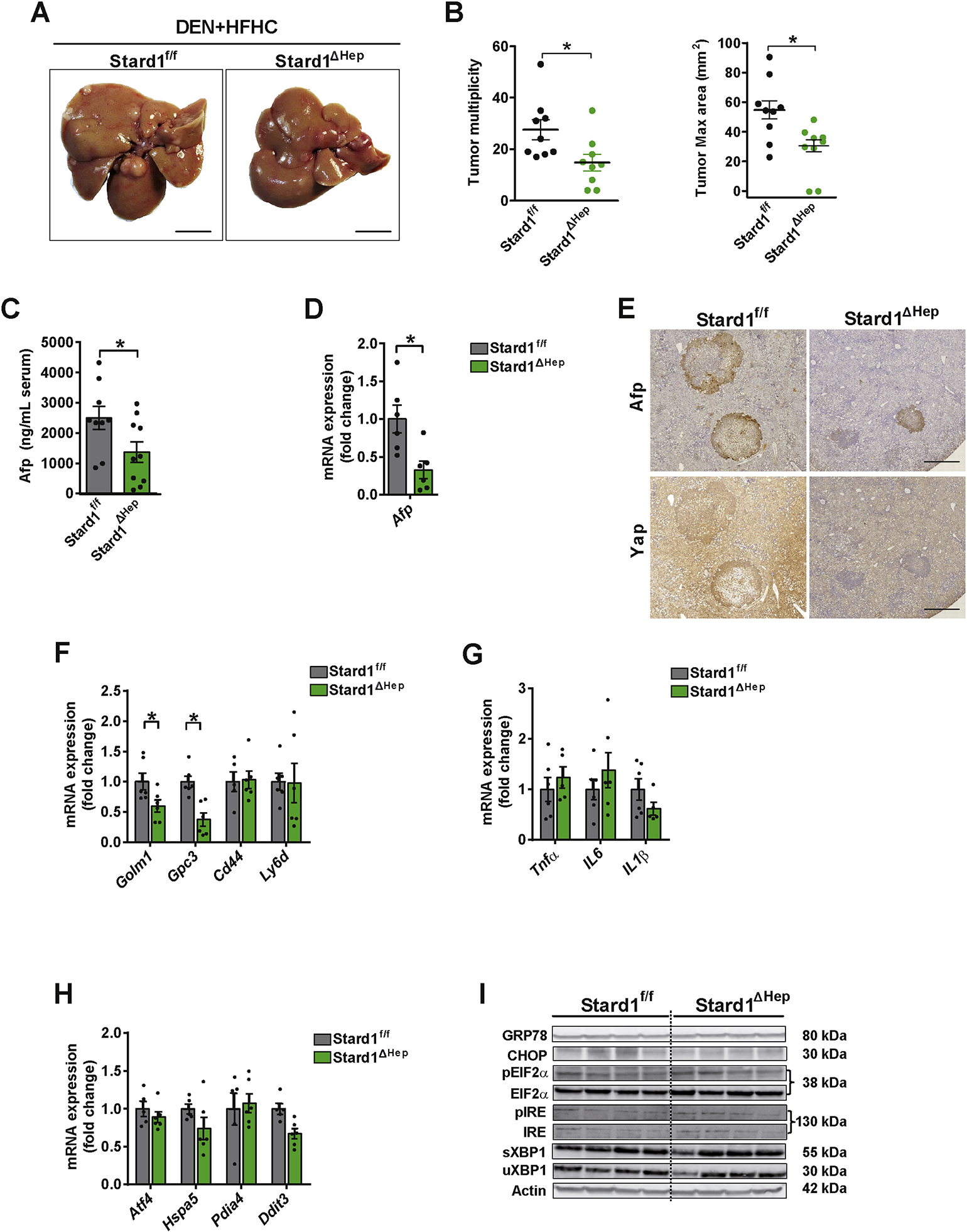
A-B) Macroscopic images of livers from Stard1f/f (N=9) and Stard1ΔHep mice (N=9) treated with DEN and fed HFHC diet for 24 weeks, with quantification of tumor multiplicity and maximal area.
C-D) Serum and mRNA expression levels of Afp from Stard1f/f (N=9) and Stard1ΔHep mice (N=9) treated with DEN and fed HFHC.
E) Immunohistochemical expression of Afp and Yap of consecutive liver sections from Stard1f/f and Stard1 ΔHep mice.
F-G) mRNA levels tumor markers and inflammation genes of whole liver tissue from Stard1f/f and Stard1ΔHep mice.
H) mRNA levels of ER stress markers of whole liver tissue from Stard1f/f (N=6) and Stard1ΔHep mice (N=6).
I) Western blot of ER stress markers as in H).
All values are mean ± SEM. *p<0.05, denote statistically significant differences respect to MUP-uPAStard1f/f or Stard1f/f mice in Student’s t test.
STARD1 overexpression exacerbates DEN plus HFHC diet-driven HCC
To further investigate the contribution of STARD1 in NASH-driven HCC, Stard1 was overexpressed in DEN+HFHC-fed wild type mice by injection with adenovirus bearing the cDNA of Stard1 (AD-Stard1) 5 weeks before sacrifice (Figure 5A), which resulted in 15-fold increase in liver Stard1 expression (mRNA and protein levels) as compared to control mice injected with empty control vector (AD-control) (Figure 5A–C). This outcome potentiated DEN+HFHC-mediated liver tumor multiplicity, although maximal area of tumors did not significantly change (Figure 5D, E). Extent of expression of HCC markers Afp and Yap in tumors was greater in AD-Stard1 group (Figure 5F). Consistent with these findings, expression of tumor markers (Afp, Yap, Golm1 or Krt19) increased upon Stard1 overexpression (Figure 5G) and this outcome was accompanied by enhanced expression of inflammatory-related and hypoxia-regulated genes (Figure 5H–I). In addition, liver oxidative stress, as measured by dihydroethidium staining of liver sections from DEN+HFHC-fed mice overexpressing Stard1, was significantly higher than that measured in AD-control group (Figure 5J). Moreover, overexpression of Stard1 in subcutaneous tumors induced by TICs in immunodeficient mice resulted in induction of genes involved in pluripotency and stemness (Figure 5K), which paralleled the increase in tumor growth compared to TICs expressing Gfp control vector (Figure 5L). The tumor promoting effect of STARD1 required dietary cholesterol feeding (2% cholesterol, HC) (Figure 5M), as indicated by the findings that STARD1 overexpression in DEN+regular diet-fed mice or feeding HC diet alone for 24 weeks did not lead to HCC development (Figure 5N, O). Overall, these findings indicate that STARD1 and dietary cholesterol synergize to promote HCC development.
Figure 5. Stard1 overexpression increases DEN+HFHC-driven HCC.
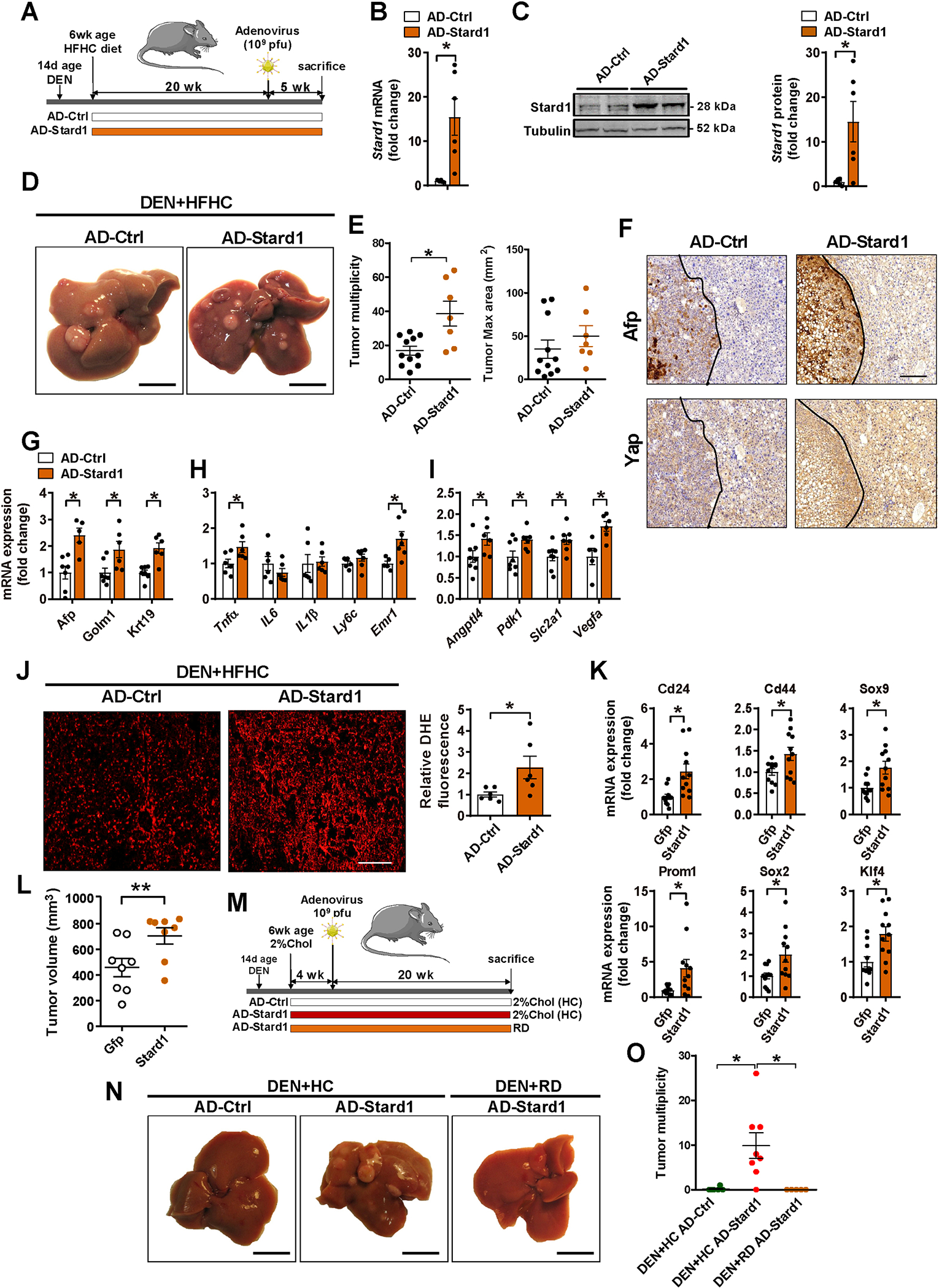
A) Schematic illustration of the experimental design used to overexpress Stard1 in wild type mice 5 months after DEN+HFHC treatment.
B) Adenoviral-induced overexpression of mouse Stard1 in liver of mice determined by qPCR. N=6 for each group.
C) Quantification of Stard1 overexpression by immunoblot densitometry and representative image of a Western blot for Stard1. N=6 for each group.
D-E) Representative images of livers and quantification of macroscopic liver tumor multiplicity and maximum size in animals after 5 weeks of recombinant adenovirus injection. N=11 for AD-Ctrl and N=7 for AD-Stard1.
F) Immunohistochemistry of consecutive sections showing the same tumor (T, delimited with a dotted line) and parenchyma stained for Afp, or Yap. Size bar 500 μmeter.
G, H, I) qPCR quantification of mRNA of HCC markers, fibrogenesis, inflammation and Hif1a target genes. N=6 per group.
J) ROS production measured and quantified in cryosections of liver tissue stained with DHE, (N=6).
K) mRNA levels of stemness genes measured in subcutaneous tumors induced by TICs with or without Stard1 overexpression. Values are mean ± SEM relative to the Gfp-expressing tumors (N=12). * denotes statistically significance in paired Student’s t test respect the matched Gfp-expressing tumors.
L) Subcutaneous tumor volume in nude mice induced by TICs transfected with control Gfp or Stard1. Values are mean ± SEM relative to the Gfp-expressing tumors (N=8). * denotes statistically significance in paired Student’s t test respect the matched Gfp-expressing tumors.
M) Schematic experimental design for adenoviral-mediated Stard1 overexpression (AD-Star) in DEN-treated wild type mice followed by feeding a regular diet (RD) (N=5) or a diet enriched in cholesterol (2%, HC) (N=8) or AD-Ctrl on HC diet (N=6).
N, O) Macroscopic images of livers from DEN-treated mice and quantification of tumor multiplicity.
All values are mean ± SEM. *p<0.05 denote statistically significant differences respect to Ad-Ctrl in a student’s t test.
STARD1 regulates the profile of hepatic BAs in NASH-driven HCC.
As BAs have been linked to NASH progression and HCC development 14, 17–19, we next addressed whether STARD1 regulates the profile of hepatic BAs in NASH-driven HCC. We performed mass spectrometry analyses of the hepatic molecular species of BAs in WT mice with STARD1 overexpression (AD-Stard1) and Stard1ΔHep mice. The total content of BAs in liver increased in AD-Stard1 mice with respect to AD-Ctrl mice (Figure 6A). These quantitative changes reflected the increase in unconjugated BAs, such as βMCA and CA and their tauroconjugated derivatives Tα/βMCA and TCA in AD-Stard1 mice, whose levels are one order of magnitude higher than those of TUDCA and TCDCA (Figure 6B, C). In contrast, the total liver BAs burden in Stard1ΔHep mice significantly decreased compared to Stard1f/f mice, with lower levels of TCA, βMCA and CA (Figure 6B, C). The amount of minor unconjugated BAs, i.e. UDCA, CDCA, DCA and HyoDCA, remained unchanged regardless of the status of Stard1 expression (Figure 6B). A similar decrease in the levels of βMCA, Tα/βMCs and TCA was observed in MUP-uPA-Stard1ΔHep mice with respect to MUP-uPA-Stardf/f mice (Figure S5). The levels of oxysterols 24S-hydroxycholesterol (24S-OH-Chol) and 27-hydroxycholesterol (27-OH-Chol), which are intermediates of BA synthesis in the acidic pathway 30, 31, did not change in AD-Stard1 or Stard1ΔHep mice (Figure S6A). Interestingly, the expression of Cyp7a1, Cyp8b1, Cyp27a1 and Cyp7b1 as well as Cyp27a1 and Cyp7a1 in AD-Stard1 or Stard1ΔHep mice remained unaltered (Figure S6B–E). In addition, while expression of FXR (Nr1h4) as well as that of Nr0b2 and Abcd11 decreased (50–60%) in AD-Stard1 mice overexpressing Stard1, the levels of Nr1h4 and its target genes Nr0b2, Abcb11 and Abcb4 in DEN plus HFHC-fed Stard1ΔHep mice was similar to DEN plus HFHC Stard1f/f mice (Figure S6F), suggesting the the regulatory role of Stard1 in BAs synthesis and HCC development is independent of FXR. These findings indicate that a significant proportion of hepatic BAs generated during HCC development were regulated by STARD1.
Figure 6. Molecular species of BAs in NASH-HCC models and their impact in expression of genes involved in self-renewal, stemness and inflammation.
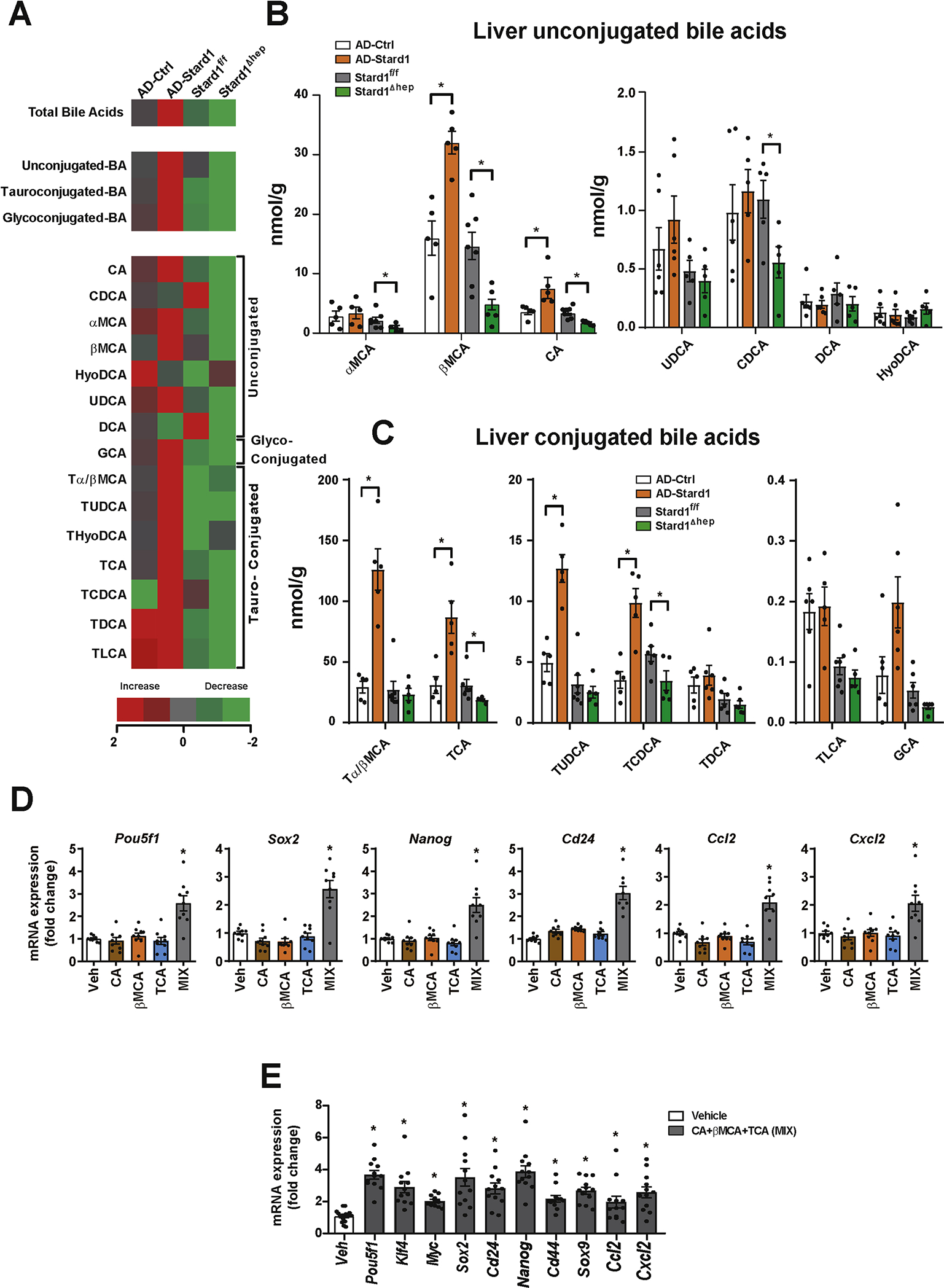
A) Heatmap of individual species of BAs measured in livers from AD-Stard1 mice and Stard1 ΔHep mice following DEN treatment and HFHC feeding, showing an increase (red) or a reduction (green) respect to the mean of AD-control and Stard1f/f mice. Values are Log2 of the fold change.
B) Quantification of the unconjugated BAs in liver tissue from each group of animals. (N=5 for AD-Ctrl/AD-Stard1 and N=6 for Stard1f/f and Stard ΔHep mice.
C) Quantification of tauroconjugated BAs in liver tissue from each group of animals.
D) mRNA levels of genes involved in self-renewal, stemness and inflammation in TICs following incubation with CA (50 μM), βMCA (50 μM) and TCA (200 μM) for 48 hours. Values are mean ± SEM. N=3 independent experiments performed in triplicates.
E) Effect of the combination of CA, βMCA and TCA in PMH for 24 hours on the mRNA levels of genes involved in self-renewal, stemness and inflammation.
Values are mean ± SEM. N=3 independent experiments performed in quadruplicates.
BAs induce the expression of genes involved in self-renewal, stemness and inflammation
To establish the link between STARD1-mediated regulation of BAs and HCC, we examined the impact of the profile of BAs regulated by STARD1 on the expression of transcription factors involved in self-renewal and pluripotency, which are of relevance for HCC pathogenesis 39, 51. TICs (CD133+/CD49f+) have been previously characterized and isolated from murine HCC models and shown to exhibit oncogenic activity and tumorigenicity 39, 52, 53. Treatment of TICs with the combination of CA, TCA and βMCA at a concentration mimicking the levels observed in AD-Stard1 mice overexpressing STARD1 increased the expression of Yamanaka transcription factors Sox2 and Pouf51 as well as the stemness markers Nanog and Cd24 and the inflammatory chemokines Ccl2 and Cxcl1 (Figure 6D). Quite interestingly, the level of expression of pluripotency and early differentiation genes in mature liver has been reported to be similar to those found in fetal liver and iPSC-derived hepatocyte-like cells 54. In line with previous findings 14 the incubation of PMH with CA, TCA and βMCA significantly increased the expression of Sox2, Myc, Klf4 and Pouf51, as well as the stemness-related and cancer stem cell markers Cd24, Cd44, Sox9 and Nanog, and the inflammatory genes Ccl2 and Cxcl2 (Figure 6E). Although CDCA and secondary BAs, DCA and LCA were cytotoxic to TICs, lower concentrations (10μM) of these BAs induced the expression of genes involved in self-renewal, stemness and inflammation (Figure S6G).
DISCUSSION
The NASH-driven HCC subset is a growing public health burden as is expected to increase worldwide due to its association with obesity and type II diabetes. Extending previous observations on the alterations of cholesterol homeostasis in human NASH 33, 34, we show here an upregulation in the expression of STARD1 in patients with NASH-derived HCC. Importantly, increasing or decreasing the expression of STARD1 in mice results in the stimulation or attenuation, respectively, of liver cancer, indicating that the induction of STARD1 in patients can be a cause of NASH-driven HCC. The physiological role of STARD1 in the liver is to provide cholesterol to the mitochondrial inner membrane for its biotransformation into BAs; however, the contribution of this pathway to NASH-driven HCC has not been explored so far. We provide evidence that STARD1 expression determines the level and composition of hepatic BAs in models of NASH-driven HCC, and establish a link whereby STARD1 promotes HCC by stimulating the synthesis of BAs through the mitochondrial alternative pathway.
The metabolism of cholesterol within mitochondria begins by its hydroxylation at position 27 by CYP27A1 yielding 27-OH-Chol, which then feeds the BAs synthesis via CYP7B1 to generate CDCA. Previous evidence has shown that STARD1 rather than CYP27A1 is the rate-limiting step in the alternative pathway of BAs synthesis. As shown in primary hepatocytes or HepG2 cells, the overexpression of STARD1 resulted in a 5-fold increase in the rate of BAs synthesis, while transfection with CYP27A1 upregulated BAs synthesis by 2-fold 55, 56. In line with this notion, the impact of modulating STARD1 expression in NASH-driven HCC development parallels the generation of BA species, with increased or decreased total BA pool in mice overexpressing STARD1 or Stard1ΔHep mice, respectively, while the levels of oxysterols 24S-OH-Chol and 27-OH-Chol remained unaltered. Moreover, these effects of STARD1 expression in BAs synthesis through the alternative pathway are not dependent on the status of Cyp27a1/Cyp7b1 expression, which remained unchanged regardless of STARD1 levels, further establishing the crucial role of STARD1 in regulating cholesterol biotransformation into BAs. These findings imply that although the classical pathway regulated by CYP7A1 is considered the predominant route of cholesterol-mediated BAs synthesis in hepatocytes, the STARD1-dependent BAs synthesis through the alternative pathway may take over the classical pathway in diseased states in which both cholesterol and STARD1 are induced, such as NASH-driven HCC. In line with this possibility, ER stress, a crucial player in NASH-HCC development, has been identified as a new mechanism that regulates BAs synthesis by decreasing the expression of CYP7A1 57. CA is the predominant BA synthesized through the classical pathway regulated by CYP7A1, which requires the action of CYP8B1 to add the 12α-hydroxylation characteristic of CA. In contrast, the other primary BA, i.e. CDCA, is predominantly synthesized through the alternative pathway via CYP27A1 and CYP7B1. While in humans, CDCA is further metabolized by intestinal bacteria to LCA, in rodents CDCA is biotransformed by hepatocytes through what can be considered a surrogate of the alternative pathway of BA synthesis into the trihydroxylated BA αMCA in positions 3α, 6β and 7α and its 7β-epimer βMCA 15, 30–32. Accordingly, our data indicate that modulation of STARD1 expression in mice by its overexpression or deletion in hepatocytes results in increased or curtailed levels of βMCA and its tauroconjugated form, TβMC, a potent FXR antagonist that relieves the FXR-mediated downregulation of CYP7A1 but not of CYP27A1 58, 59. The lack of change in the expression of CYP7A1 in the NASH-driven HCC may reflect the counterbalance between the indirect stimulating effect of TβMCA via antagonism of FXR and the suppressing action of chronic ER stress 57. Intriguingly, we show an unanticipated STARD1-dependent modulation of CA and its subsequent TCA generation in NASH-driven HCC. In line with this link, it has been recently described that in addition to the conversion of 7α-hydroxycholest-4-en-3-one to 7α, 12α-dihydroxycholest-4-en-3-one CYP8B1 can also biotransform CDCA itself into CA 60.
To address whether the tumor promoter role of STARD1 in NASH-driven HCC is linked to the regulation of the alternative pathway of BAs synthesis, we examined the impact of BAs (CA, βMCA and TCA) mimicking the profile regulated by STARD1 in the expression of self-renewal and stemness genes involved in HCC 39, 51. This profile of BAs induced the expression of genes involved in self-renewal, stemness and inflammation in TICs and PMH. Of interest, the findings in PMH are in line with previous reports indicating a similar level of expression of pluripotency and early differentiation genes in mature liver versus fetal liver or iPSC-derived hepatocyte-like cells 14, 54. Moreover, the direct link between STARD1 and BAs synthesis through the alternative pathway in HCC pathogenesis is consistent with the recognized role of BAs in promoting NASH progression and HCC development 16–19. Feeding WT mice with a CA-enriched diet increased hepatic BA pool and potentiated liver carcinogenesis 14. Furthermore, the spontaneous development of HCC in Fxr−/− mice has been shown to be reversed by decreasing BAs levels by cholestyramine 17, 19, which is reminiscent of the outcome of Stard1ΔHep mice.
The characterization of the molecular BAs species directly regulated by STARD1 expression has been limited to mice. While in human NASH-driven HCC samples we observed a correlation between STARD1 expression and increased total hepatic BAs pool, the full characterization of the individual BAs generated would require increased sample size to perform a mass spectrometry analysis. In this regard, we undertook an initial approach to address the role of secondary BAs DCA plus LCA, which in addition to their cytotoxic effects in TICs induced the expression of genes involved in self-renewal, stemness and inflammation. Another intriguing finding that deserves further research is the role of TUDCA in the STARD1-dependent HCC development. While exogenous administration of TUDCA has been shown to protect against liver tumorigenesis due to its anti-ER stress effects 7, 8, the levels of TUDCA generated in the AD-Stard1 mice were one order of magnitude lower than TMCAs and TCA.
Supplementary Material
HIGHLIGHTS.
Human NASH-driven HCC tissue specimens exhibit increased STARD1 expression
STARD1 overexpression promotes whereas STARD1 ablation curtails NASH-driven HCC
STARD1 stimulates BAs synthesis through activation of the alternative mitochondrial pathway
BAs stimulate pluripotency, stemness and inflammation related genes in tumor-initiating stem-like cells and hepatocytes
ACKNOWLEDGMENTS:
MUP-uPA transgenic mice were kindly provided by Eric P. Sandgren from the University of Wisconsin-Madison, (USA). We are grateful to Keigo Machida (University of Southern California, Los Angeles) for the generous gift of TICs. We are indebted to the Biobank core facility of the Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS) for technical help. This work was developed at the Centre Esther Koplowitz, Barcelona, Spain. We want to thank Dr. Fabián Arenas for help with graphic design.
Grant Support
We acknowledge the support from grants SAF-2015–69944R, and SAF2017–85877R and SAF2015–73579-JIN from Plan Nacional de I+D funded by the Agencia Estatal de Investigación (AEI) and the Fondo Europeo de Desarrollo Regional (FEDER) and from the CIBEREHD; the center grant P50AA011999 Southern California Research Center for ALPD and Cirrhosis funded by NIAAA/NIH; as well as support from AGAUR of the Generalitat de Catalunya SGR-2017–1112, European Cooperation in Science & Technology (COST) ACTION CA17112 Prospective European Drug-Induced Liver Injury Network, the “ER stress-mitochondrial cholesterol axis in obesity-associated insulin resistance and comorbidities”-Ayudas FUNDACION BBVA and the Red Nacional 2018–102799-T de Enfermedades Metabólicas y Cáncer. We also want to thank the support from the “Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III”, Spain (PI16/00598, co-funded by European Regional Development Fund/European Social Fund, “Investing in your future”); “Centro Internacional sobre el Envejecimiento” (OLD-HEPAMARKER, 0348_CIE_6_E), Spain. We also acknowledge support from R01 CA2344128 and U01 AA022614 grants to M.K.
ABBREVIATIONS:
- BA
Bile acids
- βMCA
7β-epimer β-muricholic acid
- CA
Cholic acid
- CDCA
Chenodeoxycholic acid
- DEN
Diethylnitrosamine
- EZE
Endoplasmic reticulum, ER Ezetimibe
- GST-PFO
GST-perfringolysin
- 27-OH-chol
24S-hydroxycholesterol, 24S-OH-chol 27-hydroxycholesterol
- HFD
High fat diet
- HFHC
High fat high cholesterol diet
- NASH
Nonalcoholic steatohepatitis
- PBC
Primary biliary cholangitis
- PMH
Primary mouse hepatocytes
- TG
Triglycerides
- STARD1
Steroidogenic acute regulatory protein 1
- (TICs)
Tumor-initiating stem-like cells
Footnotes
Conflict of Interest: The authors disclose no conflicts
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability:
data that support the study findings are available upon reasonable request from the corresponding authors (CGR, VR, JCFC). Detailed information on experimental protocols may also be shared on reasonable request.
REFERENCES
- [1].Bianchini F, Kaaks R, Vainio H. Overweight, obesity, and cancer risk. The lancet oncology 2002;3:565–574. [DOI] [PubMed] [Google Scholar]
- [2].Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. The New England journal of medicine 2003;348:1625–1638. [DOI] [PubMed] [Google Scholar]
- [3].Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet 2018;391:1301–1314. [DOI] [PubMed] [Google Scholar]
- [4].Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016;150:835–853. [DOI] [PubMed] [Google Scholar]
- [5].Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nature reviews Gastroenterology & hepatology 2013;10:34–42. [DOI] [PubMed] [Google Scholar]
- [6].Marin JJG, Herraez E, Lozano E, Macias RIR, Briz O. Models for Understanding Resistance to Chemotherapy in Liver Cancer. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer cell 2014;26:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab 2019;29:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Che L, Chi W, Qiao Y, Zhang J, Song X, Liu Y, et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2020;69:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liang JQ, Teoh N, Xu L, Pok S, Li X, Chu ESH, et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat Commun 2018;9:4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bakiri L, Hamacher R, Grana O, Guio-Carrion A, Campos-Olivas R, Martinez L, et al. Liver carcinogenesis by FOS-dependent inflammation and cholesterol dysregulation. The Journal of experimental medicine 2017;214:1387–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu D, Wong CC, Fu L, Chen H, Zhao L, Li C, et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Science translational medicine 2018;10. [DOI] [PubMed] [Google Scholar]
- [14].Sun L, Beggs K, Borude P, Edwards G, Bhushan B, Walesky C, et al. Bile acids promote diethylnitrosamine-induced hepatocellular carcinoma via increased inflammatory signaling. Am J Physiol Gastrointest Liver Physiol 2016;311:G91–G104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gadaleta RM, van Mil SW, Oldenburg B, Siersema PD, Klomp LW, van Erpecum KJ. Bile acids and their nuclear receptor FXR: Relevance for hepatobiliary and gastrointestinal disease. Biochim Biophys Acta 2010;1801:683–692. [DOI] [PubMed] [Google Scholar]
- [16].Puri P, Daita K, Joyce A, Mirshahi F, Santhekadur PK, Cazanave S, et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018;67:534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007;28:940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, Portmann BC, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006;44:478–486. [DOI] [PubMed] [Google Scholar]
- [19].Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 2007;67:863–867. [DOI] [PubMed] [Google Scholar]
- [20].Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab 2006;4:185–198. [DOI] [PubMed] [Google Scholar]
- [21].Solsona-Vilarrasa E, Fucho R, Torres S, Nunez S, Nuno-Lambarri N, Enrich C, et al. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox biology 2019;24:101214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basanez G, et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res 2008;68:5246–5256. [DOI] [PubMed] [Google Scholar]
- [23].Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell death and differentiation 2008;15:484–493. [DOI] [PubMed] [Google Scholar]
- [24].Christenson E, Merlin S, Saito M, Schlesinger P. Cholesterol effects on BAX pore activation. Journal of molecular biology 2008;381:1168–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Smith B, Land H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell reports 2012;2:580–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ribas V, Garcia-Ruiz C, Fernandez-Checa JC. Mitochondria, cholesterol and cancer cell metabolism. Clinical and Translational Medicine 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Alpy F, Tomasetto C. START ships lipids across interorganelle space. Biochimie 2014;96:85–95. [DOI] [PubMed] [Google Scholar]
- [28].Clark BJ. The mammalian START domain protein family in lipid transport in health and disease. The Journal of endocrinology 2012;212:257–275. [DOI] [PubMed] [Google Scholar]
- [29].Elustondo P, Martin LA, Karten B. Mitochondrial cholesterol import. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862:90–101. [DOI] [PubMed] [Google Scholar]
- [30].Pandak WM, Kakiyama G. The acidic pathway of bile acid synthesis: Not just an alternative pathway(). Liver Res 2019;3:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kakiyama G, Marques D, Takei H, Nittono H, Erickson S, Fuchs M, et al. Mitochondrial oxysterol biosynthetic pathway gives evidence for CYP7B1 as controller of regulatory oxysterols. J Steroid Biochem Mol Biol 2019;189:36–47. [DOI] [PubMed] [Google Scholar]
- [32].Vaz FM, Ferdinandusse S. Bile acid analysis in human disorders of bile acid biosynthesis. Molecular aspects of medicine 2017;56:10–24. [DOI] [PubMed] [Google Scholar]
- [33].Caballero F, Fernandez A, De Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol 2009;50:789–796. [DOI] [PubMed] [Google Scholar]
- [34].Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab 2012;15:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. The New England journal of medicine 1996;334:693–699. [DOI] [PubMed] [Google Scholar]
- [36].Torres S, Baulies A, Insausti-Urkia N, Alarcon-Vila C, Fucho R, Solsona-Vilarrasa E, et al. Endoplasmic Reticulum Stress-Induced Upregulation of STARD1 Promotes Acetaminophen-Induced Acute Liver Failure. Gastroenterology 2019. [DOI] [PubMed] [Google Scholar]
- [37].Weglarz TC, Degen JL, Sandgren EP. Hepatocyte transplantation into diseased mouse liver. Kinetics of parenchymal repopulation and identification of the proliferative capacity of tetraploid and octaploid hepatocytes. Am J Pathol 2000;157:1963–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Baulies A, Montero J, Matias N, Insausti N, Terrones O, Basanez G, et al. The 2-oxoglutarate carrier promotes liver cancer by sustaining mitochondrial GSH despite cholesterol loading. Redox biology 2018;14:164–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, et al. Reciprocal regulation by TLR4 and TGF-beta in tumor-initiating stem-like cells. J Clin Invest 2013;123:2832–2849. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [40].Arenas F, Castro F, Nunez S, Gay G, Garcia-Ruiz C, Fernandez-Checa JC. STARD1 and NPC1 expression as pathological markers associated with astrogliosis in post-mortem brains from patients with Alzheimer’s disease and Down syndrome. Aging 2020;12:571–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, et al. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018;175:133–145 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yang Z, Qin W, Chen Y, Yuan B, Song X, Wang B, et al. Cholesterol inhibits hepatocellular carcinoma invasion and metastasis by promoting CD44 localization in lipid rafts. Cancer letters 2018;429:66–77. [DOI] [PubMed] [Google Scholar]
- [43].Zhao Z, Zhong L, He K, Qiu C, Li Z, Zhao L, et al. Cholesterol attenuated the progression of DEN-induced hepatocellular carcinoma via inhibiting SCAP mediated fatty acid de novo synthesis. Biochemical and biophysical research communications 2019;509:855–861. [DOI] [PubMed] [Google Scholar]
- [44].Lee YL, Li WC, Tsai TH, Chiang HY, Ting CT. Body mass index and cholesterol level predict surgical outcome in patients with hepatocellular carcinoma in Taiwan - a cohort study. Oncotarget 2016;7:22948–22959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Carr BI, Giannelli G, Guerra V, Giannini EG, Farinati F, Rapaccini GL, et al. Plasma cholesterol and lipoprotein levels in relation to tumor aggressiveness and survival in HCC patients. Int J Biol Markers 2018;33:423–431. [DOI] [PubMed] [Google Scholar]
- [46].Qin WH, Yang ZS, Li M, Chen Y, Zhao XF, Qin YY, et al. High Serum Levels of Cholesterol Increase Anti-tumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology 2020. [DOI] [PubMed] [Google Scholar]
- [47].Jang JE, Park HS, Yoo HJ, Baek IJ, Yoon JE, Ko MS, et al. Protective role of endogenous plasmalogens against hepatic steatosis and steatohepatitis in mice. Hepatology 2017;66:416–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, et al. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019;69:2241–2257. [DOI] [PubMed] [Google Scholar]
- [49].Loomba R, Sirlin CB, Ang B, Bettencourt R, Jain R, Salotti J, et al. Ezetimibe for the treatment of nonalcoholic steatohepatitis: assessment by novel magnetic resonance imaging and magnetic resonance elastography in a randomized trial (MOZART trial). Hepatology 2015;61:1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Miura K, Ohnishi H, Morimoto N, Minami S, Ishioka M, Watanabe S, et al. Ezetimibe suppresses development of liver tumors by inhibiting angiogenesis in mice fed a high-fat diet. Cancer Sci 2019;110:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annual review of cell and developmental biology 2007;23:675–699. [DOI] [PubMed] [Google Scholar]
- [52].Rountree CB, Ding W, He L, Stiles B. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem cells 2009;27:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ding W, Mouzaki M, You H, Laird JC, Mato J, Lu SC, et al. CD133+ liver cancer stem cells from methionine adenosyl transferase 1A-deficient mice demonstrate resistance to transforming growth factor (TGF)-beta-induced apoptosis. Hepatology 2009;49:1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zabulica M, Srinivasan RC, Vosough M, Hammarstedt C, Wu T, Gramignoli R, et al. Guide to the Assessment of Mature Liver Gene Expression in Stem Cell-Derived Hepatocytes. Stem Cells Dev 2019;28:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pandak WM, Ren S, Marques D, Hall E, Redford K, Mallonee D, et al. Transport of cholesterol into mitochondria is rate-limiting for bile acid synthesis via the alternative pathway in primary rat hepatocytes. J Biol Chem 2002;277:48158–48164. [DOI] [PubMed] [Google Scholar]
- [56].Ren S, Hylemon PB, Marques D, Gurley E, Bodhan P, Hall E, et al. Overexpression of cholesterol transporter StAR increases in vivo rates of bile acid synthesis in the rat and mouse. Hepatology 2004;40:910–917. [DOI] [PubMed] [Google Scholar]
- [57].Henkel AS, LeCuyer B, Olivares S, Green RM. Endoplasmic Reticulum Stress Regulates Hepatic Bile Acid Metabolism in Mice. Cell Mol Gastroenterol Hepatol 2017;3:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013;17:225–235. [DOI] [PubMed] [Google Scholar]
- [59].Worthmann A, John C, Ruhlemann MC, Baguhl M, Heinsen FA, Schaltenberg N, et al. Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nature medicine 2017;23:839–849. [DOI] [PubMed] [Google Scholar]
- [60].Fan L, Joseph JF, Durairaj P, Parr MK, Bureik M. Conversion of chenodeoxycholic acid to cholic acid by human CYP8B1. Biological chemistry 2019; 400:625–628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
data that support the study findings are available upon reasonable request from the corresponding authors (CGR, VR, JCFC). Detailed information on experimental protocols may also be shared on reasonable request.