

Abstract
Rhesus macaque is a unique nonhuman primate model for human evolutionary and translational study, but the error-prone gene models critically limit its applications. Here, we de novo defined full-length macaque gene models based on single molecule, long-read transcriptome sequencing in four macaque tissues (frontal cortex, cerebellum, heart and testis). Overall, 8 588 227 poly(A)-bearing complementary DNA reads with a mean length of 14 106 nt were generated to compile the backbone of macaque transcripts, with the fine-scale structures further refined by RNA sequencing and cap analysis gene expression sequencing data. In total, 51 605 macaque gene models were accurately defined, covering 89.7% of macaque or 75.7% of human orthologous genes. Based on the full-length gene models, we performed a human–macaque comparative analysis on polyadenylation (PA) regulation. Using macaque and mouse as outgroup species, we identified 79 distal PA events newly originated in humans and found that the strengthening of the distal PA sites, rather than the weakening of the proximal sites, predominantly contributes to the origination of these human-specific isoforms. Notably, these isoforms are selectively constrained in general and contribute to the temporospatially specific reduction of gene expression, through the tinkering of previously existed mechanisms of nuclear retention and microRNA (miRNA) regulation. Overall, the protocol and resource highlight the application of bioinformatics in integrating multilayer genomics data to provide an intact reference for model animal studies, and the isoform switching detected may constitute a hitherto underestimated regulatory layer in shaping the human-specific transcriptome and phenotypic changes.
Keywords: rhesus macaque, Iso-Seq, gene models, transcriptome evolution, polyadenylation, subcellular localization
Introduction
Rhesus macaque (Macaque mulatta), with a genome sequence highly analogous to humans, is a unique primate model for studies of human biology [1]. Compared with other distantly related model organisms, macaque represents an effective reference in comparative genomics studies of genetic changes underlying human-specific traits [1]. In addition, as recent studies have revealed high degrees of cross-species diversity on the regulation and structure of transcripts in mammals [2–4], rhesus macaque thus serves as a unique model in translational studies for verifying and generalizing findings of gene functions reported in traditional model animals. However, the applications in rhesus macaques have been limited, partially due to the incomprehensive and error-prone gene models found in rhesus macaques. Notably, due to the scarce macaque messenger RNA (mRNA) and expressed sequence tag (EST) data, macaque gene models mainly rely on ab initio or comparative genomics-guided predictions [5]. Consequently, recent studies by us have revealed defective annotations of local, fine-scale transcript structures (e.g. exon boundaries) in >30% of macaque genes [6, 7]. Moreover, because human gene structures have been used to build these putative gene models in the current macaque gene annotations, human–macaque comparative studies of human evolution with the current macaque gene models are likely to be inadequate [5].
Recently, RNA sequencing (RNA-Seq) short reads were used to refine the local structures of these putative gene models in rhesus macaque [6, 7], with the assumption that the backbones of these putative gene models are accurate and representative. However, according to a pilot study based on single-molecule long-read transcriptome sequencing (isoform sequencing, Iso-Seq) in the cerebellum of humans and macaques, many newly identified isoforms exhibited even higher expression than the previously annotated major isoforms, indicating a significantly underestimated complexity of the primate transcriptome at the full-length transcript level [2]. Thus, a protocol integrating multilayer genomics data is thus urgently needed to define an atlas of full-length macaque gene models, and further broaden the applications of this unique model in human evolutionary and translational studies.
Results
A new protocol for de novo definition of full-length macaque gene models
To define full-length macaque gene models, we first performed Iso-Seq in 15 PacBio RSII cells and 13 PacBio Sequel cells [8] to profile the poly (A)-positive RNAs derived from four macaque tissues (frontal cortex, cerebellum, heart and testis). A total of 8 588 227 poly(A)-bearing complementary DNA (cDNA) reads were generated, with a mean length of 14 106 nt (Table 1). Notably, the mean length of annotated transcripts in macaque (Ensembl, release 96) is only 1829 nt. Since the circular mode of Iso-Seq was used [9], the majority of these transcripts were sequenced several times from the 5′ end to the poly(A) tail. We thus split these raw long reads into subreads and self-aligned them to assemble reads of insert (regions of interest, ROIs) with a substantially decreased sequencing error rate. Finally, 7 983 980 ROIs were identified, with an average of 10 read-through passes.
Table 1.
Statistics of Iso-Seq data
| Species | Tissue | Source | ROIs | Mean pass number | Full-length PA-containing reads | High-quality readsa | |
|---|---|---|---|---|---|---|---|
| Count | Mean length | ||||||
| Macaque | Cerebellum | Zhang et al. | 1 128 095 | 2432 | 7 | 620 944 | 443 937 |
| Frontal cortex | This study | 2 769 148 | 1756 | 8 | 1 542 070 | 889 600 | |
| Heart | This study | 2 558 353 | 1590 | 10 | 1 584 962 | 907 278 | |
| Testis | This study | 1 528 384 | 1702 | 14 | 1 144 126 | 960 690 | |
| Human | Cerebellum | Zhang et al. | 1 179 556 | 1971 | 7 | 729 586 | 446 469 |
| Frontal cortex | PacBio | 691 846 | 3277 | 5 | 503 891 | 387 717 | |
| Heart | PacBio | 1 667 650 | 2973 | 2 | 394 347 | 221 242 | |
| Liver | PacBio | 1 789 673 | 2479 | 3 | 485 741 | 438 777 | |
| Mouse | Frontal cortex | This study | 751 620 | 2086 | 16 | 470 764 | 330 869 |
aHigh-quality reads were defined as the reads not mapped to the gap regions, mapped uniquely to one locus, mapped with pass number >1 and no internal poly(A) primes detected.
Several lines of evidence verified the high sensitivity of this Iso-Seq study in defining the gene models. First, these ROIs showed lengths comparable with those of human full-length protein-coding transcripts annotated in GENCODE (median: 1569 versus 2011 nt) (Figure 1A) and covered 89.42% of the macaque genes in Ensembl putative annotations [5] (Figure 1B). Particularly, the majority of genes with significant expression (fragments per kilobase of transcript per million mapped read, FPKM >5 as estimated by RNA-Seq) [2] were identified at the current Iso-Seq sequencing depth (94.7% in frontal cortex, 87.4% in cerebellum, 92.8% in heart and 92.6% in testis). Even at half of the current sequencing depth, most of these genes were covered (93.1% in frontal cortex, 82.6% in cerebellum, 90.2% in heart and 90.1% in testis) (Additional File 1: Supplementary Figure S1), indicating that the current sequencing depth of Iso-Seq was sufficient for constructing gene models de novo for macaque genes with significant expression.
Figure 1 .
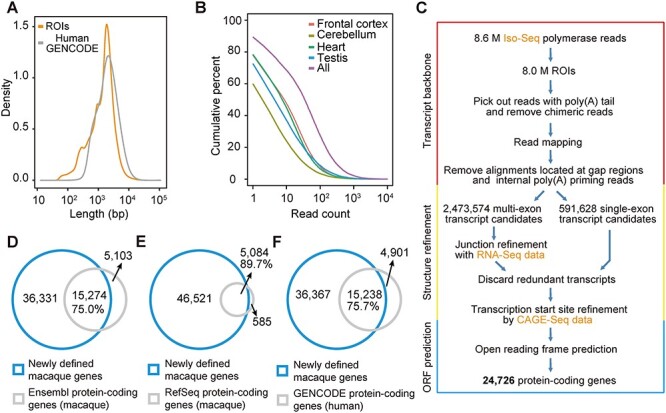
De novo definition of macaque gene models with Iso-Seq data. (A) Distribution of the lengths of Iso-Seq reads of insert (ROIs) and of human gene models annotated by GENCODE (v19) (Human GENCODE). (B) Cumulative distribution of the coverage of genes by Iso-Seq reads. All: Iso-Seq data from all four tissues. (C) The pipeline for the de novo definition of macaque gene models. (D–F) Venn diagrams showing the comparisons of gene models between the new atlas of macaque genes versus macaque protein-coding genes annotated by Ensembl (release 96) (D), RefSeq (release 94) (E) or human protein-coding genes annotated by GENCODE (F).
Although the feature of poly(A) selection ensures the accurate definition of the 3′ ends of transcripts with Iso-Seq reads, demarcating the 5′ ends is more difficult due to the underrepresented 5′ ends sequences on Iso-Seq reads. We thus integrated public macaque cap analysis gene expression sequencing (CAGE-Seq) data to define the 5′ ends of the transcripts (see Methods) [10–12]. Moreover, public RNA-Seq data [13–16] were further incorporated to refine the fine-scale local structures of these transcripts, and the exon–intron boundaries of gene models with relatively lower quality, such as those with lower Iso-Seq coverage, were revised accordingly (see Methods). Based on the refined gene models, we further introduced the 3-frame Met-to-stop translation method to define the open reading frame of each transcript (Figure 1C, Additional File 2: Supplementary Table S1).
Overall, a total of 51 605 nonredundant macaque genes were defined de novo (see Group 2, Methods), covering 89.7 or 75.0% of previously annotated macaque protein-coding genes in Reference Sequence (RefSeq) or Ensembl, respectively, or 75.7% of the human orthologous protein-coding genes (GENCODE) (Figure 1D–F). This new set of macaque gene models revealed a substantially higher complexity in the macaque transcriptome, with the mean isoform number per gene increasing from two (Ensembl, release 96) to eight.
The macaque gene models are defined with high sensitivity and specificity
The specificity of this set of newly defined macaque gene models was also verified by multiple known regulatory features of genes, such as an enrichment of active epigenetic marks [17–19] (Figure 2A) and CpG islands [20] upstream of the 5′ ends of the transcripts (Figure 2B), as well as the enrichment of poly(A)-Seq signals upstream of the poly(A) tails [21] (Figure 2C). Moreover, unambiguous sequence motifs, consistent with previously reported splicing motifs [22], could be detected at the exon–intron boundaries defined by the new gene models (Figure 2D). Notably, 75.7% of these newly defined open reading frames corresponded to known proteins, among which 89.0% are with the sequences of coding regions showing >80% overlap with known proteins in human and rhesus macaque (Figure 2E). As the human proteome is annotated in relatively higher quality, we further compared the newly defined open reading frames with human annotated open reading frames. The comparison revealed that the majority of these newly defined macaque gene models express comparable or even longer coding sequences (CDS) (Figure 2F, 79.9%). Considerable extents of protein sequence alignment (Figure 2G, 89.2% aligned gene models with >80% matched alignment) and protein sequence similarity (Figure 2H, 92.8% aligned gene models with >80% sequence identity) to translated human proteins were also detected. Viewed together, these lines of evidence suggested that the macaque gene models are accurately defined at the full-length transcript level.
Figure 2 .

Evaluation of the newly defined macaque gene models. (A–C) Regulatory attributes associated with the new macaque gene models: aggregate plots showing the H3K4me3 signals around the TSS of all genes (A). Densities of CpG islands around TSS of genes with CpG islands (nearby 200 bp) (B) and densities of poly(A)-Seq-identified PA sites around TTS of all genes (C). Newly defined macaque gene models; Ensembl, macaque gene models annotated by Ensembl (release 96). (D) Sequence motifs flanking splice sites of the newly defined macaque gene models. (E) Bar plots showing the distribution of genes with different coverage of protein-coding sequence, which was defined as the percentage of the newly defined macaque CDS covered by human/macaque protein sequences annotated by GenBank (release 223) and RefSeq (release 77). (F) Pie chart showing the comparisons of the CDS length between the newly defined macaque genes and human genes annotated by RefSeq (release 94). Only orthologous gene pairs in humans and macaques were considered in the comparison; Hg, the length of the CDS for human gene as annotated by RefSeq; Ma, the length of CDS for macaque orthologous gene as annotated by the new macaque gene models. (G and H) Bar plots showing the distributions of the proportion of alignment (G) and sequence identity (H) for the aligned regions between human and macaque protein sequences, which were translated from human RefSeq genes and the newly defined macaque genes, respectively.
As a proof of principle, an example of the newly defined gene models with supporting evidence was shown (Figure 3). Briefly, the DDX20 gene is a putative RNA helicase implicated in the alteration of RNA secondary structure [23, 24], and its disruption is linked to motor neuron degenerative disease and cancer [25, 26]. Previous annotations in rhesus macaque failed to annotate the complete gene model of this important gene, whereas the newly defined gene models provided full-length structural annotations of DDX20 (Figure 3).
Figure 3 .
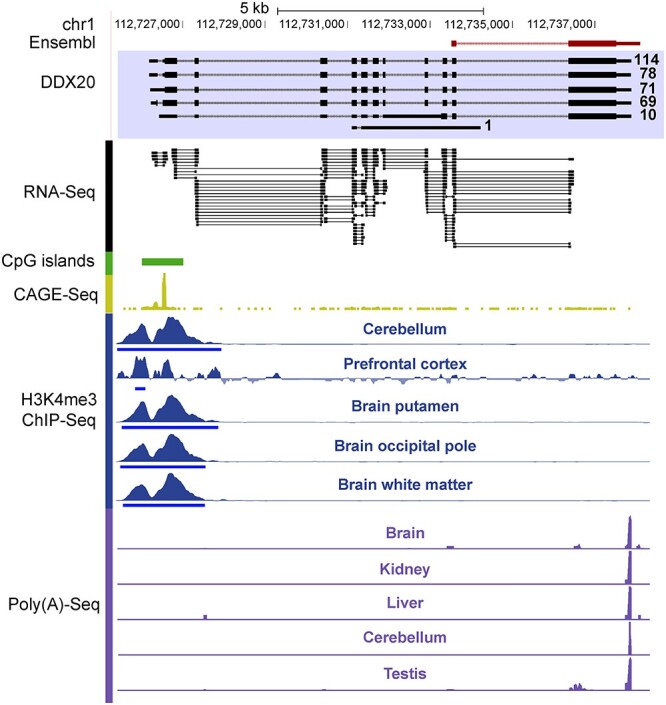
An example of improved macaque gene model. Both the previously annotated gene model (Ensembl) and a newly defined gene model are shown. The numbers of supporting reads for each transcript are shown along with the Iso-Seq reads. The positions of RNA-Seq junction reads (RNA-Seq) and CpG islands, as well as the signals of CAGE-Seq, H3K4me3 ChIPSeq and poly(A)-Seq, were presented accordingly.
Overall, this atlas of de novo defined, full-length macaque gene models should facilitate the use of this unique model in human evolutionary and translational studies, especially from the perspective of previously underestimated regulatory levels such as polyadenylation (PA) regulation.
PA profile in rhesus macaque shaped by both cis- and trans-regulations
Notably, the macaque gene models were defined with definitive 3′ ends, providing informative data to investigate the PA regulation. We then used these newly defined gene models to profile the PA sites in macaque tissues and further characterized the features of this significant posttranscriptional regulation [27]. The PA sites are determined by specific cis-regulatory motifs recognized by the cleavage and PA machinery, most notably the core polyadenylation signal (PAS). This sequence motif of AAUAAA and its variants typically located 10–40 nt upstream of the cleavage site [28]. In accordance with this distribution, PA sites determined in this study showed the characteristic sequence features [29, 30] (Additional File 1: Supplementary Figures S2 and S3).
To quantitatively examine the potential correlation between PAS type and PA efficiency, we further introduced a parameter of ‘PA usage’ to measure the efficiency of PA (see Methods). Although traditional approaches, such as poly(A)-Seq and RNA-Seq, might introduce false positive signals in PA site identification, the quantification of intact PA sites represents an accurate readout due to the deep sequencing coverage of the PA regions [21, 31, 32]. We thus evaluated the feasibility of Iso-Seq in the quantification of the PA events by cross-referencing with poly(A)-Seq data. Notably, for PA sites defined by both Iso-Seq and poly(A)-Seq data, the PA usage determined by the two approaches was well correlated (Additional File 1: Supplementary Figure S4), indicating the dual advantages of Iso-Seq in accurate definition and quantification of PA events at the full-length transcript level.
For genes with more than one PA site, the selection of PA sites during transcription might be regulated by both cis-elements and trans-factors [27]. To quantitatively explore the contributions of the two types of regulations in PA site selection, we estimated the average PA strength of a group of PA sites (PA strength) on the basis of the fraction of sites with the AAUAAA motif (Figure 4A, Additional File 1: Supplementary Figure S5; see Methods). We then made the following observations: first, consistent with other reports [27], we found that the distal PA sites showed significantly higher PA strength than proximal PA sites on the same gene (Figure 4B, Additional File 1: Supplementary Figure S6, Fisher’s exact test, P < 2.2e-16). In addition, the highest PA usage was observed for distal PA sites carrying PAS of AAUAAA, with the corresponding proximal PA sites carrying no previously reported PAS (Figure 4C, Additional File 1: Supplementary Figure S7A–C). Notably, during the transcription process, the proximal PA sites are selected first. The higher PA strength of distal PA sites may thus increase their competition with proximal PA sites, facilitating the emergence of alternative PA events.
Figure 4 .
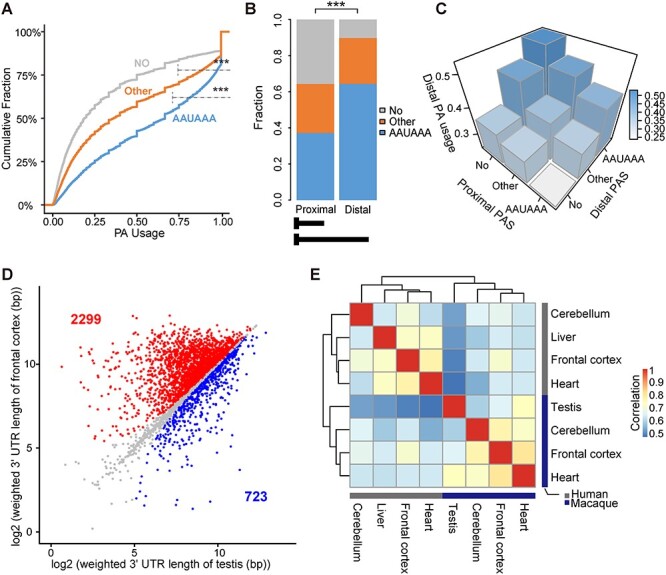
Cis- and trans-regulation of PA site selection. (A) Cumulative plots showing the distributions of PA usage in the macaque frontal cortex for three groups of PA sites with different PAS. AAUAAA, PA sites with the canonical PAS AAUAAA; Other, PA sites with other PAS variants excluding AAUAAA; No, PA sites without known PAS. (B) For genes undergoing alternative polyadenylation in the macaque frontal cortex, the proportions of the three groups of PA sites (AAUAAA, Other, No) are shown for the proximal and distal PA sites, respectively. (C) 3D histograms showing average PA usage in the macaque frontal cortex in groups with different combinations of PAS (AAUAAA, Other, No) at proximal and distal PA sites. (D) For each gene expressed in both the macaque frontal cortex and testis, pairwise comparison of the weighted length of the 3′ UTR was performed and shown. The genes with a cross-tissue difference in weighted length of >30 bp are highlighted in red (2299 genes, longer 3′ UTR in frontal cortex) or blue (723 genes, longer 3′ UTR in testis). (E) Hierarchical clustering diagram showing the Spearman correlation coefficients of pairwise comparisons of the PA usage in multiple tissue samples between human and macaque. NS, not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Second, in addition to the contributions of cis-elements, trans-regulation also contributes to the process of PA selection. In the frontal cortex and testis tissues of the same macaque animal, in which the cis-regulation is identical, we found that the weighted length of the 3′ untranslated region (UTR) is significantly longer in the frontal cortex than in the testis (Figure 4D, Wilcoxon rank-sum test, P < 2.2e-16) due to the preferentially use of distal PA sites (Additional File 1: Supplementary Figure S7D), implying the substantial contributions of tissue-biased regulators in the process of PA selection.
Human-specific PAs originated through the strengthening of the distal PA sites
As both cis- and trans-regulation could contribute to the PA site selection, we next investigated their implications in the cross-species differences in PA usage during the primate evolution. When comparing the PA usage across multiple tissues of human and macaque (see Methods), we found a more shared use of PA in different tissues of the same species, than that within the same tissue across species (Figure 4E), which indicates that the cis-element may act as the primary factor driving the cross-species PA evolution in primates.
To further clarify how these cis-elements might contribute to the cross-species PA site selection, we identified 97 distal PA sites (on 89 genes) in human but not macaque on the basis of Iso-Seq and RNA-Seq profiles in human and macaque tissues (Figure 5A and B; see Methods). To investigate whether these distal PA sites are newly originated in human, rather than representing recent PA loss events in rhesus macaque, we further introduced mouse as an outgroup species. By performing Iso-Seq for poly(A)-positive RNAs derived from mouse brain, we generated 758 884 poly(A)-bearing cDNA reads. Mouse RNA-Seq data from 58 tissue samples were also analyzed to retrieve the reads distribution information across the full-length mouse transcripts (Additional File 2: Supplementary Table S1; see Methods). On the basis of these profiles, as well as the gene models annotated in GENCODE and RefSeq, we removed the candidate human PA sites detectable also in mouse, and finally identified 79 newly originated, human-specific distal PA events on 73 genes (see Methods; Figure 5A, Additional File 3: Supplementary Table S2). To further account for the variability among human populations, we checked the existence of these candidate PA events using Genotype–Tissue Expression (GTEx) RNA-Seq data from multiple human individual [33]. Overall, >83.78% of these PA events were detected in at least two human individuals (Figure 5C, Additional File 1: Supplementary Figure S8; 93.24% for brain cortex tissues from 101 individuals; 94.69% for cerebellum tissues from 128 individuals; 83.78% for heart tissues from 225 individuals). Therefore, population-level variability should not have a strong effect in defining these human-specific distal PA events, and a large proportion of these events should represent bona fide human-specific regulations.
Figure 5 .
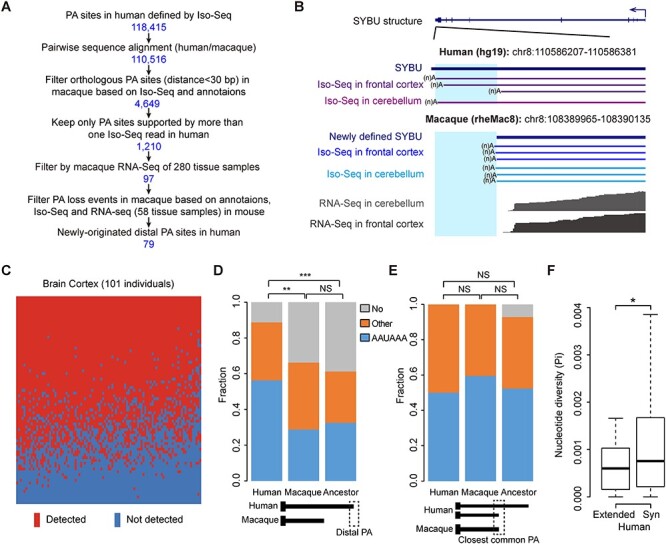
Cis-regulation is primarily attributed to cross-species PA site selection. (A) A flow chart depicting the identification of distal PA sites in human but not macaque. The number of human PA sites retained in each step is shown. (B) Track view of one example gene, SYBU, with a specific distal PA site in human but not macaque. The structure of the SYBU transcript is shown, with the 3′ UTR regions of the human (middle panel) and macaque (bottom panel) gene loci shown in the magnified view. The extended 3′ UTRs in human are highlighted in the shaded regions. (C) Heat map showing the expression of extended exonic regions in human cortex samples. Horizontal red bars indicate the expression of the extended regions in the tissue sample of the corresponding individual (FPKM >0.2). (D and E) For genes with human–macaque differences in PA site selection, the proportions of the three groups of PAS (AAUAAA, Other, No; as defined in Figure 4) are shown for the newly originated human PA sites (D) and the closest common PA sites shared by human and macaque (E). (F) Boxplots of nucleotide diversity for human-specific extended regions corresponding to 79 newly originated distal PA sites in human and all synonymous sites of human genes (Syn). NS, not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Using this list, we then traced the formation of these young distal PA sites. Presumably, three models exist for the origination of these distal PA sites after the divergence of human and macaque—the strengthening of the distal sites in human but not macaque, the weakening of the proximal sites, and both the strengthening of the distal sites and the weakening of the proximal sites. Through comparing the PA strength for distal PA sites in human but not macaque, we found significantly stronger PAS in humans than in both macaques and their ancestral sequences (Figure 5D, Fisher’s exact test, human versus macaque, P = 0.0002; human versus ancestor, P = 0.0001; macaque versus ancestor, P = 0.54), whereas the associated common proximal PA sites showed no significant difference among humans, macaques and their ancestral sequences (Figure 5E, Fisher’s exact test, human versus macaque, P = 0.51; human versus ancestor, P = 0.25; macaque versus ancestor, P = 0.29). These findings thus supported the model that the strengthening of the distal PA sites, rather than the weakening of the proximal sites, predominantly contributes to the origination of these human-specific PA events. Notably, the causal relationship between the strengthening of these distal PA sites and the origination of these young distal PA sites still need further investigations.
Human transcriptome evolution through PA site selection
To investigate whether these newly originated distal PA events in human represent functional regulations, rather than neutral transcription noises, we performed population genetics analyses to test whether the extended exonic regions through the origination of the new PA event are selectively constrained, on the basis of whole-genome sequencing data from 103 human individuals [34]. Notably, for the regions specifically transcribed in human corresponding to the 79 distal PA events, the nucleotide diversity of these regions is significantly lower than that of synonymous sites on all human annotated genes (Figure 5F, Wilcoxon rank-sum test, P = 0.02). This finding thus indicates that the human-specific isoform with new PA sites are selectively constrained in general, implying their functionality in human evolution.
We then investigated whether these selectively constrained, newly originated human PA sites may contribute to the evolution of the human transcriptome. We first checked whether these new PA events may change the protein sequences of the corresponding genes, and found only one PA-related exonic region encoding nine amino acids on TAP2 gene, involved in antigen presentation and bare lymphocyte syndrome. As TAP2 has been reported to participate in the transport of antigens from the cytoplasm to the endoplasmic reticulum, through interacting with multiple proteins such as TAP binding protein (TAPBP), transporter 1 (TAP1) and beta-2-microglobulin (B2M) [35], the inclusion of an additional nine amino acids may regulate its functions through the rewirement of the protein–protein interaction network. Notably, as most of the extended regions did not contain protein CDS, these events might largely be implicated in gene regulations, other than direct modifications of the CDS.
As alternative PA site selection is known to impact the length of 3′ UTRs, we then investigated whether they may further change the regulation and stability of the corresponding transcripts via alternative binding of miRNAs or RNA-binding proteins (RBP) [36–38]. Specifically, the distal PA sites specific in human may contribute to the gene expression reduction in humans by the inclusion of additional miRNA-binding sites [39]. In line with this hypothesis, we found that the human genes with human-specific distal PA sites showed significantly reduced expression in comparison with their orthologous genes in macaques (Figure 6A, Wilcoxon rank-sum test, P = 0.02). Considering the regulated tissue expression distribution of miRNAs [40], it is plausible that these genes encoding longer 3′ ends specific to human may also have divergent tissue expression profiles between human and rhesus macaque. Interestingly, when examining the tissue expression distributions of these genes, we found significantly lower tissue correlation coefficients for these genes in comparison with genes with shared 3′ ends between human and macaque (Figure 6B and C, Wilcoxon rank-sum test, P = 0.01).
Figure 6 .
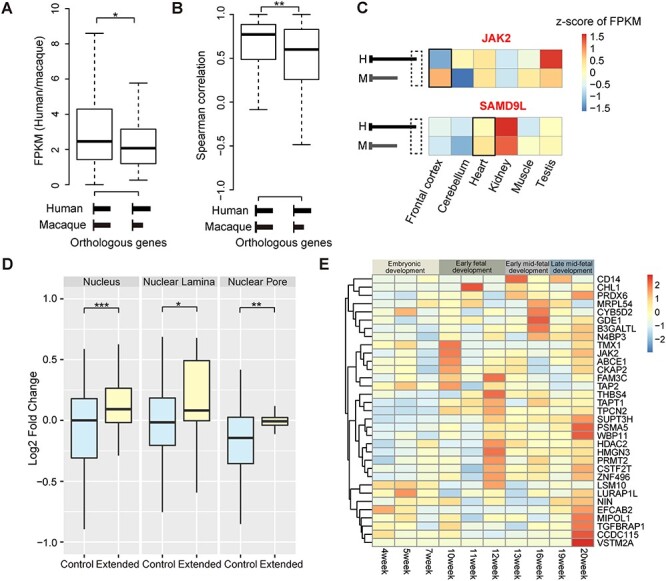
Transcriptome evolution in primates through PA site selection. (A) The ratios of FPKM between human and macaque are shown and compared for orthologous genes with (right boxplot) or without (left boxplot) distal PA sites specific in human but not macaque. (B) Boxplots showing Spearman correlation coefficients of tissue expression profiles in six tissues (frontal cortex, cerebellum, heart, kidney, muscle and testis) of human and macaque for orthologous genes with (right boxplot) or without (left boxplot) specific distal PA sites in human but not macaque. (C) Examples of genes with specific distal PA sites in human but not macaque that also exhibit divergence of tissue expression profiles. The specific tissues in which the human-specific distal PA site was initially identified were labeled by a black box in the heat maps; H, human; M, macaque. (D) Boxplots of the APEX-Seq fold changes for the extended 3′ UTR regions specific to human (Extended) and the constitutive exonic regions of the corresponding genes (Control); NS, not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. (E) Heatmap showing the relative levels of the isoforms with newly originated distal PA sites in human, during the human fetal brain development.
Especially, as previous case studies have indicated that the isoforms with different UTRs have different subcellular localizations [41–43], we investigated whether these genes encoding longer 3′ UTRs specific to human may show some specific profile of RNA subcellular localization, on the basis of ascorbic acid peroxidase sequencing (APEX-Seq) data in human cell lines [44]. We found that the isoforms with extended 3′ UTR specific to human are significantly enriched in nuclear structures, such as nucleus, nuclear lamina and nuclear pore, in comparison with the constitutive exons of the corresponding gene (Figure 6D, Wilcoxon rank-sum test, P < 2.2e-16 for nucleus, P = 0.02 for nuclear lamina and P = 0.01 for nuclear pore). The isoform switching from shorter to longer 3′ UTR may thus represent an efficient way to control for the expression of these genes on protein level, through the stronger nuclear retention of mRNAs. Notably, the relative levels of these new isoforms with distal PA sites are significantly regulated in human glioblastoma multiforme in contrast to normal brain tissues (Additional File 1: Supplementary Figure S9). It is thus possible that the retained mRNAs may play essential functions in stress conditions, such as in cancer cells, with a similar function of the formation of stress granules.
Taken together, it is plausible that this posttranscriptional PA regulation might contribute to the tissue- or stage-specific reduction of gene expression, through the tinkering of previously existed mechanisms of nuclear retention and miRNA regulation. The isoform switching through PA selection may thus constitute a hitherto underestimated regulatory layer in shaping the human-specific transcriptome and phenotypic changes.
Discussion
Currently, the incomplete and error-prone gene models in the rhesus macaque genome critically limited the use of this unique nonhuman primate model animal in evolutionary studies and potential extension to human translational research. Here, we developed a comprehensive protocol for de novo definition of the full-length macaque gene models through integration of single-molecule sequencing, RNA-Seq, CAGE-Seq and proteomics data. The catalogued full-length macaque gene models provides an accurate reference for molecular and translational studies in rhesus macaque, as well as a viable resource for evaluating the validity and generality of genetic findings from other model animals [1]. Notably, the standard computational pipeline we developed for de novo definition of full-length gene models with multilayer omics data integration should also be applicable to other model animals in facilitating the fine-scale molecular studies.
With the precise structural information, these full-length macaque gene models could be implemented to interrogate gene regulatory events associated with primate evolution. To this end, we have thoroughly profiled and delineated several transcript attributes, such as PA regulation, single-exon genes and gene fusions, which might not be adequately annotated by the traditional approaches. As a proof-of-concept example, we found that the PA regulation acts as an underestimated, yet flexible contributor to the cross-species divergence of transcriptome. This wealth of information should complement previous genetic and molecular studies of human phenotypic evolution [45–47].
Although we found that the human genes with longer 3′ UTRs showed relatively lower expression levels in contrast to their macaque orthologs, understanding this pattern from the perspective of primate evolution is not straightforward. Especially, when the new isoform with longer tails only account for a small portion of the isoform atlas encoded by this gene (Additional File 1: Supplementary Figure S10), it is unknown whether this mechanism could efficiently decrease the gene expression to the degree enough for the cross-species phenotypic changes. Notably, when investigating the RNA expression profiles of these genes in human fetal brain development, we found that the proportion of these human-specific isoforms with longer 3′ UTRs could increase substantially in stage-specific manner (Figure 6E; see Methods). For example, in week 10 to week 12, a period in human early fetal brain development characterized by the proliferation and migration of neurons, multiple key genes in neurogenesis, such as JAK2 [48], were identified to encoding more isoforms with human-specific longer tails (Figure 6E). In week 20 of late mid-fetal development characterized by the formation of synapse in the neocortical plate, most of these genes encode higher levels of isoforms with human-specific longer tails in comparison with other developmental stages (Figure 6E), such as PRDX6 previously reported to inhibits the neurogenesis of neural precursor cells [49], and N4BP3 reported to be a crucial modulator of axonal and dendritic branching [50, 51]. The isoform switching through PA selection may thus contribute substantially to human transcriptome evolution in fetal brain development and further to the species-specific phenotypic changes through reshaping the transcription profiles.
In terms of alternative PA events and differential 3′ UTR lengths, we assumed that these RNA isoforms would encode identical proteins and thus have similar functional outcome on the protein level. The combinatorial effect of the introduction of the longer transcript region through PA regulation and the preferential degradation and nuclear retention of that isoform is thus relatively mild. However, isoforms with different UTRs may exhibit variable functions as a consequence of altered mRNA localization [41–43], which is an especially significant regulatory determinant in highly polarized cells such as neurons [52, 53]. In this capacity, previous studies have found that long 3′ UTR transcripts of some genes are preferentially targeted to dendrites or distal axons to regulate neuronal morphology and functions [54, 55]. Notably, RBP have been implicated in the altered mRNA localization [56]. By analyzing 184 cross-linking immunoprecipitation sequencing (CLIP-Seq) datasets corresponding to 122 RBPs from Encyclopedia of DNA Elements project (ENCODE project) project [57], we identified 37 RBPs that could bound to these extended regions in human, such as cleavage stimulation factor subunit 2 (CSTF2) (Additional File 1: Supplementary Figure S11) involved in PA processing [58, 59]. Among these RBPs, three RBPs, such as CSTF2, heterogeneous nuclear ribonucleoprotein C (HNRNPC) and TIA1 cytotoxic granule associated RNA binding protein (TIA1), preferentially bound to these human-specific extended regions in comparison with randomly selected intergenic regions (Additional File 1: Supplementary Table S3). These trans-regulations could have contributed to the altered subcellular localization of these RNA isoforms.
Besides PA-related extended exonic regions, our previous study also reported events of human exonization (splicing-related internal exons) through differential nucleosome occupancy [15]. As we obtained full-length gene models in both human and macaque here, we repeated the study to identify the human-specific, splicing-related internal exons on the basis of the full-length gene models and identified 32 additional human-specific exons (Additional File 3: Supplementary Table S2). The nucleotide diversity of these exonic regions is also significantly lower than that of synonymous sites of all human annotated genes, indicating that the human-specific internal exons are selectively constrained (Supplementary Figure S12). These findings thus indicate that the exonizations through PA-related and splicing-related mechanisms should both play adaptive roles in human evolution.
Methods
RNA extraction, library preparation and deep sequencing
Total RNA from tissue samples (frontal cortex, heart and testis) of one rhesus macaque animal and frontal cortex sample from one mouse animal was extracted using TRIzol reagent (Invitrogen, catalog #15596018) and analyzed on an Agilent 2100 Bioanalyzer to assess quality (Additional File 1: Supplementary Table S4). The quantity of total RNA was measured by Qubit Invitrogen. cDNA was then synthesized using the SMARTer PCR cDNA Synthesis Kit (Clontech, catalog #634925) and PrimeSTAR GXL DNA Polymerase (Clontech, catalog #R050B). Single-molecule real-time (SMRT) bell libraries were generated by using an SMRTbell™ Template Prep Kit 1.0-SPv3 (PN 100-991-900) and subsequently sequenced by using Sequel Binding Kit 2.0, Sequel Sequencing Kit 2.1 and Sequel™ SMRT® Cell 1 M v2 Tray on the PacBio Sequel System (Pacific Biosciences). The Iso-Seq of cerebellum samples was performed as in our previous study [2] in which the sequencing was carried out on a real-time sequencer (Pacific Biosciences) with C4 sequencing reagents.
Sequencing data processing
All Iso-Seq and RNA-Seq data were processed and evaluated by following SMRTLink guidance and previously published pipelines [2]. Briefly, for Iso-Seq data, ROIs were extracted and primer sequences flanking ROIs were trimmed. Primer-trimmed reads were further processed to scan the PA tail and trimmed to obtain PA-trimmed reads (reads with a PA tail trimmed). PA-trimmed reads were then mapped to the reference genome (hg19 for human, rheMac8 for macaque) by GMAP (Genomic Mapping and Alignment Program) [60], and low-quality reads (reads mapped to gap regions, reads mapped to more than one locus, reads with pass number of <1 or internal poly(A) priming reads) were filtered to obtain the final processed alignments.
For RNA-Seq and chromatin immunoprecipitation sequencing (ChIP-Seq) data, raw reads were filtered to obtain high-quality reads, which were then aligned to the corresponding reference genome using TopHat2 [61] (v2.1.1) for RNA-Seq data and Burrows–Wheeler Aligner [62] (0.7.13-r1126) for ChIP-Seq data, respectively. CAGE-Seq data [11, 12] were analyzed by following the standard FANTOM pipeline [63].
De novo definition of macaque gene models with Iso-Seq
A new pipeline of gene model definition was developed on the basis of the RefSeq Eukaryotic Genome Annotation Pipeline [64, 65]. Briefly, Iso-Seq long reads were used to define the backbone of isoforms, based on which the redundant isoforms were discarded. If necessary, the junction reads of RNA-Seq were further used to refine the splicing sites of each isoform [2]. Next, we refined the transcription start site (TSS) of those truncated isoforms by elongating them to the nearest potential TSS marked by CAGE-Seq data, if the site was within 1 kb from the 5′ end of the isoform. Single-exon isoforms with no TSS marked by CAGE-Seq data were then discarded. Finally, on the basis of the refined gene models, we further introduced the 3-frame Met-to-stop translation method to define the open reading frames of each transcript, and the nucleic acid sequences were then translated into peptide sequences.
We further divided the gene models into three groups according to their qualities. (Additional File 1: Supplementary Table S5). Briefly, Group 1 represents genes with the most reliable gene models. In this category, for multiple exon genes, the full-length transcript structure was supported by poly(A)-bearing Iso-Seq reads, with each splice junction supported by RNA-Seq junction reads; for single-exon genes, the transcript structure was supported by poly(A)-bearing Iso-Seq reads, with the TSS supported by CAGE-Seq evidence. In Group 2, in addition to the genes in Group 1, all other multiple exon gene models defined by poly(A)-bearing Iso-Seq reads or refined by the RNA-Seq or CAGE-Seq data were included. Finally, in addition to the genes in Group 1 and Group 2, all other Ensembl (release 96) gene models not covered by our Iso-Seq data were also included in Group 3, which represents a comprehensive atlas of macaque gene models. The GTF (Gene Transfer Format, GTF2.2) format files (Mmul_8.0.1/rheMac8), FASTA format files for CDS sequences and protein sequences for these gene models can be downloaded from http://rhesusbase.cbi.pku.edu.cn/download/Full-length_macaque_gene_models.jsp. In this study, only genes in Group 2 were used for subsequent analyses of PA regulations.
Evaluation of the newly defined macaque gene models
To evaluate the quality of the splice junctions of the newly defined gene models, sequence motifs flanking the donor/acceptor splice sites defined by the gene models were calculated and visualized using WebLogo (v2.8) [66]. H3K4me3 ChIP-Seq dataset [17], CpG island elements and poly(A)-Seq dataset [67] were also processed and used to verify the 5′ ends and 3′ ends of these gene models (Additional File 2: Supplementary Table S1). Briefly, the aggregated H3K4me3 ChIP signals (read coverage in ChIP sample subtracted by that in input sample) across TSSs of newly defined or Ensembl annotated (release 96) macaque genes were calculated and compared. Then, genes with CpG islands located within 200 bp of TSS were used to evaluate the enrichment of CpG islands across TSS. We also calculated the poly(A)-Seq-based PA site density across transcription termination sites (TTSs) to evaluate the 3′ boundaries of these gene models. To assess the quality of the predicted open reading frames, we compared the length of open reading frames defined by gene models of macaque genes and their orthologous genes in humans (RefSeq, release 94). Pairwise sequence alignments were then performed using blastp [68] (v.2.2.28, -outfmt 6 -evalue 1e-5) between annotated protein sequences of macaque or human (GenBank or RefSeq) and the putative protein sequences translated from the newly defined macaque gene models. The extent of the aligned proportion and identity of the orthologous pairs were calculated to quantitatively evaluate similarity.
Identification and quantification of PA regulation
To identify PA sites using Iso-Seq reads, PA-containing reads were first assigned to annotated genes. The 3′ ends of these reads were then clustered if they were located within 30 bp, with the site of highest coverage being defined as the representative PA site [2]. Only PA sites with at least two supporting reads were considered for the following analyses.
For each macaque gene, the usage of a specific PA site was defined as the proportion of poly(A)-bearing reads encompassing the corresponding PA site. To identify the human orthologous PA sites for these macaque counterparts, the liftOver tool was used for the cross-species alignments. Human PA sites located within 30 bp of the orthologous regions of the macaque PA site were considered conserved PA sites and further subjected to the following clustering analyses. For the conserved PA sites, the cross-species and cross-tissue Spearman correlation coefficients of the PA usage were calculated, and the samples were grouped using the hierarchical clustering method.
To assess nucleotide composition around cleavage sites, nucleotide distributions were determined around all cleavage sites. For the core PAS identification, we searched for the canonical hexamer (AAUAAA) in a 50-nt window upstream of the cleavage site. If this was not identified, we then searched for the other hexamer variants (AUUAAA, AGUAAA, UAUAAA, CAUAAA, GAUAAA, AAUAUA, AAUACA, AAUAGA, ACUAAA, AAUAAA, AAUGAA) [69]. When comparing PA efficiency between different types of PAS, we found PA sites of the canonical AAUAAA showing the strongest PA efficiency (Figure 4A, Additional File 1: Supplementary Figure S5, Wilcoxon rank-sum test, P < 2.2e-16), a finding consistent with previous reports [30]. For effective estimation of the average strength of PAS, we calculated the fraction of PA sites with the AAUAAA motif in the corresponding group in the following analyses.
The weighted 3′ UTR length was calculated by following previously published method [30]. Briefly, transcript with the furthest 3′ transcript end and the most distal 5′ coding region end was chosen to get the 3′ UTR region of each gene. We additionally removed genes that contain annotated introns in the 3′ UTR regions. Then, the weighted 3′ UTR length was calculated as the average of all 3′ UTR lengths per gene weighted by the contribution of each isoform’s usage as measured by the PA usage.
Identification of distal PA sites newly originated in human
To identify distal PA sites specific to human but not macaque, we began with all PA sites supported by at least two Iso-Seq reads. Orthologous human PA sites detected in rhesus macaque were removed when cross-referencing to the macaque Iso-Seq data and annotated TTS (located within 30 bp). For human-specific 3′ UTR regions defined by candidate human-specific PA sites, another filter was applied based on macaque RNA-Seq data from 280 tissue samples (Additional File 2: Supplementary Table S1): if the average FPKM level (RSeQC (RNA-Seq quality control), v2.6.3) [70] was >0.2 in the corresponding macaque tissue [71], the PA site was discarded. Then, mouse was served as an outgroup to verify these PA sites are newly originated in human. Briefly, orthologous human PA sites detected also in mouse were removed from the initial candidate list for human-specific PA sites, when cross-referencing to the mouse Iso-Seq data (Table 1), mouse RNA-Seq data from 58 tissue samples (Additional File 2: Supplementary Table S1) and annotated TTS (located within 30 bp).
Identification of splicing-related internal exons newly originated in human
To identify newly originated internal exons in human, we began with human annotated internal exons (GENCODE v19) that were not overlapped with macaque exons based on the Group 3 annotated gene models and supported by human Iso-Seq reads. Then for orthologous candidate human-specific exons in rhesus macaque and mouse, filters were applied based on macaque RNA-Seq data from 280 tissue samples and mouse RNA-Seq data from 58 tissue samples (Additional File 2: Supplementary Table S1): if the FPKM level (RSeQC, v2.6.3) [70] in any of these samples was >0.2 [71], the candidate new exon was removed from the list.
Ancestral sequence definition
To define the common ancestral state of human and macaque at each site, multiple sequence alignment data were downloaded from the UCSC (University of California, Santa Cruz) Genome Browser [72], from which the human–macaque–marmoset aligned regions were extracted and analyzed as previously described [15, 73, 74].
Population genetics analyses
On the basis of the polymorphism data of 103 human individuals from the RhesusBase PopGateway [34], we measured the nucleotide diversity for the regions specifically transcribed in human but not macaque corresponding to these young PA sites [34]. The nucleotide diversity for synonymous sites on all human annotated genes was served as the control. The Wilcoxon one-tail test was performed to determine the significance of the nucleotide diversity differences between those specifically transcribed regions and the synonymous sites on all annotated genes.
Gene expression profile analyses
For the similarity of tissue expression profiles, RNA-Seq data from six paired tissues in human and macaque (frontal cortex, cerebellum, heart, kidney, muscle and testis) were integrated for the quantification of gene expression levels (Additional File 2: Supplementary Table S1). The Spearman correlation coefficients between the orthologous genes in the two species were calculated to measure the cross-species similarity in tissue expression profiles.
For the profile of RNA subcellular localization, public APEX-Seq data [44] of nine subcellular fractions (nucleus, nucleolus, nuclear lamina, nuclear pore, cytosol, endoplasmic reticulum membrane, endoplasmic reticulum lumen, outer mitochondrial membrane and mitochondrial matrix) were used to calculate the RNA subcellular localization of isoforms. For isoforms with distal PA sites in human but not macaque, the extended 3′ UTR region specific to human were extracted as representative regions, which were then used to estimate the expression levels of the isoforms with human-specific PA sites. The expression levels of the constitutive exonic regions of these genes were used as the control. Following the previously published strategy [44], we further calculated the degree of enrichment using DESeq2 [75].
For the expression profiles in different developmental stages, RNA-Seq data of human fetal brain development were obtained from https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6814/ [76] (Additional File 2: Supplementary Table S1). Prenatal human forebrain samples with fragmentation score [6] >0.885 (qualified samples) were used for the following analyses to exclude RNA-Seq datasets with potential 3′ sequencing bias. If two or more qualified samples exist for a specific developmental stage, only the sample with the highest fragmentation score was kept for the following analyses. For the expression profiles in human glioblastoma multiforme and normal brain tissues, RNA-Seq data were downloaded from the TCGA (The Cancer Genome Atlas) (https://portal.gdc.cancer.gov) and GTEx portal (https://www.gtexportal.org/home) (Additional File 2: Supplementary Table S1) [33, 77]. Then, RSeQC (v2.6.3) [70] was used for FPKM calculation of the constitutive exons, as well as the extended regions corresponding to the newly originated, distal human PA events. Finally, the percentage of the expression for the isoform with the distal PA site was estimated as the ratio of the FPKM of extended regions and the FPKM of the constitutive exons. Genes with the constitutive exon’s FPKM <0.2 were excluded from the analyses.
CLIP-Seq and RBP binding analyses
Human CLIP-Seq data were downloaded from the ENCODE data portal (www.encodeproject.org). Only peaks supported by both of the two biological replicates were retained, resulting in 184 sets of RBP-binding peaks. Then, two kinds of overlaps were generated using bedtools [78]: the overlaps between the 184 peak sets and the extended regions corresponding to the newly originated distal PA events in human; and the overlaps between the 184 peak sets and the randomly selected intergenic regions as the control. The statistical significance of the two resulting overlaps was tested with the Fisher’s exact test.
Statistical analyses
All statistical analyses were performed in R (version 3.4.0), with the corresponding tests described in the main text or figure legends. Fisher’s exact test was performed across the contingency table. For data represented in boxplots, the nonparametric Wilcoxon rank-sum test was performed to compare the two groups.
Code Availability
All the codes used to generate results can be found at GitHub via URL https://github.com/xihuimeijing/Macaque-transcriptome.
Key Points
A comprehensive protocol integrating multilayer genomics data to de novo define full-length gene models.
A comprehensive protocol to define full-length macaque gene models with high accuracy.
Human–macaque differences in PA regulation.
Human transcriptome evolution through PA site selection.
Supplementary Material
Acknowledgments
The authors thank Dr Yong E. Zhang at the Institute of Zoology, Chinese Academy of Science, Dr Tim Qing-Rong Liu at the National Institutes of Health and Dr Jia-Yu Chen at University of California for insightful suggestions. The authors thank Dr Shi-Jian Zhang at Peking University for providing part of the scripts for Iso-Seq data analyses. The data used for the analyses described in this manuscript were obtained from dbGaP (database of Genotypes and Phenotypes) accession number phs000424.v8.p2. The results shown here are in whole or part based upon data generated by the TCGA Research Network (https://www.cancer.gov/tcga) and The Genotype-Tissue Expression (GTEx) Project.
Conflict of interest
The authors declare that they have no conflicts of interests.
Yumei Li has received the PhD degree from the Institute of Molecular Medicine, Peking University.
Qing Sunny Shen has received the PhD degree from the Institute of Molecular Medicine, Peking University.
Qi Peng is a PhD student in the Institute of Molecular Medicine, College of Future Technology, Peking University.
Wanqiu Ding is an assistant investigator in the Institute of Molecular Medicine, College of Future Technology, Peking University.
Jie Zhang is a PhD student in the Institute of Molecular Medicine, College of Future Technology, Peking University.
Xiaoming Zhong has received the PhD degree from the Institute of Molecular Medicine, Peking University.
Ni A. An has received the PhD degree from the Institute of Molecular Medicine, Peking University. She is a post-doctoral investigator in the Institute of Molecular Medicine, College of Future Technology, Peking University.
Mingjun Ji is a PhD student in the Institute of Molecular Medicine, College of Future Technology, Peking University.
Wei-zhen Zhou is an assistant investigator of Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College.
Chuan-Yun Li is a professor and the principal investigator of laboratory of bioinformatics and genomic medicine, Institute of Molecular Medicine, Peking University. His research focuses on elucidating the molecular basis underlying human-specific traits, from the perspective of newly-originated, human-specific genes and regulatory events.
We developed a comprehensive protocol to de novo define the full-length macaque gene models and performed a comparative analysis on polyadenylation regulation to elucidate its contributions to the human–macaque differences.
Contributor Information
Yumei Li, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China.
Qing Sunny Shen, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China.
Qi Peng, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China; College of Future Technology, Peking University, Beijing, China.
Wanqiu Ding, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China; College of Future Technology, Peking University, Beijing, China.
Jie Zhang, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China; College of Future Technology, Peking University, Beijing, China.
Xiaoming Zhong, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China.
Ni A An, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China; College of Future Technology, Peking University, Beijing, China.
Mingjun Ji, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China; College of Future Technology, Peking University, Beijing, China.
Wei-Zhen Zhou, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, Beijing, China.
Chuan-Yun Li, Laboratory of Bioinformatics and Genomic Medicine, Institute of Molecular Medicine, Peking University, Beijing, China; College of Future Technology, Peking University, Beijing, China.
Data Availability
All sequencing data in this study are available at the NCBI (National Center for Biotechnology Information) GEO (Gene Expression Omnibus) repository under accession numbers GSE158668 and at the Genome Sequence Archive [79] in BIG Data Center [80], Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession numbers CRA001722.
Authors’ Contributions
C.Y.L. conceived the idea and designed the study. Y.L., Q.S. and Q.P. analyzed the data and performed most of the computational analyses. N.A.A., J.Z., X.Z. and W.Z.Z. performed part of the computational analyses. N.A.A. and M.J. performed the wet experiments. C.Y.L., Q.S., Y.L. and Q.P. wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the Ministry of Science and Technology of China (National Key Research and Development Program of China, 2018YFA0801405 and 2019YFA0801801), the National Natural Science Foundation of China (31871272, 31801103) and the Chinese Institute for Brain Research (Beijing) (2020-NKX-XM-11).
Ethics Approval and Consent to Participate
The macaque animals used to build the transcriptome atlas were from the Chinese population of rhesus macaques. The samples used in this study were obtained from the Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility at the Institute of Molecular Medicine, Peking University.
References
- 1. Rhesus Macaque Genome Sequencing and Analysis Consortium, Gibbs RA, Rogers J, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 2007;316:222–34. [DOI] [PubMed] [Google Scholar]
- 2. Zhang SJ, Wang C, Yan S, et al. Isoform evolution in primates through independent combination of alternative RNA processing events. Mol Biol Evol 2017;34:2453–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Merkin J, Russell C, Chen P, et al. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 2012;338:1593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang ET, Sandberg R, Luo SJ, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008;456:470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hubbard T, Barker D, Birney E, et al. The Ensembl genome database project. Nucleic Acids Res 2002;30:38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang SJ, Liu CJ, Shi M, et al. RhesusBase: a knowledgebase for the monkey research community. Nucleic Acids Res 2013;41:D892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang SJ, Liu CJ, Yu P, et al. Evolutionary interrogation of human biology in well-annotated genomic framework of rhesus macaque. Mol Biol Evol 2014;31:1309–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McCarthy A. Third generation DNA sequencing: Pacific Biosciences’ single molecule real time technology. Chem Biol 2010;17:675–6. [DOI] [PubMed] [Google Scholar]
- 9. Travers KJ, Chin CS, Rank DR, et al. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res 2010;38:e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kodzius R, Kojima M, Nishiyori H, et al. CAGE: cap analysis of gene expression. Nat Methods 2006;3:211–22. [DOI] [PubMed] [Google Scholar]
- 11. Liu S, Wang Z, Chen D, et al. Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expression in rhesus macaque brain. Genome Res 2017;27:1608–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Francescatto M, Lizio M, Philippens I, et al. Transcription start site profiling of 15 anatomical regions of the Macaca mulatto central nervous system. Sci Data 2017;4:170163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen JY, Peng Z, Zhang R, et al. RNA editome in rhesus macaque shaped by purifying selection. PLoS Genet 2014;10:e1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xie C, Zhang YE, Chen JY, et al. Hominoid-specific de novo protein-coding genes originating from long non-coding RNAs. PLoS Genet 2012;8:e1002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li YM, Li C, Li SX, et al. Human exonization through differential nucleosome occupancy. Proc Natl Acad Sci U S A 2018;115:8817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. An NA, Ding WQ, Yang XZ, et al. Evolutionarily significant A-to-I RNA editing events originated through G-to-A mutations in primates. Genome Biol 2019;20:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y, Han D, Han Y, et al. Ab initio identification of transcription start sites in the Rhesus macaque genome by histone modification and RNA-Seq. Nucleic Acids Res 2011;39:1408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lagarde J, Uszczynska-Ratajczak B, Carbonell S, et al. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat Genet 2017;49:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guenther MG, Levine SS, Boyer LA, et al. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007;130:77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A 2006;103:1412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Derti A, Garrett-Engele P, Macisaac KD, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res 2012;22:1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim LP, Burge CB. A computational analysis of sequence features involved in recognition of short introns. Proc Natl Acad Sci U S A 2001;98:11193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rocak S, Linder P. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 2004;5:232–41. [DOI] [PubMed] [Google Scholar]
- 24. Curmi F, Cauchi RJ. The multiple lives of DEAD-box RNA helicase DP103/DDX20/Gemin3. Biochem Soc Trans 2018;46:329–41. [DOI] [PubMed] [Google Scholar]
- 25. Chen W, Zhou P, Li X. High expression of DDX20 enhances the proliferation and metastatic potential of prostate cancer cells through the NF-kappaB pathway. Int J Mol Med 2016;37:1551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shin EM, Hay HS, Lee MH, et al. DEAD-box helicase DP103 defines metastatic potential of human breast cancers. J Clin Invest 2014;124:3807–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol 2017;18:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tian B, Graber JH. Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip Rev RNA 2012;3:385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang RJ, Zheng DH, Yehia G, et al. A compendium of conserved cleavage and polyadenylation events in mammalian genes. Genome Res 2018;28:1427–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanfilippo P, Wen J, Lai EC. Landscape and evolution of tissue-specific alternative polyadenylation across Drosophila species. Genome Biol 2017;18:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sheppard S, Lawson ND, Zhu LHJ. Accurate identification of polyadenylation sites from 3′ end deep sequencing using a naive Bayes classifier. Bioinformatics 2013;29:2564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nam DK, Lee S, Zhou GL, et al. Oligo(dT) primer generates a high frequency of truncated cDNAs through internal poly(A) priming during reverse transcription. Proc Natl Acad Sci U S A 2002;99:6152–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. GTEx Consortium . The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhong X, Peng J, Shen QS, et al. RhesusBase PopGateway: genome-wide population genetics atlas in rhesus macaque. Mol Biol Evol 2016;33:1370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matoulkova E, Michalova E, Vojtesek B, et al. The role of the 3' untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol 2012;9:563–76. [DOI] [PubMed] [Google Scholar]
- 37. Neve J, Burger K, Li WC, et al. Subcellular RNA profiling links splicing and nuclear DICER1 to alternative cleavage and polyadenylation. Genome Res 2016;26:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao W, Blagev D, Pollack JL, et al. Toward a systematic understanding of mRNA 3′ untranslated regions. Proc Am Thorac Soc 2011;8:163–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Behm-Ansmant I, Rehwinkel J, Izaurralde E. MicroRNAs silence gene expression by repressing protein expression and/or by promoting mRNA decay. Cold Spring Harb Symp Quant Biol 2006;71:523–30. [DOI] [PubMed] [Google Scholar]
- 40. Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007;129:1401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin KC, Ephrussi A. mRNA localization: gene expression in the spatial dimension. Cell 2009;136:719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Andreassi C, Riccio A. To localize or not to localize: mRNA fate is in 3' UTR ends. Trends Cell Biol 2009;19:465–74. [DOI] [PubMed] [Google Scholar]
- 43. Berkovits BD, Mayr C. Alternative 3' UTRs act as scaffolds to regulate membrane protein localization. Nature 2015;522:363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fazal FM, Han S, Parker KR, et al. Atlas of subcellular RNA localization revealed by APEX-Seq. Cell 2019;178:473–490.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khaitovich P, Enard W, Lachmann M, et al. Evolution of primate gene expression. Nat Rev Genet 2006;7:693–702. [DOI] [PubMed] [Google Scholar]
- 46. Necsulea A, Kaessmann H. Evolutionary dynamics of coding and non-coding transcriptomes. Nat Rev Genet 2014;15:734–48. [DOI] [PubMed] [Google Scholar]
- 47. Gilad Y, Oshlack A, Smyth GK, et al. Expression profiling in primates reveals a rapid evolution of human transcription factors. Nature 2006;440:242–5. [DOI] [PubMed] [Google Scholar]
- 48. Wang G, Zhou D, Wang C, et al. Hypoxic preconditioning suppresses group III secreted phospholipase A2-induced apoptosis via JAK2-STAT3 activation in cortical neurons. J Neurochem 2010;114:1039–48. [DOI] [PubMed] [Google Scholar]
- 49. Yeo IJ, Park MH, Son DJ, et al. PRDX6 inhibits neurogenesis through downregulation of WDFY1-mediated TLR4 signal. Mol Neurobiol 2019;56:3132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmeisser MJ, Kuhl SJ, Schoen M, et al. The Nedd4-binding protein 3 (N4BP3) is crucial for axonal and dendritic branching in developing neurons. Neural Dev 2013;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kiem LM, Dietmann P, Linnemann A, et al. The Nedd4 binding protein 3 is required for anterior neural development in Xenopus laevis. Dev Biol 2017;423:66–76. [DOI] [PubMed] [Google Scholar]
- 52. Takano T, Xu CD, Funahashi Y, et al. Neuronal polarization. Development 2015;142:2088–93. [DOI] [PubMed] [Google Scholar]
- 53. Kislauskis EH, Singer RH. Determinants of mRNA localization. Curr Opin Cell Biol 1992;4:975–8. [DOI] [PubMed] [Google Scholar]
- 54. Yudin D, Hanz S, Yoo S, et al. Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron 2008;59:241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. An JJ, Gharami K, Liao GY, et al. Distinct role of long 3' UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 2008;134:175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Muller-McNicoll M, Neugebauer KM. How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat Rev Genet 2013;14:275–87. [DOI] [PubMed] [Google Scholar]
- 57. Davis CA, Hitz BC, Sloan CA, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 2018;46:D794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takagaki Y, Manley JL. RNA recognition by the human polyadenylation factor CstF. Mol Cell Biol 1997;17:3907–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yao C, Choi EA, Weng L, et al. Overlapping and distinct functions of CstF64 and CstF64tau in mammalian mRNA 3′ processing. RNA 2013;19:1781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005;21:1859–75. [DOI] [PubMed] [Google Scholar]
- 61. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Noguchi S, Arakawa T, Fukuda S, et al. FANTOM5 CAGE profiles of human and mouse samples. Sci Data 2017;4:170112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. O’Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 2016;44:D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Françoise Thibaud-Nissen AS, Murphy T, DiCuccio M, et al. Eukaryotic Genome Annotation Pipeline. https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/; last accessed April 18, 2021.
- 66. Crooks GE, Hon G, Chandonia JM, et al. WebLogo: a sequence logo generator. Genome Res 2004;14:1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987;196:261–82. [DOI] [PubMed] [Google Scholar]
- 68. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol 1990;215:403–10. [DOI] [PubMed] [Google Scholar]
- 69. Sheets MD, Ogg SC, Wickens MP. Point mutations in AAUAAA and the poly (A) addition site: effects on the accuracy and efficiency of cleavage and polyadenylation in vitro. Nucleic Acids Res 1990;18:5799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang LG, Wang SQ, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 2012;28:2184–5. [DOI] [PubMed] [Google Scholar]
- 71. Chen JY, Shen QS, Zhou WZ, et al. Emergence, retention and selection: a trilogy of origination for functional de novo proteins from ancestral lncRNAs in primates. PLoS Genet 2015;11:e1005391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chiaromonte F, Yap VB, Miller W. Scoring pairwise genomic sequence alignments. Pac Symp Biocomput 2002;115–26. [DOI] [PubMed] [Google Scholar]
- 73. Chen XS, Chen ZD, Chen H, et al. Nucleosomes suppress spontaneous mutations base-specifically in eukaryotes. Science 2012;335:1235–8. [DOI] [PubMed] [Google Scholar]
- 74. Prendergast JGD, Semple CAM. Widespread signatures of recent selection linked to nucleosome positioning in the human lineage. Genome Res 2011;21:1777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cardoso-Moreira M, Halbert J, Valloton D, et al. Gene expression across mammalian organ development. Nature 2019;571:505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang Y, Song F, Zhu J, et al. GSA: Genome Sequence Archive. Genomics Proteomics Bioinformatics 2017;15:14–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. BIG Data Center Members . Database resources of the BIG Data Center in 2019. Nucleic Acids Res 2019;47:D8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data in this study are available at the NCBI (National Center for Biotechnology Information) GEO (Gene Expression Omnibus) repository under accession numbers GSE158668 and at the Genome Sequence Archive [79] in BIG Data Center [80], Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession numbers CRA001722.