

Abstract
Non-small cell lung cancer (NSCLC) is characterized by relatively rapid response to systemic treatments yet inevitable resistance and predisposed to distant metastasis. We thus aimed at performing sequencing analysis to determine genomic events and underlying mechanisms concerning drug resistance in NSCLC. We performed targeted sequencing of 40 medication-relevant genes on plasma samples from 98 NSCLC patients and analyzed impact of genetic alterations on clinical presentation as well as response to systemic treatments. Profiling of multi-omics data from 1024 NSCLC tissues in public datasets was carried out for comparison and validation of identified molecular events implicated in resistance. A genetic association of CYP2D6 deletion with drug resistance was identified through circulating tumor DNA (ctDNA) profiling and response assessment. FCGR3A amplification was potentially involved in resistance to EGFR inhibitors. We further verified our findings in tissue samples and focused on potential resistance mechanisms, which uncovered that depleted CYP2D6 affected a set of genes involved in EMT, oncogenic signaling as well as inflammatory pathways. Tumor microenvironment analysis revealed that NSCLC with CYP2D6 loss manifested increased levels of immunomodulatory gene expressions, PD-L1 expression, relatively high mutational burden and lymphocyte infiltration. DNA methylation alterations were also found to be correlated with mRNA expressions and copy numbers of CYP2D6. Finally, MEK inhibitors were identified by CMap as the prospective therapeutic drugs for CYP2D6 deletion. These analyses identified novel resistance mechanisms to systemic NSCLC treatments and had significant implications for the development of new treatment strategies.
Keywords: NSCLC, systemic treatment, resistance, genomic alteration, drug target
INTRODUCTION
Lung cancer, a serious global health issue, has an estimated 40.9 million disability-adjusted life-years in 2017, imposing a prominent social and economic burden on human beings [1]. As the most prevalent form of lung cancer (accounting for approximately 87%) [2], non-small cell lung carcinoma (NSCLC) is frequently diagnosed with advanced stage and predisposed to distant metastasis. While systemic NSCLC treatments including standard platinum-based chemotherapy, tyrosine kinase inhibitor (TKI) therapies targeting oncogenic alterations of EGFR, ALK, RET or ROS1 and combinatorial therapies have improved the management of NSCLC patients, most inevitably experienced drug resistance and poor overall survival (OS) after an initial response. It is thus an urgent need to investigate potential biological mechanisms responsible for resistance to further determine novel therapeutic targets, which is beneficial for overcoming drug resistance and making optimal treatment decision.
The development and progression of NSCLC are complex processes affected by a number of genomic elements, such as genetic variance, transcriptional dysregulation [3] and immunosuppression [4], as revealed in multiple studies. Several recurrent somatic mutations, gene fusions as well as important amplified genes, such as EGFR, MET, TERT and deleted genes including CDKN2A have recently been nominated to be driver events and therapeutically tractable targets for NSCLC in comprehensive targeted sequencing and whole-exome sequencing (WES) studies [5–8]. In spite of many identified driver variations for targeted therapies, a large proportion of NSCLC patients do not carry such frequent genetic aberrations. In addition, resistance almost invariably occurred in a majority of patients after receiving existing treatments [9]. KRAS and NRAS mutations were mentioned as resistance mechanisms to BRAF/MEK inhibitors in patients with BRAF-mutant NSCLC [10, 11]. EGFR C797S was identified in NSCLC patients with acquired resistance after 3rd-generation EGFR TKIs [12–14], and the drug-resistant patterns also appear to differ in them [15, 16]. These studies indicated a critical need to detect additional drivers for developing novel therapeutics or modifying response to existing drugs.
Thompson et al. demonstrated that circulating tumor DNA (ctDNA) next-generation sequencing (NGS) can be utilized for detecting therapeutically targetable drive and resistance mutations in NSCLC patients by comparing ctDNA with tissue sequencing [17]. Particularly for patients with unusable or insufficient tumor tissue, ctDNA detection may be the first choice to detect driving and resistant mutations, especially for EGFR variants [15, 17]. More recently, a variety of studies have used panels of commonly mutated driver genes to detect multiple genomic alterations that might be linked with activity of certain approved treatments or clinical trial [18–20]. And genetic test on genes that harbor drug sensitivity implications could also be helpful for optimizing drug therapy. In this current study, utilizing a noninvasive approach of liquid biopsies, a panel that comprised of 40 clinical medication–relevant genes was sequenced on plasma samples, and we further evaluated ctDNA profile of 98 NSCLC patients with comprehensive data on clinical features, treatments and follow-ups collected. Additionally, 1024 NSCLC tissues from The Cancer Genome Atlas (TCGA) were included, and we performed integrative analyses of WES, RNA-sequencing (RNA-seq), proteomic and epigenetic profile as well as clinicopathological characteristics. We focused on describing and analyzing clinical and genetic characterization of NSCLC, and assessing drug responsiveness or adaptive resistance on systemic treatments. This study aimed at determining prospective biological mechanisms entailed in drug resistance and identifying novel molecular signatures for NSCLC patients. The results uncovered a deleted gene, CYP2D6, as well as its impact on clinical outcome. Further, the potentially activated downstream signaling pathways and immune tumor microenvironment (TME) were revealed for NSCLC patients with CYP2D6 copy number loss. Eventually, therapeutic compounds were selected to identify potential drug targets and molecular mechanisms that might contribute to developing new and more effective therapeutic approaches.
MATERIALS AND METHODS
NSCLC patient samples
As for training cohort, a total of 98 histologically proven NSCLC patients who underwent systemic treatment between 17 November 2014 and 30 June 2019 were recruited from the Chongqing University Cancer Hospital (Chongqing, China), and from whom blood draws were collected and assayed. Liquid biopsies were obtained at diagnosis and/or at radiological progression, whose ctDNA analyses were conducted blind to clinicopathological information. Staging and restaging were evaluated according to tumor imaging review (including CT and MRI scans in all patients) and following 8th edition of lung cancer staging defined by AJCC staging system. Treatment outcome followed Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST V1.1). Details on baseline characteristics and treatment modalities were retrieved through patients’ medical records at the end of follow-up. This study was approved by the Medical Ethics Committee of the Hospital. Written informed consent from all patients was obtained for plasma specimen collection and data analysis. The cutoff date for patient follow-up was 12 September 2020. In validation cohort, tumor tissue samples of 1024 NSCLC patients from public TCGA database were included. The clinical characteristics and survival data of TCGA cohort were downloaded from https://gdc.xenahubs.net. Besides, the detailed drug information for 323 TCGA NSCLC patients was available at https://portal.gdc.cancer.gov/.
DNA extraction and target deep sequencing
BD Vacutainer R K2 EDTA tubes were used to collect peripheral blood, and the supernatant was immediately centrifuged at 2000 × g for 10 min. Then, the supernatant plasma was collected after centrifugation at 8000 × g for 10 min, and it would be stored at −80°C if the plasma couldn’t be used immediately. Whole blood DNA was extracted from plasma with TIANamp Genomic DNA Kit (TIANGEN, Beijing, China) according to manufacturer’s protocols. Purified DNA was qualified by Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) and quantified using Qubit 2.0 Flurometer following the manufacturer’s recommendations. Samples that passed DNA quality check (ctDNA ≥ 50 ng) were further processed for DNA sequencing. Sequencing libraries were prepared using the NEBNext DNA Library Prep Reagent Set (New England BioLabs, Ipswich, MA, USA). All exons of the 40 genes relating to clinical medication and gene fusion-involved introns of ALK, RET and ROS1 were captured using Agilent SureSelect XT (Agilent, Santa Clara, CA, USA). The libraries were sequenced using paired-end 150-bp reads on the Illumina NovaSeq 6000 and Hiseq X Ten platform (Illumina, San Diego, California, USA). The targeted average sequencing depth for blood control and cell-free DNA (cfDNA) samples was ~100× and 3000×, respectively. The 40-gene panel included genes related to the efficacy of targeted therapy (EGFR, KRAS, NRAS, HRAS, PIK3CA, ALK, ROS1, BRAF, ERBB2, RET, MET, FGFR1, FGFR2, AKT1, PTEN, SMO, KIT, PDGFRA, DDR2, GNA11, TSC1, MAP2K1, GNAQ) and chemotherapy (C8orf34, CBR3, CYP2D6, DPYD, ERCC1, FCGR3A, GSTP1, MTHFR, MTRR, SOD2, TP53, TPMT, UGT1A1, UGT1A9, UMPS, XPC, XRCC1). All types of genomic variations including SNVs, indels and SCNA could be detected for these 40 genes.
Sequence data processing, somatic mutation calling and annotation
We firstly assessed quality control for FASTQ files using FastQC (version 0.11.9) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and Samtools (version 1.9) [21]. Low-quality samples (total read counts <0.5 million, percentage of properly paired reads <95% and percentage of on-target reads <99.5%) were removed. FASTQ files were mapped to the human reference genome (version 38, hg38) using BWA-men (version 0.7.15) [22] with parameters -t 8 -T 0. Local realignment around indels and base quality recalibration of BAM files were performed with Genome Analysis Toolkit (GATK, version 4.0.4.0) [23]. We applied MuTect2 (version 4.1.0.0) [24] with default settings to detect somatic single nucleotide variants (SNVs) and small indels in a tumor-only mode (by generating a panel of normals [PoN] with eight unmatched normal samples). We applied FilterMutectCalls with default parameters on Mutect2 for selecting the most reliable variant calls. We annotated somatic variants using ANNOVAR [25], and functional impact prediction of mutations was carried out by the Variant Effect Predictor (VEP) from Ensembl. For both somatic SNVs and indels, the minimum variant allelic fraction was set to 0.4%. Also, indels that were annotated in genome repeat regions were removed. In addition, possible germline variants present in dbSNP database and common variants with AF over 5% reported by the 1000 Genomes Project (1000G) and the Exome Aggregation Consortium (ExAC) database [26, 27] were further filtered out. Besides, somatic mutation data (WES data) of TCGA that we employed for analysis consisted of somatic variant calls (using MuTect2 variant calling pipeline) in TCGA-LUAD (n = 514) and TCGA-LUSC (n = 491) cohorts, and was retrieved from the UCSC Xena database (https://gdc.xenahubs.net). Furthermore, tumor mutation burden was evaluated by computing nonsynonymous somatic alterations in coding region for each tumor sample.
Copy number alteration calling and gene fusion analysis
For cfDNA samples, CNVkit (version 0.9.7) was applied for identifying somatic copy number alterations (SCNAs) in the aligned sequence reads (sorted BAM files) [28]. Using the circular binary segmentation algorithm (CBS), we inferred the discrete copy number segments. Genomic Identification of Significant Targets in Cancer (GISTIC, version 2.0.23) [29] was then utilized for detection of genes targeted by SCNAs and producing gene-level estimates. Moreover, GeneFuse (version 0.6.1) [30] was employed for detection of gene fusions using the fusion file containing experimentally verified cancer-related fusion genes from COSMIC database. For another, the copy number profile of TCGA NSCLC tissue samples (n = 1017) we used in this study was gistic2 estimate combined from TCGA-LUAD and TCGA-LUSC datasets (Affymetrix SNP 6.0 array data), which was available at https://gdc.xenahubs.net.
GISTIC2 real-valued copy number aberrance described in a gene-level of table (‘all_data_by_gene.txt’) was used for implementing unsupervised hierarchical clustering analysis of SCNA at 40 genes, generating the heatmap of SCNAs and performing subsequent correlation analysis. Based on continuous GISTIC2 SCNA data, amplifications and deletions for genes were defined as log2[(table-value + 2)/2] > 0.1 and < −0.1, respectively.
RNA-seq data and reverse phase protein array profiling
RNA sequencing (RNA-seq) level 3 expression data (normalized read counts) of 1019 TCGA NSCLC tissues were obtained from https://tcga.xenahubs.net. The edgeR package was utilized for mRNA expression data analysis. Gene-level CPM values were calculated and log2 transformed by edgeR package, and then employed for subsequent analysis. Differentially expressed genes between two NSCLC subgroups divided according to genetic variation status were identified by the limma package using a threshold of adjusted P < 0.05 and |log2 fold change| > 1. Functional enrichment analysis was conducted by the clusterprofiler package [31] for identifying Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were enriched for differentially expressed genes (DEGs). The significantly enriched pathways with P value < 0.05 were filtered out and then visualized by GOplot package. To further assess protein levels of NSCLC samples, we downloaded protein expression profile (reverse phase protein array [RPPA] data) of TCGA cohort (including 362 LUAD and 325 LUSC samples) from MD Anderson (http://app1.bioinformatics.mdanderson. org/tcpa/_design/basic/index.html).
Methylation data analysis and cell-type fraction estimation
DNA methylation level 3 data of TCGA lung cohort (Illumina Infinium HM450K Array) were retrieved from https://tcga.xenahubs.net. DNA methylation levels were measured by beta values for each CpG probe, which ranged from 0 (completely unmethylated) to 1 (completely methylated). TCGA DNA methylation profile was parsed by limma function from the limma package to identify significant differentially methylated positions (DMPs) between 830 TCGA NSCLC tumors and 75 normal lungs. We defined DMPs as those containing CpGs that yielded a Benjamini–Hochberg (BH) adjusted P < 0.05 and without ‘NA’ for the average beta in each group. The 450 k data were annotated by R package ‘IlluminaHumanMethylation450kanno.ilmn12.hg19’. On the basis of TCGA DNA methylation data, a primary reference containing fibroblasts and epithelials, and a secondary reference that includes seven immune cell types (B-cells, NK cells, CD4+ and CD8+ T-cells, monocytes, neutrophils and eosinophils), the HEpiDISH method from the R package ‘EpiDISH’ was applied to infer individual cell type proportions for NSCLC patients. In addition, relative abundance of lymphocytes were also estimated based on gene expression profiles of TCGA NSCLC tissues using xCell package [32].
Gene set enrichment analysis
Based on mRNA expression data of TCGA cohort, using R package ‘clusterprofiler’ [31], we associated a set of 50 hallmark signatures with the identified genetic variation by performing Gene Set Enrichment Analysis (GSEA). Spearman’s correlation coefficients were calculated between the target gene and other genes, which were then used for GSEA to identify biological pathways most related to the gene alteration. Gene signatures with adjusted P < 0.05 were considered significantly enriched. The reference gene sets were downloaded from the Molecular Signature Database (MSigDB): http://software. broadinstitute.org/gsea/msigdb/index.jsp.
Potential drug targets prediction
The Connectivity Map (CMap) online tool (https://clue.io/) [33] of Broad Institute (https://portals.broadinstitute.org/cmap/) was employed for predicting candidate compounds that might be activate or inhibit pathways associated with a given biological state according to the similarity with gene expression changes from 9 cancer cell lines induced by 2429 compounds. The DEGs between two genetic variation subgroups of NSCLC patients were split into up-regulated and down-regulated genes and then uploaded separately to interrogate the CMap touchstone database. Connectivity scores (ranging from −100 to 100) that assigned on those compounds described the similarity of molecular aberrations with gene expression signatures of this database. And more positive and negative scores corresponded to the compounds that gave rise to more similar and opposing expression changes, respectively, with a connectivity score smaller than −95 suggesting inhibition of potential drugs for genetic alterations.
Statistical analysis
All statistical analyses were performed with R software (version 3.6.2). Unsupervised hierarchical clustering and heatmaps of SCNA were conducted and visualized by R package ‘ComplexHeatmap’. Visualization and summarization for MAF files were implemented by package ‘maftools’. The detailed translational effect of genetic mutations was visualized by lollipop-diagrams using R package ‘g3viz’. Wilcoxon rank sum test, Chi-squared (χ2) test or Fisher exact test and Spearman’s correlation analysis were used for assessing associations of clinical characteristics, DNA methylation, mRNA expression and protein abundance with gene variations, which were implemented and visualized by ggplot2, ggstatsplot and ggpubr packages. For evaluating implications of genetic aberrations on systemic treatment (including chemotherapy and/or targeted molecular therapy) and drug resistance, we conducted survival analysis on both NSCLC cohorts. Overall survival (OS) was defined as date of treatment initiation to date of death or last follow-up. Time to disease progression (TTP) was defined as time from treatment initiation to the disease progression or end of current follow-up. Kaplan–Meier curve analyses and log-rank tests were performed by package ‘survminer’. Clinicopathological parameters and identified gene variations were combined in a multivariable Cox proportional hazards regression model for survival analysis by the survival package. For all statistical tests, two-tailed P < 0.05 denoted statistical significance, which is indicated by *, P < 0.05, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001.
RESULT
Patients and clinical characteristics
Blood samples from 98 NSCLC patients included in CQ cohort were evaluated for ctDNA profiling. The corresponding clinicopathological characteristics were described in Table 1. Of 98 NSCLC patients, 19 without detailed therapeutic information were excluded for response assessment. All remaining patients but six who received radiation therapy alone underwent systemic NSCLC treatment, such as platinum-based chemotherapy and/or TKI therapies, bevacizumab combined with chemotherapy and so on. According to tumor response evaluation by clinical imaging, among these patients, 55 developed progressive disease (PD) after systemic therapy initiation, while 2 patients had a partial response (PR) and 22 exhibited stable disease (SD).
Table 1.
Clinicopathological features and treatment modalities of 98 patients in CQ cohort
| Characteristics | Number (%) |
|---|---|
| Sex | |
| Female | 37 (38%) |
| Male | 61 (62%) |
| Age | |
| <65 | 41 (42%) |
| ≥65 | 57 (58%) |
| Pack-years smoked | |
| <30 | 67 (68%) |
| ≥30 | 31 (32%) |
| Histology | |
| Adenocarcinoma | 88 (90%) |
| Large cell neuroendocrine carcinoma | 1 (1%) |
| Squamous cell carcinoma | 9 (9%) |
| Stage | |
| II | 1 (1%) |
| III | 11 (11%) |
| IV | 86 (88%) |
| T stage | |
| T1/T2 | 40 (41%) |
| T3/T4 | 51 (52%) |
| Tx | 7 (7%) |
| N stage | |
| N0 | 15 (15%) |
| N1 ~ N3 | 80 (82%) |
| Nx | 3 (3%) |
| M stage | |
| M0 | 11 (11%) |
| M1 | 86 (88%) |
| Mx | 1 (1%) |
| Therapy types | |
| Chemotherapy | 18 (18%) |
| Radiation | 6 (6%) |
| Targeted molecular therapy | 34 (35%) |
| Targeted molecular + chemotherapy | 21 (21%) |
| Unknown | 19 (19%) |
| Targeted molecular therapy | |
| A, EGFR-TKI | 1 (1%) |
| EGFR-TKI | 27 (28%) |
| Cri | 1 (1%) |
| A | 5 (5%) |
| Targeted molecular + chemotherapy | |
| Pem, C, EGFR-TKI | 8 (8%) |
| Tax, C, EGFR-TKI | 3 (3%) |
| Tax, C, Pem, EGFR-TKI | 3 (3%) |
| Tax, C, Pem, A | 1 (1%) |
| Pem, C, A | 1 (1%) |
| Tax, C, Cri | 1 (1%) |
| Pem, C, B | 3 (3%) |
| Tax, C, B | 1 (1%) |
| Chemotherapy | |
| C | 1 (1%) |
| Pem | 1 (1%) |
| Pem, C | 5 (5%) |
| Tax, C | 8 (8%) |
| Tax, C, Pem | 3 (4%) |
| Others | |
| Unknown | 25 (26%) |
| TTP status | |
| PD | 55 (56%) |
| PR | 2 (2%) |
| SD | 22 (22%) |
| Unknown | 19 (19%) |
| OS status | |
| Alive | 44 (45%) |
| Dead | 52 (53%) |
| Unknown | 2 (2%) |
Note: Therapeutic drugs used here were anlotinib (A), crizotinib (Cri), gefitinib/erlotinib/icotinib/osimertinib (EGFR-TKI), pemetrexed (Pem), nedaplatin/cisplatin/carboplatin (C), paclitaxel/docetaxel (Tax) and bevacizumab/endostar (B)
For TCGA cohort, a total of 1024 NSCLC tissue samples were involved in the analysis, with detailed clinical features summarized in Table S1, available online at https://academic.oup.com/bib. With respect to treatment modalities received for these cases, first-line medication data of 323 NSCLC patients were extracted and sorted for this study. All except three patients that were recruited in clinical trial of antigen-specific cancer immunotherapy (ASCI) received systemic first-line therapy. Among these cases, 298 underwent chemotherapy (including platinum-based doublets, taxol analogues and so on), 12 received EGFR-TKI therapy and 10 patients were treated with the combination of targeted agents (bevacizumab or erlotinib) and chemotherapeutic drugs. Of 320 patients who underwent systemic treatment, 136 cases developed PD, 36 and 6 showed SD and PR, respectively, and 142 achieved complete response (CR) as the best outcome.
Genomic alterations in ctDNA and tumor tissue specimens from NSCLC patients
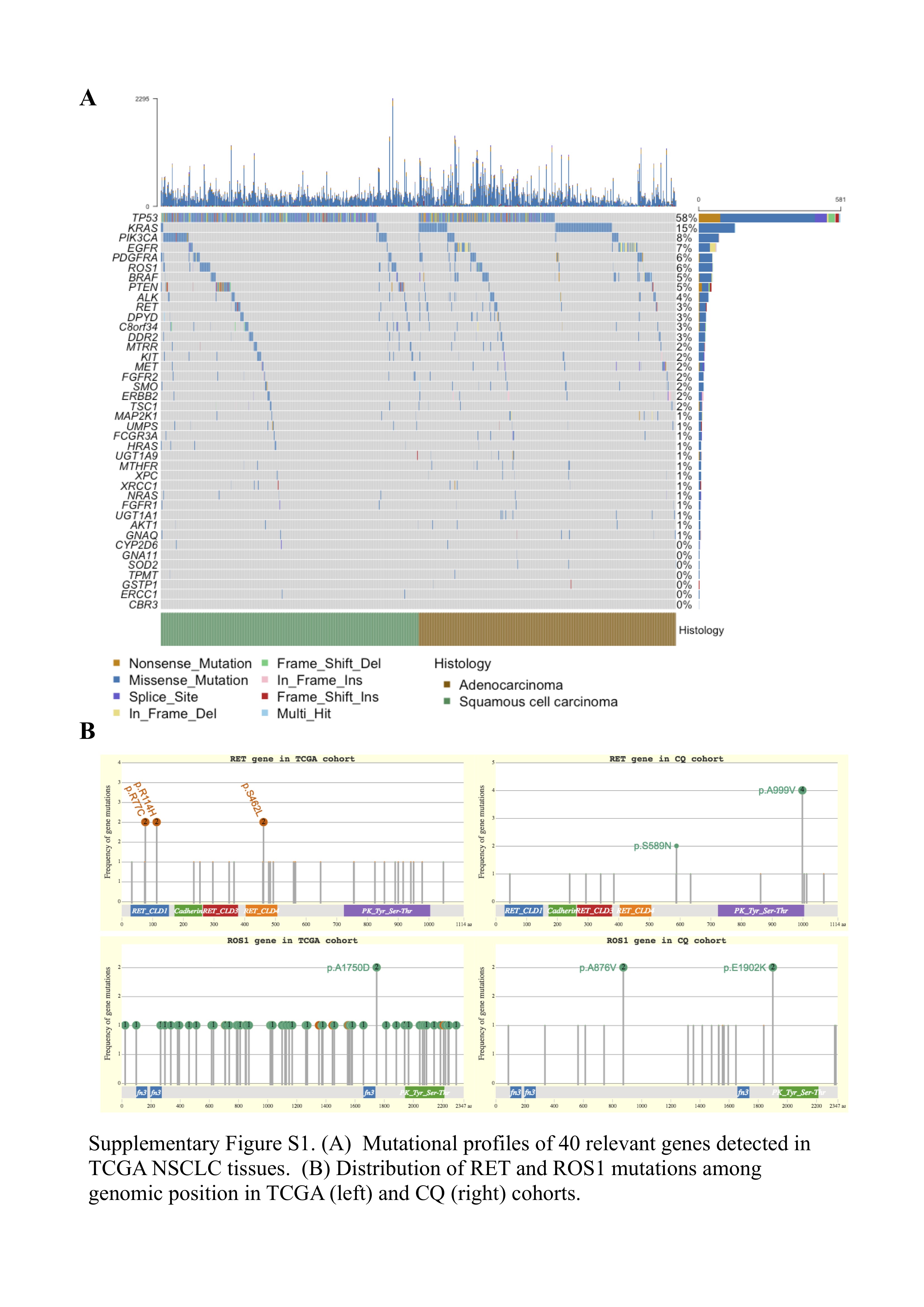
We sequenced 40 genes of interest from liquid biopsies of CQ cohort to assess molecular aberrations of NSCLC patients in targeted NGS. Meanwhile, genomic variations of TCGA cohort were also evaluated using somatic variants and SCNAs identified by the GDC variant calling and copy number variation analysis pipelines. The genomic landscapes of detected somatic mutations in two cohorts were presented in Figure 1A and Figure S1A, available online at https://academic.oup.com/bib, which showed that TP53, EGFR, ALK, RET and ROS1 were the most frequently altered genes (top 10) in both cohorts. Interestingly, we found novel mutation subtypes of RET and ROS1 in CQ cohort that differed from those in TCGA cohort. Among them, the most common SNV was RET A999V mutation (n = 4), and it had not been previously reported in studies of lung cancer Figure S1B available online at https://academic.oup.com/bib). While in TCGA NSCLC tissues, multiple different variants were spread over the whole RET and ROS1 length, and no hotspot mutants could be detected (Figure S1B available online at https://academic.oup.com/bib). Unsupervised clustering was performed on 98 cfDNA samples using SCNAs that were estimated through GISTIC2, which demonstrated frequent chromosome aberrations, including losses in 1p, 17p and 19p and gains in 1q, 5p and 7p (Figure 1B). The detected fusion events (Table S2 available online at https://academic.oup.com/bib) were also indicated in Figure 1B. The copy number analysis on TCGA tissue samples replicated aforementioned aneuploid events, including recurrent copy number gains at MTRR (5p15.31), FCGR3A (1q23.3) and EGFR (7p11.2) and losses at TP53 (17p13.1) and GNA11 (19p13.3) (Figure S2 available online at https://academic.oup.com/bib).
Figure 1 .
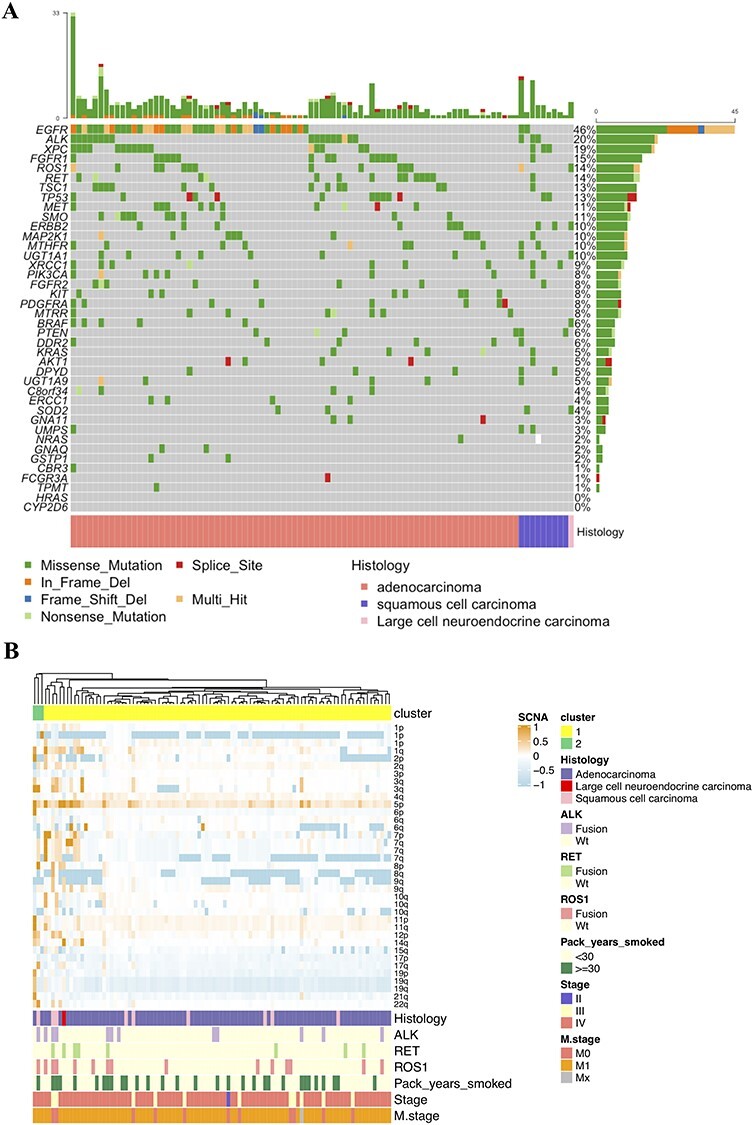
(A) The landscape of somatic mutations detected in 98 plasma samples. (B) Unsupervised clustering of SCNAs as determined by GISTIC2 analysis on 98 cfDNA samples. Clinical and molecular features include pack-years smoked, histology, stage, distant metastasis and gene fusions. Copy number gains are colored in orange and losses in light blue.
Associations of molecular aberrations with clinical features and their impact on outcome of NSCLC patients receiving systemic therapy
We then assessed the relationship between the identified genetic variations and clinical characteristics of NSCLC patients. In CQ cohort, amplifications at MTRR and FCGR3A were observed for NSCLC cases with lymphatic metastasis, and the DPYD copy number and ROS1 mutation were prominently correlated with tumor size (Figure 2A). For TCGA patients, significant FCGR3A and MET amplifications, GNA11 and CYP2D6 deletions were detected in NSCLC samples at advanced stage (Figure 2B). The copy number of FCGR3A, CYP2D6 and GNA11 was also found to be higher or lower in NSCLC cases with distant metastasis than those without (Figure 2B). Additionally, the amplification of EGFR and MET was significantly related to lymphatic metastasis (Figure 2B). We then linked identified molecular alterations to treatment response of patients who underwent between first and third lines of chemotherapies and/or TKIs. And notably, SCNAs of DPYD, CYP2D6 and GNA11 were observed to associate with response to systemic therapies in TCGA cohort (Figure 2C).
Figure 2 .
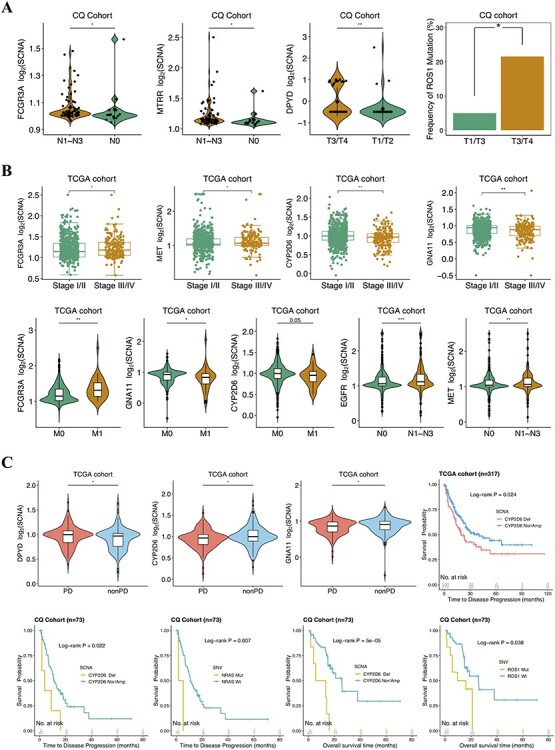
Selected clinical features associated with the implicated molecular alterations in CQ cohort (A) and TCGA cohort (B). (C) The detected copy numbers of DPYD, CYP2D6 and GNA11 at progression disease (PD) and nonprogression disease (non-PD) for TCGA (top left). Kaplan–Meier survival curves showing TTP analysis for CYP2D6 deletion in TCGA (top right) and CQ cohort (bottom left) as well as NRAS mutation in CQ cohort (bottom left); OS analysis for CYP2D6 deletion and ROS1 mutation (bottom right) in CQ cohort. Del: deletion; Nor/Amp: normal/amplification; Mut: mutant; Wt: wild-type.
Next, we sought to evaluate the impact of genetic aberrations on TTP and OS of NSCLC. Intriguingly, the depleted SCNA at CYP2D6 in CQ cohort (6/73, 8.22%) and TCGA cohort (100/319, 31.35%) was found to significantly correlate with tumor progression (Figure 2C), suggesting that deleted CYP2D6 might contribute to drug resistance in NSCLC. In addition, the NRAS mutation (n = 2) detected in CQ NSCLC patients also suggested rapid progression under systemic therapies (Figure 2C). What is more, CYP2D6 deletion and ROS1 mutation (n = 14) identified in CQ cohort showed noticeable correlation to poor OS (Figure 2C). Further, a multivariable Cox regression was then performed for adjusting potential compounding factors. As a result, CYP2D6 deletion harbored remarkable effect on inferior outcomes for NSCLC patients and independent of other clinicopathological covariates. Particularly, relative to patients in CYP2D6 normal/amplification group, hazard ratios for tumors with CYP2D6 deletion were 4.24 (P = 0.033, CI95% 1.12–15.98, TTP) and 3.76 (P = 0.048, CI95% 1.01–13.98, OS) (Figure 3). Of note, a subset of patients receiving EGFR inhibitors, even third-generation of TKI, continued to experience short survivals despite the demonstration of initial promising response rate. We thus concentrated more on molecular events underlying EGFR-TKIs’ resistance in CQ cohort. Among these patients analyzed, 30 EGFR-mutant cases (with classic and/or rare EGFR mutations) were treated with first-/second-/third-generation EGFR-TKIs (Table S3 available online at https://academic.oup.com/bib). And FCGR3A amplification (4/30, 13.33%) was observed to be implicated in EGFR-TKI resistance (Figure S3 available online at https://academic.oup.com/bib).
Figure 3 .
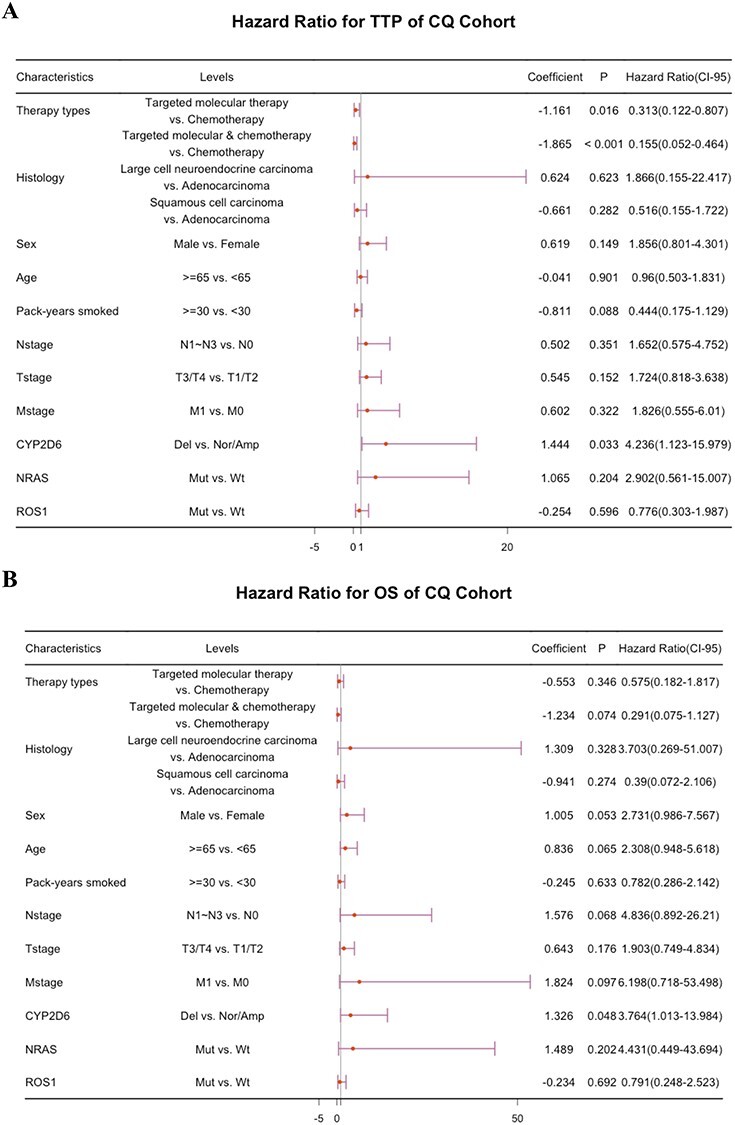
Multivariate Cox regression analyses of TTP (A) and OS (B) for CQ cohort with combinations of detected genetic variations and clinicopathological factors. TTP: time to disease progression; OS: overall survival; Nor/Amp: normal/amplification; Del: deletion.
Molecular mechanism of CYP2D6 deletion in NSCLC
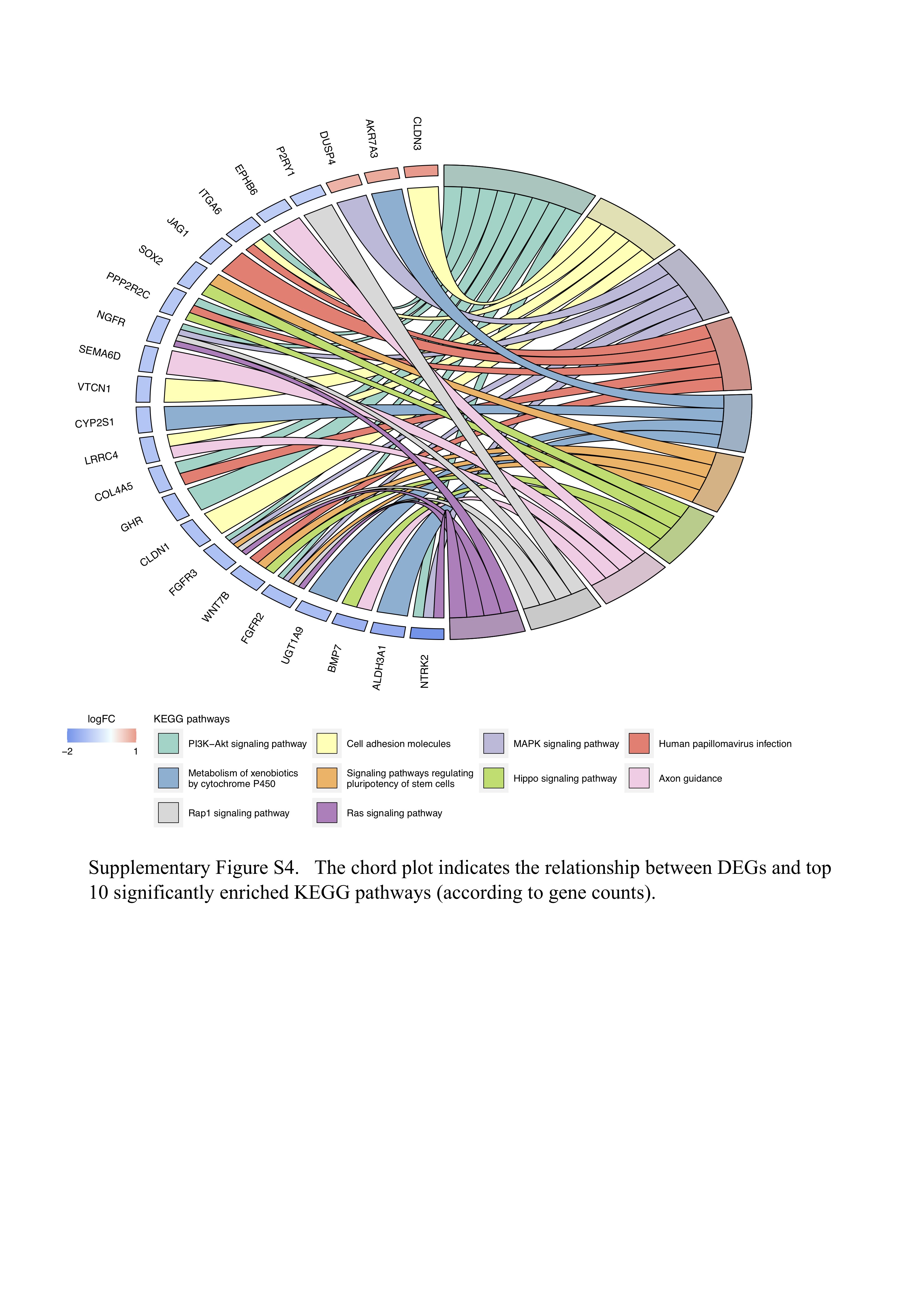
Noting that a strong correlation between CYP2D6 deletion and drug resistance was observed in both cohorts as mentioned above, we performed further molecular profiling to explore the underlying mechanism hidden behind current associations. By integrating transcriptomic data of TCGA NSCLC, we explored the effects of CYP2D6 deletion on gene expression. KEGG enrichment analysis demonstrated several signaling pathways that had relatively strong relation with DEGs between CYP2D6-depleted and CYP2D6-wild/CYP2D6-amplified samples, including ‘PI3K-Akt signaling pathway’, ‘MAPK signaling pathway’, ‘metabolism of xenobiotics by cytochrome P450’, ‘Hippo signaling pathway’ and ‘Ras signaling pathway’ (Figure S4 available online at https://academic.oup.com/bib). Additionally, we found that CYP2D6 expression level in SCNA deletion group was markedly lower than that in normal/amplification group (Figure 4A). GSEA revealed significant enrichment of epithelial–mesenchymal transition (EMT) markers and gene signatures relating to cell proliferation, such as MYC, G2M checkpoint, tumor necrosis factor alpha (TNF-α), transforming factor-β (TGF-β), KRAS signaling up as well as PI3K/AKT/MTOR signaling in depleted CYP2D6 (Figure 4B and C). We then examined relevant protein expression levels in key pathways based on TCGA RPPA data. The deleted CYP2D6 displayed notable mTOR pathway activation given significant correlations between SCNA loss and STK11-inactivating mutation, low LKB1 abundance as well as inactivation of AMPK (Figure 4C). Moreover, high levels of P38MAPK, p-P90RSK, p-MEK1 and p-JNK were detected in decreased copy number of CYP2D6 (Figure 4C), indicating its higher MAPK pathway activity [6].
Figure 4 .
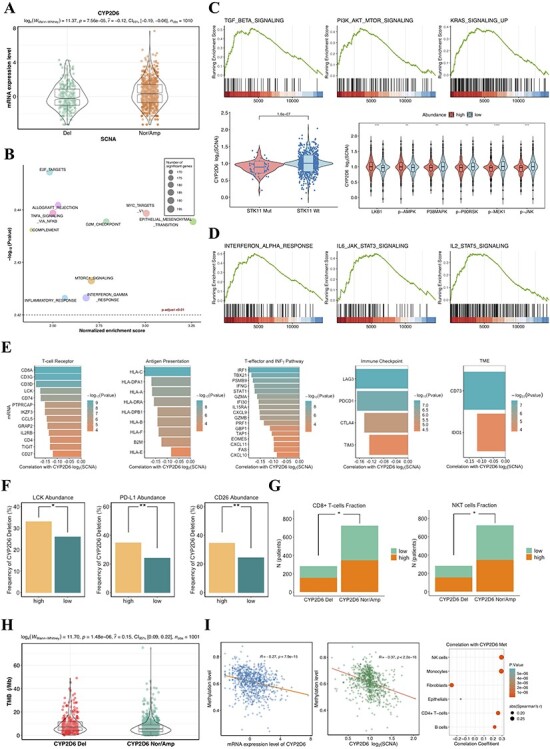
(A) The SCNA of CYP2D6 significantly associated with mRNA expression. (B) The top 10 of prominently enriched pathways for CYP2D6 loss (sorted by normalized enrichment score). (C) Top, three representative GSEA plots for carcinogenesis pathways that were significantly enriched (adjusted P < 0.01). Bottom left, copy number of CYP2D6 related to mutational status of STK11; Bottom right, proteomic analysis showing correlation between CYP2D6 SCNA and the abundance of relevant proteins involving MTOR and MAPK pathways. (D) Three representative GSEA plots for immunomodulatory pathways that correlated with CYP2D6 loss (adjusted P < 0.01). (E) Associations of CYP2D6 SCNA with mRNA expression of 48 immune-related genes. CYP2D6 deletion correlated to PD-L1, LCK, CD26 protein expressions (F), lymphocyte abundance (G) and TMB (H). (I) Relationships between methylation levels and mRNA expressions (left) as well as copy number of CYP2D6 (middle). Tumors with hypermethylated CYP2D6 presenting higher fractions of immune cells (right). Del: deletion; Nor/Amp: normal/amplification; SCNA: somatic copy number alteration; Met: methylation.
In addition to oncogenic signaling pathways mentioned above, immune-regulatory gene markers were also prominently enriched in CYP2D6 deletion subgroup (Figure 4B and D). We thus evaluated CYP2D6 SCNA loss for NSCLC in the context of immune tumor microenvironment (TME). To investigate its effect on TME, we analyzed the correlations between SCNA of CYP2D6 and mRNA expression of 58 immunomodulatory genes (Table S4 available online at https://academic.oup.com/bib) based on TCGA NSCLC database. And we found that, the increased expression levels of 48 genes were significantly correlated to CYP2D6 copy number loss (Figure 4E), implicating activation of immunity within samples that harbored deleted CYP2D6. Besides, NSCLC patients with higher frequency of CYP2D6 deletion manifested higher levels of LCK, PD-L1 and CD26 protein expression (Figure 4F). Since the lymphocyte infiltration was reported as a crucial TME variable in tumor immune response [28], we linked the SCNA of CYP2D6 to estimated immune cell fraction. Our results showed that CYP2D6 deletion significantly associated with high proportions of CD8+ T cells and natural killer T cells (Figure 4G). Furthermore, dramatically higher TMB was also detected in CYP2D6 deletion group versus normal/amplification group (Figure 4H).
Given that DNA methylation loss had been proposed to associate with chromosome instability and predict poor responses to immunotherapy [34], we next inspected the methylation level of CYP2D6 in TCGA NSCLC samples. Four significant differentially methylated CpG probes nearby CYP2D6 (cg04692870, cg07016288, cg20046859, cg20195005) were identified in tumors when compared with normal samples. Interestingly, the mean methylation level for four DMPs was found to correlate highly with mRNA expression as well as the copy number of CYP2D6 (Figure 4F). Positive relationships between CYP2D6 methylation and abundance of B cells, CD4+ T cells, NK cells and monocytes were also observed in TCGA NSCLC samples (Figure 4F), demonstrating augmented lymphocytes infiltration and improved immunogenicity in tumors with high CYP2D6 methylation levels.
Identification of potential inhibitors targeting CYP2D6 deletion
By querying CMap touchstone database, we sought to search for candidate therapeutic compounds that might be involved in pathways related to SCNA loss of CYP2D6. The identified compounds with negative connectivity scores below −95 were shown in Table 2. Inhibitors of MEK, IKK, PDGFR receptor and JNK were the most representative pharmaceutical perturbagens among predicted drugs. And five perturbagens (U-0126, selumetinib, PD-0325901, PD-184352, AS-703026) shared the mode-of-action as MEK inhibition, suggesting prospective compounds inducing antagonistic effect on CYP2D6 deletion.
Table 2.
Significant therapeutic drugs predicted by CMap
| Drug name | Description | Target | CMap Score |
|---|---|---|---|
| Nafadotride | Dopamine receptor antagonist | DRD3, DRD2, HTR1A | −99.22 |
| U-0126 | MEK inhibitor | AKT1, CHEK1, GSK3B, JAK2, LCK, MAP2K1, MAP2K2, MAP2K7, MAPK1, MAPK11, MAPK12, MAPK14, MAPK8, PRKCA, RAF1, ROCK1, RPS6KB1, SGK1 | −98.63 |
| Selumetinib | MEK inhibitor | MAP2K1, MAP2K2 | −98.22 |
| BMS-345541 | IKK inhibitor | IKBKB, CHUK | −98.07 |
| Linifanib | PDGFR receptor inhibitor | CSF1R, KDR, PDGFRB, FLT1, FLT3, FLT4, CSF1, KIT, PDGFRA, RET, TEK | −97.67 |
| PD-0325901 | MEK inhibitor | MAP2K1, MAP2K2 | −97.36 |
| PD-184352 | MEK inhibitor | MAP2K1, MAP2K2, MAP3K1, MAP3K2 | −97.22 |
| ZG-10 | JNK inhibitor | MAPK8 | −96.85 |
| AS-703026 | MEK inhibitor | MAP2K1, MAP2K2 | −96.11 |
DISCUSSION
Systemic NSCLC treatments targeting oncogenic variations achieved enormous success due to their high sensitivity and superior efficacy, especially various TKI inhibitors, whereas for the vast majority of patients, sustained clinical response developed inevitably into disease progression. Although the introduction of immunotherapy had shifted the therapeutic paradigms, durable benefit was limited to a small number of NSCLC patients. On account of substantial heterogeneity in genomic alteration of NSCLC, investigation into additional driver events and emerging mechanisms of therapeutic resistance is warranted for development of more effective treatments.
In an era of growing importance in liquid biopsy, clinically relevant insights into genomic variations were obtained through targeted NGS on ctDNA. Recent studies have proposed that plasma ctDNA was quite useful for evaluating biological complexity of cancer formation and maintenance as well as monitoring response to targeted therapy in NSCLC [35, 36]. In this study, we exploited the liquid biopsy approach to sequence 98 NSCLC samples from CQ cohort and assessed tumor responses or progression on systemic treatment after drug exposure. In addition, utilizing 1024 tissue samples from TCGA database, we characterized the genomic landscape of NSCLC and identified genetic abnormalities that were indicative of poor response to systemic NSCLC therapy. CYP2D6, the key molecule identified in this study, is one of the cytochrome P450 superfamily of genes that play critical role in drug metabolism and has been regarded valuable for guiding clinical drug selections. The genetic polymorphism in CYP2D6 was revealed to be associated with the increased risk of recurrence or death for breast cancer patients receiving tamoxifen treatment [37, 38]. The structural variation of CYP2D6 has also been reported in previous studies, with CYP2D6 deletion being referred to as CYP2D6*5 that led to a poor metabolizer [39, 40], whereas its influence on drug resistance of NSCLC is not yet described. Here, we found that both blood and tissue samples with SCNA loss of CYP2D6 exhibited shorter TTP when received systemic treatments. By integrating multi-omics data of TCGA tumors, potential molecular mechanisms for CYP2D6 deletion was further investigated. Functional enrichment analysis showed DEGs associated with CYP2D6 depletion in NSCLC were enriched in oncogenic and drug-metabolizing pathways, implying that CYP2D6 played a pivotal role in the development of cancer and drug resistance [41]. Interestingly, copy number deletion strikingly correlated with decreased mRNA expression of CYP2D6, on which our analysis revealed EMT and oncogenicity-associated signaling (exemplarily, TNF-α, TGF-β, KRAS and PI3K/AKT/MTOR) as well as inflammatory pathways were involved in and likely to result in resistance clones and poor prognosis. The proteomes data also demonstrated activation of related pathways in tumors with deleted CYP2D6. Further, we observed CYP2D6 loss was significantly associated with high expressions of immunomodulatory gene signatures and enriched compositions of immune cells in TME. More intriguingly, CYP2D6 deletion was also responsible for increased TMB as well as PD-L1 expression levels. It’s well-established that elevated PD-L1 expression, tumor-infiltrating lymphocytes and relatively higher TMB were predictive factors to the identification of responders to PD-1 blockade [42, 43]. Our findings above suggested a possibility that CYP2D6 copy number loss underlay resistance mechanisms through affecting oncogenic signaling including PI3K and MAPK activation. Besides, CYP2D6 depletion was associated with antitumor immune activity and involved in regulating immune biomarkers, which induced a responsive TME with adaptive immune resistance and improved immunity, and such tumors were thus expected to be more susceptible to immunotherapy and render durable clinical benefit.
It’s noteworthy that DNA methylation status related remarkably to both mRNA expressions and SCNAs of CYP2D6. As was reported, in most tumor types, global methylation levels highly correlated with chromosomal SCNAs, and demethylation that was caused by mitotic cell division suppressed immune gene expressions, decreased tumor immunity and promoted immune evasion [34]. CYP2D6 methylation also displayed significant associations with higher immune cell fractions, indicating preexisting immunity within CYP2D6 hypermethylation NSCLC tissues. CMap analysis identified potential therapeutic compounds that were efficacious for drug resistance of patients with CYP2D6 deletion. Specific targeted agents were predicted to act on several signaling pathways, which include MAPK and PDGFR, with MEK inhibitors well-represented among these drugs. These results suggested by CMap database were in line with our observations for pathway enrichment of CYP2D6 loss. As was reported, the combination of MEK inhibitors with other drugs, such as EGFR-TKIs, BRAF inhibitors, immune checkpoint inhibitors or chemotherapy drugs, pronouncedly increases the therapeutic effects and causes delay on occurrence of resistance for patients with lung cancer [44]. Our study implied that systemic treatments plus MEK inhibitors were promising strategies that can improve the clinical efficacy for NSCLC patients harboring CYP2D6 deletion.
Of note, our analysis on CQ cohort also uncovered FCGR3A amplification was predicative of resistance to EGFR-TKI treatments. Copy number alterations in FCGR3A contributed to pathogenesis of a number of disorders [45], and amplified FCGR3A was reported to correlate with the risk of gout [46]. To the best of our knowledge, this is the first report of FCGR3A amplification entailed in resistance to EGFR-TKIs. Nevertheless, it still remains unclear how this genomic alteration interacts with EGFR inhibitors. Our study also had several limitations. Firstly, targeted NGS on ctDNA using limited panel (40-gene panel) could not detect additional pivotal coexisting alterations, and plasma NGS had less detection sensitivity compared with tissue biopsy. Moreover, sample size of CQ cohort analyzed here was small; besides, robust treatment information was retrospective and available for the relatively small number of patients, which conferred limited statistical power on associations of genomic events with clinical phenotypes as well as prognostic predictive effect. Lastly, multidimensional analysis was restricted to data from TCGA portal as multi-omics data analysis was highly costly and difficult to achieve in clinical practice. Also, no laboratory experimental validation of the identified biological mechanisms that engender drug resistance was performed. Overall, in spite of aforementioned drawbacks, our study revealed previously undiscovered molecular mechanisms involving resistance to systemic NSCLC treatments by utilizing the noninvasive approach of plasma genotyping, which was further supported in tissues from public datasets. These findings might act as a foundation and advocate for development of novel therapeutic strategies in NSCLC. It’s conceivable that promising combination therapies based on molecularly targeted therapy and even possibly coupled with immunotherapy will overcome resistance and generate remarkable clinical benefit.
Key Points
Identification of novel molecular events predicting NSCLC therapeutic response through liquid biopsy–based NGS and tissue-based WES.
Multidimensional analysis on CYP2D6 gene revealed the copy number loss potentially affected oncogenic pathways and immune response in NSCLC, which might be responsible for resistance to systemic treatment.
Discovery of the genomic feature (FCGR3A amplification) potentially involved in resistance to EGFR inhibitors by ctDNA profiling in NSCLC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are grateful to all patients and families who contributed to this study.
Ruihan Luo is an MSc student at Department of Bioinformatics, The Basic Medical School of Chongqing Medical University, China. Her main interests lie in bioinformatics, genomics and cancer informatics. She conceived the study, performed all data analyses and interpretation and wrote the manuscript.
Chuang Ge is a supervising technician in the Clinical Laboratory of Chongqing University Cancer Hospital, China. His research interests include genomics and translational medicine. He helped with clinical samples consent, collection and cohort data generation and interpretation.
Xiao Xiao is an M.Med. student at Department of Surgery, The First Affiliated Hospital of Chongqing Medical University, China. His current research interests include translational medicine and resistance to molecular targeted therapies. He assisted with samples response assessment, clinical information gathering and data interpretation.
Jing Song is a PhD student in Molecular and Tumor Research Center, Chongqing Medical University, China. His research interests include bioinformatics and systemic biology. He helped with data analysis and interpretation.
Shiqi Miao is an MSc student in Molecular and Tumor Research Center, Chongqing Medical University, China. Her main interests lie in genomics and bioinformatics. She reviewed the manuscript.
Yongyao Tang is an MSc student in Molecular and Tumor Research Center, Chongqing Medical University, China. His main interests lie in genomics and bioinformatics. He reviewed the manuscript.
Jiayi Lai is an MSc student at Department of Bioinformatics, The Basic Medical School of Chongqing Medical University, China. Her main interests include bioinformatics. She reviewed the manuscript.
Weiqi Nian is the chief physician and director of the Phase 1 Clinical Trial Center of Chongqing University Cancer Hospital, China. He provided the clinical specimens, useful suggestions and discussions.
Fangzhou Song is a professor in Molecular and Tumor Research Center, Chongqing Medical University, China. He works on applying experimental and computational methods that help decipher genomic complexity to untangle the causes of cancer. He reviewed the manuscript.
Longke Ran is a professor at Department of Bioinformatics, The Basic Medical School of Chongqing Medical University, China. His research interests include medical genomics, bioinformatics and cancer informatics. He conceived and supervised the study.
Contributor Information
Ruihan Luo, Department of Bioinformatics, The Basic Medical School of Chongqing Medical University, China.
Chuang Ge, Clinical Laboratory of Chongqing University Cancer Hospital, China.
Xiao Xiao, Department of Surgery, The First Affiliated Hospital of Chongqing Medical University, China.
Jing Song, Molecular and Tumor Research Center, Chongqing Medical University, China.
Shiqi Miao, Molecular and Tumor Research Center, Chongqing Medical University, China.
Yongyao Tang, Molecular and Tumor Research Center, Chongqing Medical University, China.
Jiayi Lai, Department of Bioinformatics, The Basic Medical School of Chongqing Medical University, China.
Weiqi Nian, Phase 1 Clinical Trial Center of Chongqing University Cancer Hospital, China.
Fangzhou Song, Molecular and Tumor Research Center, Chongqing Medical University, China.
Longke Ran, Department of Bioinformatics, The Basic Medical School of Chongqing Medical University, China.
Data Availability
| Data resource | Source | Identifier |
|---|---|---|
| TCGA clinical and survival information, somatic mutation and copy number data | UCSC Xena database | https://gdc.xenahubs. net |
| TCGA RNA-seq and DNA methylation data | UCSC Xena database | https://tcga.xenahubs. net |
| Drug information for TCGA NSCLC | Genomic Data Commons | https://portal.gdc. cancer.gov/ |
| RPPA data for TCGA NSCLC | RPPA Core Facility, MD Anderson Cancer Center | http://app1. bioinformatics.mdanderson.org/ tcpa/_design/basic/ index.html |
| Raw sequence data for CQ NSCLC | NCBI Sequence Read Archive database | https://trace.ncbi. nlm.nih.gov/Traces/ study/?acc=SRP310820 |
Funding
The Intelligence Medicine Project of Chongqing Medical University (No. ZHYX2019015 to Longke Ran and No. YJSZHYX202013 to Ruihan Luo) supported by Medical Data Science Academy; Pioneer Natural Science Foundation of Chongqing (cstc2019jcyj-xfkxX0003); Special Performance Incentive and Guidance for Research Institutes of Chongqing (cstc2018jxjl130057).
References
- 1. Fitzmaurice C, Abate D, Abbasi N, et al. . Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the global burden of disease study. JAMA Oncol 2019;5:1749–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morgensztern D, Ng SH, Gao F, et al. . Trends in stage distribution for patients with non-small cell lung cancer: a national cancer database survey. J Thorac Oncol 2010;5:29–33. [DOI] [PubMed] [Google Scholar]
- 3. Chen C-Y, Jan Y-H, Juan Y-H, et al. . Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc Natl Acad Sci USA 2013;110:630–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carbone DP, Gandara DR, Antonia SJ, et al. . Non-small-cell lung cancer: role of the immune system and potential for immunotherapy. J Thorac Oncol 2015;10:974–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weir BA, Woo MS, Getz G, et al. . Characterizing the cancer genome in lung adenocarcinoma. Nature 2007;450:893–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cancer Genome Atlas Research N . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu P, Morrison C, Wang L, et al. . Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 2012;33:1270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soda M, Choi YL, Enomoto M, et al. . Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6. [DOI] [PubMed] [Google Scholar]
- 9. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer 2017;17:637–58. [DOI] [PubMed] [Google Scholar]
- 10. Abravanel DL, Nishino M, Sholl LM, et al. . An acquired NRAS Q61K mutation in BRAF V600E-mutant lung adenocarcinoma resistant to Dabrafenib plus Trametinib. J Thorac Oncol 2018;13:e131–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Niemantsverdriet M, Schuuring E, Elst AT, et al. . KRAS mutation as a resistance mechanism to BRAF/MEK inhibition in NSCLC. J Thorac Oncol 2018;13:e249–51. [DOI] [PubMed] [Google Scholar]
- 12. Thress KS, Paweletz CP, Felip E, et al. . Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015;21:560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang S, Tsui ST, Liu C, et al. . EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol 2016;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Piotrowska Z, Niederst MJ, Karlovich CA, et al. . Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov 2015;5:713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chabon JJ, Simmons AD, Lovejoy AF, et al. . Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 2016;7:11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee J, Kim HS, Lee B, et al. . Genomic landscape of acquired resistance to third-generation EGFR tyrosine kinase inhibitors in EGFR T790M-mutant non-small cell lung cancer. Cancer 2020;126:2704–12. [DOI] [PubMed] [Google Scholar]
- 17. Thompson JC, Yee SS, Troxel AB, et al. . Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next-generation sequencing of cell-free circulating tumor DNA. Clin Cancer Res 2016;22:5772–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paweletz CP, Sacher AG, Raymond CK, et al. . Bias-corrected targeted next-generation sequencing for rapid, multiplexed detection of actionable alterations in cell-free DNA from advanced lung cancer patients. Clin Cancer Res 2016;22:915–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Facchinetti F, Lacroix L, Mezquita L, et al. . Molecular mechanisms of resistance to BRAF and MEK inhibitors in BRAFV600E non–small cell lung cancer. Eur J Cancer 2020;132:211–23. [DOI] [PubMed] [Google Scholar]
- 20. Abbosh C, Birkbak NJ, Wilson GA, et al. . Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H, Handsaker B, Wysoker A, et al. . The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van der Auwera GA, Carneiro MO, Hartl C, et al. . From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cibulskis K, Lawrence MS, Carter SL, et al. . Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 2015;10:1556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lek M, Karczewski KJ, Minikel EV, et al. . Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Auton A, Brooks LD, Durbin RM, et al. . A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seed G, Yuan W, Mateo J, et al. . Gene copy number estimation from targeted next-generation sequencing of prostate cancer biopsies: analytic validation and clinical qualification. Clin Cancer Res 2017;23:6070–7. [DOI] [PubMed] [Google Scholar]
- 29. Mermel CH, Schumacher SE, Hill B, et al. . GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen S, Liu M, Huang T, et al. . GeneFuse: detection and visualization of target gene fusions from DNA sequencing data. Int J Biol Sci 2018;14:843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu G, Wang L-G, Han Y, et al. . clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Subramanian A, Narayan R, Corsello SM, et al. . A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017;171:1437–1452.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jung H, Kim HS, Kim JY, et al. . DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat Commun 2019;10:4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Siravegna G, Marsoni S, Siena S, et al. . Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 2017;14:531–48. [DOI] [PubMed] [Google Scholar]
- 36. Guibert N, Jones G, Beeler JF, et al. . Targeted sequencing of plasma cell-free DNA to predict response to PD1 inhibitors in advanced non-small cell lung cancer. Lung Cancer 2019;137:1–6. [DOI] [PubMed] [Google Scholar]
- 37. Ahmed JH, Makonnen E, Fotoohi A, et al. . CYP2D6 genotype predicts plasma concentrations of tamoxifen metabolites in Ethiopian breast cancer patients. Cancer 2019;11:1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Madlensky L, Natarajan L, Tchu S, et al. . Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin Pharmacol Ther 2011;89:718–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hicks JK, Bishop JR, Sangkuhl K, et al. . Clinical pharmacogenetics implementation consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther 2015;98:127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liao H-W, Tsai IL, Chen G-Y, et al. . Simultaneous detection of single nucleotide polymorphisms and copy number variations in the CYP2D6 gene by multiplex polymerase chain reaction combined with capillary electrophoresis. Anal Chim Acta 2013;763:67–75. [DOI] [PubMed] [Google Scholar]
- 41. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 2011;32:81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rizvi NA, Hellmann MD, Snyder A, et al. . Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang H, Wang Y, Chlewicki LK, et al. . Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell 2016;29:285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han J, Liu Y, Yang S, et al. . MEK inhibitors for the treatment of non-small cell lung cancer. J Hematol Oncol 2021;14:1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen J-Y, Wang C-M, Chang S-W, et al. . Association of FCGR3A and FCGR3B copy number variations with systemic lupus erythematosus and rheumatoid arthritis in Taiwanese patients. Arthritis Rheumatol 2014;66:3113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dong Z, Li Y, Zhou J, et al. . Copy number variants of ABCF1, IL17REL, and FCGR3A are associated with the risk of gout. Protein Cell 2017;8:467–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
| Data resource | Source | Identifier |
|---|---|---|
| TCGA clinical and survival information, somatic mutation and copy number data | UCSC Xena database | https://gdc.xenahubs. net |
| TCGA RNA-seq and DNA methylation data | UCSC Xena database | https://tcga.xenahubs. net |
| Drug information for TCGA NSCLC | Genomic Data Commons | https://portal.gdc. cancer.gov/ |
| RPPA data for TCGA NSCLC | RPPA Core Facility, MD Anderson Cancer Center | http://app1. bioinformatics.mdanderson.org/ tcpa/_design/basic/ index.html |
| Raw sequence data for CQ NSCLC | NCBI Sequence Read Archive database | https://trace.ncbi. nlm.nih.gov/Traces/ study/?acc=SRP310820 |