

Abstract
Viral infection involves a large number of protein–protein interactions (PPIs) between human and virus. The PPIs range from the initial binding of viral coat proteins to host membrane receptors to the hijacking of host transcription machinery. However, few interspecies PPIs have been identified, because experimental methods including mass spectrometry are time-consuming and expensive, and molecular dynamic simulation is limited only to the proteins whose 3D structures are solved. Sequence-based machine learning methods are expected to overcome these problems. We have first developed the LSTM model with word2vec to predict PPIs between human and virus, named LSTM-PHV, by using amino acid sequences alone. The LSTM-PHV effectively learnt the training data with a highly imbalanced ratio of positive to negative samples and achieved AUCs of 0.976 and 0.973 and accuracies of 0.984 and 0.985 on the training and independent datasets, respectively. In predicting PPIs between human and unknown or new virus, the LSTM-PHV learned greatly outperformed the existing state-of-the-art PPI predictors. Interestingly, learning of only sequence contexts as words is sufficient for PPI prediction. Use of uniform manifold approximation and projection demonstrated that the LSTM-PHV clearly distinguished the positive PPI samples from the negative ones. We presented the LSTM-PHV online web server and support data that are freely available at http://kurata35.bio.kyutech.ac.jp/LSTM-PHV.
Keywords: LSTM, word2vec, human-virus protein–protein interaction, deep learning, SARS-CoV2
Introduction
Viral infections are one of the major causes of human health, as we can see from the current status of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that raises a global pandemic. As of February 2021, more than 110 million people infected and nearly 2.4 million deaths have been reported worldwide for the coronavirus disease 2019 (COVID-19) disease [1]. Viruses achieve their own life cycle and proliferate their clones by hijacking and utilizing the functions of their hosts. To carry out these processes, viruses interact with host proteins to control cell cycles and apoptosis and to transport their own genetic material into the host nucleus [2, 3]. Therefore, it is important to identify human-virus protein–protein interactions (HV-PPIs) and to understand the mechanisms of viral infections and host immune responses to find new drug targets. However, compared to intraspecies PPIs, few interspecies PPIs have been identified. To identify the PPIs, experimental methods such as yeast-to-hybrid and mass spectrometry have been widely used [4, 5], but they are time-consuming and laborious. For this reason, it is difficult to apply experimental methods for all protein pairs. Therefore, the computational approach is a preliminary treatment prior to the experimental method.
The use of amino acid sequence information is promising in the prediction of PPIs because the experimental data of PPIs and sequence information of proteins are abundant. Machine learning (ML)-based approaches are very attractive [6] that use the amino acid binary profiles [7], evolutionary properties [8], physicochemical properties [9] and structural information [10]. Zhou et al. [11] integrated different encoding methods, such as relative frequency of amino acid triplets, frequency difference of amino acid triplets and amino acid composition to construct a SVM-based PPI predictor [11]. Yang et al. [12] have employed a position-specific scoring matrix (PSSM) to build a convolutional neural network (CNN) for PPI prediction. Recently, promising encoding schemes have been proposed to capture the sequence patterns of proteins, including the conjoint triad [13], auto covariance [14] and autocorrelation [15].
Human-virus PPIs involve not only the various properties of amino acid sequences but also the distributions of 20 amino acid residues in the context of whole protein sequences. While many predictors have focused on the former features, the latter context-based information is suggested to be effective in predicting HV-PPIs. To capture the context information of amino acid sequences as much as possible, word/document embedding techniques have recently been proposed. Yang et al. [16] combined the doc2vec encoding schemes with a random forest method to predict PPIs.
To utilize the amino acid sequence context as words effectively, we have proposed the long short-term memory (LSTM) model [17] with the word2vec embedding method that predicts the PPIs between human and virus, named LSTM-PHV. To the best of our knowledge, this is the first application of the LSTM with the word2vec to sequence-based PPI prediction. Interestingly, use of the sequence context as words presented remarkably accurate prediction of the interactions between human and unknown virus proteins.
Materials and methods
Benchmark dataset construction
The PPIs datasets were downloaded from the Host-Pathogen Interaction Database 3.0 (HPIDB 3.0) [18]. The retrieved HV-PPIs were further selected in the following process. First, to ensure interactions with a certain level of confidence, the PPIs with an MI score of below 0.3 were removed. The MI score is the confidence score assigned to each PPI from IntAct [19] and VirHostNet [20]. Second, redundant PPIs were excluded by using CD-HIT with an identity threshold of 0.95 [21]. Third, the PPIs that contained the proteins consisting of standard amino acids only and the proteins with a length of more than 30 residues and less than 1000 residues were selected. Finally, 22 383 PPIs from 5882 human and 996 virus proteins were considered as positive samples.
To the best of our knowledge, there is no gold standard for generating negative samples. Many previous studies used a random sampling method. Pairs of the human and virus proteins that do not appear in the positive PPI dataset are randomly sampled as negative data. However, the random sampling method may incorrectly assign many positive samples to negative ones [9, 22]. To address this problem, the dissimilarity negative sampling method was developed [9], which used a sequence similarity-based method to explore the protein pairs that are unlikely to interact. We employed the dissimilarity-based negative sampling method as follows. We calculated the sequence similarities of all pairs of virus proteins in positive samples with the Needleman–Wunsch algorithm of BLOSUM30 and defined a similarity vector for each virus protein. Subsequently, we excluded the virus proteins showing lower sequence similarities than 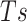 for more than half of the total virus proteins as outliers.
for more than half of the total virus proteins as outliers.  was calculated by:
was calculated by:
 |
(1) |
where  and
and  are the first quartile and quartile range of the similarity scores for the
are the first quartile and quartile range of the similarity scores for the  -th virus protein
-th virus protein  , respectively. By setting the maximum and minimum values of the similarity scores to 0 and 1, respectively, the similarity score was normalized and converted into the distance.
, respectively. By setting the maximum and minimum values of the similarity scores to 0 and 1, respectively, the similarity score was normalized and converted into the distance.
The human proteins that consisted of the standard amino acids and whose residue length was longer than 30 and shorter than 1000 were retrieved from the UniProtKB/Swiss-Prot database [23]. Then, the human proteins that interacted with the virus proteins showing a distance from viral protein 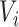 of less than distance threshold T were removed, considering that they were likely to interact with virus protein
of less than distance threshold T were removed, considering that they were likely to interact with virus protein  . The remaining pairs of the human and virus proteins were regarded as negative samples. According to the previous study [9], the threshold
. The remaining pairs of the human and virus proteins were regarded as negative samples. According to the previous study [9], the threshold  was set to 0.8. We randomly sampled from the candidates so that the ratio of positive to negative samples was 1:10. The positive and negative samples were labeled 1 and 0, respectively. The resultant dataset was divided into training data and independent test data at a ratio of 8:2.
was set to 0.8. We randomly sampled from the candidates so that the ratio of positive to negative samples was 1:10. The positive and negative samples were labeled 1 and 0, respectively. The resultant dataset was divided into training data and independent test data at a ratio of 8:2.
SARS-CoV-2 PPIs dataset construction
We downloaded the datasets of SARS-CoV-2 from BioGRID (COVID-19 Coronavirus Project 4.3.195) [24] and extracted the PPIs between human and SARS-CoV-2. We removed the PPIs having the proteins whose amino acid sequences were not registered in the UniProtKB database [23]. We further removed the PPIs that contained nonstandard amino acids and the proteins with a length of fewer than 30 residues and more than 1000 residues. Finally, we obtained 7373 PPIs that consisted of 2943 human proteins and 11 SARS-CoV-2 proteins as the positive samples. The negative samples were generated by the dissimilarity negative sampling method. We randomly selected negative samples so that the ratio of positive to negative samples was 1:10. The resultant dataset was divided into training data and independent test data at a ratio of 8:2.
Nonviral pathogen PPIs dataset construction
In order to investigate whether our model can be extended to pathogens other than viruses, we constructed the datasets of PPIs between human and nonviral pathogens. The PPIs having virus proteins were excluded from the PPI dataset of HPIDB. The PPIs were prepared in the same manner as the benchmark dataset construction mentioned above. Subsequently, 8412 PPIs consisting of 3317 human proteins and 3068 virus proteins were selected as positive data. The negative datasets were generated by the dissimilarity negative sampling method. We randomly selected negative samples so that the ratio of positive to negative samples was 1:10. The resultant dataset was divided into training data and independent test data at a ratio of 8:2.
Embedding of protein sequences by word2vec
In the field of natural language processing, embedding methods such as word2vec [25] and doc2vec [26] were developed to obtain the distributed representation of words and documents, respectively. In word2vec, the weights in a neural network learn the context of words to provide the distributed representation that encodes different linguistic regularities and patterns [27]. There are two methods for learning the context of words: Continuous Bag-of-Words Model (CBOW) and the Continuous Skip-Gram Model (Skip-Gram). CBOW predicts the current word based on the context, while Skip-Gram predicts the context from the current word. Skip-gram is more efficient with less training data, while CBOW learns faster and more frequent words. At present, computational biology used these methods [28, 29].
The amino acid sequences of human and virus proteins registered as positive and negative samples were encoded as matrixes using the word2vec method. The k-mers (k consecutive amino acids) in amino acid sequences were regarded as a single word (unit) and each amino acid sequence was represented by multiple k-mers. For example, given an amino acid sequence MAEDDPYL, the units of the 4-mers are MAED, AEDD, EDDP, DDPY and DPYL (Figure 1). We trained a CBOW-based word2vec model to learn the appearance pattern of k-mers from the computational speed standpoint by using the genism of the python package [30]. Here, k-mers and protein sequences correspond to words and sentences in natural language. Human and virus proteins in positive samples and nonredundant proteins in the UniProtKB/Swiss-Prot database [23] were used to train the word2vec model. The nonredundant proteins were collected by applying CD-HIT to all proteins with an identity threshold of 0.9. The k-mers up to three neighbors of a specific k-mer are considered as the peripheral k-mers, and training was iterated 1000 times. The trained word2vec model produced 128-dimensional embedding vectors in each k-mer and they were concatenated to produce the embedding matrixes of proteins. Since 4-mer provided the largest AUC by 5-fold cross-validation in a previous study [16], we set k to 4.
Figure 1 .

Embedding of amino acid sequences in a case of 4-mer. Amino acid sequences were represented by 4-mers and embedded as a matrix by training the word2vec model. The matrixes were generated by concatenating the vectors of 4-mers in a row.
Construction of LSTM-PHV
Neural networks such as CNN and recurrent neural network (RNN), in particular, are very powerful and have been applied to difficult problems such as speech recognition and visual object recognition [31]. The RNN learns time or step dependencies in sequence data and enables training on variable-length data. The LSTM solves the gradient explosion and gradient disappearance problems of RNNs, enabling long-term time-dependent learning.
The LSTM-PHV is composed of three sub-networks, as shown in Figure 2. The two, upstream networks with the same structure transformed the human and virus proteins-embedding matrixes into two fixed-length vectors. The third network used their concatenated fixed-length vectors to predict the PPIs. They are referred to as ‘concatenated vectors’. The amino acid sequence column vectors in the embedding matrixes are inputted to each step of the LSTM units. The LSTM units were expanded in both the N- to C-terminus and the C- to N-terminus directions. The 64 × 2-dimensional vectors generated from one LSTM unit were concatenated in a row. The dimensions of the vectors generated through the three layers decreased in the order of 64, 32 and 1. In the first two layers, the rectified linear unit (ReLU) function with a dropout rate of 0.3 was used as an activation function. The scalar values generated from the third layer were lined up into a vector, which was provided to the softmax function. A fixed-length vector was generated by summarizing the weighted vectors in all the steps.
Figure 2 .

Network structure of LSTM-PHV. The human and virus protein matrices were transformed into the two fixed-length vectors by the two upstream neural networks with the LSTM, respectively. The networks were surrounded by blue and red lines. Feature vectors of each 4-mer in protein matrices were inputted into the LSTM unit at each step. Scalar values were generated by applying three fully connected layers to output from the LSTM unit at each step. The broadcasted scalar values and output from the LSTM unit were multiplied. The fixed-length vectors were produced by adding the multiplied vectors and concatenating the vectors in human and virus. Final outputs were obtained by four fully connected layers surrounded by the purple line.
The fixed-length vectors for the human and virus proteins were concatenated in line and propagated into the final network. The final network consists of three layers and an output layer. The dimensions of the generated vectors from each layer decreased in the order of 200, 100, 40 and 1. The ReLU function with a dropout rate of 0.3 was applied to the output of the three layers. To obtain a final output with a value between 0 and 1, the sigmoid function was used as an activation function at the output layer. The construction and learning of the neural networks were performed using the PyTorch [32] of the python package.
Training of imbalanced data
A 5-fold cross-validation was applied on the training dataset, while conserving the ratio of positive and negative samples at each subset. We set the learning rate to 0.001, used the rectified adam (RAdam) optimizer [33] as the optimization function, and set a mini-batch learning size to 1024. To train the model on imbalanced data, we weighted a binary cross-entropy loss function in the manner reported by Cui et al. (2019) [34]. The loss functions used are shown below.
 |
(2) |
where  is the correct label,
is the correct label,  is the model-predicted probability of interaction,
is the model-predicted probability of interaction, 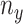 is the number of data whose label is
is the number of data whose label is 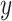 in the mini-batch and
in the mini-batch and  is the hyperparameter.
is the hyperparameter.  was set to 0.99. To prevent overlearning, the training process was terminated when the maximum accuracy in the validation data was not updated for consecutive 20 epochs. To prevent the weight of the loss function from being 0, we set an approximately equal ratio of labels for all the mini-batches.
was set to 0.99. To prevent overlearning, the training process was terminated when the maximum accuracy in the validation data was not updated for consecutive 20 epochs. To prevent the weight of the loss function from being 0, we set an approximately equal ratio of labels for all the mini-batches.
Measures
To evaluate the prediction performance, seven statistics measures were used: sensitivity (SN; recall), specificity (SP), accuracy (ACC), Matthews correlation coefficient (MCC), positive predictive value (PPV), F1-score (F1), area under the curve (AUC) and area under the precision-recall curve (AUPRC). MCC, F1-score and AUPRC are effective in assessments of imbalanced data. The measures other than the AUC and AUPRC are given by:
 |
(3) |
 |
(4) |
 |
(5) |
 |
(6) |
 |
(7) |
 |
(8) |
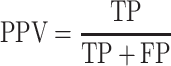
where TP, FP, TN and FN are the numbers of the correctly predicted positive samples, incorrectly predicted positive samples, correctly predicted negative samples and incorrectly predicted negative samples, respectively. The threshold for a determination of whether protein pairs interact or not was set to a predicted probability of 0.5. AUC and AUPRC are the areas beneath the ROC curve and PR curve, respectively. These measures were calculated by the scikit-learn of the python package [35].
Visualization of positive and negative samples
To visualize the concatenated vectors, we reduced the dimensionality of the concatenated vector from 256 to 2 using uniform manifold approximation and projection (UMAP) [36]. UMAP is the nonlinear dimensionality reduction approach [36], which can preserve not only local patterns but also global patterns in low-dimensional space. We set the number of neighbors in the k-neighbor graph to 50, and set a minimum distance between points in the low-dimensional space to 0. The distances between any points were calculated by the Euclidean distance. The optimization was implemented up to 500 epochs with a learning rate of 1.0.
Results and discussion
Predictive performance of LSTM-PHV
We evaluated the predictive performance of LSTM-PHV via 5-fold cross-validation on the training dataset and test it on the independent dataset, as shown in Figure 3 and Table S1. Out of the five subset models, the model with the highest AUC was used to predict the independent dataset. The AUCs were 0.976 and 0.973 on the training and independent datasets, respectively; the ACCs were 0.984 and 0.985 on the training and independent datasets, respectively. The MCC, F1 and AUPRC also presented high scores on both the datasets. MCC has been used in many previous studies in bioinformatics as an evaluation measure for imbalance data [16, 37, 38]. A high MCC indicated that the LSTM-PHV was able to effectively learn the imbalanced data. LSTM-PHV provided remarkable performance of PPI prediction.
Figure 3 .

Performance of the LSTM-PHV via 5-fold cross-validation on the training dataset. The measures of the five subset models in 5-fold cross-validation were averaged.
Performance comparison with state-of-the-art existing machine learning models
To characterize the performance of LSTM-PHV, we compared it with an RF model with Doc2vec, named Yang’s model [16], on our independent test data, as shown in Figure 4 and Table S2. The source code and trained model were provided by Yang et al. with their recommended three threshold values. As in our case, Yang et al. built the imbalanced data that contained 10 times more negative samples than positive samples, while generating negative samples by the dissimilarity-based negative sampling method. The LSTM-PHV presented higher values than Yang’s model not only for AUC and ACC but also for MCC, F1-score and AUPRC (Figure 4). The LSTM-PHV was able to learn the imbalanced data better than Yang’s model. Particularly, LSTM-PHV takes an advantage in the high MCC value, because learning of imbalanced data is not evitable. At present the number of known PPIs is very small compared to the total number of protein pairs. Thus, negative samples are typically produced much more than positive ones in the absence of golden standard of generating negative samples.
Figure 4 .

Performance comparison of LSTM-PHV with Yang’s RF model with doc2vec using our independent test. Thresholds at SP of 0.90, 0.95 and 0.99 in Yang’s study were used according to their suggestion.
The LSTM-PVM that combined LSTM with word2vec outperformed the latest state-of-the-art model of Yang’s RF model, probably because LSTM was able to efficiently capture the context of amino acid sequence patterns. Interestingly, we revealed that learning of only sequence contexts as words presented remarkably high performances without any biochemical properties.
To assess whether the LSTM-PHV is applicable to unknown virus species, we compared LSTM-PHV with a SVM model with commonly used encoding methods, named Zhou’s model [11]. We employed Zhou’s dataset that consisted of the four training datasets: PPIs between human and any virus except Influenza A virus subtype H1N1 (H1N1) (TR1), PPIs between human and any virus except Ebola virus (TR2), PPIs between any host and any virus except H1N1 (TR3), PPIs between any host and any virus except Ebola virus (TR4) and two test datasets: PPIs between human and H1N1 virus (TS1) and PPIs between human and Ebola virus (TS2). In training the LSTM-PHV, we set a batch size to 256 and used the normal binary cross-entropy loss function, because Zhou’s datasets were much smaller than our dataset and it was balanced data. As shown in Figure 5 and Table S3, we compared the LSTM-PHV with Zhou’s SVM model on the four datasets. We trained the LSTM-PHV by the same datasets of TR1 and TR2 that do not include the PPIs between human and H1N1 and Ebola virus, respectively. Compared to Zhou’s model, the LSTM-PHV predicted TS1 and TS2 with high ACC and AUC. When the predictors were trained on multiple host protein-including TR3 and TR4, the LSTM-PHV also presented higher ACC and AUC to predict TS1 and TS2. The AUCs on the four datasets were significantly different between LSTM-PHV and Zhou’s model (two-sample t-test; P < 0.05). LSTM-PHV learnt host and virus protein sequence contexts more efficiently than the SVM model.
Figure 5 .

Prediction performance of LSTM-PHV with existing state-of-the-art predictors. We employed the four datasets that combine the four training data with two test data according to Zhou’s study. The four datasets containing human–virus interactions (TR1-TS1 and TR2-TS2) and multiple host–virus interactions (TR3-TS1 and TR4-TS2) were applied to LSTM-PHV. The performances of the Zhou’s model are from table 5 of their paper.
Very recently, Yang et al. [12] have proposed a CNN-based PPI predictor with a PSSM. They generated the PSSM by applying PSI-BLAST to amino acid sequences, and then inputted the PSSM to a CNN as a feature matrix. CNNs take in an advantage in learning the features of noncontinuous data such as image data, while LSTMs (RNN-type models) effectively learn the long-term memory features. Our LSTM-PHV achieved high prediction performance of human-virus PPIs. Differing from Yang’s model, we used LSTM to intensively read the contexts of the whole amino acid sequences.
Prediction of SARS-CoV-2 and nonviral pathogens PPIs
To demonstrate the applicability of LSTM-PHV to other species, we applied it to PPIs between human and SARS-CoV-2. A 5-fold cross-validation was performed on the training dataset. The subset model providing the highest AUC for all the five was employed as the final model and tested on the independent dataset. LSTM-PHV achieved AUCs of 0.955 and 0.956 in 5-fold cross-validation and independent test, respectively (Table S4). Our method was found available to prediction of the human and SARS-CoV-2 PPIs.
Furthermore, to investigate whether LSTM-PHV can be extended to nonviral pathogens, we applied it to PPIs between human and nonviral pathogens. A 5-fold cross-validation was carried on the training dataset. The subset model providing the highest AUC for all the five was employed as the final model and tested on the independent dataset. LSTM-PHV achieved AUCs of 0.920 and 0.922 in 5-fold cross-validation and independent test, respectively (Table S5). These results demonstrated that LSTM-PHV can be extended to predict the PPIs between human and nonviral pathogens.
Superiority of dissimilarity negative sampling method
To demonstrate the superiority of the dissimilarity negative sampling method, we compared it with a random negative sampling method, as shown in Figure 6 and Table S6. A 5-fold cross-validation and the independent test were used. The AUC and AUPRC in the dissimilarity negative sampling method were higher than those in the random sampling method. The AUCs and AUPRCs by the 5-fold cross-validation were significantly different (two-sample t-test; P < 0.0001) between the two methods. These results suggest some of the randomly generated-negative samples impair the prediction as noisy data.
Figure 6 .

Comparison of the dissimilarity negative sampling method with a random sampling method. A 5-fold cross-validation was applied to the training data generated by the dissimilarity negative sampling method and by a random sampling method. The bars and error bars indicate the mean and standard deviation of AUC and AUPRC in the five subset models.
Visualization of positive and negative samples
The two upstream neural networks with the LSTM generated the fixed-length vectors (Figure 2). To examine how these neural networks extract PPI-related information, we drew the UMAP map of their concatenated vectors on the training and independent datasets of our benchmark dataset (Figure 7). In addition, we made the t-SNE map of them (Figure S1). In both the UMAP and T-SNE maps, multiple clusters were generated, and positive samples were distinguished from negative samples within the clusters. The false negative and false positive samples were located between the true negative and true positive samples. The numbers of the false negative and false positive samples were small. These results suggested that the upstream neural networks extract critical information responsible for predicting PPIs from the amino acid sequences of each protein. The UMAP accumulated the true positive samples more densely than t-SNE, which corresponded to the previous suggestion [39] that UMAP preserves not only local structure but also the global structure. The LSTM-PHV showed almost similar distributions between the training and independent datasets in both the UMAP and t-SNE, demonstrating the robustness of LSTM-PHV to an independent dataset or to changes in datasets.
Figure 7 .

UMAP map of the positive and negative samples of our benchmark dataset. (A) The concatenated vectors provided in the 5-fold cross-validation, which showed the highest value of AUC for all the five, were projected. (B) The concatenated vectors provided in the independent test were projected. The true positive, false positive, false negative and true negative samples were visualized.
Webserver implementation
We used apache (2.4.18), python (3.8.0) and flask (1.1.2) to build a web server application of LSTM-PHV at http://kurata35.bio.kyutech.ac.jp/LSTM-PHV. The users can either input or upload the amino acid sequences of human and virus proteins in FASTA format to evaluate the PPIs with prediction scores. In addition, the attention weights and transformed vectors generated during prediction are provided. A threshold to determine whether the inputted proteins interact was set to 0.5. To facilitate the community, we provide the datasets used in the present study, which can be downloaded from our website. For other overviews, refer to the help of the website.
Conclusions
To accurately predict PPIs between human and virus, we proposed the LSTM-PHV that combined LSTM with the word2vec embedding method by considering the whole sequence context of amino acid residues. The word2vec is able to preserve the information about patterns of the local amino acid residues. Interestingly, the method does not use any biochemical properties of amino acid residues, while existing models intensively used their biochemical properties. The LSTM further learns the amino acid patterns in the whole sequence contexts. The LSTM-PHV learnt highly imbalanced data and was able to accurately predict the interaction of a human protein to an unknown virus protein, compared to existing state-of-the-art models. Interestingly, it could be extended not only to the PPIs between SARS-CoV-2 and human but also to the PPIs between nonviral pathogens and human. On the other hand, our method requires more memory or computational cost compared to existing models, because the number of elements in the feature matrix increases with an increase in the length of the sequence. Use of the LSTM-PHV enhances the screening of drug targets that inhibit human-virus PPIs and definitely contributes to advances in remedies of infectious diseases including COVID-19.
Key Points
The LSTM-based model with word2vec (LSTM-PHV) efficiently learns highly imbalanced training data to accurately predict PPIs between human and virus.
Learning of amino acid sequence contexts as words without any biochemical properties is sufficient for PPI prediction.
UMAP visualizes that positive samples are clearly distinguished from negative samples.
LSTM-PHV is applied to prediction of the human and SARS-CoV-2 PPIs and human and nonviral pathogens PPIs.
Supplementary Material
Sho Tsukiyama is a student of Department of Interdisciplinary Informatics in the Kyushu Institute of Technology, Japan. His main research interests include machine learning and computational biology.
Md Mehedi Hasan is currently a postdoctoral fellow at the Tulane University, USA. Before his current position, he worked as a Japan Society for the Promotion of Science international PD fellow in the Kyushu Institute of Technology, Japan. He also worked as a researcher at the Chinese University of Hong Kong. He received his PhD degree in bioinformatics from the China Agricultural University, Beijing, in 2016. His main research interests include protein structure prediction, machine learning, data mining, computational biology and functional genomics.
Satoshi Fujii is an assistant professor of Department of Bioscience and Bioinformatics in the Kyushu Institute of Technology, Japan. His research interests include machine learning, clinical data analysis and biomedical design.
Hiroyuki Kurata is a professor of Department of Bioscience and Bioinformatics in the Kyushu Institute of Technology, Japan. His research interests primarily focus on systems biology, synthetics biology, functional genomics, machine learning and their applications.
Contributor Information
Sho Tsukiyama, Department of Interdisciplinary Informatics in the Kyushu Institute of Technology, Japan.
Md Mehedi Hasan, Tulane University, USA.
Satoshi Fujii, Department of Bioscience and Bioinformatics in the Kyushu Institute of Technology, Japan.
Hiroyuki Kurata, Department of Bioscience and Bioinformatics in the Kyushu Institute of Technology, Japan.
Acknowledgement
This work was supported by the Grant-in-Aid for Scientific Research (B) (19H04208) and partially supported by the Grant-in-Aid for JSPS Research Fellow (19F19377) from Japan Society for the Promotion of Science (JSPS).
References
- 1. World Health Organization . Coronavirus disease (covid-19) situation dashboard. https://covid19.who.int/(21 February 2021, date last accessed).
- 2. Yang S, Fu C, Lian X, et al. Understanding human-virus protein-protein interactions using a human protein complex-based analysis framework. mSystems 2019;4:e00303–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dyer MD, Murali TM, Sobral BW. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog 2008;4(2):e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. Part I. experimental techniques and databases. PLoS Comput Biol 2007;3(3):e42–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ito T, Chiba T, Ozawa R, et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci 2001;98(8):4569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khatun MS, Shoombuatong W, Hasan MM, et al. Evolution of sequence-based bioinformatics tools for protein-protein interaction prediction. Curr Genomics 2020;21(6):454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang YA, You ZH, Chen X, et al. Sequence-based prediction of protein-protein interactions using weighted sparse representation model combined with global encoding. BMC Bioinformatics 2016;17(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hamp T, Rost B. Evolutionary profiles improve protein-protein interaction prediction from sequence. Bioinformatics 2015;31(12):1945–50. [DOI] [PubMed] [Google Scholar]
- 9. Eid FE, ElHefnawi M, Heath LS. DeNovo: virus-host sequence-based protein-protein interaction prediction. Bioinformatics 2016;32:1144–50. [DOI] [PubMed] [Google Scholar]
- 10. Neuvirth H, Raz R, Schreiber G. ProMate: a structure based prediction program to identify the location of protein-protein binding sites. J Mol Biol 2004;338:181–99. [DOI] [PubMed] [Google Scholar]
- 11. Zhou X, Park B, Choi D, et al. A generalized approach to predicting protein-protein interactions between virus and host. BMC Genomics 2018;19:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang X, Yang S, Lian X, et al. Transfer learning via multi-scale convolutional neural layers for human-virus protein-protein interaction prediction. bioRxiv 2021; 4314202021. doi: 10.1101/2021.02.16.431420. [DOI] [PMC free article] [PubMed]
- 13. Wang J, Zhang L, Jia L, et al. Protein-protein interactions prediction using a novel local conjoint triad descriptor of amino acid sequences. Int J Mol Sci 2017;18:2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo Y, Yu L, Wen Z, et al. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res 2008;36(9):3025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khatun MS, Hasan MM, Mollah MNHet al. SIPMA: A Systematic Identification of Protein-Protein Interactions in Zea mays Using Autocorrelation Features in a Machine-Learning Framework. In: 2018 IEEE 18th International Conference on Bioinformatics and Bioengineering (BIBE). Taichung, Taiwan: IEEE, 2018, 122–5.
- 16. Yang X, Yang S, Li Q, et al. Prediction of human-virus protein-protein interactions through a sequence embedding-based machine learning method. Comput Struct Biotechnol J 2020;18:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput 1997;9(8):1735–80. [DOI] [PubMed] [Google Scholar]
- 18. Ammari MG, Gresham CR, McCarthy FM, et al. HPIDB 2.0: a curated database for host-pathogen interactions. Database (Oxford) 2016;2016:baw103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kerrien S, Aranda B, Breuza L, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res 2012;40(D1):D841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guirimand T, Delmotte S, Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res 2015;43(D1):D583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012;28(23):3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dey L, Chakraborty S, Mukhopadhyay A. Machine learning techniques for sequence-based prediction of viral-host interactions between SARS-CoV-2 and human proteins. Biom J 2020;43(5):438–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. The UniProt Consortium . UniProt: the universal protein knowledgebase. Nucleic Acids Res 2017;45(D1):D158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stark C, Breitkreutz B-J, Reguly T, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006;34(90001):D535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mikolov T, Chen K, Corrado G, et al. Efficient estimation of word representations in vector space. arXiv 2013; 1301.3781.
- 26. Le Q, Mikolov T. Distributed representations of sentences and documents. International Conference on International Conference on Machine Learning 2014;31:1188–96. [Google Scholar]
- 27. Mikolov T, Sutskever I, Chen K, et al. Distributed representations of words and phrases and their compositionality. 2013arXiv:1310.4546; October 18, 2013preprint: not peer reviewed.
- 28. Hamid MN, Friedberg I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics 2019;35(12):2009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu C, Gao R, Zhang Y, et al. PTPD: predicting therapeutic peptides by deep learning and word2vec. BMC Bioinformatics 2019;20(1):456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Řehůřek R, Sojka P. Software Framework for Topic Modelling with Large Corpora. In Proceedings of LREC 2010 Workshop on New Challenges for NLP Frameworks. Malta: University of Malta, 2010, 45–50.
- 31. Sutskever I, Vinyals O, Le QV. Sequence to sequence learning with neural networks. arXiv 2014; 1409.3215.
- 32. Paszke A, Gross S, Chintala S, et al. Automatic Differentiation in PyTorch. In: NIPS 2017 Workshop on Autodiff, Long Beach, California, USA, 2017.
- 33. Liu L, Jiang H, He P, et al. On the variance of the adaptive learning rate and beyond. arXiv 2019; 1908.03265.
- 34. Cui Y, Jia M, Lin T-Y, et al. Class-balanced loss based on effective number of samples. 2019, arXiv:1901.05555.
- 35. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikitlearn: machine learning in python. J Mach Learn Res 2012;12:2825–30. [Google Scholar]
- 36. McInnes L, Healy J, Saul N, et al. UMAP: uniform manifold approximation and projection for dimension reduction. J. Open Source Softw 2018;3:861. [Google Scholar]
- 37. Lin W, Xu D. Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics (Oxford, England) 2016;32(24):3745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Z, Xiao X, Qiu WR, et al. iDNA-methyl: identifying DNA methylation sites via pseudo trinucleotide composition. Anal Biochem 2015;474:69–77. [DOI] [PubMed] [Google Scholar]
- 39. Becht E, McInnes L, Healy J, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2019;37(1):38–44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.