

Abstract
Each patient’s cancer consists of multiple cell subpopulations that are inherently heterogeneous and may develop differing phenotypes such as drug sensitivity or resistance. A personalized treatment regimen should therefore target multiple oncoproteins in the cancer cell populations that are driving the treatment resistance or disease progression in a given patient to provide maximal therapeutic effect, while avoiding severe co-inhibition of non-malignant cells that would lead to toxic side effects. To address the intra- and inter-tumoral heterogeneity when designing combinatorial treatment regimens for cancer patients, we have implemented a machine learning-based platform to guide identification of safe and effective combinatorial treatments that selectively inhibit cancer-related dysfunctions or resistance mechanisms in individual patients. In this case study, we show how the platform enables prediction of cancer-selective drug combinations for patients with high-grade serous ovarian cancer using single-cell imaging cytometry drug response assay, combined with genome-wide transcriptomic and genetic profiles. The platform makes use of drug-target interaction networks to prioritize those combinations that warrant further preclinical testing in scarce patient-derived primary cells. During the case study in ovarian cancer patients, we investigated (i) the relative performance of various ensemble learning algorithms for drug response prediction, (ii) the use of matched single-cell RNA-sequencing data to deconvolute cell population-specific transcriptome profiles from bulk RNA-seq data, (iii) and whether multi-patient or patient-specific predictive models lead to better predictive accuracy. The general platform and the comparison results are expected to become useful for future studies that use similar predictive approaches also in other cancer types.
Keywords: toxic effects, combination synergy, ovarian cancer, network visualization, precision oncology, machine learning, drug combinations
Introduction
Combinatorial cancer treatments may lead to therapeutic benefits both by enhancing treatment efficacy and by avoiding monotherapy resistance [1]. Furthermore, individual drugs used in the combinatorial treatments may sometimes be administered at lower doses than when used as monotherapies, thus reducing the risk of treatment toxicity and other side-effects. High-throughput drug screening (HTS) of the phenotypic effects of drug combinations in preclinical cancer models is often used for unbiased exploration of candidate drug combinations. However, even with automated HTS instrumentation, systematic screening of drug combinations quickly becomes impractical, both in terms of time and patient specimens required for the combinatorial testing, due to combinatorial explosion of the number of potential combinations. Furthermore, the pathways that drive cancer progression or treatment resistance are often highly variable between individual patients even with the same cancer type, hence leading to further experimental challenges, as the panels of combinations need to be tested in cells of each individual patient. Therefore, we and others have developed computational approaches to guide the discovery of most potent combinations to be prioritized in HTS for further testing [2–5].
Most computational and experimental combinatorial discovery strategies aim to identify drug combinations that are more effective when combined, compared to the single-agent responses when used as monotherapies, hence leading to synergistic effects [6]. At the same time, the successful combinations should show minimal toxicity to the non-malignant cells. However, most preclinical screening efforts emphasize merely the combination synergy as key determinant of the drug combination performance [7], even though cancer cell selectivity is critical for the clinical success of combinatorial therapies [8]. This leads to the translational challenge that requires careful assessment of potential toxic effects along with synergistic efficacy, as there is a fundamental trade-off between treatment efficacy and tolerable toxicity [9]. To date, there has been a lack of computational approaches that could address these experimental and translational challenges: (i) identifying among the massive number of potential drug combinations those that simultaneously show both maximal therapeutic potential and cancer selectivity, and (ii) bridging the gap to the clinical practice to enable real-world applications in translational studies and to establish their potential utility in clinical decision-making process.
We recently developed a two-phase machine learning (ML) and interactive visual-evaluation strategy for efficient in silico prioritization of combinations for individual cancer patients [10]. In the first phase, the ComboPred algorithm predicts patient-customized drug combinations by integrating single-drug responses and molecular profiles of the primary patient cells from ex vivo cell cultures. To explore the massive combinatorial search spaces among potential combinations, the algorithm uses drug-target interaction networks combined with Random Forest algorithm to identify cancer-selective and synergistic combinations as safe and effective treatment options. The toxic effects were estimated using differential single-drug sensitivity profiles between patient cells and healthy controls. In the next phase, PatientNet web-application allows for interactive visualization of patient-customized co-vulnerability networks using the baseline genomic and molecular profiles of individual patients to guide further testing of the patient-specific combination and clinical translation phases. The platform was initially piloted in a hematological cancer, T-cell prolymphocytic leukemia (T-PLL), where it successfully identified distinct combinations for T-PLL patients, each presenting with different resistance patterns and synergy mechanisms [10].
In the present case study, we show how the same approach can be modified to predict cancer-selective drug combinations also for solid tumors, here in cells from patients with high-grade serous ovarian carcinoma (HGSOC). Instead of using bulk drug response profiling, as in the pilot study, we make use of single-cell data from imaging cytometry drug response assays to make combination predictions at the level of tumor cell subpopulations. The subpopulation-level analysis also avoids the need of healthy individuals as controls, as the non-malignant cells of each patient can serve as her own control. We investigate a number of ensemble ML algorithms that enable learning from sparse and heterogeneous data sources (drug response data combined with genomic data), with the aim to provide accurate patient-tailored response predictions. The platform prioritizes those combinations that warrant further pre-clinical testing in scarce patient-derived primary cells. During the case study on HGSOC patient cells, we made several important observations, related to both computational and experimental analyses, which we expect to become useful for others using similar precision oncology approaches also in other cancer types that are accessible for bulk or single-cell RNA-sequencing and drug sensitivity testing.
Material and Methods
This case study made use of four genetically-validated HGSOC patient samples that had functional single-drug response profiles, as well as genome-wide molecular and genomic data available from the same cell cultures in the HERCULES study (Table 1). Two of the patients (EOC0939 and EOC1103) had rapidly progressing platinum resistant disease, whereas one patient (EOC1107) had platinum sensitive disease. Two of the patient samples (EOC0939_pAsc and EOC1107_pAsc) contain cells from primary ascites of the newly-diagnosed patients 0939 and 1107, where ‘primary’ means that the sample is taken before chemotherapy (i.e. treatment-naive). The other two samples (EOC1103_pOme1 and EOC1103_pPer1) were sampled from omental and peritoneal tumor sites, respectively, i.e. primary cells from surgically removed tumors from a newly diagnosed treatment-naive patient 1103. In each patient sample, we considered two cell subpopulations: cells that are either positive for the HGSOC lineage marker PAX8 [11] (PAX8+ cells) with a TP53 mutation (malignant ‘cancer cells’), or PAX8- cells without a TP53 mutation (non-malignant ‘normal cells’). The single-drug responses were profiled for both of the cell populations in the ex vivo cell cultures (see below), and these were used as outcome vectors for the population-level ML predictions, while the genome-wide transcriptomic and genetic profiles were obtained at the sample-level (so-called ‘bulk’ assays). Fresh dissociated tissue specimens were available from all the four samples, where `fresh' means that the dissociation protocol is started after surgery without freezing of the tumor cells, important for genomic analyses. Genome-wide single-cell transcriptomic data were available from only one of the ex vivo patient cell cultures (from cultured cryopreserved dissociated tissue of EOC0939).
Table 1.
Profiling data available from the 4 HGSOC patient cell cultures ex vivo
| Data | Level | Note |
|---|---|---|
| Single-drug response profiling (DSS) | Subpopulation (PAX8+/−) | Imaging cytometry-based cell population viability assay |
| Bulk RNA-sequencing (RNA-seq) | Sample | PRISM decomposition for subpopulation-specific profiles |
| Bulk whole-genome sequencing (WGS) | Sample | Assuming no somatic mutations in PAX8- subpopulations |
| Single-cell RNA-sequencing (scRNA-seq) | Sample | EOC0939_pAsc before and after 1 week of culture ex vivo |
These data come from the patients profiled in the HERCULES project (https://www.project-hercules.eu/).
For high-throughput ex vivo drug sensitivity and resistance testing in patient-derived HGSOC cultures, we performed imaging cytometry-based analysis of the tumor cell subpopulation responses [12]. The technology offers single-cell resolution that captures heterogeneous cell behavior in response to multiple drug treatments (i.e. changes in number of cells expressing a specific marker protein). Dissociated tumor cell samples were pre-cultured for one week in the optimal medium and seeded at 1000 cells per well in 384-well plates with pre-added drugs. A total of 528 approved and investigational drugs were tested over five different concentrations covering a 10 000-fold concentration range, where the dose range was individually optimized for each drug to cover relevant concentrations [13]. The cells were incubated with the drugs for 7 days. To detect the surviving cells of different subpopulations at the end of drug treatment, the cells were fixed with 4% paraformaldehyde and immunostained with polyclonal anti-PAX8 rabbit antibody (Peprotech) using automated liquid handling. Imaging cytometry used automated microscopy to determine the numbers of PAX8-expressing HGSOC cells and PAX8-negative non-cancerous cells (stromal and normal epithelial cells). These raw cell counts were used for calculation of the PAX8+/− subpopulation-specific drug sensitivity scores (DSS) [14], based on the dose–response curve fitting in the web-based interactive application Breeze (https://breeze.fimm.fi/) [15]. The DSS values were determined for the two subpopulations, the PAX8-positive HGSOC cells and the PAX8-negative non-cancerous cells, based on the cell viability values for each dose. The cell viability was calculated as a percent of cells of each subpopulation after each drug treatment, normalized to the number of cells of the respective population in the DMSO control.
Since our focus was on molecularly targeted drugs that should lead to more selective responses and less toxic effects, we used 352 single-drugs targeting 423 proteins (Figure 1; Supplementary Table S1), and excluded standard cytotoxic chemotherapeutics and compounds with undefined molecular targets. Each of the 352 drugs were tested on each of the eight cell subpopulation cultures ex vivo (4 patient samples, each with PAX8+/− subpopulations). The 423 targets were extracted based on the bioactivity dose–response measurements from our crowdsourcing bioactivity data platform DrugTargetCommons (DTC) [16] (https://drugtargetcommons.fimm.fi/), similar to the original study [10]. Briefly, for each drug-target pair, we compared the median level of available dose–response bioactivity measurements in DTC (log-transformed Kd, Ki or IC50 endpoints), and classified as potent targets all the proteins within log-fold change ≤ 2 from the smallest bioactivity value among all the profiled targets of the drug (so-called nominal or primary target). This drug-target binary interaction matrix was then subjected to further examination by an expert (K.W.), who manually excluded non-potent off-targets and included known potent targets of the drugs that were missed by the target profiling studies available in DTC (Supplementary Table S1). Since the combination prediction model makes use of the drug-target interaction profiles (Supplementary Figure S1), the aim was to collect as comprehensive target profiles as possible among proteins that are expected to contribute to the mode-of-action of the drugs. We also excluded drugs with extreme monotherapy responses (DSS ≥ 40), since it would become challenging to find a partner drug to gain synergistic effect if one of the drugs already alone is showing an extreme response. Many of the extreme responses also originated from broadly toxic non-targeted chemotherapeutics.
Figure 1 .
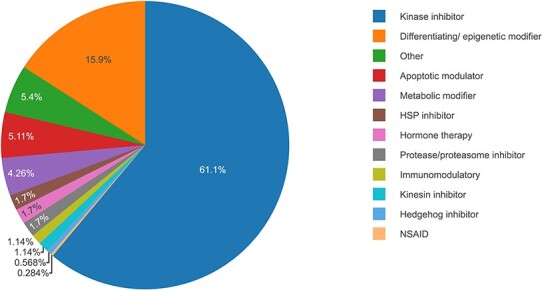
Molecularly targeted drug classes among the 352 single-agents used in the predictive modeling. HSP, heat shock protein. NSAID, nonsteroidal anti-inflammatory drugs.
The computational methodology present in our earlier study [10] was extended to combine the imaging cytometry-based ex vivo drug response profiling of HGSOC patient cell cultures with corresponding sequencing data to investigate cancer cell subpopulation-specific responses to treatments, both monotherapies and combinatorial therapies, and to identify potential mechanisms involved in drug combination synergy (Supplementary Figure S1). Due to the lack of scRNA-seq data from each of the HGSOC patient cultures (Table 1), we decomposed the sample-level bulk RNA-seq data into PAX8+ and PAX8- subpopulation profiles using a latent statistical framework PRISM [17]. PRISM utilized the scRNA-seq data available from the fresh dissociated tissues as reference to infer the expression profiles for the PAX8+ and PAX8- subpopulations from each individual bulk sample (Supplementary Figure S2). PRISM has been shown to provide an accurate estimation of both the cell composition and expression profiles for cancer, stromal, and immune cells based on 214 HGSOC samples from the HERCULES study using only eight samples that had both bulk and scRNA data [17]. This is because it adapts the expression profiles both to each individual bulk data and to the single-cell reference. The original combination prediction platform combined multi-omics data from all the patients to train a single model [10]. In addition to such a multi-patient model, we also trained separate models for each patient case using patient-specific data as an alternative way of modeling drug responses. While the original study used random forest (RF) algorithm [18], we also explored in this study other ensemble learning methods, namely, gradient boosting (GB) [19, 20] and XGBoost [21], to investigate what is an optimal ML algorithm to obtain robust and reliable predictions in such a small sample size learning task (i.e. n = 1 for patient-specific model and n = 4 for multi-patient model).
Results
Predictive modeling and cross-validation setups
As inputs for the prediction algorithms, we used binary variables for both 423 drug-targets and 110 point mutations, as detected from the four HGSOC patient cell cultures using the WGS data (Supplementary Figure S1), combined with the continuous expression levels of 698 cancer genes (Supplementary Table S1). The mutation detection was focused only on exonic and splicing variants (mutation frequency > 0.2, and combined annotation dependent depletion (CADD) score [22] > 10). The expression levels were measured with reads per kilobase of transcript, per million mapped reads (RPKM). The cancer genes combined both ovarian and pan-cancer markers. The ovarian cancer markers come from the overexpressed genes, calculated based on the differential gene expression of 76 HGSOC samples from the HERCULES study (Wilcoxon test, adjusted P < 0.01 and log fold-change > 0.2). The pan-cancer markers are genes associated with cancer development, tumor suppressors, and drug sensitivity or resistance from the best performer teams in AstraZeneca-Sanger Drug Combination Prediction DREAM Challenge [4].
We used 10-fold cross validation (CV) for tuning the model parameters and for selecting the best performing models. In the leave-drug-out CV setup (Figure 2), we used 90% of the data as training data, and the remaining 10% was used as test data to evaluate how well the models generalize to new drug responses. For each subpopulation, the same drugs were left out for testing (10% of the total drugs), and therefore the drugs in the test dataset were not seen in the training data. We used this setup to investigate the effect of various experimental and computational factors on the accuracy of predicting monotherapy response DSS values in the eight subpopulations (see next sections). Spearman correlation was used in the fine-tuning of the model parameters in CV, due to its generally robust behavior, and the estimated models were also evaluated using Pearson correlation, mean-squared error and mean absolute error (Supplementary Figures S3–S7). The optimized prediction models were then used to make combinatorial predictions for each patient (see Drug combination predictions and network visualizations section), similar to the original study [10].
Figure 2 .
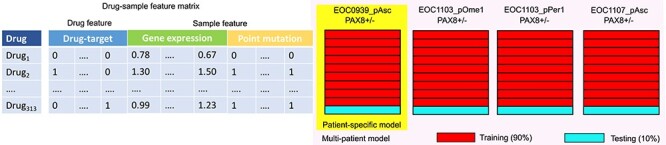
Construction and validation of the multi-patient and patient-specific predictive models. For a given sample, each drug was associated with a feature vector corresponding both to its drug-target profile and to the gene expression profile (decomposed from bulk RNA-seq) and point mutation detections (extracted from WGS) of the particular sample (left). In each iteration of 10-fold CV, 90% of the drug-sample feature matrix were used for training and the remaining 10% was used for testing of the monotherapy prediction accuracy, either using the PAX8+ and PAX8- samples from a single patient case (right, Patient-specific model), or all the patient samples (multi-patient model).
Effect of using scRNA-seq data on prediction accuracy
We first investigated whether the availability of the scRNA-seq data from one of the patient cultures could improve the accuracy of deconvolution of the bulk RNA-seq data into subpopulation-specific gene expression profiles, and hence the prediction of population-level monotherapy responses. We observed that the use of scRNA-seq from the EOC0939_pAsc sample slightly improved the predictions of the patient EOC0939 responses, as expected, but when considering all the samples, the use scRNA-seq data from one of the samples did not affect the monotherapy predictions, regardless whether using the multi-patient or patient-specific models (Figure 3). This result indicates that there is no advantage in performing scRNA-seq for each patient culture ex vivo, provided scRNA-seq data are available from the fresh tumors for the subpopulation deconvolution. However, this result could change if scRNA-seq data were available from each individual patient culture.
Figure 3 .
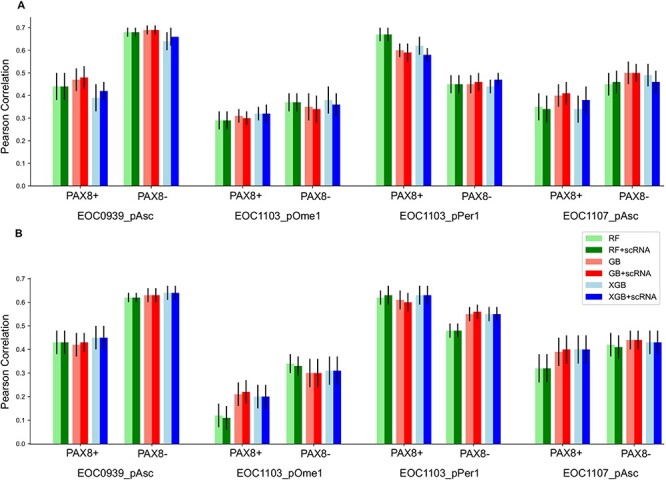
Comparison of different models for monotherapy response predictions with/without scRNA-seq data from EOC0939_pAsc sample. (A) Multi-patient models, (B) patient-specific models. The bar heights show the mean accuracies over the 10 CV folds, and the error bars mark the standard error of the mean (SEM). RF, random forest; GB, gradient boosting; XGB, XGBoost; +scRNA, with scRNA from the EOC0939 sample.
When comparing the multi-patient models against the patient-specific models (Figure 3), we observed that in four out of the eight patient subpopulations (EOC0939_pAsc PAX8-, EOC1103_pOme1 PAX8+, EOC1103_pOme PAX8-, EOC1107_pAsc PAX8-) a single multi-patient model had slightly better performance compared to that of using the patient-specific models (see also Supplementary Figure S7). In the remaining samples, the multi-patient model led to similar performance as the patient-specific models. This result suggests that leveraging information from the other patient samples boosts predictive power to some degree in such a n of 1 personalized medicine prediction task. However, we note that also the multi-patient model makes patient-specific predictions, even though it was trained also using the data from the other samples (Figure 2). The multi-patient model was used also in the original study [10], but there a leave-one-drug-out CV was used instead of a 10-fold leave-drug-out CV.
Even though there were no large differences in the predictive accuracy among the ensemble learning approaches, the GB algorithm combined with scRNA-seq data provided overall accurate and robust predictive behavior (Figure 3). In comparison to the methodological factors (e.g. use of matched scRNA-seq data, multi-patient or patient-specific models, or different ML algorithms), there appeared to be much higher inter-individual differences in the accuracy of predicting monotherapy responses; in particular, the sample EOC1103_pOme1 was the most difficult to predict, followed by EOC1107_pAsc, EOC1103_pPer1 and EOC0939_pAsc.To study whether the location of the specimen affects prediction accuracies would require large sample cohorts. While these conclusions were made based on Pearson correlation, the other evaluation metrics (Spearman correlation, mean squared error and mean absolute error) generally showed the same trend (Supplementary Figures S3 and S4).
Effect of drug response data on predictive accuracy
We next investigated whether there would be any factors related to the drug information and dose–response data that could explain the variability in the accuracy of predicting monotherapies across the eight subpopulations. In particular, we investigated the effect of (i) the number of other drugs with the same protein target(s) than the drug whose response was being predicted, (ii) filtering out unreliable dose–response curve fits (i.e. bad quality monotherapy outcome data based on expert visual examination of the dose–response curve shapes, IC50 values, and variability between responses to drugs of the same classes of mechanism of action), and (iii) various drug and target classes (Figure 1). Among these factors, we observed that the use of drugs with reliable curve fittings improved predictive performance in most of the samples (Figure 4A), and this improvement was even more consistent in terms of Spearman correlation (Figure 4B); one notable exception was EOC1103_pOme PAX8+ subpopulation, which had only 98 drugs left after the drug filtering. This result indicates that the inter-individual differences in prediction performance were mainly related to the drug response data (i.e. the outcome variable).
Figure 4 .
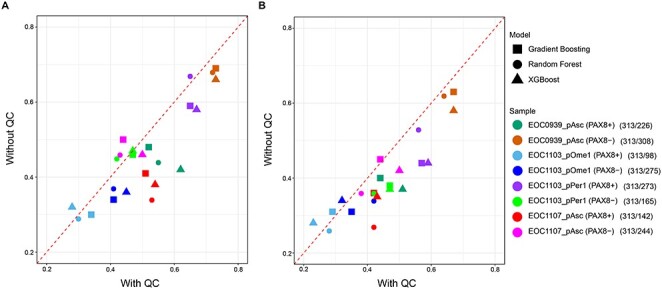
Monotherapy prediction accuracy of the multi-patient model after keeping only reliable curve fittings based on visual quality control (QC). (A) Pearson correlation, (B) Spearman correlation. The numbers in parentheses show the number of drugs used for predictive modeling before/after a visual inspection of the dose–response curve fittings by an expert (D.B.).
When investigating the patient sample EOC1103_pOme that had the lowest prediction accuracy, we noticed it had largest number of experimentally measured DSS values close to zero (i.e. no efficacy, Figure 5). Even though it is quite expected that many of the targeted drugs do not show high DSS values, if they do not target the cancer driving oncoproteins or pathways of the particular patient, such zero-peaked outcome distributions may pose challenges for the predictive modeling algorithms. More specifically, the total number of drugs with DSS ≤ 0.2 values in the four patient samples was: EOC1103_pOme1, 192 (31%); EOC1107_pAsc, 148 (24%); EOC1103_pPer1, 145 (23%); EOC0939_pAsc, 116 (19%), when combining the PAX8 +/− populations. We therefore tested whether excluding all the drugs that show no significant monotherapy efficacy (DSS ≤ 5) before the monotherapy modeling phase would improve the predictive accuracy. Even though there were inter-sample differences, we observed that keeping the drugs even with low efficacy generally improved the model performance (Figure 6). This result is likely due to the increased number of drugs available for model training and CV.
Figure 5 .
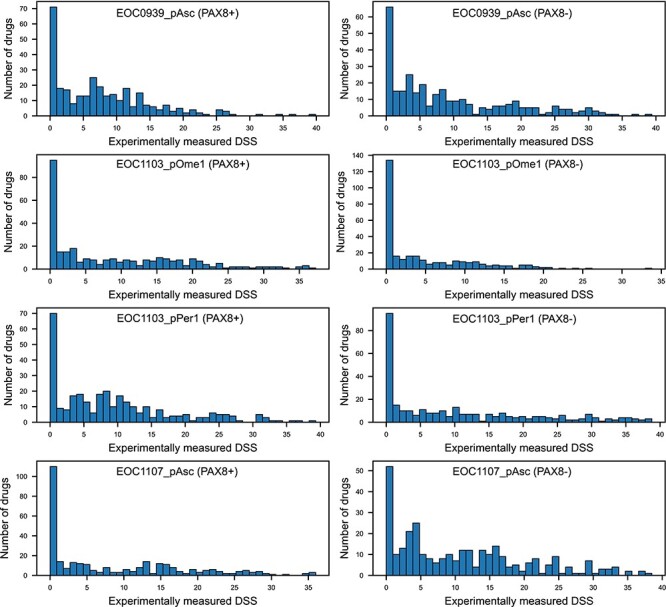
Distribution of experimentally measured DSS values for the eight subpopulations. The total number of overlapping drugs in the multi-patient model was 313. Note: the y-axis range varies between the panels.
Figure 6 .
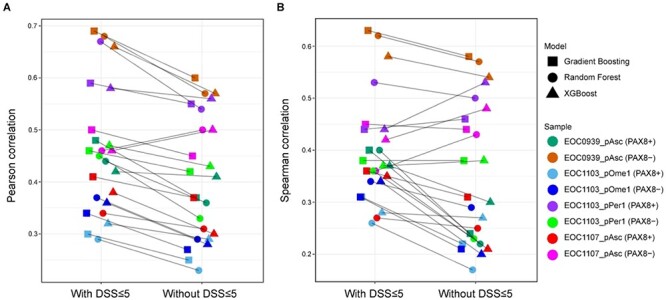
Monotherapy prediction accuracy of the multi-patient model with/without removing drugs with low efficacy (DSS ≤ 5). (A) Pearson correlation, (B) Spearman correlation.
Drug combination predictions and network visualizations
We made combination predictions for each patient sample using the prediction model that led to the generally best and relatively robust accuracy for monotherapy predictions (XGBoost using only drugs with reliable curve fittings and scRNA-seq from EOC0939 culture, see Supplementary Table S2). To identify combinations that show selective efficacy and synergy mostly in the PAX8+ tumor cell population, and that avoid severe co-inhibition of the non-malignant PAX8- cells, we used the highest single-agent (HSA) synergy score to rank the pairwise combinations, similar to the original study [10]. More specifically, HSA score uses both the measured and predicted DSS values, i.e. HSA = predicted combination DSS – max(measured drug1 DSS, measured drug2 DSS). In the present study, however, all the DSS values corresponded specifically to the PAX8+ cell population. When selecting the combinations for further consideration, we also required that the HSA score in the PAX8+ population must be positive (i.e. showing selective synergy in the tumor cells), and that HSA score in the PAX8- population must be negative (i.e. non-synergistic co-inhibition in the non-malignant cells). In the combination shortlisting, we also required that the measured DSS values for both of the single-agents must be between 5 and 20 in the PAX8+ cells to exclude single-agents with no efficacy at all (that are prone to experimental noise), and those with extreme potency already as monotherapy (for which it is difficult to find partner drugs to boost the synergy).
Based on the top-20 model-ranked combinations (Supplementary Tables S3–S6), the domain experts selected one combination for each of the four patient samples based on the robustness of the drug response observed in the drug testing experiments, translational potential of the combination, previous success of the single-agents of the combination in clinical trials, and the known mechanistic interactions of the drug targets. We then used the PatientNet R/Shiny web-application to visualize the patient-customized co-vulnerability networks using the baseline genomic and molecular profiles of individual patients that may provide additional support for the combination discoveries for the clinical translation phase. The PatientNet application uses as input the patient-specific somatic mutations from whole-genome or exome-sequencing, transcriptional changes form whole-genome RNA-seq data, along with the single-agent responses and target annotations to highlight the most plausible target pathways and networks. The PatientNet algorithm finds the shortest paths that connect the potent drug targets of the predicted combinations to the patient-specific genetic aberrations and molecular changes, including mutated or dysregulated genes, through comprehensive cancer signaling networks [23]. We implemented in this case study a new version of the algorithm that allows for the user to specify the maximum path length from the drug targets to the mutations to simplify the network visualizations. The patient-customized network can be visualized either in a web browser or in Cytoscape network analysis software [24].
For the patient sample EOC0939_pAsc, we selected the combination between vistusertib (mTOR inhibitor) and A1155463 (BCL2L1 inhibitor), since these two agents when used alone showed rather modest efficacy in the EOC0939 PAX8+ cells (DSS of 11.4 and 11.8, Supplementary Table S3), whereas their predicted combination effect became relatively high (DSS of 21.6), leading to high synergy score (HSA of 9.7). In this sample, BCL2L1 and MTOR showed also overexpression in the PAX8+ cells, compared to PAX8- cells (Figure 7A), further supporting the dual inhibition of these two cancer survival pathways (metabolic PI3K/AKT/mTOR pathway and apoptotic Bcl-2/Bcl-xL signaling pathway). For the EOC1107_pAsc sample, we selected the combination between AZD-5363 (AKT inhibitor) and Panobinostat (HDAC inhibitor), which also targets the PI3K/AKT/mTOR pathway, but this time combined with HDAC signaling pathway for this particular patient sample, in which both AKT2 and HDAC9 showed overexpression in the PAX8+ tumor cells (Figure 7D). For the EOC1103_pPer1 sample, we selected combination between verdinexor and AZD-8186, since PIK3CA was mutated in this patient sample, hence providing additional support for its pharmaceutical targeting (Figure 7C). We also note that verdinexor was combined with six other agents among the top-20 combinations in this particular sample, supporting its importance and patient-specificity (Supplementary Table S5). For EOC1103_pOme1, we selected cobimetinib-BMS777607 combination, since this combination co-targets many of the overexpressed proteins in this particular patient sample (Figure 7B).
Figure 7 .
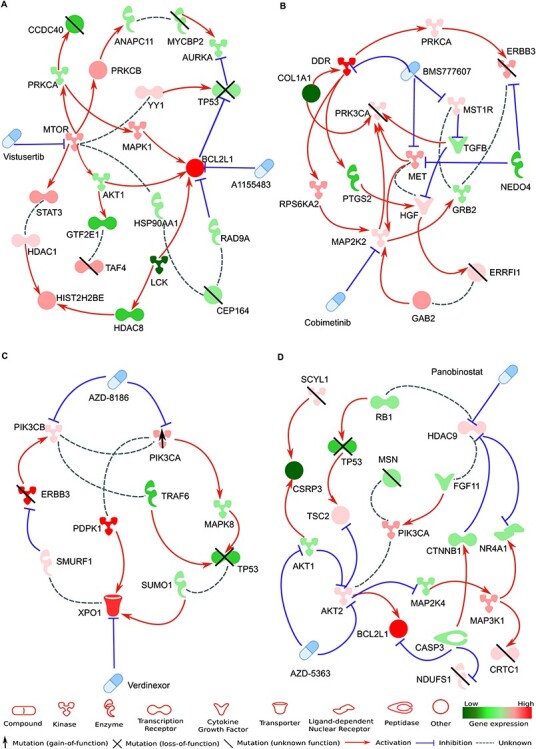
Patient-customized co-vulnerability networks for the selected drug combination for each patient case. (A) Combination between vistusertib (mTOR inhibitor) and A1155463 (BCL2L1 inhibitor) for EOC0939_pAsc, (B) combination between cobimetinib (MAP2K2 inhibitor) and BMS777607 (DDR1, MET and MERTK inhibitor) for EOC1103pOme1, (C) combination between verdinexor (XPO1 inhibitor) and AZD-8186 (PIK3C inhibitor) for EOC1103_pPer1, (D) combination between AZD-5363 (AKT1/2 inhibitor) and panobinostat (HDAC9 inhibitor) for EOC1107_pAsc. The maximum path length in the PatientNet algorithm from the protein targets to differentially expressed or mutated genes was set to 3 in panels (B-D), whereas for panel (A) the network was simple enough without further filtering of pathways and nodes. The patient-specific cancer vulnerability network allows for a visual investigation of the mechanisms of action of the selected drug combinations in the patient’s cellular context, hence providing further support for the tumor-selective combination effects, with possibilities to identify potential biomarkers for the synergistic responses.
Comparison of cultured cells against fresh tumors
We next explored how similar the ex vivo cultured cells and the fresh dissociated tissue of the patients are in terms of transcriptomic signatures by investigating how accurately the deconvoluted RNA-seq profiles of the patient cell cultures capture the PAX8 subpopulation-specific marker genes, originally identified using the scRNA-seq data from the fresh tumor tissues (Wilcoxon test, adjusted P < 0.01 and log fold-change > 1). Using the same statistical cutoffs in the scRNA-seq from cultured cryopreserved dissociated tissue, we observed that the accuracy of the marker detection was ≥91% for the PAX8+ markers and ≥ 57% for the PAX8- markers (Table 2). Interestingly, the usage of the scRNA-seq data available for the EOC0939_pAsc sample improved the detection of PAX8- subpopulation markers (≥78%), while it slightly decreased the accuracy of detecting PAX8+ subpopulation markers (≥85%). The differences in the detection accuracies were relatively similar across all the patient samples.
Table 2.
Coverage of PAX8 marker gene detection using deconvoluted RNA-seq data
| Patient sample | PAX8+ marker coverage | PAX8- marker coverage | ||
|---|---|---|---|---|
| with scRNA data | without scRNA data | with scRNA data | without scRNA data | |
| EOC0939 pAsc | 87% | 94% | 81% | 58% |
| EOC1103 pOme1 | 87% | 94% | 82% | 64% |
| EOC1103 pPer1 | 87% | 91% | 82% | 70% |
| EOC1107 pAsc | 85% | 91% | 78% | 57% |
Coverage was defined as the number of overlapping markers divided by the number of fresh samples markers, where the markers were identified in the fresh tumor samples and after ex vivo culturing using the same marker selection criteria (Wilcoxon test, adjusted P < 0.01 and log fold-change > 1 cutoffs). The total number of PAX8+ and PAX8- markers was 46 and 74, respectively, as detected from the scRNA-seq data of the fresh dissociated tissues.
When investigating the scRNA-seq profiles of EOC0939_pAsc cells before and after the ex vivo culturing for 1 week in terms of the PAX8 marker signatures, we observed that the cells from the cryopreserved dissociated tissue (before culture) were rather different from cultured cryopreserved dissociated tissue cells (after culture), and also from the fresh tumor single-cell dissociate sample (Figure 8). This result might be attributed to the negative selection of a specific subset of tumor cells by the cell culture conditions. Collectively, these results indicate that the deconvolution of the bulk RNA-seq data using the PRISM algorithm into the PAX8 positive and negative profiles was able to capture relatively accurately the marker genes detected based on the scRNA-seq data of the fresh tumor samples, and this was further improved by using the available scRNA-seq data from the ex vivo cultures. However, the cultured cells remained rather distant from the fresh tumors, especially for the PAX8- markers, which needs to be taken into account in the eventual clinical applications.
Figure 8 .
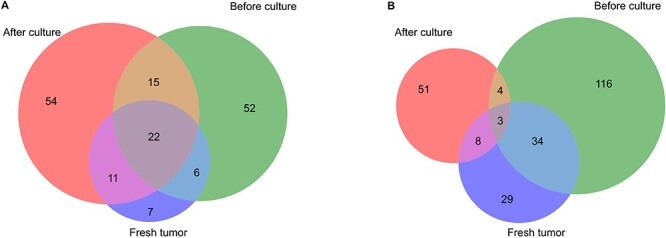
Overlap of marker genes detected in EOC0939_pAsc scRNA-seq data versus fresh tumor samples. (A) PAX8+ markers, (B) PAX8- markers. The marker genes were detected from the scRNA-seq data with Wilcoxon test, using adjusted P < 0.01 and log fold-change > 1 cutoffs. Fresh tumor, scRNA-seq from fresh dissociated tissue; before culture, scRNA-seq from cryopreserved dissociated tissue; after culture, scRNA-seq from cultured cryopreserved dissociated tissue after 1 week in culture.
Discussion
To the best of our knowledge, this is the first computational network-guided approach to tailor personalized combinatorial regimens in solid tumors that takes into account both the molecular heterogeneity of cancer cells and the possible nonselective effects of the drug combinations. Comprehensive drug-target interaction networks were used both in the prediction phase, with the aim to improve both combination efficacy and tolerability, and in the combination interpretation phase, together with genomic and molecular aberrations to construct patient-specific co-vulnerability networks. Such in silico prediction approach is expected to reduce the need for systematic HTS combinatorial screening that requires extensive resources and automatic instrumentation, beyond the capability of most academic laboratories. Testing of hundreds of drug combinations is also impossible in limited numbers of primary cells from patients. The computational prediction models are therefore expected to enormously increase cost- and time-efficacy, since the drug screening efforts can be targeted to verifying the most promising drug combinations only, with maximal cancer-selectivity, using more advanced cancer models that are not accessible for HTS. For instance, the fact that the ex vivo cultured ovarian cancer cells were relatively distant from the fresh tumor samples in terms of their transcriptomic signatures indicates that there are unique molecular-level changes that are not well represented in the cultured cells, as well as new changes that appeared during the ex vivo cultures (Figure 8). Therefore, the predicted combinations need to be further validated in ex vivo or in vivo tumor models with higher levels of inner heterogeneity, such as 3D organoids or patient-derived xenografts [25, 26], which better represent the complexity of the patient tumors, before clinical translation.
A wide range of computational models have been developed to prioritize the most potential drug combinations for experimental testing [1–3, 27, 28]. The recent AstraZeneca-Sanger Drug Combination DREAM Challenge benchmarked a variety of such prediction methods, and observed that the winning methods incorporated prior knowledge of drug-target interactions [4]. Predictive models learned on rich data available from in vitro cancer cell lines enable the training of more advanced machine learning algorithms, such as tensor learning [5] or deep learning [29]. However, translating the results from the established cell lines to individual cancer patients is not straightforward [30], whereas testing of multiple combinations in patient-derived cells is limited by the scarcity of patient specimens [31]. Therefore, a recent study made use of ex vivo high-throughput screening of cancer biopsies using a microfluidic assay, combined with logic-based modeling of signaling pathways to generate patient-specific dynamic models for predicting personalized combinatorial treatments with limited number of cells from pancreatic cancer patients [32]. However, all of these models predict the combination effects on tumor cells only, while not considering the nonselective toxic effects on non-malignant cells. In our original study [10], we estimated the toxic effects using differential ex vivo single-drug sensitivity profiles between patient cells and healthy controls to make sample-level combination predictions for hematological cancer patients. In the present case study, we made subpopulation-level treatment predictions to identify cancer-selective drug combinations for patients with solid tumors. The RF algorithm that was used in the original study provided also relatively accurate predictions in the current case study (Supplementary Table S2), whereas the overall best-performing multi-patient model was based on the XGBoost algorithm [21].
A primary standard chemotherapy treatment for HGSOC is based on the combination of platinum drugs (carboplatin and cisplatin) with taxanes (paclitaxel and docetaxel), and in rare cases, platinum is combined with other cytotoxic chemotherapeutics (gemcitabine and doxorubicine) [33]. Most of the recurring HGSOC tumors are subjected to re-treatment under the same chemotherapeutic regimen. In advanced disease, combining anti-angiogenic drug bevacizumab with chemotherapy has improved progression free survival as maintenance therapy. Targeted therapeutics, including inhibitors of poly (ADP-ribose) polymerase (PARP) protein family, have been increasingly used both as front-line and recurrent cancer therapy, typically as maintenance therapy for platinum sensitive cases [34]. However, the rapid chemotherapy resistance development and lack of sensitivity to PARP inhibitors in half of the HGSOC cases urge the need to find targeted drug combinations to eliminate HGSOC cells more effectively and selectively. Novel combinatorial regimens, including combinations of immunotherapy and anti-angiogenesis agents, may further change the current treatment landscape [33]. Furthermore, new biomarker-driven drug approvals indicate that women may benefit from somatic molecular testing of BRCA and other genes [34]. From computational point of view, there is a need for mechanistic-agnostic prediction models that will be applicable also to non-targeted treatments, including cytotoxic chemotherapeutics and non-specific immunotherapies, which modulate their effects through tumor-agnostic mechanisms. To gain the best trade-off between treatment efficacy and side effects, predictive models should be applied in the context of physiologically relevant disease models to identify combinations that target multiple malignant cell populations that drive the cancer or treatment resistance, including also cancer stem cells, while avoiding co-inhibition of non-malignant cells, including immune cells, which trigger the immune system to destroy cancer cells.
Planned future developments for the computational prediction platform include the implementation of regularized feature selection approaches and the use of sparse modeling approaches for more explainable models that may also enable more systematic identification of omics marker combinations for synergy prediction. More comprehensive drug-target interaction networks could be extracted from community efforts toward bioactivity data collection and harmonization [16, 35], or from predictive models for target activities [36]. For selected drug combinations, the use of copy number variation (CNV) might boost the predictive power, and CNV data could be used as additional features in the prediction model (Supplementary Figure S1). In addition, more fine-grained modeling of the genetic events could be used as categorical features, instead of treating point mutations as binary variables. Similarly, the use of gene isoform-level features from bulk RNA-seq data [37], or scRNA-seq data from multiple time points [38], is also expected to lead to more predictive and selective longitudinal models. In particular, single-cell data provides high-resolution information about the different cell subpopulations present in the complex samples. We recently demonstrated in a leukemia case study how the XGBoost algorithm enables combining scRNA-seq data with ex vivo drug response profiles for accurate prediction of patient-specific combinations that resulted not only in synergistic cancer cell co-inhibition, but were also capable of selectively targeting of specific leukemic cell subpopulations that emerge in differing stages of disease pathogenesis or treatment regimens with close to real-time clinical timeframe [38]. The predictive approach is widely applicable to various cancer types, where ex vivo monotherapy profiling can be done, and it may significantly accelerate the future design and testing of combination therapies, as well as increase their success rates in pre-clinical and clinical studies. The generic approach is also applicable beyond cancer research, e.g. finding drug combinations that synergistically inhibit virus replication, with minimal effects on non-infected host cells.
Key Points
The scRNA-seq transcriptomic profiles obtained before and after cell culture were relatively different from each other, and also from the fresh dissociated tissue, in terms of differentially expressed marker genes.
The use of scRNA-seq data even from one of the primary patient cell cultures partially improved marker coverage of the deconvoluted profiles, but not so much the predictive accuracy of the monotherapy responses.
The multi-patient models across samples led to better prediction accuracies when compared to the patient-specific models, suggesting that leveraging information from multiple patient samples boosts predictive power.
Based on our systematic evaluations, the overall best-performing multi-patient model was based on XGBoost algorithm that included only drugs with reliable curve fittings and scRNA-seq data from all the samples available.
The drug-combination predictions showed wide heterogeneity in terms of both drugs and targets, even in distinct tissue origins of the same patient, highlighting the need for tailored approaches for combination optimization.
Supplementary Material
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme (under grant agreement No 667403 for HERCULES), the Academy of Finland (Projects Nos 289059, 319243 to A.V.H.; No 322927 to A.H.; No 317680 to J.T.; No 310507, 313267, 326238 and 344698 to T.A.), Helse Sør-Øst (grant 2020026 to T.A.), ERANET PerMed Co-Fund (projects JAKSTAT-TARGET and CLL-CLUE to T.A.), the Sigrid Jusélius Foundation (T.A. and J.T.), and the Cancer Foundation Finland (T.A. and L.H.), the European Research Council (ERC) starting grant DrugComb (No. 716063 to J.T.), Orion Research Foundation sr (L.H.), the Novo Nordisk Foundation Center for Stem Cell Biology, DanStem (grant no NNF17CC0027852 to K.W.).
Liye He is a postdoctoral researcher at Institute for Molecular Medicine Finland (FIMM), where he develops machine learning approaches to identify personalized combination therapies.
Daria Bulanova is a postdoctoral researcher at Biotech Research & Innovation Centre (BRIC) at the University of Copenhagen (UC). She develops high-throughput phenotypic profiling assays to study mechanisms of resistance in ovarian cancer.
Jaana Oikkonen is geneticist at ONCOSYS Research Program in UH. Her research interests include genetic analyses on cancer evolution and chemoresistance in ovarian cancer.
Antti Häkkinen is an academy postdoctoral fellow at University of Helsinki (UH), where he focuses on the development of statistical methods for analysis and integration of high-throughput data to understand drug resistance in ovarian cancer patients.
Kaiyang Zhang is a PhD student at ONCOSYS Research Program in UH, where she focuses on transcriptomics data and gene regulatory networks to understand drug resistance in ovarian cancer.
Wenyu Wang is a PhD student at ONCOSYS Research Program in UH. His research topic is integration of genetics and functional genetics data for drug target discovery
Shuyu Zheng is a PhD student at ONCOSYS Research Program in UH. Her research topic is integration of multi-omics and drug screening data for personalized medicine
Erdogan Pekcan Erkan is a postdoctoral researcher at ONCOSYS Research Program in UH.
Olli Carpén is a group leader at ONCOSYS Research Programme in UH. His research group focuses on prognostic tissue biomarkers in ovarian cancer.
Johanna Hynninen is a senior consultant in Gynecologic oncology in Turku University Hospital. Her research group focuses on ovarian cancer clinical research.
Kaisa Huhtinen is a senior researcher at ONCOSYS Research Program in UH and in University of Turku (UTU).
Sampsa Hautaniemi is a group leader at ONCOSYS Research Programme in UH. His research group focuses on characterizing and overcoming drug resistance in cancers.
Anna Vähärautio is a group leader at ONCOSYS Research Programme in UH. Her laboratory focuses on single cell transcriptomics of ovarian cancer to gain high-resolution understanding of emerging treatment resistance.
Jing Tang is a group leader at ONCOSYS Research Programme in UH. His research group focuses on network pharmacological approaches for understanding personalized drug combinations.
Krister Wennerberg is a group leader at Biotech Research & Innovation Centre (BRIC) at UC. His group uses phenotypic profiling of primary cancer cells to discover cancer precision therapies.
Tero Aittokallio is a group leader at FIMM and OCBE/OUH. His groups use network-centric and machine learning-based approaches to predicting optimal treatment regimens for cancer patients.
Contributor Information
Liye He, Institute for Molecular Medicine Finland (FIMM), Helsinki, Finland.
Daria Bulanova, Biotech Research & Innovation Centre (BRIC) at the University of Copenhagen (UC), Helsinki, Finland.
Jaana Oikkonen, ONCOSYS Research Program in UH, Helsinki, Finland.
Antti Häkkinen, University of Helsinki (UH), Helsinki, Finland.
Kaiyang Zhang, ONCOSYS Research Program in UH, Helsinki, Finland.
Shuyu Zheng, ONCOSYS Research Program in UH, Helsinki, Finland.
Wenyu Wang, ONCOSYS Research Program in UH, Helsinki, Finland.
Erdogan Pekcan Erkan, ONCOSYS Research Program in UH, Helsinki, Finland.
Olli Carpén, ONCOSYS Research Program in UH, Helsinki, Finland.
Titta Joutsiniemi, Gynecologic oncology in Turku University Hospital, Helsinki, Finland.
Sakari Hietanen, ONCOSYS Research Program in UH and in University of Turku (UTU), Helsinki, Finland.
Johanna Hynninen, ONCOSYS Research Programme in UH, Helsinki, Finland.
Kaisa Huhtinen, ONCOSYS Research Programme in UH, Helsinki, Finland.
Sampsa Hautaniemi, ONCOSYS Research Programme in UH, Helsinki, Finland.
Anna Vähärautio, ONCOSYS Research Programme in UH, Helsinki, Finland.
Jing Tang, ONCOSYS Research Programme in UH, Helsinki, Finland.
Krister Wennerberg, Biotech Research & Innovation Centre (BRIC), Helsinki, Finland.
Tero Aittokallio, FIMM and OCBE/OUH, Helsinki, Finland.
Data availability
The ex-vivo data used in the work are available from the corresponding author upon reasonable request. The clinical and other sensitive data of the patient samples originates from the HERCULES project (https://www.project-hercules.eu/Contact.html).
References
- 1. Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol 2012;30:679–92. [DOI] [PubMed] [Google Scholar]
- 2. Bulusu KC, Guha R, Mason DJ, et al. . Modelling of compound combination effects and applications to efficacy and toxicity: state-of-the-art, challenges and perspectives. Drug Discov Today 2016;21:225–38. [DOI] [PubMed] [Google Scholar]
- 3. Ianevski A, Giri AK, Gautam P, et al. . Prediction of drug combination effects with a minimal set of experiments. Nature Machine Intelligence 2019;1:568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Menden MP, Wang D, Mason MJ, et al. . Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat Commun 2019;10:2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Julkunen H, Cichonska A, Gautam P, et al. . Leveraging multi-way interactions for systematic prediction of pre-clinical drug combination effects. Nat Commun 2020;11:6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vlot AHC, Aniceto N, Menden MP, et al. . Applying synergy metrics to combination screening data: agreements, disagreements and pitfalls. Drug Discov Today 2019;24:2286–98. [DOI] [PubMed] [Google Scholar]
- 7. Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 2017;171:1678–1691.e1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boshuizen J, Peeper DS. Rational cancer treatment combinations: an urgent clinical need. Mol Cell 2020;78:1002–18. [DOI] [PubMed] [Google Scholar]
- 9. Pulkkinen OI, Gautam P, Mustonen V, et al. . Multiobjective optimization identifies cancer-selective combination therapies. PLoS Comput Biol 2021;16:e1008538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He L, Tang J, Andersson EI, et al. . Patient-customized drug combination prediction and testing for T-cell Prolymphocytic leukemia patients. Cancer Res 2018;78:2407. [DOI] [PubMed] [Google Scholar]
- 11. Cheung HW, Cowley GS, Weir BA, et al. . Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A 2011;108:12372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Snijder B, Vladimer GI, Krall N, et al. . Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: interim results from a single-arm, open-label, pilot study, the lancet. Haematology 2017;4:e595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pemovska T, Kontro M, Yadav B, et al. . Individualized systems medicine (ISM) strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov 2013;3:1416–29. [DOI] [PubMed] [Google Scholar]
- 14. Yadav B, Pemovska T, Szwajda A, et al. . Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep 2014;4:5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Potdar S, Ianevski A, Mpindi J-P, et al. . Breeze: an integrated quality control and data analysis application for high-throughput drug screening. Bioinformatics (Oxford, England) 2020;36:3602–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang J, Tanoli Z-U-R, Ravikumar B, et al. . Drug target commons: a community effort to build a consensus knowledge base for drug-target interactions. Cell Chemical Biology 2018;25:224–229.e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Häkkinen A, Zhang K, Alkodsi A, et al. . PRISM: recovering cell type specific expression profiles from individual composite RNA-seq samples. Bioinformatics 2021. doi: 10.1093/bioinformatics/btab178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Breiman L. Random forests. Mach Learn 2001;45:5–32. [Google Scholar]
- 19. Friedman JH. Stochastic gradient boosting. Comput Stat Data Anal 2002;38:367–78. [Google Scholar]
- 20. Friedman JH. Greedy function approximation: a gradient boosting machine. Ann Stat 2001;29:1189–232. [Google Scholar]
- 21. Chen T, Guestrin C. XGBoost: a scalable tree boosting system. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, 785–94.
- 22. Rentzsch P, Witten D, Cooper GM, et al. . CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47:D886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zaman N, Li L, Jaramillo Maria L, et al. . Signaling network assessment of mutations and copy number variations predict breast cancer subtype-specific drug targets. Cell Rep 2013;5:216–23. [DOI] [PubMed] [Google Scholar]
- 24. Shannon P, Markiel A, Ozier O, et al. . Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kopper O, Witte CJ, Lõhmussaar K, et al. . An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat Med 2019;25:838–49. [DOI] [PubMed] [Google Scholar]
- 26. Liu JF, Palakurthi S, Zeng Q, et al. . Establishment of patient-derived tumor Xenograft models of epithelial ovarian Cancer for preclinical evaluation of novel therapeutics. Clinical Cancer Res Off J Am Assoc Cancer Res 2017;23:1263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adam G, Rampášek L, Safikhani Z, et al. . Machine learning approaches to drug response prediction: challenges and recent progress, NPJ precision. Oncology 2020;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ling A, Huang RS. Computationally predicting clinical drug combination efficacy with cancer cell line screens and independent drug action. Nat Commun 2020;11:5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuenzi BM, Park J, Fong SH, et al. . Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell 2020;38:672–684.e676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schätzle L-K, Hadizadeh Esfahani A, Schuppert A. Methodological challenges in translational drug response modeling in cancer: a systematic analysis with FORESEE. PLoS Comput Biol 2020;16:e1007803–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tanoli Z, Vähä-Koskela M, Aittokallio T. Artificial intelligence, machine learning, and drug repurposing in cancer. Expert Opin Drug Discovery. Published online: 12 Feb 2021;1–13. doi.org/10.1080/17460441.2021.1883585. [DOI] [PubMed] [Google Scholar]
- 32. Eduati F, Jaaks P, Wappler J, et al. . Patient-specific logic models of signaling pathways from screenings on cancer biopsies to prioritize personalized combination therapies. Mol Syst Biol 2020;16:e9690–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buechel M, Herzog TJ, Westin SN, et al. . Treatment of patients with recurrent epithelial ovarian cancer for whom platinum is still an option. Ann Oncol 2019;30:721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kurnit KC, Fleming GF, Lengyel E. Updates and new options in advanced epithelial ovarian Cancer treatment. Obstet Gynecol 2021;137:108–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Freshour SL, Kiwala S, Cotto KC, et al. . Integration of the drug-gene interaction database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res 2021;49:D1144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cichońska A, Ravikumar B, Allaway RJ, et al. . Crowdsourced mapping of unexplored target space of kinase inhibitors. Nat Commun 2021;12:3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Safikhani Z, Smirnov P, Thu KL, et al. . Gene isoforms as expression-based biomarkers predictive of drug response in vitro. Nat Commun 2017;8:1126–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ianevski A, Lahtela J, Javarappa KK, et al. . Patient-tailored design for selective co-inhibition of leukemic cell subpopulations. Sci Adv 2021;7:eabe4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ex-vivo data used in the work are available from the corresponding author upon reasonable request. The clinical and other sensitive data of the patient samples originates from the HERCULES project (https://www.project-hercules.eu/Contact.html).