

Abstract
NGS long-reads sequencing technologies (or third generation) such as Pacific BioSciences (PacBio) have revolutionized the sequencing field over the last decade improving multiple genomic applications like de novo genome assemblies. However, their error rate, mostly involving insertions and deletions (indels), is currently an important concern that requires special attention to be solved. Multiple algorithms are available to fix these sequencing errors using short reads (such as Illumina), although they require long processing times and some errors may persist.
Here, we present Accurate long-Reads Assembly correction Method for Indel errorS (ARAMIS), the first NGS long-reads indels correction pipeline that combines several correction software in just one step using accurate short reads. As a proof OF concept, six organisms were selected based on their different GC content, size and genome complexity, and their PacBio-assembled genomes were corrected thoroughly by this pipeline. We found that the presence of systematic sequencing errors in long-reads PacBio sequences affecting homopolymeric regions, and that the type of indel error introduced during PacBio sequencing are related to the GC content of the organism. The lack of knowledge of this fact leads to the existence of numerous published studies where such errors have been found and should be resolved since they may contain incorrect biological information.
ARAMIS yields better results with less computational resources needed than other correction tools and gives the possibility of detecting the nature of the found indel errors found and its distribution along the genome. The source code of ARAMIS is available at https://github.com/genomics-ngsCBMSO/ARAMIS.git
Keywords: error correction, next-generation sequencing, homopolymer, long read, genome assembly
Introduction
Next-generation sequencing (NGS) technologies have improved over the last years and have had a big impact in genomics research, due to their decreasing cost and higher efficiency than the Sanger sequencing method. Nowadays, NGS allows for a more comprehensive analysis of the structure and content of genomes than was previously possible [1].
Nonetheless, these advancements are not without limitations since sequencing technologies may be affected by different sources of bias, such as palindromic sequences, large homopolymeric regions, highly repetitive sequences or AT/GC-rich genomes. Third-generation sequencing technologies, as single-molecule real-time (SMRT) sequencing, developed by Pacific BioSciences (PacBio) and Oxford Nanopore Technologies (ONT) have provided new methods to overcome some of those sources of bias by generating reads that are substantially longer than those of second-generation sequencing technologies (such as Illumina). However, the gain in sequence length is often traded for quality, with low (~85%) accuracy of base calls resulting in high raw read error rates (higher than 10%) [2, 3]. These errors appear to be mostly small insertions and deletions (indels) assumed to be randomly distributed within each read; hence, a high coverage should theoretically be enough to overcome the high error rate [4]. Even though the existence of an error pattern of indels occurring predominantly in homopolymers in long-read technologies has been reported [5, 6], errors are still assumed to be randomly distributed. Nonetheless, some of these technologies offer ways to overcome these single-pass errors. PacBio SMRT sequencing results are generated through consensus analysis, averaging the sequence information from multiple reads for each reference position. While it is true that single-pass sequence reads in SMRT sequencing are more error-prone, it does not prohibit the determination of a high-quality consensus exceeding 99.999% accuracy (https://www.pacb.com/).
In this work, we observed sequence systematic errors that ended on apparently truncated proteins, some of them likely essential for cell viability, in different de novo PacBio-only assemblies in spite of their high coverage. As a paradigmatic example, the essential protein DNA gyrase in T. thermophilus genome essential for bacterial DNA replication was truncated at three points at the available genome assembly based on 361.03× coverage of PacBio reads. However, alignment of Illumina short reads identified three single-base deletions introduced systematically by PacBio reads. This observation suggested to us that high coverage in PacBio was not enough to overcome the apparent systematic sequencing errors, mostly consisting of single-base indels. This type of inaccuracy has been reported before, with long-read technologies introducing frameshifts and premature stop codons, which have the potential to critically affect the interpretation and prediction of protein sequences [7].
Multiple algorithms have been recently developed to fix or remove sequencing errors in long reads, even as an additional step included in de novo long reads assembly pipelines, such as Canu [8] and HGAP [9]. Several articles dealt with comparisons among hybrid methods, which require accurate short reads [10–12] or between hybrid and non-hybrid methods, which take a self-correction approach [13, 14]. Although software that use the self-correction strategy (e.g., LoRMA [15], FLAS [16]) have good efficiency, according to Zhang et al. [14] the hybrid methods show better performance than non-hybrid ones in terms of correction quality and computing resource usage, especially when handling low coverage reads. Within the hybrid methodologies, there are two main approaches: alignment of short reads to long reads using a variety of aligners to achieve maximum accuracy (e.g., HECIL [17]); or to perform firstly an assembly with short reads and then to align against it the long reads to correct them (e.g., HALC [18]). In the first case, almost all methods have high computational requirements so the correction process is slower. The second case solves these issues although with the handicap of adding the need to generate a short-read-based assembly, which could not be attained for genomes rich in repeated sequences. There are a small number of programs such as Pilon [19] or Racon [20] that can be used as polishing tools after the assembly with either Illumina data or data produced by third generation of sequencing. Nonetheless, those tools may show bias in cases of low-coverage data or the presence of highly repetitive regions in the genome [21].
Here, we describe ARAMIS, a new pipeline to detect and correct sequence errors in long reads (e.g. PacBio)-only genome assemblies using accurate short reads (e.g. Illumina) in just one step. It combines and benefits from the power of several software for genome assembly improvement, enhancing the confidence of the results and reducing the number of false positives. Moreover, it requires less computing time than other hybrid and non-hybrid correction methods.
As a feasibility study, a total of six organisms were selected to test ARAMIS based on their GC content and their complexity: Plasmodium falciparum strain 3D7, which has a complex genome structure with low GC content; Escherichia coli str. K-12 substr. MG1655 as an example of bacteria whose GC content is near 50%; Leishmania infantum JPMC5 as a GC-rich complex genome and Thermus thermophilus strain NAR1, Mycolicibacterium hassiacum DSM 44199 and Tessaracoccus sp. strain T2.5–30 as GC-rich simple genomes.
P. falciparum
P. falciparum is a unicellular protozoan parasite that causes malaria in humans being responsible for the majority of malaria-related deaths. The canonical reference of P. falciparum strain 3D7 was published in 2002 for the first time [22] but given the complexity of the genome the reference sequence is continually updated. The assembly with the information of PacBio long reads was reported in 2019 as the final assembly for P. falciparum strain 3D7 achieving a complete genome (with 14 chromosomes) of 23.4 Mbp with a gene density of 32% and a low GC content of 19.33% [23].
E. coli
E. coli, which is an enteric bacterium typically present in the lower intestine of humans, is one of the most diverse microbial species containing both pathogenic and non-pathogenic strains. E. coli str. K-12, a non-pathogenic strain, is the most studied and commonly used as a reference, with a gene density of 89.5% and a GC content of 50.79%. A complete genome assembly of around 4.6 Mbp of E. coli str. K-12 substr. MG1655 with PacBio long-reads technology was reported in 2015 [24].
L. infantum
L. infantum is a causative agent of visceral leishmaniasis (VL) in the Mediterranean basin, the Middle East and Latin America [25]. Obtaining a reliable genomic sequence is essential for molecular studies leading to the development of leishmaniasis control strategies. However, the existence of a large number of repetitive sequences, the high presence of homopolymeric regions, and the high GC content (59.74%) of this species affect the accuracy of the assembly. Since the publication of the L. infantum JPCM5 strain (MCAN/ES/98/LLM-724) fragmented and incomplete genome in 2007, transcriptomics and proteomics studies have been accomplished using this reference genome. In 2017, the complete sequence of L. infantum assembly in its 36 chromosomes (around 32.8 Mb) using long PacBio reads and Illumina information for joining fragmented chromosomes was published [26]. The gene density of this genome is around 48%.
T. thermophilus
T. thermophilus a polyploid Gram-negative extremely thermophilic bacterium with a very high GC content (around 68.4%), a gene density of 96% and a very efficient natural competence system that contributes to the genomic plasticity observed in thermal environments. The genome of T. thermophilus strain NAR1 [27] was sequenced in 2007 and de novo assembly of the complete chromosome (2 Mpb) was achieved in 2019 [28]. In addition, three more contigs were obtained corresponding to plasmid sequences [29].
M. hassiacum
The genus Mycolicibacterium includes a group of Gram-positive bacteria in the Actinobacteria phylum [30]. M. hassiacum DSM 44199 has the ability to grow at temperatures up to 65°C due to its very high GC content (69.29%), which makes it the most thermophilic of all the mycobacteria. A recent study (2019) reported the 5.2-Mbp complete genome sequence of this singular organism that was assembled into a single circular chromosome using PacBio long reads [31]. The gene density of this genome is 91%.
Tessaracoccus sp.
The genus Tessaracoccus was characterized as Gram-positive non-spore-forming facultative anaerobic bacteria and classified within the Actinobacteria phylum [32]. Tessaracoccus sp. strain T2.5–30 was isolated from the subsurface of the Iberian Pyritic Belt (IPB, Peña de Hierro, Spain). In 2017, the complete genome sequence of Tessaracoccus sp. strain T2.5–30, which consists of a chromosome with 3.2 Mbp was achieved by PacBio sequencing [33] and performing a PacBio-only assembly. This organism shows the highest GC content (70.4%) of the organisms studied in this article, with a gene density of 93% [34].
The pipeline
ARAMIS can be downloaded at the github repository: indicated in the abstract. An installation guide, usage instructions and all test data used can also be found at the above platform. The pipeline that we developed includes two principal steps to carry out the analysis. The correction step is performed with the customized correction.sh bash script that corrects the long-reads-only assembly file. In addition, the statistical step is run with the customized indel_analysis.sh bash script, which includes all the functions to generate coverage, GC skew and homopolymers statistics and their correspondent plots (Figure 1). Once the pipeline is finished, all intermediate, final and statistical files and figures will be accessible in each directory created.
Figure 1 .
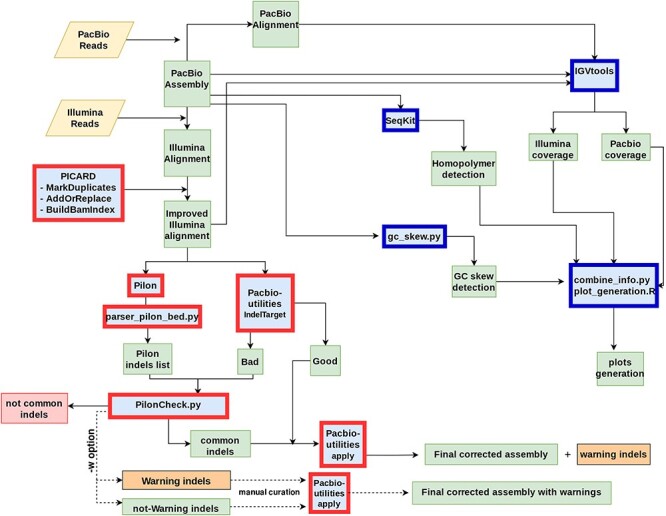
Schematic overview of the workflow leading to the correction of PacBio-only genome assembly. Input files (Raw Reads) are represented as yellow rhomboids. All the different software is shown in blue boxes. Those tools implemented in the Correction_step of ARAMIS are shown with red borders. Dark-blue borders show the set of tools used by the Statistical_step of ARAMIS. Those common indels in which the pipeline is unable to determine the correct sequence are flagged as warnings (orange boxes) for posterior manual curation (−w option). This additional step, outside the main pipeline, is represented with a dashed line. Output files are represented in green boxes. Discarded data are shown in red boxes.
The pipeline: correction step
In order to run ARAMIS, it is needed to start with a long-read genome assembly. The corresponding long (PacBio) raw reads and additional accurate short (Illumina) reads sequences must be aligned against the assembly. We recommend using a BWT-based aligner, like BWA-mem [35], because they show good accuracy and better efficiency, both in time and memory, than other algorithms [36]. We recommend filtering out those reads with quality phred score less than 20 (which indicates a 99% probability of the base being called correctly) to reduce the number of errors. No quality filter was needed for the six samples used in this study. The indel fraction, or minimum fraction of reads supporting the identified error for each position, should also be provided, so ARAMIS will not report indels below the selected indel fraction.
With this information, the pipeline removes PCR duplicates to mitigate potential biases on alignment (see Figure 1), add read groups information corresponding to individual samples and creates index files with Picard’s MarkDuplicates, AddOrReplace and BuildBamIndex tools (http://broadinstitute.github.io/picard/). This improved alignment is then used to detect indels that must be corrected with the indel fraction selected using PacBio-utilities indelTarget (https://github.com/douglasgscofield/PacBio-utilities), which generates files with indel targets for correction labelled as ‘good’ or ‘bad’ based on whether the target passes quality criteria or not (at least 10× of coverage discarding multi-hit reads).
At the same time, another correction step is carried out using a customized Python script called PilonCheck.py, which uses the output generated by the Pilon tool to find those indels appearing in both the ‘bad’ PacBio-utilities files and in the Pilon indels list (converted to BED format using the customized parser_pilon_bed.py Python script). Indels that are found only by one software (not-common indels are discarded at this point). Those common indels detected by both tools in which the pipeline is unable to determine the correct sequence are flagged as warnings and saved in a file for posterior manual curation.
Later, the common indels between ‘bad’ PacBio-utilities and Pilon together with ‘good’ PacBio-utilities indels are joined in one final list of targets to correct. This list is finally introduced to the PacBio-utilities indel-apply command to perform the final correct assembly. Furthermore, if a manual correction of indel errors flagged as warning is performed by the users, the pipeline offers an additional second step (—warning; −w) to correct this new list of target positions.
The pipeline: statistical step
In order to analyze in depth the assembly correction, optional steps were included in the pipeline allowing to study the principal features of the detected sequence errors thoroughly and to visualize them across the complete genome.
Firstly, the sequencing coverage of both Illumina and PacBio alignments is computed through the function count of the IGVtools utility [37]. Then, the toolkit for FASTA/FASTQ manipulation SeqKit [38] is used to locate any region in the non-corrected genome assembly containing a homopolimer (repetitions tracks of length equal or higher than two bases).
Last, the Python script gc_skew.py, from the set of tools IRep [39] is used to calculate GC skew (a measure of the strand asymmetry in the distribution of guanines and cytosines) across the whole genome. All the obtained information is combined through combine_info.py an in-house Python script, which additionally calculates the indel fraction for each alignment of both technologies. It computes, as well, the percentage of homopolymeric regions affected, indicating the homopolymeric nucleotide and the length of each region.
Finally, three different plots are generated by means of a customized R script (plot_generation.R) to represent the results: a barplot of the length and nucleotide of the affected homopolymeric tracks, a Kernel density estimation (KDE) plot of the location of the indels detected across the whole genome combined with the GC skew information, and a scatter plot showing the indel fraction calculated for each position for both technologies.
Proof of concept
To confirm the initial hypothesis about the systematic errors found in long-reads sequencing and to test ARAMIS, we ran the pipeline in six different organisms selected based on their GC content and their complexity. In this study, only PacBio-based assemblies were used in order to obtain comparable results, although ARAMIS can be used on any long-read genome assembly aside from technology. Besides, the pipeline can be used in larger genomes as observed when it was tested with Homo sapiens data. However, due to the size of the results, human analysis was not included in the Proof OF Concept, but a summary can be seen in Supplementary File 1.
The workflow relies on the combination of existing tools to achieve better results in detection and correction of indel errors efficiently. On the other hand, specific cases that required additional curation of indel errors were corrected using the additional second step of the pipeline. Figure 1 shows the pipeline followed to achieve the final assemblies (further technical details have been provided in The Pipeline section).
In order to analyze the efficiency of the pipeline, all the correction processes described in this article have been performed with the same parameters and an indel fraction of 0.5, so at least a 50% of the accurate short reads supports the presence of an indel error. However, in cases of very complex organisms such as L. infantum, we recommend using stricter parameters with an indel fraction of 0.8. In fact, in the case of diploids or even polyploids genomes, we suggest using a higher value to consider phased haplotype regions.
ARAMIS can also automatically generate plots that show the distribution of the detected indels across the assemblies and their corresponding indel fraction from both Illumina and PacBio reads aligned against it. As an example, in Figure 2, in most indel positions, PacBio alignment shows a low indel fraction indicating that the majority of the long reads do not support the existence of the detected indels across the assembly. Moreover, high accurate short Illumina reads do support the presence of an insertion or deletion with an indel fraction equal or higher than the selected cut-off (see Figure 2). This behavior proves that according to PacBio long-reads, there are no errors in the genome assembly despite the high coverage, whereas Illumina short-reads support the existence of an indel error.
Figure 2 .
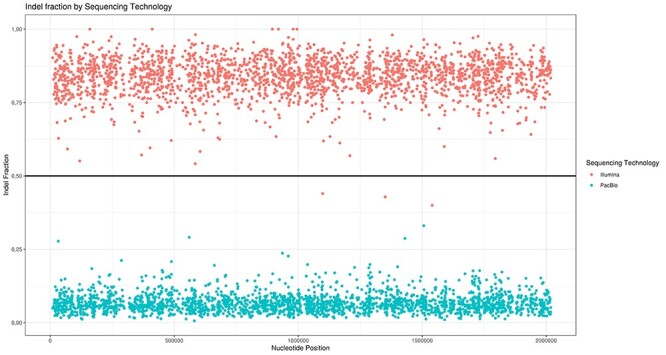
Indel distribution across T. thermophilus chromosome based on indel fraction calculated for both sequencing technologies. Blue and red dots show the indel fraction and position according to PacBio and Illumina reads alignment, respectively. The indel fraction threshold used is pointed with a horizontal line.
KDE plots of the location of the indels detected across each chromosome combined with the GC skew information is also shown through ARAMIS. All the generated figures are available at the specific Github repository. Moreover, ARAMIS is also capable of generating KDE plots that show the relationship between the presence of indel errors and a high GC or AT content in the whole genome. Thereby, the detected indels in the four organisms with higher GC content are located in GC-rich regions whereas those in P. falciparum genome with a GC content lower than 20% are located in AT-rich regions (Figure 3).
Figure 3 .
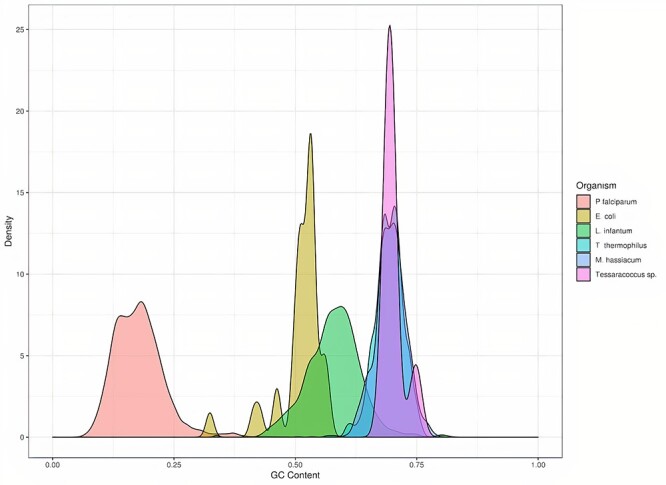
KDE plot of indels detected based on the genome GC content. P. falciparum, E. coli, L. infantum, T. thermophilus, M. hassiacum and Tessaracoccus sp. are represented in red, yellow, green, light-blue, dark-blue and pink curves, respectively.
Additional analysis
Hypergeometric test
In order to check whether indels error distribution is directly related to the presence of homopolymers, a hypergeometric test was performed with the null hypothesis (H0) being that the probability of introducing an indel error in homopolymers is due to chance. All statistical tests were calculated with R software (http://www.r-project.org).
PacBio subreads length and fragment size analysis
The analysis of PacBio long-reads features was performed with R. In Pacific Biosciences RS II instrument model, which was used to sequence all the studied organisms, each polymerase read is fragmented into one or more subreads, which contain sequence from a single pass of a polymerase on a single strand of an insert within an SMRTbell template. The consensus sequence from the alignment between subreads yields a circular consensus sequence (CCS) read from multiple passes of a single template. Theoretically, this method produces higher accuracy since it reduces significantly random errors in individual reads. The length and accuracy of CCS reads are limited by the original insert size, the number of passes and the overall read length of the sequencing platform.
Both reads and subreads lengths were calculated and analyzed based on their relation to the original DNA fragment size and the number of passes of the polymerase.
Comparison with other correction software
The performance of ARAMIS was compared against two different correction softwares: LoRMA, a self-correction method for long reads, and RACON that uses short reads to correct long-read-based assemblies, using the six organisms. Canu software was used in order to assemble the corrected reads by LoRMA.
Corrected assemblies obtained with RACON and ARAMIS were compared against the non-corrected ones using dnadiff [40], which provides detailed information on the differences between two genomes. All the differences classified as SNP or below coverage 10× were filtered out in order to make the results comparable. Furthermore, these LoRMA and RACON new assemblies were corrected again using ARAMIS tool to check the quality of the correction process.
All tools were run on a machine with four Intel(R) Xeon(R) E5–2670 @ 2.60 GHz CPUs (32 cores in total) and 64 GB of memory. RACON failed to finish with these characteristics in L. infantum dataset; hence, we run it at the Center for Scientific Computing (CCC-UAM). For all results, the default parameters are used.
Results
Proof of concept results
Table 1 summarizes the main results of the correction process in the six assemblies, including the results of both steps (Correction and Indel analysis). It indicates the number of errors detected by each software and how many of them were finally corrected. It also shows the type of error detected, the number of homopolymers affected and the number of affected genes.
Table 1.
Summary of the correction process and the subsequent analysis of the indels characteristics
| Correction step | Indel analysis step | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Organism | Genome Size (bp) | %GC | Indels detected by Pilon | Indels detected by PacBioutilities | Final corrected errors | Type of error detected | Number of homopolymer | Number of affected Genes | ||||||
| Good | Bad | Insertion | Deletion | Total | Affected by errors | GC | AT | |||||||
| Common | Not Common | |||||||||||||
| P. falciparum | 23,617,102 | 19.35 | 9441 | 4492 | 5037 | 1355 | 9529 | 9427 | 102 | 4,394,067 | 9349 | 17 | 9332 | 740 |
| E. coli | 4,636,831 | 50.79 | 37 | 33 | 4 | 0 | 37 | 8 | 29 | 904,567 | 32 | 25 | 7 | 16 |
| L. infantum | 32,802,969 | 59.74 | 1230 | 585 | 694 | 325 | 1279 | 393 | 886 | 1,310,260 | 1148 | 831 | 317 | 98 |
| T. thermophilus NAR1 | 2,476,708 | 68.39 | 2923 | 2674 | 254 | 101 | 2928 | 1 | 2927 | 598,509 | 2914 | 2909 | 5 | 1297 |
| M. hassiacum | 5,268,611 | 69.29 | 457 | 369 | 114 | 11 | 483 | 0 | 483 | 1,039,182 | 481 | 481 | 0 | 216 |
| T. lapidicaptus | 3,212,699 | 70.36 | 7 | 5 | 2 | 35 | 7 | 1 | 6 | 609,842 | 5 | 4 | 1 | 3 |
In the case of E. coli all indel errors identified by the pipeline were corrected, with no additional manual correction required (see Figure 4B). In the rest of the organisms both good and bad indels were detected through the pipeline (see Figure 4A, C-F). Only those bad indels detected by more than one software were corrected though not-common indels can be consulted in the intermediate files for subsequent manual correction (See The Pipeline section). As can be seen, combinations of several correction software enable the correction of more indel errors than if these tools were used independently.
Figure 4 .
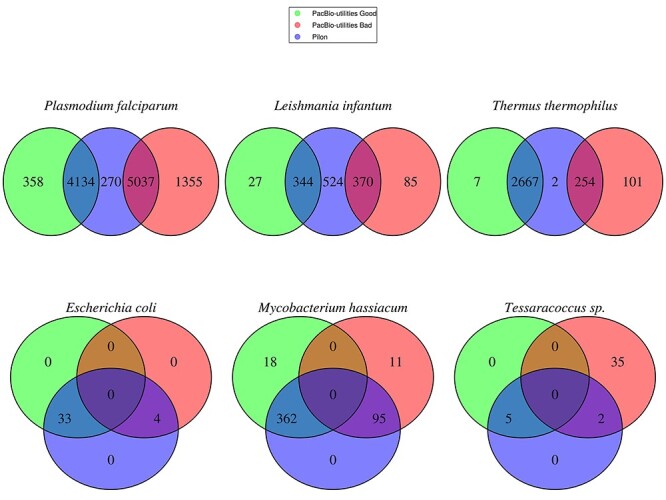
Venn diagram of indels detected by PacBio-utilities and Pilon software. PacBio-utilities indels flagged as Good (green), PacBio-utilities indels flagged as Bad (red), and indels detected by Pilon (blue) are shown. Panels A–F show the indels detected in the six organisms studied.
Gene analysis revealed that in all the studied bacteria a high number of genes were affected by PacBio errors in relation to the total number of indels detected (see Table 1). This result highlights the importance of a good method for correcting PacBio-only assemblies in order to achieve accurate genome annotations. On the other hand, the coding density in parasites such as P. falciparum and L. infantum is much lower than in bacteria. Thus, despite the fact that the analysis of genes revealed that 740 genes were affected by these errors in P. falciparum and 98 genes in L. infantum, which turns into the possibility of annotating truncate proteins, this only represent a 7% of the total amount of indel errors detected in both genomes.
Surprisingly, despite the fact that Tessaracoccus has the highest GC content of all the studied organisms, exceeding 70%, only seven indel errors were detected (although only three genes were affected) and corrected (Table 1).
All six organisms show a similar percentage of homopolymers across the genome (Table 2), except for T. thermophilus, which presents near 60%. In spite of this high fraction, the number of homopolymers affected is not very large in any of the organisms studied (less than 0.2%) but >70% of the indel errors of the PacBio reads (P-value >2.83e-02), are located in homopolymer regions. This proves that the sequencing error is systematic, though is not present in all the homopolymeric sequences of the genome.
Table 2.
Statistical analysis results
| Organism | % Homopolymers in the genome | % Indels in homopolymers | P-value |
|---|---|---|---|
| P. falciparum | 53.74 | 98.11 | 0 |
| E. coli | 45.78 | 86.49 | 4.02e-08 |
| L. infantum | 39.91 | 84.62 | 0 |
| T. thermophilus NAR1 | 59.88 | 99.52 | 0 |
| M. hassiacum | 44.24 | 98.95 | 0 |
| T. lapidicaptus | 43.12 | 71.43 | 2.83e-02 |
In agreement with the low GC content of P. falciparum genome the majority of the indel errors detected, which mostly affect homopolymeric regions, came from insertions introduced by the PacBio sequencing corresponded to single-base thymine nucleotides (T) and adenine nucleotides (A) (Figure 5A). The total number of homopolymers affected is distributed as a Gaussian-like distribution where the majority of the homopolymers detected correspond to repetitive nucleotides of T or A and the most of them have a length between 10 to 16 bases (Figure 5A).
Figure 5 .
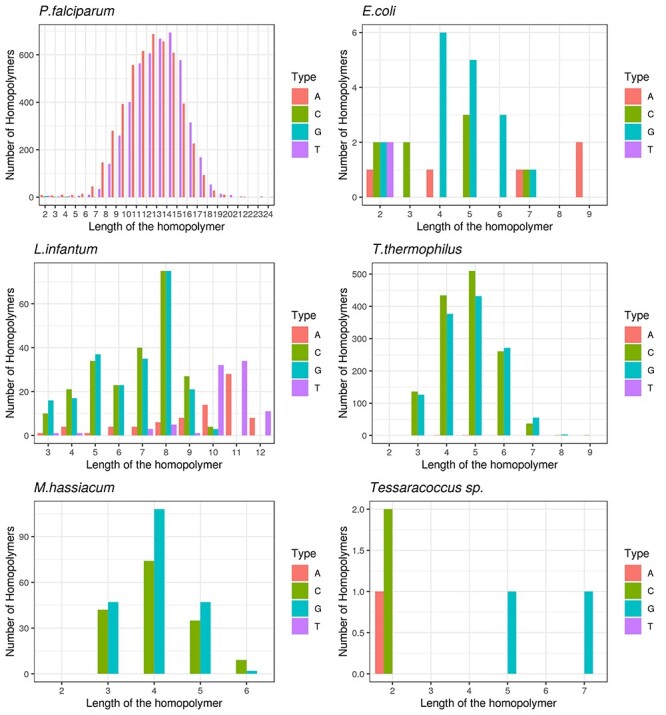
Variant distribution in homopolymers based on length and nucleotide. Red, green, blue and purple bars correspond to A, C, G and T affected homopolymers, respectively. Panels A–F show the type of homopolymers affected in each organism.
In contrast, the majority of the indel errors detected in the rest of the organisms came from deletions introduced by the PacBio sequencing, mostly corresponding to guanine (G) or cytosine (C) single-base nucleotides (Figure 5). The length of the homopolymers ranged from four to six nucleotides long both in T. thermophilus and M. hassiacum. In the case of E. coli, the total number of affected homopolymers has not a homogeneous distribution presenting indels in A, T, C and G homopolymeric tracks with different lengths. L. infantum, due to its complexity presents the highest variability of homopolymers affected with size range between 2 and 16 nucleotides being most of them eight nucleotides in length. In spite of the low number of indel errors detected in Tessaracoccus genome most of them are located in homopolymeric regions, supporting the relation between the errors and the presence of homopolymers even with a low number of indels (Figure 5F).
In order to elucidate the odd results obtained for Tessaracoccus sp. compared to the rest of the studied bacteria, a graphical analysis of the PacBio subreads length was performed. Figure 6 shows the distribution and range of subreads length in all the organisms studied. As can be seen, Tessaracoccus sp., the organism with the lowest number of indel errors, shows the shortest subread length (below 10,000 bp) while T. thermophilus (with the highest number of errors), and E. coli have a wide length range with subreads above 20,000 bp. In M. hassiacum case, the subread lengths are higher than Tessaraccocus sp. (below 20,000 bp). The number of indel errors in the protists P. falciparum and L. infantum are high; however, the subread length range is not comparable with the studied bacteria due to their genome complexity. Additionally, in Table 3, all the long-reads characteristics of all the organisms studied can be seen. It is important to emphasize that Tessaracoccus sp., has the highest number of subreads and the largest coverage of all the bacteria. On the contrary, despite the high coverage of T. thermophilus, it has the lower number of subreads.
Figure 6 .
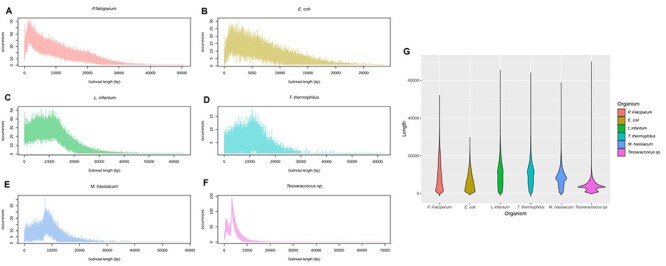
Frequency distribution of PacBio subread lengths in the six organisms studied. The length distribution of the datasets is shown as a frequency plot (Panels A–F) and as a violin plot (Panel G).
Table 3.
PacBio Subread length and fragment size results
| Organism | Polymerase read mean length (kb) | Number of reads | Fragment mean length (kb) | Number of subreads | Subread mean length (kb) | Subread min/max length (bp) |
|---|---|---|---|---|---|---|
| P. falciparum | 12.3 | 525,996 | 9.5 | 578,188 | 9.6 | 1–50,720 |
| E. coli | 6.8 | 131,520 | 5.7 | 142,673 | 6.2 | 1–28,647 |
| L. infantum | 17.0 | 311,471 | 11.7 | 445,620 | 10.3 | 35–64,072 |
| T. thermophilus NAR1 | 16.5 | 60,929 | 11.3 | 98,247 | 10.2 | 35–62,446 |
| M. hassiacum | 17.0 | 69,152 | 9.4 | 141,382 | 8.3 | 35–57,888 |
| T. lapidicaptus | 14.7 | 107,224 | 4.9 | 382,405 | 4.0 | 1–69,567 |
Time and memory requirements
Figure 7 shows the processing time of the three software in relation to the genome size of the organisms (see Additional Supplementary File 2 for detailed data). As it was expected, LoRMA as the non-hybrid method is the slowest tool, whereas ARAMIS and RACON show similar time requirements for almost all the organisms studied though RACON processing time is slightly higher.
Figure 7 .
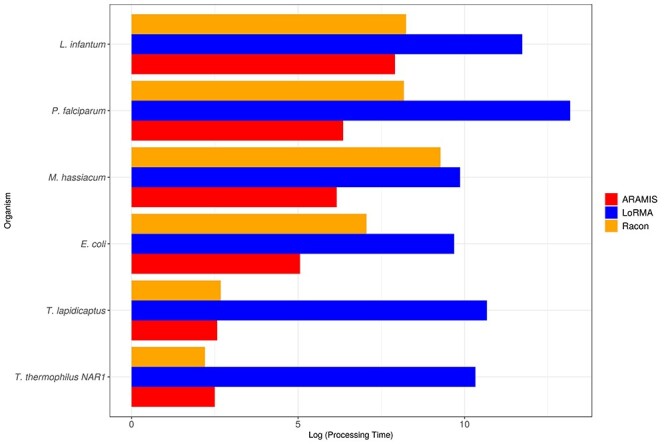
Processing time (minutes) of the correction process with each correction software in relation to genome size (normalized on a logarithmic scale). Red, orange and blue bars represent ARAMIS, RACON and LoRMA performance, respectively.
Figure 8 shows the maximum memory usage (in Gigabytes) for each process. Even though RACON and ARAMIS are the most memory-efficient tools, the increase in genome size results in a higher memory usage by RACON compared to ARAMIS. Again, LoRMA has the highest memory requirements in almost all the organisms.
Figure 8 .
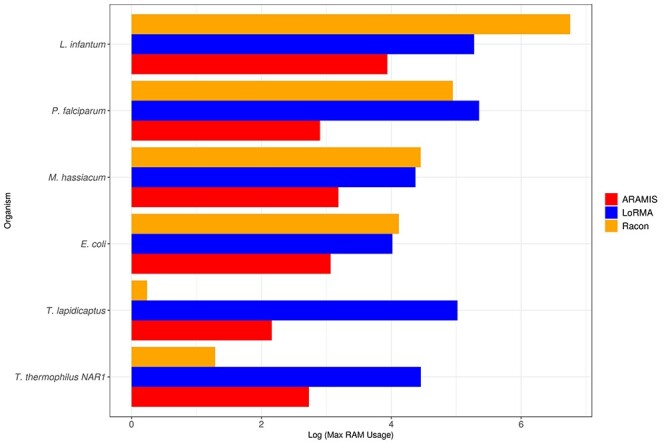
Maximum RAM memory usage (GB) used by each correction software in relation to genome size (normalized on a logarithmic scale). Red, orange and blue bars represent ARAMIS, RACON and LoRMA performance, respectively.
In both requirements, there is a great difference depending on the genome size of the organism. The non-hybrid method needs a large amount of time and memory to correct the bigger and more complex genomes as P. falciparum and L. infantum. ARAMIS and RACON, both hybrid methods, share a more stable performance along all six correction processes, though with genomes bigger than 4 Mbp RACON needs both more memory and processing time and, in some cases, uses even more RAM memory than the non-hybrid method.
Evaluation comparison
Since LoRMA does not provide the error rate of the corrected reads, a new assembly was made to compare with the other tools evaluated. However, due to the high fragmentation of the new assembly, the total number of errors detected with both tools is not comparable. Nevertheless, ARAMIS was run over this assembly and it detected the same type of errors: insertion of A and T, and deletion of G and C near homopolymeric regions (see Supplementary File 3).
On the other hand, since both RACON and ARAMIS are assembly-based correction tools, it was possible to do a complete comparison between them. Like LoRMA, RACON does not provide base-by-base output, making it more challenging to compare corrections. Therefore, the comparison between both software was made calculating the differences between the corrected assemblies and the non-corrected genome assemblies.
More than 90% of indel errors corrected by ARAMIS were also corrected by RACON. Despite the similar results between both softwares, we have seen that a significant percentage of errors called by RACON were not supported by Illumina coverage (see Supplementary File 3). In fact, when ARAMIS was used over RACON assembly, these positions introduced by RACON were rectified.
Regarding the correction process itself, the main differences between both hybrid methods come from how they manage short-read alignment coverage. ARAMIS controls the minimum number of reads to call an error (at least 10×) though RACON does not. Also, multihit reads are discarded by ARAMIS but not by RACON.
Conclusions
Third-generation sequencing technologies such as PacBio have been enhanced over the last few years providing the possibility of sequencing and assembling a wide range of different genomes more efficiently. As a great advantage, they provide long reads allowing to overcome repetitive regions, palindromic sequences and other difficult regions to sequence. However, these emerging technologies have one major drawback: a high error rate dominated by indels. Therefore, the correction of assemblies generated with long reads has been reported as an essential step in genomic studies.
Although the errors in PacBio reads are reported as randomly distributed and easily overcome by high coverage, we observed several truncated predicted proteins in different high coverage de novo PacBio-only assemblies. This annotation problem escalates dramatically across the genome and leads to misprediction of multiple gene products.
In this study, we demonstrated a direct relationship between indel presence, homopolymers and GC content. The errors detected in the selected genomes with high GC content corresponded to deletions of nucleotides of C and G introduced by PacBio, whereas P. falciparum chosen in this study as an example of low GC content organism, shows insertion errors of A and T. In genomes with an intermediate percentage of GC content such as E. coli, the existence of both insertion errors of A and T, and deletion errors of G and C were detected. Therefore, we can conclude that the GC content of the studied organism is remarkably related to the type of indel error that is going to be introduced in PacBio sequencing.
The results showed that more than 70% of indels detected were located in homopolymeric tracks in all organisms. Moreover, a statistical study performed to discover if indel errors are randomly distributed along the genome showed that the probability of finding an indel error within a homopolymeric region is higher than by chance in all studied organisms. It is remarkable that T. thermophilus, being the organism with the highest percentage of homopolymers (around 60%), also has the highest number of indels in relation to its genome length, which could be related with its extremophilic characteristics. Contrary to expectations, Tessaracoccus sp., the organism with the highest GC content, shows an odd behavior with regard to the indel presence. Taking into account the similar coverage (around 370x) in these two organisms, these results are not expected. However, the study of the read features revealed low number of indel errors detected in Tessaracoccus sp., may be explained by important differences in the sequencing process. Since each PacBio subread contains sequence information for one polymerase pass, the total number of subreads—and therefore the accuracy of the final sequence—is inversely proportional to the DNA fragment size sequenced. Likewise, the length of the subreads is also related to the original insert size. Thus, the shorter length of the Tessaracoccus sp. PacBio subreads along with its shorter fragment size (4.9 kb) compared to the other bacteria could explain the lower number of errors introduced during the sequencing. On the contrary, T. thermophilus assembly shows more indel errors, since the length of the reads and fragment size (around 11 kb) generated are much higher. All these results indicate a greater effectiveness of PacBio consensus approach to improve accuracy when working with subreads below 10 kb and short-fragment sizes.
Despite the clear tendency of systematic indel distribution in homopolymeric areas, it should be noted that only a minor fraction of the total number of homopolymers in the genome are affected by indels error. Besides, larger homopolymers do not translate into more presence of indel errors indicating that errors during sequencing process are not caused by homopolymers length. In fact, L. infantum presents the highest variability of homopolymer types and sizes with a maximum of 70 nucleotides, but all the affected homopolymer regions have a size range between 2 and 12 nucleotides. Likewise, the rest of the studied organisms show the same wide length range of homopolymers affected by the errors introduced by PacBio.
Due to the high gene density present in bacteria, more than 88% of homopolymers tracks are located in gene sequences. Moreover, most of the genes (more than 90%) of the studied organisms include one or more homopolymer sequences (even in P. falciparum and L. infantum whose gene density and percentage of homopolymers present in genes is less than 50%). However, in most cases, more than one indel error can be located in the same gene, which translates into a low but non-negligible number of genes affected by PacBio sequencing errors, which is somewhat higher in bacteria. By introducing these errors, the predicted protein sequences can be altered, leading to a negative impact on the interpretation of the translated regions.
In addition, we performed a comparison of the performance of our pipeline against other well-known correction software, both hybrid (RACON) and non-hybrid (LoRMA). Among the three tools, ARAMIS and RACON are the fastest and memory efficient. Even though in the three cases memory usage and the time of the process increase with higher genome size, we prove that ARAMIS requires low memory usage even when correcting more complex and rich GC genomes.
Regarding the results, after correcting with LoRMA we still detected indel errors that remained uncorrected. This fact supports the idea of PacBio high coverage not being enough to overcome these sequencing errors. On the other hand, RACON corrected almost all the indel errors detected by ARAMIS. However, it is important to highlight that some of the errors call by RACON were not supported by Illumina reads. Due to this type of coverage issues, one of the most used methods is to further polish the sequences applying software such as RACON for more iterations or multiple polishing software. ARAMIS removes the requirement of using assembly polishing algorithms multiple times as it allows using multiple software in one step. In addition, the results showed that ARAMIS do a more precise and controlled correction process filtering out variants in positions below the coverage threshold and those supported by multihit reads.
Here, we describe a new pipeline, which combines different already published software with in house scripts to optimize the correction process of PacBio-only assembled genomes adding information from high accurate Illumina short reads. In contrast to other error-correction tools, this workflow corrects directly the PacBio assembly, which provides excellent results in a user-friendly manner. Six Pacbio-only genome assemblies with different levels of complexity were successfully corrected with this pipeline. Despite the existence of other tools specialized in assembly polishing, ARAMIS was designed for easy installation and use reducing the number of steps during the correction process and minimizing the bias related to coverage. As far as our knowledge goes, this is the only published pipeline that includes more than one correction software in one step reducing false positives and increasing confidence in the generated corrected assembly.
In summary, our results show that PacBio sequencing is appropriate for achieving an effective assembly of complete genomes, but high-accuracy short-reads data of sequencing is essential nowadays to correct sequence errors within homopolymeric regions in the assemblies generated with long reads (as PacBio). This correction process is especially important not only in cases with low-depth sequencing but also in those with a high-fragment mean length, as they showed a higher error rate. Moreover, we demonstrated that short reads from public databases are ideally suited for correcting this type of genome assembly errors and that already available PacBio-only assemblies in public databases can be polished through this pipeline.
In the future, it would be necessary to improve both the sequencing methods and the bioinformatics tools developed by data analyst specialists in order to achieve even more accurate assemblies.
Data availability statement
All data used for the manuscript have been downloaded from the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/) (See Table 4). The PacBio-only genome assemblies of the Tessaracoccus sp. Strain T2.5–30, L. infantum (JPCM5 strain), T. thermophilus NARI, and M. hassiacum DSM 44199 organisms were performed at the Genomics and NGS Core Facility at the Centro de Biología Molecular Severo Ochoa (GENGS-CBMSO, CSIC-UAM). In order to facilitate the double check of the performed analysis, all PacBio-only and new corrected genomes from all organisms studied here will be available at https://github.com/genomics-ngsCBMSO/ARAMIS.git.
Table 4.
Test data summary
| Type of data | P. falciparum | E. coli | L. infantum | T. thermophilus NAR1 | M. hassiacum | T. lapidicaptus |
|---|---|---|---|---|---|---|
| Illumina Reads | PRJEB2649 | PRJNA30551 | PRJEB20254 | PRJEB29203 | PRJEB25261 | PRJEB30798 |
| PacBio reads | PRJNA313199 | PRJNA237120 | PRJEB20254 | PRJEB29203 | PRJEB25261 | PRJEB30798 |
| Pacbio-only assembly | PRJNA313199 | PRJNA237120 | Github Aramis | Github Aramis | Github Aramis | CP019229 |
| Pacbio-only assembly-Corrected | Github Aramis | Github Aramis | PRJEB20254 | PRJEB29203 | PRJEB25261 | PRJEB30798 |
| PacBio-only Assembler | HGAP3[a] | Celera Assembler v. 8.2[b] | HGAP3[a] | HGAP3[a] | HGAP3[a] | HGAP3[a] |
| Illumina reads Aligner tool | BWA-mem [c] | BWA-mem [c] | BWA-mem [c] | BWA-mem [c] | BWA-mem [c] | BWA-mem [c] |
| PacBio reads Aligner tool | Pbalign[d] | Pbalign[d] | Pbalign[d] | Pbalign[d] | Pbalign[d] | Pbalign[d] |
Key points
Current algorithms available to fix NGS long-reads sequencing errors require long processing times and some errors may persist.
ARAMIS, a novel indel error correction pipeline, was designed for friendly user installation and for offering a new correction process using more than one software in just one step, reducing false positives, increasing confidence in the generated corrected assembly and minimizing the bias related to coverage.
The pipeline involves two steps: the correction and the statistical analysis of the results, which correct indel errors in the genome assembly and explore the nature and distribution of the indel errors along the genome, respectively.
The detected sequencing indel errors are mostly located in homopolymeric regions and the type of indel is directly related to the AT/GC content of the organism and the characteristics of the sequencing process.
Supplementary Material
Acknowledgements
The authors appreciate the advice and helpful comments received from members of the Genomics and NGS Core Facility (GENGS, http://genomics-ngs.cbm.uam.es/) and in special to Adrián Gómez Repollés, at the Centro de Biología Molecular Severo Ochoa (CBMSO, CSIC-UAM), which is part of the CEI UAM + CSIC, Madrid, Spain. Genome sequence data from European Nucleotide Archive (ENA) were invaluable for this work, and their provision in the public domain is gratefully acknowledged. The authors thank all the staff members of Dr Berenguer, Dr Requena and Dr Amils laboratories at the CBMSO, especially Dr Alba Blesa, Dr Mercedes Sanchez, Esther Camacho and Jose Manuel Martínez. Computational time from the Centro de Computación Científica (CCC) of Universidad Autónoma de Madrid is also gratefully acknowledged.
Funding
The GENGS receives institutional funding from the CBMSO (CSIC-UAM). The “Programa de Empleo Juvenil (PEJ)” funded contracts to E.S-H (PEJD-2018-PRE/BMD-9388) Community of Madrid, and to S.G-F (PEJD-2017-PRE/BMD-4828) Comunidad de Madrid and (PEJ2018-005067-P) Spanish Ministery of Science and Innovation, all within the European Youth Employment Initiative (YEI). The “Subprograma Personal Técnicos de Apoyo (PTA)” from the Spanish Ministery of Science and Innovation funded a contract to R.P-P (PTA2017-14628-I). The CBMSO receives institutional grants from the Fundación Ramón Areces and from the Fundación Banco de Santander.
Sacristán-Horcajada, E. is an NGS data analyst at the Genomic and NGS Facility (GENGS) at the Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM).
González-de la Fuente, S. is an NGS data analyst at the Genomic and NGS Facility (GENGS) at the Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM).
Peiró-Pastor, R. was an NGS data analyst at the at the Genomic and NGS Facility (GENGS) at the Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM). Current address: Animal Breeding Department Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA).
Carrasco-Ramiro, F. is the technical supervisor at the Genomic and NGS Facility (GENGS) at the Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM).
Amils, R. is the principal investigator of molecular ecology of extreme environments group at the Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM).
Requena, JM. is the principal investigator of the regulation of gene expression in Leishmania group at the Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM).
Berenguer, J. is the principal investigator of biotechnology and genetics of extreme thermophiles group at the Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM).
Aguado, B. is the scientific supervisor at the Genomic and NGS Facility (GENGS) at the Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM).
Contributor Information
E Sacristán-Horcajada, Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
S González-de la Fuente, Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
R Peiró-Pastor, Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
F Carrasco-Ramiro, Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
R Amils, Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
J M Requena, Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
J Berenguer, Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
B Aguado, Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
References
- 1. Djik EL, Jaszczyszyn Y, Daquin D, et al. The third revolution in sequencing technology. Trends Genet 2018;34(9):666–81. doi: 10.1016/j.tig.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 2. Ardui S, Ameur A, Vermeesch JR, et al. Single molecule real time (SMRT) sequencing comes of age: applications and utilities for medical diagnostic. Nucleics Acid Research 2018;46(5):2159–68. doi: 10.1093/nar/gky066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weirather JL, Cesare M, Wang Y, et al. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore technologies and their applications to transcriptome analysis. F1000Research 2017;6:100. doi: 10.12688/f1000research.10571.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 2016;17(6):333–51. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitsuhashi S, Frith MC, Mizuguchi T, et al. Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol 2019;20(1):58. doi: 10.1186/s13059-019-1667-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 2015;13(5):278–89. doi: 10.1016/j.gpb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watson M, Warr A. Errors in long-reads assemblies can critically affect protein prediction. Nat Biotechnol 2019;37(2):124–6. doi: 10.1038/s41587-018-0004-z. [DOI] [PubMed] [Google Scholar]
- 8. Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 2017;27(5):722–36. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chin C, Alexander DH, Marks P, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 2013;10(6):563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 10. Fu, Wang A, Au KF. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol 2019;20(1):26. doi: 10.1186/s13059-018-1605-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin HH, Liao YC. Evaluation and validation of assembling corrected PacBio long reads for microbial genome completion via hybrid approaches. PLoS One 2015;10(12):e0144305. doi: 10.1371/journal.pone.0144305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laehnemann D, Borkhardt A, McHardy AC. Denoising DNA deep sequencing data-high-throughput sequencing errors and their correction. Brief Bioinform 2016;17(1):154–79. doi: 10.1093/bib/bbv029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mahmoud M, Zwicki M, Twardowski T, et al. Efficiency of PacBio long read correction by 2nd generation Illumina sequencing. Genomics 2019;111(1):43–9. doi: 10.1016/j.ygeno.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 14. Zhang H, Jain C, Aluru S. A comprehensive evaluation of long read error correction methods. bioRxiv 2019;519330. doi: 10.1101/519330January 13, 2019, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salmela L, Walve R, Rivals E, et al. Accurate self-correction of errors in long reads using de Bruijn graphs. Bioinformatics 2017;33(6):799–806. doi: 10.1093/bioinformatics/btw321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bao E, Xie F, Song C, et al. FLAS: fast and high throughput algorithm for PacBio long read self-correction. Bioinformatics 2019;35(20):3953–60. doi: 10.1093/bioinformatics/btz206. [DOI] [PubMed] [Google Scholar]
- 17. Choudhury O, Chakrabarty A, Emrich SJ. HECIL: a hybrid error correction algorithm for long reads with iterative learning. Sci Rep 2018;8(1):9936. doi: 10.1038/s41598-018-28364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bao E, Lan L. HALC: high throughput algorithm for long read error correction. BMC bioinformatics 2017;18(1):204. doi: 10.1186/s12859-017-1610-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant Detectionand genome assembly improvement. PLoS One 2014;9(11):e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vaser R, Sović I, Nagarajan N, et al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 2017;27(5):737–46. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Browne PD, Nielsen TK, Kot W, et al. GC bias affects genomic and metagenomic reconstructions, underrepresenting GC-poor organisms. GigaScience 2020;9(2):giaa008. doi: 10.1093/gigascience/giaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gardner MJ, Hall N, Fung E, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002;419(6906):498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Korhonen PK, Hall RS, Young ND, et al. Common workflow language (CWL)-based software pipeline for de novo genome assembly from long- and short-read data. GigaScience 2019;8(4):giz014. doi: 10.1093/gigascience/giz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berlin K, Koren S, Chin CS, et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 2015;33(6):623–30. doi: 10.1038/nbt.3238. [DOI] [PubMed] [Google Scholar]
- 25. Lukes J, Mauricio IL, Schönian G, et al. Evolutionary and geographical history of the Leishmania donovani complex with a revision of current taxonomy. Proc Natl Acad Sci U S A 2007;104(22):9375–80. doi: 10.1073/pnas.0703678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. González-de la Fuente S, Peiró-Pastor R, Rastrojo A, et al. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci Rep 2017;7(1):18050. doi: 10.1038/s41598-017-18374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cava F, Laptenko O, Borukhov S, et al. Control of the respiratory metabolism of Thermus thermophilus by the nitrate respiration conjugative element NCE. Mol Microbiol 2007;64(3):630–46. doi: 10.1111/j.1365-2958.2007.05687.x. [DOI] [PubMed] [Google Scholar]
- 28. Blesa A, Sánchez M, Sacristán-Horcajada E, et al. Into the Thermus Mobilome: presence, diversity and recent activities of insertion sequences across Thermus spp. Microorganisms 2019;7(1):25. doi: 10.3390/microorganisms7010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sánchez-Costa M, Blesa A, Berenguer J. Nitrate respiration in Thermus thermophilus NAR1: from horizontal gene transfer to internal evolution. Genes 2020;11:1308. doi: 10.3390/genes11111308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gupta RS, Lo B, Son J. Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front Microbiol 2018;9:67. doi: 10.3389/fmicb.2018.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sánchez M, Blesa A, Sacristán-Horcajada E, et al. Complete genome sequence of Mycolicibacterium hassiacum DSM 44199. Microbiology resource announcements 2019;8(4):e01522–18. doi: 10.1128/MRA.01522-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maszenan AM, Seviour RJ, Patel BKC, et al. Tessaracoccus bendigoensis gen. Nov., sp. nov., a gram-positive coccus occurring in regular packages or tetrads, isolated from activated sludge biomass. Int J Syst Bacteriol 1999;49:459–68. doi: 10.1099/00207713-49-2-459. [DOI] [PubMed] [Google Scholar]
- 33. Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science 2009;323(5910):133–8. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 34. Leandro T, Costa MS, Sanz JL, et al. Complete genome sequence of Tessaracoccus sp. strain T2.5-30 isolated from 139.5 meters deep on the subsurface of the Iberian Pyritic Belt. Genome Announc 2017;5(17):e00238–17. doi: 10.1128/genomeA.00238-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Canzar S, Salzberg SL. Short read mapping: an algorithmic tour. Proc IEEE Inst Electr Electron Eng 2017;105(3):436–58. doi: 10.1109/JPROC.2015.2455551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013;14(2):178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen W, Le S, Li Y, et al. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 2016;11(10):e0163962. doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown CT, Olm MR, Thomas BC, et al. Measurement of bacterial replication rates in microbial communities. Nat Biotechnol 2016;34(12):1256–63. doi: 10.1038/nbt.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used for the manuscript have been downloaded from the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/) (See Table 4). The PacBio-only genome assemblies of the Tessaracoccus sp. Strain T2.5–30, L. infantum (JPCM5 strain), T. thermophilus NARI, and M. hassiacum DSM 44199 organisms were performed at the Genomics and NGS Core Facility at the Centro de Biología Molecular Severo Ochoa (GENGS-CBMSO, CSIC-UAM). In order to facilitate the double check of the performed analysis, all PacBio-only and new corrected genomes from all organisms studied here will be available at https://github.com/genomics-ngsCBMSO/ARAMIS.git.
Table 4.
Test data summary
| Type of data | P. falciparum | E. coli | L. infantum | T. thermophilus NAR1 | M. hassiacum | T. lapidicaptus |
|---|---|---|---|---|---|---|
| Illumina Reads | PRJEB2649 | PRJNA30551 | PRJEB20254 | PRJEB29203 | PRJEB25261 | PRJEB30798 |
| PacBio reads | PRJNA313199 | PRJNA237120 | PRJEB20254 | PRJEB29203 | PRJEB25261 | PRJEB30798 |
| Pacbio-only assembly | PRJNA313199 | PRJNA237120 | Github Aramis | Github Aramis | Github Aramis | CP019229 |
| Pacbio-only assembly-Corrected | Github Aramis | Github Aramis | PRJEB20254 | PRJEB29203 | PRJEB25261 | PRJEB30798 |
| PacBio-only Assembler | HGAP3[a] | Celera Assembler v. 8.2[b] | HGAP3[a] | HGAP3[a] | HGAP3[a] | HGAP3[a] |
| Illumina reads Aligner tool | BWA-mem [c] | BWA-mem [c] | BWA-mem [c] | BWA-mem [c] | BWA-mem [c] | BWA-mem [c] |
| PacBio reads Aligner tool | Pbalign[d] | Pbalign[d] | Pbalign[d] | Pbalign[d] | Pbalign[d] | Pbalign[d] |
Key points
Current algorithms available to fix NGS long-reads sequencing errors require long processing times and some errors may persist.
ARAMIS, a novel indel error correction pipeline, was designed for friendly user installation and for offering a new correction process using more than one software in just one step, reducing false positives, increasing confidence in the generated corrected assembly and minimizing the bias related to coverage.
The pipeline involves two steps: the correction and the statistical analysis of the results, which correct indel errors in the genome assembly and explore the nature and distribution of the indel errors along the genome, respectively.
The detected sequencing indel errors are mostly located in homopolymeric regions and the type of indel is directly related to the AT/GC content of the organism and the characteristics of the sequencing process.