

Abstract
Genomic epidemiology is a core component in investigating the spread of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). In this study, the efficacy of control strategies in South Korea was evaluated using genomic epidemiology based on viral genome sequences of 2,065 SARS-CoV-2 cases identified in South Korea from January 2020 to December 2020. Phylogenetic analysis revealed that the majority of viruses introduced from inbound travelers did not further spread throughout South Korea; however, four distinct subgroups (KR.1–4, belonging to B.1.497, B.1, K.1 and B.41) of viruses caused local epidemics. After the introduction of enhanced social distancing, the viral population size and daily case numbers decreased, and KR.2–4 subgroups were extinguished from South Korea. Nevertheless, there was a subsequent increase in KR.1 subgroups after the downgrading of social distancing level. These results indicate that the international traveler quarantine system implemented in South Korea along with social distancing measures efficiently reduced the introduction and spread of SARS-CoV-2, but it was not completely controlled. An improvement of control strategies will be required to better control SARS-CoV-2, its variants, and future pandemic viruses.
Keywords: SARS-CoV-2, genomic epidemiology, phylogeography, social distancing, traveler quarantine
1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has resulted in a global pandemic, leading to a total of 82,380,200 confirmed cases worldwide and 1,800,818 deaths from December 2019 to December 2020 (Wu et al., 2020; WHO, 2021). Despite the implementation of global disease control strategies, including travel restriction and social distancing, SARS-CoV-2 has spread worldwide, causing a significant change in life for human population throughout the world (Devi 2020; Fazio et al., 2021). Besides, new variants bearing multiple spike protein mutations were recently detected and have rapidly spread to other countries (Campbell et al., 2021; Davies et al., 2021; Tang, Tambyah, and Hui 2020; Tegally et al., 2021). Hence, there is a need to evaluate the viral transmission patterns and efficacy of control strategy to inform the design of improved prevention and control strategies for current or future pandemic diseases.
Previous studies have indicated that 40–45 per cent of SARS-CoV-2 infections were asymptomatic, allowing the efficient spread of the virus to other people (Gandhi, Yokoe, and Havlir 2020; Oran and Topol 2020; Yanes-Lane et al., 2020). The asymptomatic transmission and numerous multiple outbreaks have impeded contact tracing and epidemiological investigations (Gandhi, Yokoe, and Havlir 2020; Rockett et al., 2020; Seemann et al., 2020). To date, the integration of epidemiological data and genomic analysis has contributed to providing the detailed epidemiology of SARS-CoV-2 (Alpert et al., 2021; du Plessis et al., 2021). The land border of South Korea is completely blocked and all inbound travelers are required to follow 14-day quarantine protocol since 1 April 2020, including SARS-CoV-2 test at arrival and mandatory 14-day isolation at government-designated facility or their own residences (KDCA, 2020). Thus the analysis of SARS-CoV-2 epidemiology in South Korea could demonstrate the efficacy of the national disease control strategies without outside influence on disease transmission. In the present study, we analyzed the nucleotide sequences of the 2,065 SARS-CoV-2 cases identified in South Korea from January 2020 to December 2020, accounting for 3.40 per cent of the total confirmed cases (n = 61,769). The genomic epidemiology of SARS-CoV-2 in South Korea was analyzed by incorporating the travel history and location data of infected persons into phylogenetic analysis.
2. Materials and methods
2.1. Virus detection and full-genome sequencing
Nasopharyngeal and oropharyngeal swabs and sputum specimens were collected from symptomatic patients to detect SARS-CoV-2 by real-time reverse transcriptase-polymerase chain reaction (RT-PCR). RNA was extracted from 140 μL of the sample using a Qiagen viral RNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Real-time RT-PCR was performed on the extracted RNA, and the cycle threshold value of the SARS-CoV-2 target gene was determined (Kim et al., 2020).
Samples were selected for sequencing to maximize epidemiologic breadth. As such, samples were chosen based on the epidemiological links inferred from outbreak investigation data. We selected samples from sporadic cases, and we randomly selected a few representative samples from epidemiologically linked large outbreaks. Approximately 5 per cent of viruses detected from Korean residents and 10 per cent of viruses detected from inbound travelers were selected for whole-genome sequence analysis. We obtained a total of 2,065 complete genome sequences, including 2.94 per cent of Korean resident cases (1,656 sequences/56,359 cases) and 7.56 per cent of inbound traveler cases (409 sequences/5,410 cases).
For full-genome sequencing, cDNA was amplified using the ARTIC primer pools (https://artic.network/ncov-2019). Libraries were prepared using the Nextera DNA Flex Library Prep Kit (Illumina, USA), and sequencing was performed on the MiSeq instrument using a MiSeq reagent kit V2 (Illumina, USA) to obtain an average genome coverage of >1000× for all samples. The reads were trimmed and mapped to the reference genome Wuhan-Hu-1 (GenBank:MN908947.3) using CLC Genomics Workbench version 20.0.3 (CLC Bio, Denmark) (Park et al., 2021). The lineages and clades of SARS-CoV-2 sequences were assigned by PANGOLIN (Rambaut et al., 2020) (https://cov-lineages.org/) and Nextclade Beta (https://clades.nextstrain.org/).
2.2. Phylogenetic analysis to identify multiple introductions
All available SARS-CoV-2 full-genome sequences identified as of 31 December 2020, in other countries, with <1 per cent ambiguous nucleotides, were downloaded from the GISAID EpiCoV™ database (https://www.gisaid.org/) for reference sequences (n = 2,33,456). For efficient computation and better visualization without losing the major clade structure and continental distribution, we selected 3,896 representative sequences (249 from Asia; 2,108 from Europe; 1,051 from North America; 64 from South America; 169 from Africa; 255 from Oceania) based on clustering with nucleotide sequence identity at 99.7 per cent level for each continent separately using the program cd-hit (Li and Godzik 2006). The number of sequences from South Korea was reduced from 2,065 to 1,278 after excluding the identical sequences using the program cd-hit. Each sequence was aligned to the reference sequence Wuhan-Hu-1 using the Geneious Prime software (https://www.geneious.com/). The aligned sequences were manually trimmed to equal lengths (29,409 bp) from the start codon of ORF1ab to the stop codon of ORF10. The temporal signal of the dataset was examined by Tempest (Rambaut et al., 2016) using an approximately maximum-likelihood phylogenetic tree constructed by FastTree under the generalized time-reversible nucleotide substitution model with gamma-distributed rates among sites (general time reversible + gamma [GTR + γ]) (Price, Dehal, and Arkin 2009). The outlier sequences, which have >8.0 × 10–4 deviation (about 24 mutations) from the regression line in the root-to-tip regression, were excluded, and the remaining sequences were used as the final dataset (n = 5,145) (Figure S1). An approximately maximum-likelihood phylogenetic tree was reconstructed by FastTree under the GTR + γ nucleotide substitution model using the final dataset. The phylogenetic tree was visualized and annotated using iTOL (https://itol.embl.de/).
The monophyletic cluster sharing a common ancestral node with other Korean viruses with a FastTree support value of >0.7 and belonging to the same Pangolin lineage was defined as an independent viral introduction from outside of Korea. The viruses detected from distinct inbound travelers having different travel histories were defined as a distinct viral introduction. Fisher’s exact test was used to analyze the statistical significance of the number of virus introductions against the total number of viral clusters before and after the implementation of all the traveler quarantine strategies.
2.3. Time-scaled phylogenetic analysis to identify the temporal distribution of SARS-CoV-2 lineages in South Korea
To identify the temporal distribution of SARS-CoV-2 subgroups and their phylogenetic relationship in South Korea, we constructed a time-scaled phylogenetic tree using BEAST v.1.10.4 (Suchard et al., 2018). Among the 2,065 SARS-CoV-2 virus sequences from South Korea, we selected 942 sequences, after excluding the viruses detected from inbound travelers and identical sequences. The posterior phylogenetic tree distribution was estimated using Bayesian phylogenetic inference. The GTR + γ nucleotide substitution model was selected through the Bayesian phylogenetic site model averaging using the bModelTest (Bouckaert and Drummond 2017). To estimate changes in the viral population size with a flexible approach, a nonparametric tree model and Gaussian Markov random field (GMRF) Bayesian Skyride coalescent tree prior were used with an uncorrelated relaxed (UCLD) clock model. The Markov chain Monte Carlo (MCMC) was run in parallel for three chains, each with 100 million steps, and the parameters and trees were sampled every 10,000 steps. The parameters were analyzed using TRACER v1.7.1 (Rambaut et al., 2018) and burned-in 40–50 per cent of the result. Most of the parameters had an effective sample size (ESS) of >200 (Table S5). The resulting log and tree files were combined with LogCombiner v1.10.4 (https://beast.community/logcombiner), yielding a total of 16,003 parameter states and posterior trees. Time-scaled maximum clade credibility (MCC) tree was generated using TreeAnnotator (https://beast.community/treeannotator) in BEAST and visualized using FigTree 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
2.4. Phylodynamic analysis of KR.1 and KR.4 subgroups to identify the transmission dynamic in South Korea
Two major South Korean subgroups, KR.1 and KR.4, were monophyletic subgroups that do not include viruses from travelers and other countries (Fig. 1A). Both KR.1 and KR.4 were detected in multiple provinces in South Korea over a relatively long period. Thus, we selected these subgroups for within-country transmission dynamic analysis of SARS-CoV-2 in South Korea.
Figure 1.
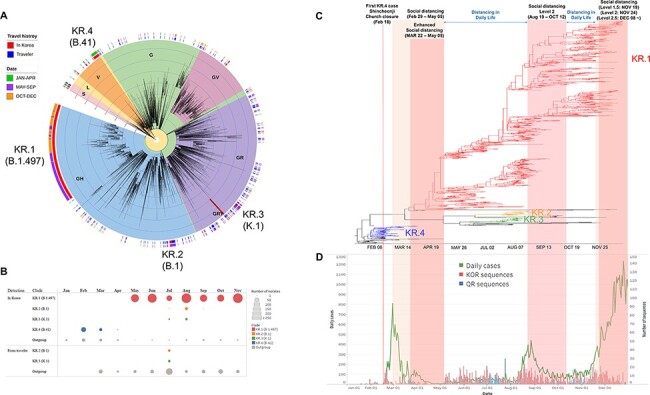
Multiple introductions of SARS-CoV-2 into South Korea. (A) Approximately maximum-likelihood phylogenetic tree of the 5,145 SARS-CoV-2 full genomes. The SARS-CoV-2 viruses detected from travelers entering South Korea or their direct contacts were annotated by blue color strips, and viruses detected from South Korean residents with no travel history were annotated by red color strips. Internal tree scales for branch lengths indicate nucleotide substitutions per site. (B) The number of sequences of each Korean subgroup in each month. The viruses that do not belong to KR.1–4 were classified as outgroup. (C) A time-scaled maximum clade credibility tree of all SARS-CoV-2 detected from South Korean residents. Branches are colored according to the Korean subgroups detected in Fig. 1A. (D) The daily case number of SARS-CoV-2 (left axis) and the number of analyzed sequences (right axis) in South Korea.
All the SARS-CoV-2 sequences belonging to the KR.4 subgroup were used for phylogeography analysis of the KR.4 subgroup. For the KR.1 subgroup, the sequence data were reduced from 1,418 to 798 sequences after excluding the identical sequences from the same geographic location. The sequences of KR.1 and KR.4 subgroups were coded into geographic locations in South Korea (Table S1). The Bayesian phylogeography in discrete space was estimated using BEAST v.1.10.4 (https://beast.community) with same substitution model (GTR + γ), clock model (UCLD) and tree prior (GMRF Bayesian Skyride). The ancestral locations were reconstructed, and the asymmetric viral exchanges between regions were estimated using the nonreversible continuous-time Markov chain model. We applied a Bayesian stochastic search variable selection procedure to identify the best-supported transitions between discrete states using the Bayes factors test, which is a feature of the SPREAD3 software v0.9.6 (Lemey et al., 2009; Bielejec et al., 2016). The migration routes were visualized using SPREAD3. A transition was identified as significant when the Bayes factor was >6 and the posterior probability was >0.5.
For the KR.4 subgroup, the MCMC method was run in parallel for three chains, each with 100 million steps, and the parameters and trees were sampled every 10,000 steps. The resulting log and tree files were combined using LogCombiner v1.10.4 (https://beast.community/logcombiner) after 10 per cent burn-in, yielding a total of 27,003 parameter states and posterior trees. The parameters were analyzed using TRACER v1.7.1, and all the parameters had an ESS of >200 (Table S5). For the KR.1 subgroup, the MCMC method was run in parallel for ten chains, each with 100 million steps, and the parameters and trees were sampled every 10,000 steps. The resulting log and tree files were combined using LogCombiner v1.10.4 (https://beast.community/logcombiner) after 40–50 per cent burn-in, yielding a total of 57,010 parameter states and posterior trees. Most of the parameters had an ESS of >200 (Table S5). An MCC trees were generated using TreeAnnotator (https://beast.community/treeannotator) in BEAST and visualized using FigTree 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
The rate and number of transitions among regions (Markov jumps) were estimated using stochastic mapping techniques implemented in the BEAST package (Minin and Suchard 2008). We analyzed posterior trees using the PACT program (http://www.trevorbedford.com/pact) to compute the number of transition events between regions through time. The posterior trees were broken up into multiple temporal sections (half a month per section). The migrations of location state at the tree nodes were counted for each time window for each posterior tree.
3. Results
3.1. Reduced virus introductions and subsequent spread in South Korea
The phylogenetic tree of the global and Korean SARS-CoV-2 sequences indicated that at least 261 phylogenetically distinct viral clusters were introduced into South Korea (Fig. 1A). However, only thirty-five viral clusters were detected in South Korean residents who had no travel history, including four clusters containing viruses detected from both inbound travelers and Korean residents. Other 226 clusters were detected from only inbound travelers without subsequent spread in South Korea (Fig. 1, Table S1). All travelers entering South Korea were subject to the 14-day quarantine protocol implemented from 1 April 2020, following the special entry procedures applied to arrivals from outbreak countries from February (KDCA, 2020). The virus introduction from travelers to Korean residents was significantly reduced after April 14 (April 14 to December 31: 21 clusters from resident/215 total introductions) compared to that before (January 01 to April 13: 14 clusters from resident/46 total introductions) (P = 0.0006) (Fig. 1, Table S1).
Among the thirty-five viral clusters detected in South Korea, only four clusters (KR.1–4) caused multiple outbreaks in South Korea for more than 2 weeks (Fig. 1). The KR.1 subgroup, belonging to GH (GISAID clade; https://www.gisaid.org/) and B.1.497 (PANGOLIN lineage; https://cov-lineages.org/), was first detected on 5 May 2020 and became the predominant subgroup (number of sequences = 1,418) in South Korea (Fig. 1). The KR.1 subgroup viruses were clustered with North American viruses in the phylogenetic tree, but any related virus was not detected from inbound travelers. The KR.2 and KR.3 subgroups have been detected in South Korea since July 2020 (Fig. 1, Table S1). The KR.2 subgroup belonged to GH clade and B.1 lineage, and the KR.3 subgroup belonged to GR clade and K.1 lineage. Both KR.2 and KR.3 subgroups were detected from close contacts of foreign ship crew members before its spread in South Korea (Table S1). The outbreaks of KR.2 and KR.3 subgroup viruses were concentrated in Busan city and have not been detected since 1 September 2020 (Fig. 1, Table S1). The KR.4 subgroup viruses belonged to V clade and B.41 lineage. The KR.4 subgroup was predominant during the early outbreak period but has not been detected since April 2020 (Fig. 1, Table S1).
3.2. KR.4 (B.41) subgroup caused large-scale outbreak but disappeared from South Korea
The KR.4 subgroup viruses were detected during February 17 to April 2 in South Korea. In the initial stage, the majority of KR.4 subgroup viruses, including its first case, were identified from Shincheonji Church members or their contacts in Daegu city (Table S1). The first confirmed case of the KR.4 subgroup was reported on February 18, but the mean times for the most recent common ancestor (tMRCA) of all KR.4 subgroup viruses detected in South Korea was determined to be 23 January 2020 (95 per cent height posterior density [HPD]: January 10 to 4 February 2020) (Fig. 2, Table S2). The results indicate that the transmissions of the KR.4 subgroup to South Korea most probably occurred ≈2–6 weeks before the initial detection (Fig. 2, Table S2). However, the possibility of the multiple introductions of KR.4 subgroup viruses during mid-February could not be excluded.
Figure 2.
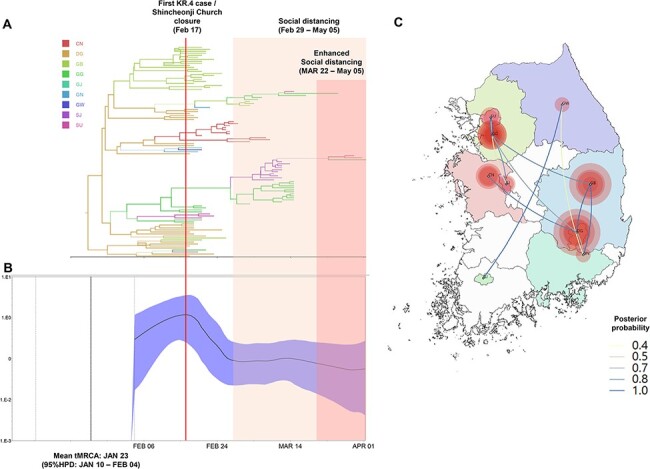
Phylogeography and effective population size analysis of KR.4 (B.41) subgroup viruses. (A) A time-scaled maximum clade credibility tree of the KR.4 subgroup SARS-CoV-2. Branches are colored according to locations in South Korea, and the thickness of branches indicates the posterior probabilities of the inferred ancestral location. (B) Gaussian Markov random field Bayesian Skyride plots indicating effective population size (relative genetic diversity) over time. (C) Spatiotemporal reconstruction of the spread of the KR.4 subgroup SARS-CoV-2 in South Korea. The diameters of the circles represent the number of branches maintaining a particular location state at each time period. The color of the transmission line represents the posterior possibility of each transmission. CN, Chungcheongnamdo; DG, Daegu; GB, Gyeongsangbukdo; GG, Gyeonggido; GJ, Gwangju; GN, Gyeongsangnamdo; GW, Gangwondo; SJ, Sejong; and SU, Seoul.
We conducted Bayesian phylogenetic analysis to determine the changes in the viral effective population size and the spatiotemporal transmission of the KR.4 subgroup by ancestral reconstruction of the geographical information (Fig. 2, Video S1). The virus population dynamic analysis revealed that the effective population size of the KR.4 subgroup peaked on 15 February 2020. After detecting a large outbreak associated with the Shincheonji Church on February 17, the Korean government forced the church to close. Moreover, social distancing and enhanced social distancing were implemented on February 29 and March 22, respectively. The population size of the KR.4 subgroup decreased after February 17 and has not been detected after 2 April 2020 (Fig. 2). The phylogeography results showed that the KR.4 subgroup viruses were disseminated from Daegu and Gyeongsangbukdo to other regions of South Korea (Fig. 2, Figure S2, Table S3, Video S1).
3.3. KR.1 (B.1.497) subgroup caused long-term outbreak in South Korea
The majority of early cases of the KR.1 subgroup were epidemiologically related to people who visited clubs and bars in Itaewon, Seoul city, during April 29 to 6 May 2020 (Fig. 3, Table S1, Table S2). We conducted Bayesian phylogeography to determine the changes in the viral effective population size and the spatiotemporal transmission of the KR.1 subgroup. The mean tMRCA of the KR.1 subgroup was determined to be May 1 (95 per cent BCI: April 23 to May 6), indicating that the viruses were immediately detected after their introduction into South Korea. The effective population size of the KR.1 subgroup had gradually increased from May and peaked on 17 August 2020 (Fig. 3B).
Figure 3.
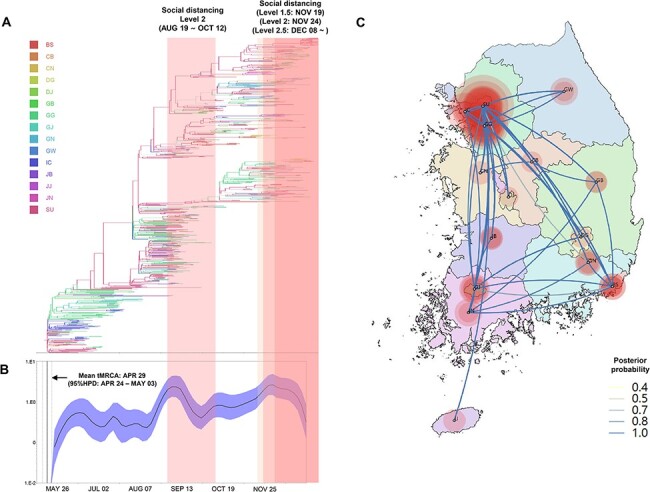
Phylogeography and effective population size analysis of KR.1 (B.1.497) subgroup viruses. (A) A time-scaled maximum clade credibility tree of the KR.1 subgroup SARS-CoV-2. Branches are colored according to locations in South Korea, and the thickness of branches indicates the posterior probabilities of the inferred ancestral location. (B) Gaussian Markov random field Bayesian Skyride plots indicating effective population size (relative genetic diversity) over time. (C) Spatiotemporal reconstruction of the spread of the KR.1 subgroup SRAS-CoV-2 in South Korea. The diameters of the circles represent the number of branches maintaining a particular location state at each time period. The color of the transmission line represents the posterior possibility of each transmission. BS, Busan; CB, Chungcheongbukdo; CN, Chungcheongnamdo; DG, Daegu; DJ, Daejeon; GB, Gyeongsangbukdo; GG, Gyeonggido; GJ, Gwangju; GN, Gyeongsangnamdo; IC, Incheon; JB, Jeollabukdo; JJ, Jejudo; JN, Jeollanamdo; GW, Gangwondo; and SU, Seoul.
The daily case numbers had increased during early August, and level 2 social distancing was introduced from 19 August 2020 in metropolitan regions and 22 August 2020 in other regions in South Korea until 12 October 2020. The viral population size had decreased during the level 2 social distancing, but it had gradually increased after the social distancing level was decreased to level 1 (Fig. 3B). As the daily number of cases increased, the social distancing level was increased from November 19 (level 1.5) to December 08 (level 2.5). The number of daily cases grew during December 2020 when the level 2.5 social distancing intervention was applied (Fig. 1D), but viral population size had decreased during this period (Fig. 3B). The number of sequences was insufficient for December to precisely determine the population size of that period (Fig. 1D). Thus, more extended monitoring and continued genome sequencing will be required to estimate the effect of level 2.5 social distancing intervention on epidemics during December.
Unlike the KR.4 subgroup, the metropolitan regions of Seoul, Gyeonggido, and Incheon were the epicenter of the KR.1 subgroup viruses, and frequent viral transmissions were detected between the metropolitan regions (Fig. 3, Figure S3, Table S4, Video S2). Based on phylogeography analysis, Seoul and Gyeonggido have been a major source of the KR.1 subgroup that spread between the regions since May 2020 (Fig. 3, Figure S3, Table S4, Video S2).
3.4. Mutation of SARS-CoV-2 in South Korea
We did not detect any new additional genetic mutation, which was fixed in KR.2–4 subgroups (Fig. 4). However, several synonymous and nonsynonymous mutations occurred in the KR.1 subgroup during circulation in South Korea (Fig. 4). A total of 13 additional nonsynonymous mutations, shared by more than 15 different sequences, were detected in the KR.1 subgroup. In particular, four nonsynonymous mutations (H1113Y, T2408N, P4223S, and A5770S) in ORF1ab were detected in 106 unique sequences identified during October–December 2020. The role of these mutations on the properties of the virus has not yet been fully determined. Additional mutations in the Spike gene were also detected in a small number of KR.1 subgroup viruses (e.g. Q52H, A222V, E556K, T716I, and A1070V), but the spread of variants bearing multiple Spike gene mutations was not detected in this study.
Figure 4.

A graphical representation of mutations of SARS-CoV-2 detected in South Korea during January to December 2020. Major nucleotide variations from the reference sequence Wuhan-Hu-1 (MN908947.3) are shown as vertical black lines and annotated by red (nonsynonymous mutations) or blue (synonymous mutations) letters. Four Korean subgroups (KR.1–4) are indicated by horizontal color stripes. An approximately maximum-likelihood phylogenetic tree of South Korean viruses is shown on the left. Syn, synonymous mutation.
4. Discussion
This study highlights that the 14-day traveler quarantine protocol is considerably effective in blocking the introduction of viruses from overseas. Various unsequenced viruses could have existed in inbound travelers considering the low sequence coverage (7.56 per cent of inbound traveler cases). However, the possibility of the presence of undetected transmission lineage in South Korea is low because we selected representative samples based on the epidemiological investigation. Thus, it is presumed that the national quarantine system blocked more introductions and subsequent spread caused by unsequenced viruses. Compared to the special entry system for only travelers from specific countries with severe outbreak situations, the quarantine system for all inbound travelers implemented from 1 April 2020 significantly reduced the number of virus introductions from travelers. Moreover, the efficacy of social distancing in decreasing the viral population size was identified in this study. In particular, three subgroups, KR.2–4, were found to have disappeared from South Korea.
Other control strategies, including wearing mask, contact tracing, and real-time alert systems, could contribute to limiting the introduction and spread of the virus (Betsch et al., 2020; Cheng, Lam, and Leung 2020). Because these policies were continuously implemented in South Korea during all outbreak periods, it is most likely that enhanced social distancing interventions dramatically affected the observed alterations in viral population size of SARS-CoV-2 in South Korea. We assume that these control measures possibly led to the extinction of three subgroups, KR.2–4 in South Korea.
Only the KR.1 subgroup has survived, expanded and mainly circulated in South Korea. SARS-CoV-2 has continuously evolved, and some new variants were considered to possess increased transmissibility, including the G clade bearing D614G mutation on the spike protein (Volz et al., 2021). Especially, the Alpha and Delta variants of concern have spread worldwide and become dominant lineages (Tang, Tambyah, and Hui 2020; Campbell et al., 2021; Davies et al., 2021; Singh et al., 2021). The KR.1 subgroup viruses that possess D614G mutation have also induced long-term outbreaks in South Korea compared to the other subgroups found in South Korea. We also assume that not only the increased viral transmissibility acquired through the D614G mutation but also the large outbreaks in metropolitan areas could be considered as a significant factor contributing to the higher and prolonged spread of KR.1 subgroup than the other subgroups.
The results of this study demonstrate that the social distancing and strict traveler quarantine systems implemented in South Korea efficiently reduced the introduction and spread of SARS-CoV-2. However, viral introductions were not completely blocked, and SARS-CoV-2 continues to evolve. Furthermore, people’s compliance to social distancing and other control strategies could be decreased through the long-term pandemic (Hoeben et al., 2021). During December, 2020, the number of daily confirmed cases did not decrease despite the implementation of the enhanced social distancing strategy. It is necessary to improve the monitoring system for travelers and develop efficient social distancing criteria to better control SARS-CoV-2, its variants, and future pandemic viruses.
Data availability
The sequences were shared through the GISAID’s EpiCoV (https://www.gisaid.org/) database. The list of accession numbers and metadata of South Korean SARS-CoV-2 sequences used in this study is available in Table S1. Other data are available in supplementary files except the BEAST XML files can be accessed at https://github.com/junghoon-kwon/SARS-CoV-2_Korea_phylodynamic.
Supplementary Material
Acknowledgements
We gratefully acknowledge the authors of the originating laboratories and the submitting laboratories who have deposited and shared genome data on GISAID’s EpiCoV (https://www.gisaid.org/) database. The GISAID acknowledgment tables for sequences used in this study are shown in Table S6.
Contributor Information
Jung-Hoon Kwon, College of Veterinary Medicine, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea.
Jeong-Min Kim, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Dong-hun Lee, Department of Pathobiology and Veterinary Science, University of Connecticut, Storrs, CT 06269, USA.
Ae Kyung Park, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Il-Hwan Kim, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Da-Won Kim, College of Veterinary Medicine, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea.
Ji-Yun Kim, College of Veterinary Medicine, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea.
Noori Lim, College of Veterinary Medicine, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea.
Kyeong-Yeon Cho, College of Veterinary Medicine, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea.
Heui Man Kim, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Nam-Joo Lee, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
SangHee Woo, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Chae Young Lee, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Jin Sun No, Division of High-Risk Pathogens, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Junyoung Kim, Division of Bacterial Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
JeeEun Rhee, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Myung-Guk Han, Division of Viral Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Gi-Eun Rhie, Division of High-Risk Pathogens, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Cheon Kwon Yoo, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Eun-Jin Kim, Division of Emerging Infectious Diseases, Bureau of Infectious Disease Diagnosis Control, Korea Disease Control and Prevention Agency, 187, Osongsaengmyeong2-ro, Osong-eup, Heungdeok-gu, Cheongju-si, Chungcheongbuk-do 28159, Republic of Korea.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This study was supported by the Korea Disease Control and Prevention Agency grant (4800-4837-301).
Conflict of interest
None declared.
References
- Alpert T. et al. (2021) ‘Early Introductions and Transmission of SARS-CoV-2 Variant B.1.1.7 In the United States’, Cell, 184: 2595–2604 e2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betsch C. et al. (2020) ‘Social and Behavioral Consequences of Mask Policies during the COVID-19 Pandemic’, Proceedings of the National Academy of Sciences, 117: 21851–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielejec F. et al. (2016) ‘SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes’, Molecular Biology and Evolution, 33: 2167–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R. R., and Drummond A. J. (2017) ‘bModelTest: Bayesian Phylogenetic Site Model Averaging and Model Comparison’, BMC Ecology Evolution, 17: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell F. et al. (2021) ‘Increased Transmissibility and Global Spread of SARS-CoV-2 Variants of Concern as at June 2021’, Eurosurveillance, 26: 2100509. doi: 10.2807/1560-7917.ES.2021.26.24.2100509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K. K., Lam T. H., and Leung C. C. (2020) ‘Wearing Face Masks in the Community during the COVID-19 Pandemic: Altruism and Solidarity’, TheLancet, S0140–6736(20): 30918–1. doi: 10.1016/S0140-6736(20)30918-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N. G. et al. (2021) ‘Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 In England’, Science, 372: eabg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi S. (2020) ‘Travel Restrictions Hampering COVID-19 Response’, TheLancet, 395: 1331–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Plessis L. et al. (2021) ‘Establishment and Lineage Dynamics of the SARS-CoV-2 Epidemic in the UK’, Science, 371: 708–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio R. H. et al. (2021) ‘Social Distancing Decreases an Individual’s Likelihood of Contracting COVID-19’, Proceedings of the National Academy of Sciences, 118: e2023131118. doi: 10.1073/pnas.2023131118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi M., Yokoe D. S., and Havlir D. V. (2020) ‘Asymptomatic Transmission, the Achilles’ Heel of Current Strategies to Control Covid-19’, New England Journal of Medicine, 382: 2158–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeben E. M. et al. (2021) ‘Social Distancing Compliance: A Video Observational Analysis’, PLoS One, 16: e0248221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. M. et al. (2020) ‘Identification of Coronavirus Isolated from a Patient in Korea with COVID-19’, Osong Public Health and Research Perspectives, 11: 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korea Disease Control and Prevention Agency (KDCA) (2020) All about Korea’s Response to COVID-19 <https://www.kdca.go.kr/upload_comm/syview/doc.html?fn=160276224199800.pdf&rs=/upload_comm/docu/0030/> accessed 14 Jun 2020.
- Lemey P. et al. (2009) ‘Bayesian Phylogeography Finds Its Roots’, PLoS Computational Biology, 5: e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., and Godzik A. (2006) ‘Cd-hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences’, Bioinformatics, 22: 1658–9. [DOI] [PubMed] [Google Scholar]
- Minin V. N., and Suchard M. A. (2008) ‘Fast, Accurate and Simulation-free Stochastic Mapping’, Philosophical Transactions of the Royal SocietyB: Biological Sciences, 363: 3985–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oran D. P., and Topol E. J. (2020) ‘Prevalence of Asymptomatic SARS-CoV-2 Infection: A Narrative Review’, Annals of Internal Medicine, 173: 362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A. K. et al. (2021) ‘Genomic Surveillance of SARS-CoV-2: Distribution of Clades in the Republic of Korea in 2020’, Osong Public Health and Research Perspectives, 12: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Dehal P. S., and Arkin A. P. (2009) ‘FastTree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix’, Molecular Biology and Evolution, 26: 1641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, Virus Evolution, 2: vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’, Systematic Biology, 67: 901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) ‘A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology’, Nature Microbiology, 5: 1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockett R. J. et al. (2020) ‘Revealing COVID-19 Transmission in Australia by SARS-CoV-2 Genome Sequencing and Agent-based Modeling’, Nature Medicine, 26: 1398–404. [DOI] [PubMed] [Google Scholar]
- Seemann T. et al. (2020) ‘Tracking the COVID-19 Pandemic in Australia Using Genomics’, Nature Communications, 11: 4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J. et al. (2021) ‘SARS-CoV-2 Variants of Concern are Emerging in India’, Nature Medicine, 27: 1131–3. [DOI] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. W., Tambyah P. A., and Hui D. S. (2020) ‘Emergence of a New SARS-CoV-2 Variant in the UK’, The Journal of Infection, 82: e27–8. doi: 10.1016/j.jinf.2020.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H. et al. (2021) ‘Detection of a SARS-CoV-2 Variant of Concern in South Africa’, Nature, 592: 438–43. [DOI] [PubMed] [Google Scholar]
- Volz E. et al. (2021) ‘Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity’, Cell, 184: 64–75 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (WHO) (2021) WHO Coronavirus Disease (COVID-19) Dashboard <https://covid19.who.int/> accessed 06 May 2021.
- Wu F. et al. (2020) ‘A New Coronavirus Associated with Human Respiratory Disease in China’, Nature, 579: 265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanes-Lane M. et al. (2020) ‘Proportion of Asymptomatic Infection among COVID-19 Positive Persons and Their Transmission Potential: A Systematic Review and Meta-analysis’, PLoS One, 15: e0241536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequences were shared through the GISAID’s EpiCoV (https://www.gisaid.org/) database. The list of accession numbers and metadata of South Korean SARS-CoV-2 sequences used in this study is available in Table S1. Other data are available in supplementary files except the BEAST XML files can be accessed at https://github.com/junghoon-kwon/SARS-CoV-2_Korea_phylodynamic.