

Abstract
Promoter annotation is an important task in the analysis of a genome. One of the main challenges for this task is locating the border between the promoter region and the transcribing region of the gene, the transcription start site (TSS). The TSS is the reference point to delimit the DNA sequence responsible for the assembly of the transcribing complex. As the same gene can have more than one TSS, so to delimit the promoter region, it is important to locate the closest TSS to the site of the beginning of the translation. This paper presents TSSFinder, a new software for the prediction of the TSS signal of eukaryotic genes that is significantly more accurate than other available software. We currently are the only application to offer pre-trained models for six different eukaryotic organisms: Arabidopsis thaliana, Drosophila melanogaster, Gallus gallus, Homo sapiens, Oryza sativa and Saccharomyces cerevisiae. Additionally, our software can be easily customized for specific organisms using only 125 DNA sequences with a validated TSS signal and corresponding genomic locations as a training set. TSSFinder is a valuable new tool for the annotation of genomes. TSSFinder source code and docker container can be downloaded from http://tssfinder.github.io. Alternatively, TSSFinder is also available as a web service at http://sucest-fun.org/wsapp/tssfinder/.
Keywords: transcription start site, promoter region, annotation of genomes, conditional random fields
Introduction
The process of gene expression is controlled by different sub-sequences of DNA called regulatory sequences [1]. Regardless of the organism, the upstream region known as the promoter region is the main DNA sequence responsible for the recognition and binding of RNA polymerase. In eukaryotes the DNA sub-sequence known as promoter region is the main regulatory sequence, presenting a diverse and complex architecture [1, 2]. It is possible to divide the promoter region into three sub-regions [2]: core, proximal and distal promoter. The core promoter is characterized by the presence of sequence motifs such as Initiator (Inr), TCT, TATA-box, downstream promoter element (DPE) and DNA recognition element. These motifs are generally positioned in a window of up to 50 nucleotides upstream or downstream of the transcription start site (TSS) [3–5]. The proximal promoter region is the region comprising the 500 nucleotides upstream of the TSS signal and can present sequence motifs such as CAAT-box, GC-box and cis-regulatory modules [2, 4]. The distal promoter region is located upstream of the proximal promoter region and contains regulatory sequences such as enhancers, isolators and silencers [2].
The core promoter has particular relevance for gene transcription because it is where the cell anchors the general transcription factors responsible for the recruitment and positioning of RNA polymerase II (RNA pol II) during gene transcription [1–4]. This makes this region of particular importance for synthetic biology as it is the primary target region for the identification of transcription factor binding sites (TFBSs) [6–8].
Based on OligoCap, CAGE, deepCAGE and PEAT experiments, TSS signals can be considered sharp (focused) or broad (dispersed). Sharp signals contain only one TSS peak, and broad signals can contain more peaks. In general, TSSs with sharp signals contain the TATA-box motif, placed at a distance of approximately 30 nucleotides upstream [1]. In contrast, TSSs with a dispersed signal are associated with large distances from the start codon, usually with the TSS a distance farther than 1000 nucleotides upstream of the gene body. In addition, in these sequences, the motifs Inr, DPE and DCE do not have a fixed position and, in general, the TATA-box motif is absent [1, 9]. For this reason, the TSS signal prediction tools and, consequently, the identification of the core promoter region is modeled using numerous features (flexibility, curvature, base stacking and duplex stability) and dependencies (TATA-box/Inr with a 30-nt distance, nucleosome-free region and epigenetic marks). Unfortunately, with this type of modeling, the procedure used to predict the TSS signal can affect the time of execution of the model and the accuracy of the results. Thus, some tools use heuristics to filter the results through prediction scores or simply choose the prediction closest to the reference point. In general the reference used is the beginning of the gene (start codon) or the annotated TSS signal [1, 2].
Originally, in silico promoter region classification was performed without the correct TSS demarcation, the most common procedure was placing it at a fixed distance from the annotated TSS [4, 9, 10]. As the size of the 5’UTR region is generally between 650 and 950 nucleotides in plant monocots and dicots [10] and 500 to 1000 nucleotides in humans and mouse [11], a postulated distance between the start codon and the TSS between 500 and 1500 is common. In procaryotes, the TSS signal is located at a distance of up to 100nt from the beginning of the gene but, in some cases, this distance may be close to 400 nt [12]. Due to the variation in the size of this distance ( 5’UTR regions and, eventually, the corresponding introns), this procedure tends to, as a rule, either include 5’UTR and intron regions or exclude parts of the core promoter [4, 9, 10, 12]. As a consequence, we can see in the literature conflicting characterizations of the core promoter regions when the analysis was performed in distinct studies for the same organism [13, 14].
In eukaryotes we can have up to three divisions of the promoter region (core, proximal and distal promoter) and two main classes (TATA-box and TATA-less). On the other hand, for prokaryotic the size of the DNA sequence is not considered as a criterion for dividing the promoter region. Furthermore, the classification of this region is defined by motifs of the 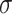 factor in the DNA sequence. So far it is possible to classify the promoter region into 8 groups:
factor in the DNA sequence. So far it is possible to classify the promoter region into 8 groups:  ,
, 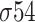 ,
, 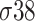 ,
,  ,
, 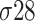 ,
,  and
and 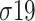 [15–17].
[15–17].
Each factor  presents peculiarities both in the location and in the consensus sequences; however, in each class, these characteristics are well defined. For example, for class
presents peculiarities both in the location and in the consensus sequences; however, in each class, these characteristics are well defined. For example, for class 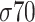 and
and 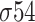 , there are two defined motifs. For the
, there are two defined motifs. For the  class, the first motif is located 10 nt upstream from the TSS and it has TATAAT consensus sequence and the second is 35 upstream from TSS with TTGACA consensus sequence. For the
class, the first motif is located 10 nt upstream from the TSS and it has TATAAT consensus sequence and the second is 35 upstream from TSS with TTGACA consensus sequence. For the 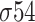 class, the first is located 12 nt upstream from the TSS and has consensus sequence TGC[AT][TA] and the second motifs is 24 nt upstream from TSS with [CT]TGGCA[CT][GA] consensus sequence [15, 18].
class, the first is located 12 nt upstream from the TSS and has consensus sequence TGC[AT][TA] and the second motifs is 24 nt upstream from TSS with [CT]TGGCA[CT][GA] consensus sequence [15, 18].
Due to these characteristics, the software used in the labeling of the prokaryotic promoter region aims to classify the DNA sequence in the different  groups in detriment to the prediction of biological signals, such as the TSS signal [16, 17, 19].
groups in detriment to the prediction of biological signals, such as the TSS signal [16, 17, 19].
Identification of TSS in vivo and in silico
The TSS signals can be determined by in vivo experiments performed on the pre-mRNA molecule using techniques such as OligoCap, CAGE, deepCAGE and PEAT [1, 9, 14, 20]. However, despite their accuracy, these methods have a high cost of execution and do not always produce satisfactory results [21].
Probably the most straightforward way to locate the TSS is by mapping full cDNA transcripts into the genome using an alignment tool such as SIM-4 [22], BLAT [23] and GMAP [24]. However, obtaining a significant percentage of all full transcripts for a given organism is not a simple task, since the beginning and end of these sequences may be missing or incomplete [25, 26]. In general, the assembly tools vary in quality from the skill in executing the technique, in the length of the sequence, and in the coverage of the untranslated region, especially in the 5’UTR region [25, 26].
An indication of the difficulties in locating the TSS signal either by mapping or by in vitro experiments is that the eukaryotic promoter database (EPD database), the main repository for experimentally confirmed eukaryotic promoters, includes promoter regions of only 15 species, most of which are model organisms [20]. The coverage of genes labeled with the TSS signal may represent around 30% of the annotated genes [20].
With the high cost of in vivo determination of the TSS location, and the low availability of full transcripts for most organisms, in silico prediction is frequently the only annotation available. To label the promoting region, there are different labeling models that can present simple models such as position weight matrix up to more complex models such as convolutional neural network [17, 27]. During the 1990s the identification of the core promoter was performed by measuring the enrichment of known motifs (PromoterScan [28]; TSSW, TSSG, TSSP and TSSP-TCM [29]) or by using hexamer frequency (PromFind [30]). At that time the core promoter analysis was less specific and the studies were not directed towards the discovery of the TSS signals. In general, these models have been used in prokaryotic and eukaryotic organisms, but only to classify the DNA sequence as promoter region or not, and not to locate the TSS location [16, 19, 31].
In the 21st century, the tools began to direct their efforts to label the core promoter through the identification of the TSS signal. The great majority of the tools was directed at eukaryotes, and a smaller number of software targeted at prokaryotes, ensemble-SVM, TSS-PREDICT and IBBP [12, 32, 33]. New approaches were presented using neural networks (DragonGSF [34]), support vector machine (ensemble-SVM [12], TSS-PREDICT [32], ARTS [35]), self-organizing maps (ProSOM [36]), convolutional neural network (iPromoter-BnCNN [17]), image processing techniques (IBBP [33]) and stacked-ensemble approach (SELECTOR [19]). Still, the accuracy of these approaches is very low: all tools predicted less than 55% of TSSs with an error smaller than 500 nt, both ARTS and ProSOM are targeted only to humans and DragonGFS is currently not available for free download. Recently, Cassiano and Silva-Rocha performed a review to assess the capacity of 10 tools for predicting the promoter region in prokaryotic. They showed that only three software presented an accuracy close to 75% [37]. For the eukaryotic promoter region, both Lai and collaborators developed iProEp [18] and Zhu and collaborators developed Depicter [38], tools with high precision in distinguishing promoter region from non-promoter region, but, still, they are not a predictor and are only able to classify a previously selected region as a promoter or non-promoter sequence. In literature we could find only a few promoter characterization applications that attempt to characterize both eukaryotic and prokaryotic sequences and, again, they are just classifiers that do not attempt to locate the TSS.
In the past 4 years three new methodologies were able to raise significantly the precision of the TSS predictions: logistic regression, neural networks and Bayes networks.
Logistic regression was used by two related tools: 3PEAT [39] and TIPR [40]. None of the tools is a TSS predictor per se, they both output, given a sequence of size 8000–10000 nucleotides, a series of signal peaks associated with the strength of the TSS signal. The two tools use the same methodology and differ in two aspects: (i) target organism [3PEAT was implemented for an (Arabidopsis thaliana model) and TIPR for a (Mus musculus model)]; and (ii) the classification of the predicted TSS sites of TSS–3PEAT (narrow, broad and weak peak) and TIPR (single and broad peak). Neural network is used by TSSPlant [4] and TransPrise [41]. TransPrise in particular uses convolutional neural networks to improve the prediction performance of the neural network-based model (TSSPlant). Bayesian networks are used by the software BayesProm [42], to model the positional density of several hexamers that are associated with the promoter region.
TSSFinder
In this work we present TSSFinder, a new approach for the TSS prediction of annotated genes using linear chain conditional random fields (LCCRFs) [43]. Linear chain conditional random fields are a restriction in the more general conditional random fiedls (CRFs) but present clear advantages in the speed of the inference algorithms [44]. Conditional random fields have been used successfully for the characterization of gene structures [44–46], the CRF-based models did not have their previous use directed to find the location of the TSS signal.
In this article, we will show that LCCRFs present higher precision than other approaches. TSSFinder is directed to find the focused TSS signal from the protein-encoding genes and into genes with the dispersed TSS signal, the TSS signal closest to the start codon. Finally, TSSFinder offers pre-trained models for plants, vertebrates, insects and fungi. Users can either perform predictions using the available model of the closest organism or can train organism-specific new models with as little as 125 full transcripts.
We compared TSSFinder’s performance in determining the most downstream TSS for a gene against TSSPlant, TransPrise and BayesProm using confirmed TSS data of A. thaliana, Oryza sativa and Homo sapiens. Similar to TSSFinder, TSSPlant, TransPrise and BayesProm are software that targets the characterization of the core promoter directly on upstream regions of annotated genes and can then be used without any previous in vitro experiments [4, 41, 42].
Additionally, we performed 5-fold cross-validation of TSSFinder in six organisms: A. thaliana, Drosophila melanogaster, Gallus gallus, H. sapiens, O. sativa and Saccharomyces cerevisiae. Finally, we used TSSFinder to predict TSS signals in more than 140 000 ENSEMBL genes with no previous TSS annotation.
Methods
Promoter model
TSSFinder uses a LCCRF [43] to model the promoter region. The architecture of the model is illustrated in Figure 1.
Figure 1.

State machine that defines features of TSSFinder’s model. In the figure, each rectangle (state) represents a signal, where rectangles with gray background refer to the main signals used in modeling the problem. The states are: i)Begin - determines the starting point of the DNA sequence; ii) Promoter, describing the  end of the promoter sequence; iii)TATA-box - the location coordinates of the nucleotides of the TATA-box site in the DNA sequence; iv) Post TATA-box, the region between the TATA-box and the TSS; v) TSS - the location coordinate of the TSS site in the DNA sequence; vi)PSC(Pre-Start Codon region), to model the region between the TSS and the start codon - PSC); vii) START CODON, modeling the ATG site of the gene. The arrows represent possible paths for the labeling process. Of note, TSSFinder can label sequences without a TATA-box and even without the TSS.
end of the promoter sequence; iii)TATA-box - the location coordinates of the nucleotides of the TATA-box site in the DNA sequence; iv) Post TATA-box, the region between the TATA-box and the TSS; v) TSS - the location coordinate of the TSS site in the DNA sequence; vi)PSC(Pre-Start Codon region), to model the region between the TSS and the start codon - PSC); vii) START CODON, modeling the ATG site of the gene. The arrows represent possible paths for the labeling process. Of note, TSSFinder can label sequences without a TATA-box and even without the TSS.
Our model covers the proximal and core promoter regions, as well as the DNA sequence separating the TSS from the start codon. Therefore, we consider the region after the TSS, even though it is not part of the promoter region as defined above. Following the input model, TSSFinder will segment the input sequence into seven parts, each one corresponding to a label: (i) begin—the start position of the promoter region; (ii) promoter: the sub-region after the beginning of the promoter region and before the TATA-box (if there is no TATA-box this state represents the whole promoter region); (iii) TATA-box: the TATA-box pattern; (iv) pos TATA-box: the sub-region between the TATA-box and the TSS; (v) TSS: the position of the TSS; (vi) PSC (pre-start codon region); (vii) Start Codon, representing the end of the modeled sequence.
CRFs in general, and LCCRFs in particular, measure the characteristics of the subject using ‘feature functions’, which are measures associated with positions in the input. The time efficiency of the model is directly related to the number of feature functions used. So, limiting the number of feature functions was important to lower computing and training. There are two types of feature functions commonly used in CRFs: binary features functions and n-grams.
We used binary features to characterize the distances from each part of our model to the start codon. We transformed distance measures into binary functions for each distance interval (0–50 nt, 51–100 nt, etc.), which values ‘1’ if the feature is in that interval and ‘0’ otherwise. As a consequence, we have a new feature function for each interval. We found that intervals of 50 nucleotides presented a good balance between precision and the number of feature functions. Other common feature functions in CRFs are n-grams [47], which measure the occurrence of a sequence of k-consecutive letters. We can modify this feature to measure the various sub-sequences of size k within a fixed window, effectively measuring the dependencies between each k nucleotides within that distance window. In our experiments, we found out that a window of size 7 was effective. However, if we used all n-grams that would mean 16 384 different features (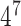 ), which would require too many training sequences and also increase training and computing time. We, therefore, opted to use all 2 grams inside the window of size 7, a total of 42 features (one for each 2 grams), which presented a good balance between precision and processing times.
), which would require too many training sequences and also increase training and computing time. We, therefore, opted to use all 2 grams inside the window of size 7, a total of 42 features (one for each 2 grams), which presented a good balance between precision and processing times.
The LCCRF models were trained, for each data set, using promoter nucleotide sequences, the position of the start codon, the confirmed TSS location and, when available, the location of the TATA-box. A more detailed description of the LCCRF model is given in the section Linear-chain CRF promoter model in Supplementary Material.
Datasets
To perform the comparison and cross-validation experiments we used datasets from six different organisms: A. thaliana, D. melanogaster, G. gallus, H. sapiens, S. cerevisiae and O. sativa. We selected s and promoter sequences for the first five organisms from the EPD Database [20]. For each organism, we downloaded all promoter sequences of genes with a TSS validated by high-throughput experiments using the cap-trapper or oligo-capping technique: 20 183 promoters of A. thaliana, 16 972 promoters of D. melanogaster, 6127 promoters of G. gallus, 21 170 promoters of H. sapiens and 5117 promoters of S. cerevisiae. Following Pachganov [41] and Shahmuradov [4], we used sequences with only one annotated TSS. Then, we randomly selected 5000 sequences for each organism, except S. cerevisiae, for which there were only 4675 available after the filtering. For O. sativa we selected 5’UTR and promoter sequences validated using full-length cDNA sequences of O. sativa ssp. japonica cv. Nipponbare (Os-Nipponbare-Reference-IRGSP-1.0), obtained from the Rice Annotation Project database, RAP-DB [48]. From these, we eliminated all gene entries with more than one annotated start codon. Table 1 describes the sizes and sources of the datasets.
Table 1.
Datasets
| Organisms | Genes | Promoters | Filtered | Final | Method/ |
|---|---|---|---|---|---|
| size | size | size | size | source | |
| Arabidopsis | 19 924 | 20 183 | 19 626 | 5000 | Cap-trapper |
| thaliana | and/or | ||||
| (v003) | oligo-capping | ||||
| (EPD) | |||||
| Drosophila | 13 399 | 16 972 | 10 922 | 5000 | Cap-trapper |
| melanogaster | and/or | ||||
| (v005) | oligo-capping | ||||
| (EPD) | |||||
| Gallus | 5632 | 6127 | 3202 | 3200 | Cap-trapper |
| gallus | and/or | ||||
| (v001) | oligo-capping | ||||
| (EPD) | |||||
| Homo | 17 892 | 21 170 | 16 349 | 5000 | Cap-trapper |
| sapiens | and/or | ||||
| (v005) | oligo-capping | ||||
| (EPD) | |||||
| Saccharomyces | 5117 | 5117 | 4675 | 4675 | Cap-trapper |
| cerevisiae | and/or | ||||
| (v002) | oligo-capping | ||||
| (EPD) | |||||
| Oryza | 29 113 | 45 641 | 28 834 | 5000 | Full-length |
| sativa | cDNA | ||||
| (RAP-DB) |
This table summarizes the process to obtain the final benchmarks used in the experiments. The first column (original size) contains the number of genes for which there were validated promoters, the second (filtered size) the number of genes with a single validated TSS, the third (final size) the number of genes randomly selected from the filtered set. The last column indicates the procedure reported for the validation of the TSS. v00x: Eukaryotic Promoter Database data version number; EPD: Eukaryotic Promoter Database; RAP-DB: Rice Annotation Project Database.
All the sequences used in the experiments are available in TSSFinder’s github page http://tssfinder.github.io in the downloads section under the heading cross-validation.
Accuracy computation
To compute accuracy we used the standard precision, recall and F-1 score measures:
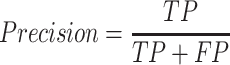 |
(1) |
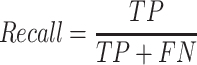 |
(2) |
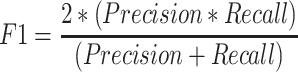 |
(3) |
We used five different distance thresholds to define a ‘true positive value’: 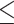 0–50nt,
0–50nt, 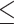 0–100 nt,
0–100 nt, 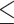 0–150 nt,
0–150 nt, 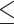 0–200 nt and
0–200 nt and 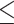 0–250 nt. Predictions within each threshold distance of the TSS were considered true positives (TPs) and predictions at a greater distance were considered false positives (FPs). False negatives (FNs) were any sequence without a TSS prediction. Sequences without a TSS were considered negative examples, and predictions in these sequences were considered false positives (FPs).
0–250 nt. Predictions within each threshold distance of the TSS were considered true positives (TPs) and predictions at a greater distance were considered false positives (FPs). False negatives (FNs) were any sequence without a TSS prediction. Sequences without a TSS were considered negative examples, and predictions in these sequences were considered false positives (FPs).
Comparing TSSFinder with other software
There are many different computer programs previously developed that can be used to characterize TSSs of a eukaryotic gene but, to the best of our knowledge, TSSPlant [4], TransPrise [41] and BayesProm [42] are currently the only freely available predictors for which more than 50% of the TSS predictions are closer than 500 nt from a confirmed TSS and that are targeted to the direct use in the genome without the need for previous in vitro experiments to locate the target regions [4, 41, 42].
As TSSPlant is targeted only to plants, we compared TSSFinder against TSSPlant and TransPrise in two plant organisms. As BayesProm was trained in humans we compared it with TSSFinder using the same organism. We have compared TSSFinder and TSSPlant in promoter sequences with experimentally validated TSSs of A. thaliana and O. sativa. For the comparison, TSSPlant was applied to sequences 1100 nt long comprising 1000 nt upstream of the start codon and 100 nt downstream of the start codon, as indicated in the original article. For consistency in the comparisons, we applied TSSFinder to the 1000 nt region upstream of the start codon (TSSFinder does not model the Coding Sequence region—CDS—after the start codon). Accordingly, in these experiments, any promoter sequence with a TSS at a distance greater than 1000 nt from the start codon was considered a negative example. To run TSSPlant we used the model provided by the developers [4]. TransPrise is not directed to any specific group of organisms, and it can be trained. However, we were not able to install the native version in our Linux systems, as some of the required libraries could not be located (contact with the authors was also fruitless). We used the Docker container that has only an O. sativa model [41]. Due to this limitation, we used the same plant organisms (A. thaliana and O. sativa) in the comparison. For consistency in the comparison, we used the region of 1000 nt upstream of the start codon as the search sequence. For both TSSFinder and TransPrise, when more than one TSS was predicted, we used the most downstream prediction (in these experiments, this choice led to better scores for TSSPlant, and roughly equivalent results in TransPrise, with differences lower than 0.3 percentage points).
BayesProm can be obtained only in a.exe file for the Microsoft Windows environment and it is pre-trained for H. sapiens. Therefore, we performed a separate comparison between TSSFinder and BayesProm using this organism. As the original article stated that the minimum size of the scanned region is 1000 nt, we opted to use region comprising 2000 nt upstream of the start codon and 500 nt downstream (excluding the downstream nucleotides incurred in much inferior results). Accordingly, genes with the TSS located more than 2000 nt upstream of the start codon were considered negative examples. When more than one TSS was predicted, we used the most downstream prediction (again, this choice led to better scores in this experiment).
Both TSSPlant, TransPrise and BayesProm were executed locally on a Linux Ubuntu laptop. For TSSPlant we used the execution script available at http://www.softberry.com/distrib/freedownload/promoter/TSSPlant/doc/TSSPlant-docs.tar.bz2. For TransPrise we used the docker system available at https://hub.docker.com/r/zarubinaa/tss-rice/. For BayesProm, we use the executable application on a Windows Desktop available at https://www.comp.nus.edu.sg/∼bioinfo/BayesProm/index.htm.
Validation
To confirm TSSFinder’s performance in a broad range of situations, we performed a series of validation experiments: (i) six 5-fold cross-validation experiments in organism-specific benchmarks; (ii), six cross-organism experiments, where TSSFinder was trained using promoter data from of one organism and evaluated in promoter regions of another organism.
Cross-validation
We performed independent 5-fold cross-validation experiments for TSSFinder in six different organisms: A. thaliana, D. melanogaster, G. gallus, H. sapiens, S. cerevisiae and O. sativa. For each evaluated organism, we divided each data set into five subsets of equal size and, on each step, one different sub-set was used for testing and four remaining sub-sets were joined to train the model. Training data consisted of the 2000 nt upstream region from the annotated start codon of each gene, the annotated TATA-signal position if it was present, and the validated TSS location. Prediction input consisted of the 2000 nt upstream region of the annotated start codon. For the promoters of A. thaliana, D. melanogaster, G. gallus, H. sapiens and S. cerevisiae the BED file of TATA-box locations included the locations recorded in the Eukaryotic Promoter Database. For O. sativa core promoters the position of the TATA-box was estimated using Motif-Suite [49] with a position weight matrix (PWM—sequence consensus G/C TA/T TA/T A/T AA/T G/A G/C C/G G/C G/C G/C G/C ) obtained from the JASPAR core polymerase II database [50]. For these, we analyzed the region of 100 nucleotides upstream of the annotated TSS. All sequences with a positive mapping of the TATA-box PWM located between nucleotides 10 and 35 upstream of the TSS signal were considered valid TATA-box, and the region registered in the BED training files [10, 20, 50].
Assessing inter-species model precision
Ideally, TSSFinder should be used with a model trained in the same organism. However, in many cases, researchers may not have the necessary validated TSSs for training. In these cases, TSSFinder can use a model trained for a related organism. To provide an initial assessment of the precision of inter-species TSS annotation, TSSs were predicted in five organisms: Apis mellifera, A. thaliana, Canis familiaris, O. sativa and Zea mays. Similarly to the validation experiment, for each organism, we used regions from all genes with experimentally confirmed TSS in EPD. The test data set sizes were, respectively 5195, 5403 and 7229 promoter regions. For the predictions, we used five different models, each trained in one of the folds of the validation experiment for the organism. Four organisms were used to train the models: A. thaliana, D. melanogaster, H. sapiens and O. sativa. Table 2 depicts the details.
Table 2.
Target organisms versus training sequence organisms for inter-species model precision
| Target | Test set | Training set | Training set |
|---|---|---|---|
| organism | size | organism | size |
| Apis | 5195 | Drosophila | 4000 |
| mellifera | melanogaster | ||
| Arabidopsis | 5000 | Oryza | 4000 |
| thaliana | sativa | ||
| Canis | 5403 | Homo | 4000 |
| familiaris | sapiens | ||
| Oryza | 5000 | Arabidopsis | 4000 |
| sativa | thaliana | ||
| Zea | 7229 | Arabidopsis | 4000 |
| mays | thaliana | ||
| Zea | 7229 | Oryza | 4000 |
| mays | sativa |
Each of the training sets consisted of one of the training datasets of the 5-fold cross-validation experiment of the corresponding organism. With the exception of Zea mays promoter sequences and annotations were selected from the benchmarks of the validation process. For Z. mays, we used promoter sequences with validated TSS in the EPD data set [20].
Learning curve
To estimate a minimum size for a training set to produce reliable predictions, we measured the accuracy versus training set size using O. sativa. We randomly selected one of the folds in the previous validation process and created training sets of sizes 4000, 2000, 1000, 500, 250 and 125 promoter sequences. On each step, we randomly selected half of the training set of the previous step. The trained models were used to predict the testing set for that fold (1000 sequences). We computed the true positive rates for the different training set sizes, considering, for each one, different target distances (errors less than 50 nt, 100 nt, 150 nt, 200 nt and 250 nt).
Annotation of new TSS sites
We used TSSFinder to predict new TSS sites for ENSEMBL genes that currently lack a TSS annotation. For this, we downloaded from ENSEMBL [51] the genomic sequences of the 2000 nt upstream regions for 14 organisms: A. thaliana, Arabidopsis lyrata, D. melanogaster, A. mellifera, G. gallus, H. sapiens, Macaca mulatta, M. musculus, Rattus norvegicus, Canis familiaris, S. cerevisiae, Oryza glaberrima, Oryza brachyantha and O. sativa.
In each genome we performed the prediction of the TSS signal as follows: for A. thaliana and A. lyrata we used the previously trained A. thaliana model; for D. melanogaster and A. mellifera we used the D. melanogaster model; for S. cerevisiae we used the S. cerevisiae model; for G. gallus we used the G. gallus model; for O. brachyantha, O. glaberrima and O. sativa we used the O. sativa model; for C. familiaris, H. sapiens, M. mulatta, M. musculus and R. norvegicus we used the H. sapiens model.
RESULTS
Comparing TSSFinder with other software
We compared TSSFinder’s performance against TSSPlant [4], TransPrise [41] and BayesProm [42]. The comparison against TransPrise and TSSPlant was performed in A. thaliana and O. sativa, and the comparison against BayesProm was performed in H. sapiens. F1-scores were computed for five different distance brackets: 0–50 nt, 0–100 nt, 0–150 nt, 0–200 nt and -250 nt. The F1-score results are summarized in Figure 2 and show that TSSFinder’s predictions present a much higher F1-score for all distance brackets.
Figure 2.

(a) Prediction of the TSS signal in Arabidopsis thaliana. (b) Prediction of the TSS signal in Oryza sativa. (c) Prediction of the TSS signal in Homo sapiens. Precision numbers indicate average of the values across the four validation datasets of the cross-validation experiment. The regions analyzed were (-1000,0) for, (-2000,500) for BayesProm, (-1000,100) for TSSPlant, (-1000,0) for TSSFinder in A. thaliana and O. sativa and (-2000,0) for TSSFinder in H. sapiens. The width represents the distance in nucleotides of the predicted TSS signal in relation to the TSS signal validated by biological experiments. The standard deviation is represented in the figure using error margins. A more detailed description of the figures can be found in Tables 1, 2 and 3 in Supplementary Material.
In A. thaliana F1-score values for TSSFinder were well above those of TSSPlant and TransPrise, in particular for predictions in the distance range of 0–50 nt (average 68.6%, 41.3% and 3.9%, respectively), with decreasing advantages in relation to TSSPlant for distances 200 nt and 250 nt, but still maintaining a significant advantage in the 250 nt range (94.1 and 84.2, respectively). For O. sativa TSSFinder presented numbers consistent with those of A. thaliana: performance advantage ranged from 53.0% versus 21.1% and 2.7% in the 50 nt range to 88.9% versus 80.1% and 5.9% in the 250 nt range. The performance of TransPrise in this last benchmark was inferior to the first one, a fact that is curious because the model used in the experiment was trained with O. sativa ssp. japonica cv. Nipponbare [41]. Of note, while TSSFinder and TSSPlant presented similar precision and recall values in each bracket, TransPrise consistently presented very low recall values, which lowered significantly the F1-score. Still, precision values were much lower than TSSFinder and TSSPlant (see in Tables 1 and 2 in Supplementary Material).
In H. sapiens, TSSFinder also presented the best F1-score values, this time compared with BayesProm. The F1-score advantage ranged from 34.3% versus 17.8% in the 0–50 nt range to 72.1% versus 55.4% in the 0–250 nt range. The full precision and recall numbers for each fold can be found in Table 3 in the Supplementary Material.
Processing times
TSSFinder also has an advantage in prediction speed compared with TSSPlant and TransPrise: using the same data set, TSSFinder performed the predictions using only between 7% and 8% of the computing time used by TSSPlant, and less than 2% of the time used by TransPrise. We used for the processing time test four datasets with 500, 1000 and 2000 sequences extracted from the A. thaliana and 500 sequences from the O. sativa benchmarks. We could not perform a uniform speed comparison against BayesProm, as this application executes only on Microsoft Windows computers (all the others run in Linux) and the same CPU was not available in Linux and Windows systems. BayesProm considerably smaller processing time, but in a faster CPU (intel I3 1.8 Ghz versus intel I5 2.5 Ghz). However, considering the timing difference (approximately seven times), we can consider BayesProm the faster alternative. The results for the set of 500 sequences are shown in Table 3.
Table 3.
Processing times
| Software | Organism | Dataset | nt | Time |
|---|---|---|---|---|
| TSSFinder | A. thaliana | 500 | 1000 | 02m45s |
| TSSPlant | A. thaliana | 500 | 1000 | 40m15s |
| TransPrise* | A. thaliana | 500 | 1000 | 3h45m41s |
| TSSFinder | O. sativa | 500 | 1000 | 03m19s |
| TSSPlant | O. sativa | 500 | 1000 | 42m20s |
| TransPrise* | O. sativa | 500 | 1000 | 3h54m50s |
| TSSFinder | H. sapiens | 500 | 2500 | 04m47s |
| BayesProm | H. sapiens | 500 | 2500 | 0m35s |
Processing times for TSSFinder, TSSPlant and TransPrise were recorded with software on an Intel(R) Core(TM) i3-3217U CPU @ 1.80 GHz and 6 Gb RAM, with the Linux Ubuntu operating system. Processing times for BayesProm were recorded with software running on an Intel(R) Cor(TM) i5p CPU @ 2.5 GHz and 6 Gb RAM, with the Microsoft Windows operating system. The tests used only one CPU, as TSSPlant cannot benefit from a multi-core architecture. *Analyses with TransPrise software were performed using the docker image (zarubinaa/tss-rice), as native installation of the software was not possible (a couple of required libraries could not be found or obtained from the original authors). nt—number of nucleotides in the DNA sequence. Data set—number of DNA sequences. Additional information in Table 4 in Supplementary Material.
Validation
Cross-validation
We also validated TSSFinder’s prediction models by performing 5-fold cross-validation experiments in six different organisms: a dicot plant (A. thaliana), a monocot plant (O. sativa), an insect (D. melanogaster), a bird (G. gallus), a mammal (H. sapiens) and a fungus (S. cerevisiae).
TSSFinder showed a high degree of precision and very low standard variation in the various steps of each validation. Table 4 summarizes the results. Of the four organism datasets, in four the majority of predictions presented an error smaller than 50 nt (68.8% for A. thaliana, 55.1% for O. sativa, 80.3% for S. cerevisiae and 57.8% for G. gallus). The small standard deviation shows the robustness of the method. The good results for O. sativa indicates that the strategy can be applied even in the absence of annotated TATA-boxes in the training set: just using a TATA-box predictor suffices. H. sapiens and D. melanogaster proved to be a harder challenge. Only 27.8% of the Drosophila predictions had a distance of less than 50 nt from the annotated TSS, but the performance increased remarkably at the 100 nt range, with 67.6% of the predictions. In both cases the vast majority of the predictions was at distance smaller than 250 nt (71% for H. sapiens and 88.2% for D. melanogaster), a result superior than previously recorded results in literature [34–36].
Table 4.
TSSFinder: Accuracy of TSS ab initio prediction*
| A. thaliana | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
|---|---|---|---|---|---|---|
| 50 nt | 67.9 | 1.6 | 67.8 | 1.7 | 67.9 | 1.6 |
| 100 nt | 83.5 | 1.1 | 83.4 | 1.2 | 83.5 | 1.1 |
| 150 nt | 89.0 | 1.1 | 88.9 | 1.2 | 89.0 | 1.1 |
| 200 nt | 91.4 | 1.1 | 91.4 | 1.1 | 91.5 | 1.1 |
| 250 nt | 91.8 | 0.9 | 93.2 | 0.9 | 93.2 | 0.9 |
| D. melanogaster | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 27.8 | 1.7 | 27.8 | 1.7 | 27.8 | 1.7 |
| 100 nt | 67.6 | 2.1 | 67.6 | 2.1 | 67.6 | 2.1 |
| 150 nt | 81.5 | 2.1 | 81.5 | 1.9 | 81.5 | 1.9 |
| 200 nt | 85.9 | 1.9 | 85.8 | 1.1 | 85.9 | 1.1 |
| 250 nt | 88.2 | 0.7 | 88.2 | 0.7 | 88.2 | 0.7 |
| G. gallus | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 61.3 | 1.5 | 61.9 | 1.7 | 61.6 | 1.6 |
| 100 nt | 80.2 | 2.0 | 80.0 | 2.0 | 80.6 | 2.0 |
| 150 nt | 87.7 | 1.2 | 88.5 | 1.0 | 88.1 | 1.1 |
| 200 nt | 91.4 | 1.2 | 92.2 | 1.2 | 91.8 | 1.2 |
| 250 nt | 93.3 | 1.3 | 94.1 | 1.2 | 93.8 | 1.3 |
| H. sapiens | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 34.0 | 2.2 | 34.0 | 2.2 | 34.3 | 2.0 |
| 100 nt | 51.7 | 1.4 | 51.6 | 1.3 | 51.5 | 1.3 |
| 150 nt | 61.2 | 1.8 | 60.9 | 1.8 | 61.2 | 1.6 |
| 200 nt | 67.4 | 1.3 | 67.3 | 1.2 | 67.4 | 1.1 |
| 250 nt | 72.2 | 1.2 | 72.1 | 1.2 | 72.1 | 1.2 |
| O. sativa | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 53.3 | 2.9 | 52.8 | 2.8 | 53.0 | 2.8 |
| 100 nt | 75.0 | 2.1 | 74.7 | 1.8 | 74.6 | 1.8 |
| 150 nt | 82.86 | 1.4 | 81,9 | 1.1 | 82.4 | 1.1 |
| 200 nt | 86.7 | 1.2 | 85.8 | 0.7 | 86.3 | 0.8 |
| 250 nt | 89.3 | 0.8 | 98.4 | 0.5 | 88.9 | 0.5 |
| S. cerevisiae | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 80.3 | 0.8 | 80.1 | 0.7 | 80.2 | 0.7 |
| 100 nt | 96.1 | 0.7 | 96.7 | 0.6 | 96.1 | 0.7 |
| 150 nt | 100 | 0.0 | 99.5 | 0.2 | 99.9 | 0.0 |
| 200 nt | 100 | 0.0 | 99.5 | 0.2 | 99.9 | 0.0 |
| 250 nt | 100 | 0.0 | 99.5 | 0.2 | 99.9 | 0.0 |
Prec.—Precision, Rec.—Recall, Stdv—Standard deviation. Precision and recall values indicate average across the five validation sets.
Approximately 5 to 10% of the eukaryotic promoter regions have the TATA-box motif [1, 10]. Accordingly, in our model of the promoter region, we have the TATA-box as one of the seven features, but modeled as optional. Analyzing the results for TATA-box and TATA-less genes we found that, with the maximum threshold of 250 nt, the results were similar for TATA-box and TATA-less genes. The only exception was D. melanogaster, where we obtained superior performance in sequences of TATA-less genes. For D. melanogaster this result is plausible since the TATA-box motif can be replaced by the DPE motif. In general, the DPE motif has a similar function to the TATA-box motif and it is positioned up to 30 nt upstream to the TSS signal. This particularity may be interfering with the performance of the TSSFinder. The TSSFinder performance both in the DNA sequences of the TATA-box promoter region and in the TATA-less sequences (not TATA-box) still manages to maintain its competitiveness with the other evaluated tools [4, 41, 42]. For more details see Table 5 in the Supplementary Material.
Assessing inter-species model precision
TSSFinder is optimized when there are complete mRNA sequences available of the same species for training. However, this may not always be the case. To estimate the accuracy of TSSFinder in less than ideal conditions we compared the performance of TSSFinder in three different organisms, but using a model trained for a different organism: A. mellifera using a model trained in D. melanogaster, C. familiaris using a model trained in H. sapiens and Z. mays using models trained in A. thaliana and O. sativa, to evaluate the performance of a closely related genome and a more distant one. The results are shown in Table 5.
Table 5.
Prediction of the TSS signal using a model trained for a different organism
| A. mellifera | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
|---|---|---|---|---|---|---|
| 50 nt | 30.8 | 0.4 | 30.6 | 0.4 | 30.7 | 0.4 |
| 100 nt | 45.0 | 0.3 | 44.7 | 0.3 | 44.9 | 0.3 |
| 150 nt | 54.0 | 0.1 | 53.7 | 0.2 | 53.9 | 0.2 |
| 200 nt | 60.6 | 0.2 | 60.2 | 0.2 | 60.4 | 0.2 |
| 250 nt | 66.7 | 0.1 | 66.3 | 0.1 | 66.5 | 0.1 |
| A. thaliana** | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 57.0 | 0.6 | 56.9 | 0.6 | 56.9 | 0.6 |
| 100 nt | 74.0 | 0.5 | 73.9 | 0.5 | 73.0 | 0.5 |
| 150 nt | 80.2 | 0.6 | 80.1 | 0.6 | 80.1 | 0.6 |
| 200 nt | 83.7 | 0.5 | 83.7 | 0.6 | 83.7 | 0.5 |
| 250 nt | 86.6 | 0.4 | 86.6 | 0.5 | 86.6 | 0.5 |
| C. familiaris | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 51.2 | 0.3 | 50.3 | 0.2 | 50.8 | 0.3 |
| 100 nt | 70.7 | 0.7 | 69.5 | 0.5 | 70.1 | 0.6 |
| 150 nt | 80.6 | 0.9 | 79.2 | 0.7 | 79.9 | 0.8 |
| 200 nt | 86.3 | 0.7 | 84.8 | 0.5 | 85.5 | 0.6 |
| 250 nt | 89.8 | 0.6 | 88.3 | 0.4 | 89.0 | 0.4 |
| O. sativa* | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 52.2 | 0.3 | 51.5 | 0.4 | 51.8 | 0.4 |
| 100 nt | 71.8 | 0.4 | 70.8 | 0.7 | 71.3 | 0.5 |
| 150 nt | 80.0 | 0.3 | 79.0 | 0.8 | 79.5 | 0.4 |
| 200 nt | 85.4 | 0.1 | 84.2 | 0.9 | 84.8 | 0.2 |
| 250 nt | 88.8 | 0.1 | 87.6 | 0.9 | 88.2 | 0.2 |
| Z. mays* | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 43.6 | 0.4 | 43.5 | 0.4 | 43.6 | 0.4 |
| 100 nt | 65.8 | 0.3 | 65.8 | 0.3 | 65.9 | 0.3 |
| 150 nt | 77.5 | 0.2 | 77.4 | 0.2 | 77.4 | 0.2 |
| 200 nt | 83.9 | 0.2 | 83.8 | 0.2 | 83.8 | 0.2 |
| 250 nt | 87.5 | 0.1 | 87.4 | 0.2 | 87.4 | 0.2 |
| Z. mays** | Prec. | Stdv | Rec. | Stdv | F1 | Stdv |
| 50 nt | 52.5 | 0.6 | 52.5 | 0.6 | 52.5 | 0.6 |
| 100 nt | 73.6 | 0.4 | 73.5 | 0.4 | 73.5 | 0.4 |
| 150 nt | 82.5 | 0.2 | 82.5 | 0.2 | 82.5 | 0.2 |
| 200 nt | 87.7 | 0.2 | 87.7 | 0.1 | 87.7 | 0.1 |
| 250 nt | 90.1 | 0.1 | 90.1 | 0.1 | 90.1 | 0.1 |
Prec.—Precision, Rec.—Recall, Stdv—Standard deviation. Precision and recall values indicate average across the five validation sets. *—A. thaliana model; **—O.sativa model
The results of the application of the TSSFinder inter-species kept the values similar to the models evaluated in the cross-validation process (intra-species).
The highlight is the O. sativa model, which presented 57.0% and 52.5% of the predictions of the TSS signal with an error smaller than 50 nt in A. thaliana and Z mays; respectively. This performance is equal the result observed in the O. sativa model performed in the cross-validation process (intra-species).
For the C. familiaris predictions using the H. sapiens model, the result was surprising: accuracy measures were superior to those obtained in the H. sapiens cross-validation experiments in all distance thresholds, with F1-score values ranging from 50.8% to 80.9% versus 34.3% to 72.1%.
Training set size
The tests performed in the previous sections suppose a situation close to ideal, with the availability of really large training sets. In real life, annotation project researchers may not have a large number of confirmed full-length cDNAs to use. It is important to know, therefore, how the precision of TSSFinder evolves with the size of the original training set. To estimate which would be the sufficient size for a training set, we performed a test to evaluate the learning curve of TSSFinder. To ensure a more realistic setting, close to that of the annotation of a new genome, we used O. sativa promoter sequences without confirmed TATA-box annotation and with TSSs determined by full transcript mapping.
We first isolated one of the training and validation sets of the cross-validation experiment. We then performed five steps: on each one we randomly selected only half of the training sequences of the previous step. Table 6 shows that, even for training sets of only 125 promoter sequences, the majority of the TSS predictions (56.5%) has an error of less than 50 nt and almost all predictions (89.4%) have an error of at most 250 nt. The numbers tend to stabilize with training sets of size 250.
Table 6.
Training set size evaluation
| Training set | 50 nt | 100 nt | 150 nt | 200 nt | 250 nt |
|---|---|---|---|---|---|
| 125 Seqs | 56.50 | 75.9 | 83.3 | 87.2 | 89.4 |
| 250 Seqs | 61.6 | 79.4 | 85.6 | 88.9 | 91.5 |
| 500 Seqs | 62.0 | 79.9 | 85.9 | 89.5 | 91.7 |
| 1000 Seqs | 62.1 | 80.4 | 86.8 | 90.1 | 91.3 |
| 2000 Seqs | 66.5 | 82.8 | 89.0 | 92.0 | 93.1 |
| 4000 Seqs | 65.8 | 83.1 | 88.7 | 92.3 | 92.8 |
Second column indicates the average percentage of predictions with an error of 50 nt or less on the five steps. Next columns indicate similar measures for 100 nt, 150 nt, 200 nt and 250 nt. Best scores are boldface. Seqs—number of DNA sequences
New TSS predictions
We applied TSSFinder to the upstream region of all ENSEMBL genes without a TSS annotation in 14 organisms, predicting 149 883 TSSs in previously uncharacterized genes. The number of predictions for each organism is shown in Table 7.
Table 7.
TSSFinder prediction results in plants, fungus and metazoans
| Organisms | TSSFinder model | New TSS annot |
|---|---|---|
| Apis mellifera | D. melanogaster | 4177 |
| Arabidopsis lyrata | A. thaliana | 11916 |
| Arabidopsis thaliana | A. thaliana | 4059 |
| Canis familiaris | H. sapiens | 3225 |
| Drosophila melanogaster | D. melanogaster | 214 |
| Gallus gallus | G. gallus | 1853 |
| Homo sapiens | H. sapiens | 659 |
| Macaca mullata | H. sapiens | 2629 |
| Mus musculus | H. sapiens | 1443 |
| Oryza brachyantha | O. sativa | 24761 |
| Oryza glaberrima | O. sativa | 29846 |
| Oryza sativa | O. sativa | 8751 |
| Rattus norvegicus | H. sapiens | 2753 |
| Saccharomyces cerevisiae | S. cerevisiae | 3146 |
| Zea mays | O. sativa | 50451 |
| Total | 149883 |
We used the O. sativa model to annotate the TSS signal in more than 75% and 85% of the Oryza brachyantha and Oryza glaberrima transcripts, respectively. The results of the application of TSSFinder in non-model organisms indicate the potential of TSSFinder to fill the gap in the annotation of the genome, especially to label the TSS signal.
In addition, for Z. mays, the O. sativa TSSFinder model made it possible to identify the TSS signal for over 50% of the transcripts available in Ensembl. This result indicates that even model or widely studied organisms still have gaps in the genome annotation and that the use of TSSFinder can be a powerful tool to fill this gap.
The results of the new predictions are available in.xlsx format in Table Supplementary Material and, as a BED file in the github repository http://tssfinder.github.io.
Discussion
TSSFinder is the first TSS prediction method described in the literature that uses a probabilistic model based on LCCRFs. TSSFinder estimates the position of the first TSS and further classifies the promoter region into TATA-box or TATA-less.
LCCRFs have been used very effectively for natural language processing; however, their use in genomics has been relatively sparse. In 2012, Liu and colleagues [52] assessed the potential of labeling CpG islands in humans, comparing three probabilistic techniques: conditional random fields (CRFs), hidden Markov models (HMMs) and maximum entropy Markov model (MEMM). In this study, CRF-based model showed greater efficiency than the other two techniques. In 2013, Wang and Zhou [53] constructed two classification models of the core promoter region (the region upstream of the TSS) using LCCRFs and multivariate hidden Markov model. Again, the LCCRF model was more efficient in classifying the regulatory regions and about 20% of the evaluated signals could not be annotated using the HMM model. However, even with these promising results, no CRF or LCCRF application for locating the TSS was developed. Of note, HMM models developed for promoter characterization are classifiers and not TSS predictors. The good performance exhibited by LCCRFs made the technique a natural candidate to characterize the TSS location.
LCCRFs can be very effective labeling models when we have extensive amounts of data available. When comparing LCCRFs with neural networks, the other technology used in TSS classification and placement, LCCRFs have the advantage of being able to incorporate more easily previous biological knowledge, such as the architecture of the promoter region, which is used in our model. This can reduce the number of parameters in the model and speed up computing times. This is evident when we compare the running times of TSSFinder against TSSPlant and TransPrise. We had also the added advantage of using a locally developed variant of the Viterbi algorithm that speeds up LCCRFs analysis. Our improved performance was achieved with very few parameters. These included a distance measure, which was fundamental in improving gene predictions in the past and which is not easily modeled by neural networks without even bigger computational cost. If we are willing to sacrifice on computing times a possible way of improving precision even further would be the use of other characteristics of the promoter regions, maybe specializing in TSS predictions of various promoter region architectures.
In our analysis, TSSFinder outperformed TSSPlant, TransPrise and BayesProm, to the best of our knowledge the most recent and best-performing TSS characterization methods in plants and humans. TSSFinder was able to perform the prediction of the TSS in 500 sequences in just a few minutes, a time that, even though much higher than BayesProm, was significantly lower than the ones for TSSPlant and TransPrise. We attribute these advantages to two fundamental characteristics of our tool: the first is the reduced number of characteristics modeled, only four (start codon position, 5’UTR region size, TATA-box composition, TSS site composition); the second is the efficient implementation of linear chain conditional random fields implemented by ToPS. TSSPlant, on the other hand, used dozens of position weight matrices as a base for their classification. TSSPlant also uses the score of different k-mers close to the TSS region and a neural network to analyze the three signal types. Of the four predictors, TransPrise had the worst results, which seems in contrast with the results in the original publication. However, the ideal dataset for TransPrise seems to be a 2000-nt region that includes the real TSS in the center, as indicated by their main benchmark. This indicates that TransPrise is more adequate to process genomic data located by imuno-precipitation experiments, where the location of the TSS is more central to the DNA segment. Finally, BayesProm uses only the positional densities of hexamers and a Bayes network, which probably explain the increased speed advantage. BayesProm performs the labeling of the promoter region through the positional density of hexamers present in the core promoter region such as TATA-box, Inr, among other motifs. However, we believe that the labeling of DNA sequences where biological signals (hexamers) do not have fixed positions can interfere in the performance of the model proposed by BayesProm.
We have not included in this article comparisons with 3PEAT and TIPR. The two tools rely on the target TSS being positioned close to the center of the subject sequence, as indicated in the benchmarks used in the original articles (TIPR and 3PEAT use regions of 8000–10000nt). This seems to indicate that, similarly to TransPrise, they are, in fact, targeted to genomic regions previously selected by immuno-precipitation experiments such as chip-seq [54, 55]. In the absence of such experiments, datasets would have to be based on the annotated position of the start codon and, due to the variation on the distance of the TSS from the 3’ end of the sequence used for prediction, heavily impact the performance of the predictors (data not shown). In preliminary studies, all these three tools exhibited poor performance when using datasets based on a fixed region upstream of the start codon (data not shown).
TSSFinder can be easily trained by the user to be applied in new organisms. Just around 125 validated TSS sites are enough for a prediction with better accuracy than other available options.
Finally, we showed that even though TSSFinder performs best when the model used is trained in the same organism, the results for predictions using a model trained in a closely related organism still compared favorably with those of TSSPlant.
This good performance motivated us to apply TSSFinder models on ENSEMBL genes without a TSS annotation in 14 different genomes. As a result, we present more than 140 000 new predictions of the TSS signal with approximately 85% of the organisms presenting numbers greater than 1400 new TSS characterizations. These results indicate that TSSFinder can be used to solve the labeling of genes that have not been previously annotated.
Conclusion
We presented TSSFinder, a software to characterize promoter sequences of Eukaryotic organisms. TSSFinder has better accuracy than previously published software and can be customized to novel organisms. We currently include in the prediction model only four types of information: putative TATA-box location, when available, distance from the TSS to the TATA-box, distance from the TSS to the start codon, and composition of the TSS region. In the future we may be able to increase accuracy with the inclusion of other promoter-related sequences, including intrinsic properties such as DNA structure or nucleosome occupancy [56–58] and extrinsic properties such as chromatin state [59] or ChIP-Seq transcription factor [54, 55].
Availability and supporting source code and requirements
TSSFinder is implemented using Python V.3.6 and a customized compiled C++ extension of the ToPS probabilistic framework [60] and consists of two scripts: the training script and the prediction script.
The training script is used to produce a new promoter model from a training set consisting of a multi-fasta file with genomic regions and a BED file [61] describing, respectively, the TSS, TATA-box and start codon locations of the promoters in the DNA sequences. This script uses ToPS to build and train the LCCRF model.
The prediction script receives a multi-fasta file with contigs/chromosomes, a BED file describing the location of the START codons, and two files representing a trained model. It then generates the configuration files necessary to run the trained ToPS model for the predictions. Results are presented using two BED files: TSS locations and TATA-box locations. The workflow is depicted in Figure 3. TSSFinder is available as source code, as a docker container and as a web service. The first two can be obtained on our github page (http://tssfinder.github.io) and the web service is available at http://sucest-fun.org/wsapp/tssfinder/.
Figure 3.

Worfklow of TSSFinder: (A) Training workflow: one input file (GENOME) in fasta format with the DNA sequences and three BED input files with the locations of the TSSs (TSS.bed), start codons (start_codon.bed) and the TATA boxes when present (TATAbox.bed). TSSFinder outputs two files: one for the emission probabilities of the model (Emission.tops) and one for the transition probabilities (Transition.tops). (B) Prediction workflow: two input files with the probabilistic model (Emission.tops and Transition.tops), one fasta input file with the genomic sequences to be analyzed, one BED input file with the location of the start codons of each gene (start_codon.bed). TSSFinder outputs two BED files, one with the TSS locations (TSS.bed) and the other with the location of the putative TATA boxes (TATAbox.bed).
Key Points
TSSFinder is available for three groups of organisms: metazoan, plants and fungi.
TSSFinder is able to predict the TSS signal in different organisms with higher accuracy than previous tools.
Using the TSSFinder tool, we were able to annotate the TSS signal on more than 140 000 new genes in different genomes deposited in Ensembl.
The output of the TSSFinder tool is in bed format. The use of this format aims to facilitate the genome annotation process or contribute with synthetic biology in the selection of target DNA sequences.
The TSSFinder tool is available in an online version (http://sucest-fun.org/wsapp/tssfinder/) and stand-alone (http://tssfinder.github.io)
Supplementary Material
Acknowledgments
We would like to thank Geraldo Cantelli for quickly building the TSSFinder online submission website.
Funding
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES) (Finance Code 001); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) Fellowship (DS-1454337to M.M.O.); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) Fellowship (DS-1560211 to I.B.); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) Productivity Fellowship (304360/2014-7 to G.M.S.); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) Productivity Fellowship (309566/2015-0 to A.M.D.); Fundação de Amparo à Pesquisa do Estado de Sào Paulo (FAPESP) (grant 2014/50921-8)—and on the eScience Network—IME/FAPESP (grant 2011/50761-20), through the computers on which all benchmarks were run.
Mauro de Medeiros Oliveira received his PhD in bioinformatics from the Universidade de São Paulo. Currently, he is a postdoctoral researcher at the Instituto Carlos Chagas Fiocruz, Paraná, Brazil. His research focuses on the genetics, phylogeny and epidemiology of the coronavirus (COVID-19).
Igor Bonadio received his PhD in computer science from the Universidade de São Paulo and is currently Tech Lead (machine learning engineering and information retrieval) at the technology company Elo7.
Alicia Lie de Melo is a PhD student in bioinformatics of the Universidade of São Paulo. Her research focuses on genomics, transcriptome and bioinformatics of sugarcane.
Glaucia Mendes Souza received his PhD in biochemistry from the Universidade of São Paulo and is currently is full professor at the Institute of Chemistry of the Universidade de São Paulo, Brazil. Her research focuses on biotechnology, genomics and bioinformatics for sugarcane.
Alan Mitchell Durham received his PhD in computer science from the University of Illinois at Urbana-Champaign and is currently an associate professor of the Universidade de São Paulo, Brazil. His research focuses on the development of probabilistic object-oriented frameworks and their application in genomics and transcriptomics bioinformatics software.
Contributor Information
Mauro de Medeiros Oliveira, Instituto Carlos Chagas, Fundação Oswaldo Cruz, Paraná, Brazil.
Igor Bonadio, Data Science, Elo7 Research Lab, São Paulo, Brazil.
Alicia Lie de Melo, Biochemistry Department, Universidade de São Paulo, São Paulo, Brazil.
Glaucia Mendes Souza, Biochemistry Department, Universidade de São Paulo, São Paulo, Brazil.
Alan Mitchell Durham, Computer Science, Universidade de São Paulo, São Paulo, Brazil.
Competing Interests
The author(s) declare that they have no competing interests.
Author’s Contributions
M.M.O., I.B. and A.M.D. developed the main concept. M.M.O. and A.L.M. performed all the texts and devised the benchmarks. I.B. implemented the probabilistic model in C++. I.B. and M.M.O. developed the original prediction architecture. A.M.D. and G.M.S. supervised the project and the strategies for validation. All authors participated in the writing and reviewing of the paper.
References
- 1. Lenhard B, Sandelin A, Carninci P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet 2012; 13(4): 233–45. [DOI] [PubMed] [Google Scholar]
- 2. Yella VR, Kumar A, Bansal M. Identification of putative promoters in 48 eukaryotic genomes on the basis of DNA free energy. Sci Rep 2018; 8(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roy AL, Singer DS. Core promoters in transcription: old problem, new insights. Trends Biochem Sci 2015; 40(3): 165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shahmuradov IA, Umarov RK, Solovyev VV. TSSPlant: a new tool for prediction of plant Pol II promoters. Nucleic Acids Res 2017; 45(8): e65–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parry TJ, Theisen JWM, Hsu J-Y, et al. The TCT motif, a key component of an RNA polymerase II transcription system for the translational machinery. Genes Dev 2010; 24(18): 2013–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hehl R. Plant Synthetic Promoters: Methods and Protocols. New York: Springer, 2016. [Google Scholar]
- 7. Liu W, Stewart Jr CN. Plant synthetic promoters and transcription factors. Curr Opin Biotechnol, 37:36–44, 2016. [DOI] [PubMed] [Google Scholar]
- 8. Engstrom MD, Pfleger BF. Transcription control engineering and applications in synthetic biology. Synth Syst Biotechnol 2017; 2(3): 176–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mejía-Guerra MK, Li W, Galeano NF, et al. Core promoter plasticity between maize tissues and genotypes contrasts with predominance of sharp transcription initiation sites. Plant Cell 2015; 27(12): 3309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumari S, Ware D. Genome-wide computational prediction and analysis of core promoter elements across plant monocots and dicots. PLoS One 2013; 8(10): e79011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grillo G, Turi A, Licciulli F, et al. Utrdb and utrsite (release 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res 38(suppl_1):D75–80, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gordon JJ, Towsey MW, Hogan JM, et al. Improved prediction of bacterial transcription start sites. Bioinformatics 2006; 22(2): 142–8. [DOI] [PubMed] [Google Scholar]
- 13. Abeel T, de Peer Y, Saeys Y. Toward a gold standard for promoter prediction evaluation. Bioinformatics 2009; 25(12): i313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Narlikar L, Ovcharenko I. Identifying regulatory elements in eukaryotic genomes. Brief Funct Genom Proteom 2009; 8(4): 215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liang Z-Y, Lai H-Y, Yang H, et al. Pro54db: a database for experimentally verified sigma-54 promoters. Bioinformatics, 33(3): 467–69, 2017. [DOI] [PubMed] [Google Scholar]
- 16. Zhang M, Li F, Marquez-Lago TT, et al. Multiply: a novel multi-layer predictor for discovering general and specific types of promoters. Bioinformatics, 35(17): 2957–65, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Amin R, Rahman CR, Ahmed S, et al. iPromoter-BnCNN: a novel branched CNN-based predictor for identifying and classifying sigma promoters. Bioinformatics 2020; 36(19): 4869–75. [DOI] [PubMed] [Google Scholar]
- 18. Lai HY, Zhang ZY, Su ZD, et al. iProEP: a computational predictor for predicting promoter. Mol Ther Nucleic Acids, 17:337–46, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li F, Chen J, Ge Z, et al. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform 2021; 22(2): 2126–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dreos R, Ambrosini G, Groux R, et al. The eukaryotic promoter database in its 30th year: focus on non-vertebrate organisms. Nucleic Acids Res 2017; 45(D1): D51–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ladunga I. Computational Biology of Transcription Factor Binding. New York: Springer, 2010. [DOI] [PubMed] [Google Scholar]
- 22. Florea L, Hartzell G, Zheng Z, et al. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res 1998; 8(9): 967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res, 12(4): 656–64, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005; 21(9): 1859. [DOI] [PubMed] [Google Scholar]
- 25. Chen N, Wang WM, Wang HL. An efficient full-length cDNA amplification strategy based on bioinformatics technology and multiplexed PCR methods. Sci Rep 2016; 5(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cartolano M, Huettel B, Hartwig B, et al. cDNA library enrichment of full length transcripts for SMRT long read sequencing. PLoS One 2016; 11(6): e0157779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pedersen AG, Baldi P, Brunak S, et al. Characterization of prokaryotic and eukaryotic promoters using hidden Markov models. Proceedings of the International Conference on Intelligent Systems for Molecular Biology. Saint Louis, Missouri, 1996; 4:182–91. PMID: 8877518. [PubMed] [Google Scholar]
- 28. Prestridge DS. Predicting Pol II promoter sequences using transcription factor binding sites. J Mol Biol 1995; 249(5): 923–32. [DOI] [PubMed] [Google Scholar]
- 29. Solovyev VV, Shahmuradov IA, Salamov AA. Identification of promoter regions and regulatory sites. In: Computational Biology of Transcription Factor Binding. New York: Springer, 2010, 57–83. [DOI] [PubMed] [Google Scholar]
- 30. Hutchinson GB. The prediction of vertebrate promoter regions using differential hexamer frequency analysis. Bioinformatics 1996; 12(5): 391–8. [DOI] [PubMed] [Google Scholar]
- 31. Zhu Y, Li F, Xiang D, et al. Computational identification of eukaryotic promoters based on cascaded deep capsule neural networks. Brief Bioinform 2020; Nov 24:bbaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Towsey M, Timms P, Hogan J, et al. The cross-species prediction of bacterial promoters using a support vector machine. Comput Biol Chem 2008; 32(5): 359–66. [DOI] [PubMed] [Google Scholar]
- 33. Wang S, Cheng X, Li Y, et al. Image-based promoter prediction: a promoter prediction method based on evolutionarily generated patterns. Sci Rep 2018; 8(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bajic VB, Seah SH. Dragon Gene Start Finder identifies approximate locations of the 5’ ends of genes. Nucleic Acids Res 2003; 31(13): 3560–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sonnenburg S, Zien A, Rätsch G. ARTS: accurate recognition of transcription starts in human. Bioinformatics 2006; 22(14): e472–80. [DOI] [PubMed] [Google Scholar]
- 36. Abeel T, Saeys Y, Rouzé P, et al. ProSOM: core promoter prediction based on unsupervised clustering of DNA physical profiles. Bioinformatics 2008; 24(13): i24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cassiano MHA, Silva-Rocha R. Benchmarking available bacterial promoter prediction tools: potentialities and limitations. bioRxiv, 2020. [DOI] [PMC free article] [PubMed]
- 38. Zhu Y, Li F, Xiang D, et al. Computational identification of eukaryotic promoters based on cascaded deep capsule neural networks. Brief Bioinform, 2020; bbaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morton T, Petricka J, Corcoran DL, et al. Paired-end analysis of transcription start sites in Arabidopsis reveals plant-specific promoter signatures. Plant Cell 2014; 26(7): 2746–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morton T, Wong W-K, Megraw M. TIPR: transcription initiation pattern recognition on a genome scale. Bioinformatics 2015; 31(23): 3725–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pachganov S, Murtazalieva K, Zarubin A, et al. Transprise: a novel machine learning approach for eukaryotic promoter prediction. PeerJ 2019; 2019(11): 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Narang V, Sung W-K, Mittal A. Computational modeling of oligonucleotide positional densities for human promoter prediction. Artif Intell Med 2005; 35(1–2): 107–19. [DOI] [PubMed] [Google Scholar]
- 43. Lafferty J, McCallum A, Pereira FCN. Conditional random fields: probabilistic models for segmenting and labeling sequence data. In: Machine Learning-International Workshop then conference, 2001, 282–9.
- 44. Vinson JP, DeCaprio MD, et al. Comparative gene prediction using conditional random fields. AdvNeural Inf Process Syst, 2006; 2017:1441–8. [Google Scholar]
- 45. Bernal A, Crammer K, Pereira F. Automated gene-model curation using global discriminative learning. Bioinformatics 2012; 28(12): 1571–8. [DOI] [PubMed] [Google Scholar]
- 46. DeCaprio D, Vinson JP, Pearson MD, et al. Gene prediction using conditional random fields. Genome Res 2007; 17(9): 1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ravikiran M, Madgula K, Saha S. Teamdl at semeval-2018 task 8: cybersecurity text analysis using convolutional neural network and conditional random fields. In: Proceedings of The 12th International Workshop on Semantic Evaluation, 2018, 868–73.
- 48. Sakai H, Lee SS, Tanaka T, et al. Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol 2013; 54(2): e6–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Claeys M, Storms V, Sun H, et al. MotifSuite: workflow for probabilistic motif detection and assessment. Bioinformatics 2012; 28(14): 1931–2. [DOI] [PubMed] [Google Scholar]
- 50. Khan A, Fornes O, Stigliani A, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2017; 46(D1): D260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res, 46(D1): D754–61, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu W, Chen H, Chen L. Identifying CPG islands in genome using conditional random fields. In: International Conference on Intelligent Computing. New York: Springer, 2012, 309–18. [Google Scholar]
- 53. Wang H, Zhou X. Detection and characterization of regulatory elements using probabilistic conditional random field and hidden Markov models. Chinese J Cancer 2013; 32(4): 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gusmao EG, Dieterich C, Zenke M, et al. Detection of active transcription factor binding sites with the combination of DNase hypersensitivity and histone modifications. Bioinformatics 2014; 30(22): 3143–51. [DOI] [PubMed] [Google Scholar]
- 55. He Y, Zhang Y, Zheng G, et al. CTF: a CRF-based transcription factor binding sites finding system. BMC Genomics 2012; 13(8): S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Friedel M, Nikolajewa S, Sühnel J, et al. DiProDB: a database for dinucleotide properties. Nucleic Acids Res 37(suppl_1): D37–40, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Il’icheva IA, Khodikov MV, Poptsova MS, et al. Structural features of DNA that determine RNA polymerase II core promoter. BMC Genomics 2016; 17(1): 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morey C, Mookherjee S, Rajasekaran G, et al. DNA free energy based promoter prediction and comparative analysis of Arabidopsis and rice genomes. Plant Physiol 2011; 156(3):1300–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tsai ZT-Y, Shiu S-H, Tsai H-K. Contribution of sequence motif, chromatin state, and DNA structure features to predictive models of transcription factor binding in yeast. PLoS Comput Biol 11(8): e1004418, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kashiwabara AY, Bonadio I, Onuchic V, et al. ToPS: a framework to manipulate probabilistic models of sequence data. PLoS Comput Biol 2013; 9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quinlan AR. Ira M Hall. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26(6): 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.