

SUMMARY
Immune checkpoint blockade (ICB) has improved outcomes in some cancers. A major limitation of ICB is that most patients fail to respond, which is partly attributable to immunosuppression. Obesity appears to improve immune checkpoint therapies in some cancers, but impacts on breast cancer (BC) remain unknown. In lean and obese mice, tumor progression and immune reprogramming were quantified in BC tumors treated with anti-programmed death-1 (PD-1) or control. Obesity augments tumor incidence and progression. Anti-PD-1 induces regression in lean mice and potently abrogates progression in obese mice. BC primes systemic immunity to be highly responsive to obesity, leading to greater immunosuppression, which may explain greater anti-PD-1 efficacy. Anti-PD-1 significantly reinvigorates antitumor immunity despite persistent obesity. Laminin subunit beta-2 (Lamb2), downregulated by anti-PD-1, significantly predicts patient survival. Lastly, a microbial signature associated with anti-PD-1 efficacy is identified. Thus, anti-PD-1 is highly efficacious in obese mice by reinvigorating durable antitumor immunity.
In brief
Pingili et al. show that breast cancer exacerbates obesity-driven immunosuppression. Anti-PD-1 reinvigorates antitumor immunity in the tumor microenvironment, mammary fat pad, and peripherally. Lamb2, downregulated by anti-PD-1 in tumors, associates with obesity and poor survival in patients. A microbial signature associated with immune checkpoint inhibitor efficacy is identified.
Graphical Abstract
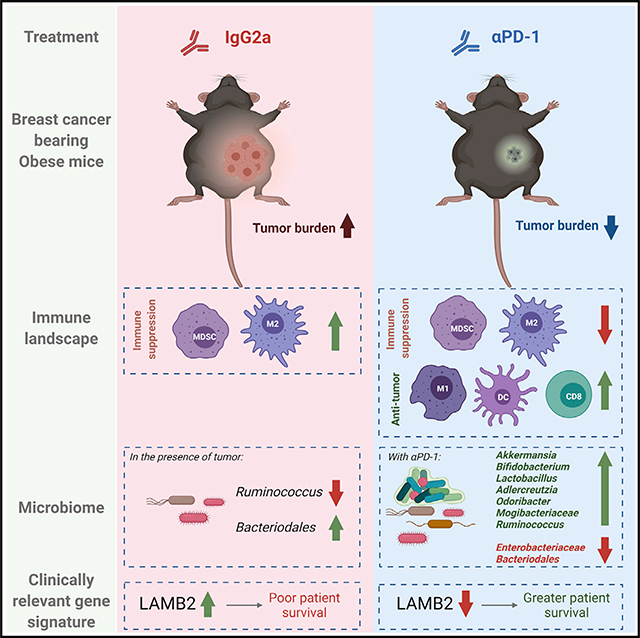
INTRODUCTION
Tumors evade the immune system primarily through overexpression of immune checkpoints that lead to T cell anergy and an impaired antitumor immune response (Chaib et al., 2020). Monoclonal antibody immune checkpoint blockade (ICB) immunotherapy has dramatically altered the landscape of cancer care. Blocking immune checkpoints such as programmed death-1 (PD-1), its ligand programmed death-ligand 1 (PD-L1), or other ligands restores T cell function with relative success and fewer side effects than chemotherapy. It is estimated that obesity and overweight status are attributable risk factors in 11% of all cancers in women (Cozzo et al., 2017). Hormones and adipokines associated with systemic changes in obesity influence cancer growth (Cozzo et al., 2017). Obesity also mediates normal and tumor microenvironment (TME) dysfunction through components including microbes, metabolites, growth factors, and immune cells (Hanahan and Weinberg, 2011; Bissell and Hines, 2011; Correia and Bissell, 2012), which accelerate tumor onset and progression (Makowski et al., 2020; Rathmell, 2021). Work by our group and others demonstrated that obesity is associated with low-grade adipose inflammation in the normal human breast and non-human primate and murine mammary gland, which influences breast cancer (BC) risk, disease outcomes, and therapeutic efficacy (Lengyel et al., 2018; Cozzo et al., 2017; Iyengar et al., 2016; Shively et al., 2018). Immunosuppressive PD-1 and PD-L1 expression, or ligand-positive immune cells, are increased by obesity (Yang et al., 2016; Del Cornò et al., 2016; Wang et al., 2019; Shirakawa et al., 2015, Circulation, abstract). Obese women have a higher risk of BC invasion, distant metastases, tumor recurrence, and impaired therapeutic response, which together increase mortality (Naik et al., 2019). Through several ICB trials, a surprising finding emerged wherein obese cancer patients responded better to immunotherapy (Woodall et al., 2020). Obesity or overweight status was shown to improve immunotherapy efficacy in melanoma, non-small cell lung cancer, and renal cell carcinoma in retrospective studies (Woodall et al., 2020). However, the impacts of ICB in obese BC patients are currently unknown. Trials suggest that ICB is a promising clinical approach in BC, including the most aggressive BC subtype, triple-negative breast cancer (TNBC), for which patients have few clinical options. For TNBC, combination therapy of nab-paclitaxel with atezolizumab (Tecentriq, anti-PD-L1) was the first ICB approved by the U.S. Food and Drug Administration (FDA) (Schmid et al., 2018b). Pembrolizumab (Keytruda, anti-PD-1) also received FDA approval (Schmid et al., 2020; Adams et al., 2019a, 2019b; Nanda et al., 2016). However, most BC patients fail to respond to ICB, because efficacy occurs in only a minority of patients (Schmid et al., 2018b; Franzoi et al., 2021), thus limiting the potential for vast improvements in clinical outcomes. There is a great need to discover additional targets to bolster ICB efficacy, as well as aid in the identification of patients who may benefit from ICB to improve outcomes overall. Therefore, we sought to determine whether obesity-associated ICB therapeutic outcome was improved in an established BC preclinical model. Our results show that although obesity accelerated tumor progression, treatment with anti-PD-1 significantly reduced obesity-associated tumor burden. Anti-PD-1 reshaped the peripheral and local TME immune landscape by decreasing immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) and increasing antitumor immune cells such as CD8+ T cells and M1-like macrophages (Macs). In addition, increasing evidence suggests that the host microbiome affects both chemotherapeutic and ICB efficacy (Cheng et al., 2020; Sepich-Poore et al., 2021; Kalaora et al., 2021). Major regulators of the gut and extra-intestinal microbiome are diet and obesity, which provide a cogent link between BC risk and poor response to therapy (Sipe et al., 2020; Parida and Sharma, 2020). We identified gut microbes that correlate with tumor size and an ICB-associated responsive microbial signature as a putative biomarker of ICB efficacy. Altogether, the findings demonstrate that the immunosuppressive milieu of BC exacerbated by obesity is not immutable but may be reinvigorated efficiently with ICB.
RESULTS
Obesity-increased tumor burden was reduced by anti-PD-1
Adult female C57BL/6J mice were randomly assigned to an obesogenic high-fat diet (HFD) or control low-fat diet (LFD) groups at eight weeks of age (schema in Figure S1A). Female mice fed an obesogenic HFD significantly increased body weight by almost 2-fold compared with lean LFD-fed littermate controls (Figure S1B). Adiposity was significantly increased by 3.6-fold in obese mice compared with lean mice (Figure S1C). Mammary gland adipocyte diameter was significantly increased with obesity (Figure S1D). After significant obesity developed at 26 weeks of age, mice were orthotopically injected with the BC model E0771 cells and treated with ICB anti-PD-1 or IgG2a isotype control twice weekly (Figure S1A). Treatment with anti-PD-1 or IgG2a did not affect body weight or fat composition of lean or obese mice (Figures S1B and S1C). However, mice treated with anti-PD-1 had significantly larger adipocytes in obese adipose compared with isotype controls (Figures S1D and S1E).
E0771 cells are known to have genetic instability and have been published with varied phenotypes (reviewed by Le Naour et al., 2020). We determined E0771 to be triple negative for nuclear hormone receptors with absent or extremely low expression for Esr1, Esr2, and PGR and with low expression of Erbb2 (Figure S1F; data not shown). Syngeneic tumor cells orthotopically implanted into the mammary fat pads (MFPs) grew comparably in lean and obese mice during the first two weeks. However, at two weeks post-implantation, tumors in obese mice progressed rapidly in the isotype control group. Tumor progression advanced significantly in obese IgG2a-treated mice compared with lean IgG2a-treated controls (Figures 1A and 1B), which was evident by the greater tumor volume (Figure 1C) and weight at the endpoint (Figure 1D). Treatment with anti-PD-1 significantly decreased obesity-induced tumor progression (Figures 1A and 1B). In obese mice treated with anti-PD-1, tumor volume was reduced by 5.7-fold (Figure 1C) and tumor weight was reduced by 4.3-fold (Figure 1D) to tumor burdens that mirrored those of lean isotype controls. Furthermore, histologic analysis of mitotic nuclei in tumors from obese mice revealed that treatment with anti-PD-1 significantly decreased cell proliferation (Figure 1E).
Figure 1. Obesity-accelerated BC progression is reduced with immune checkpoint blockade.
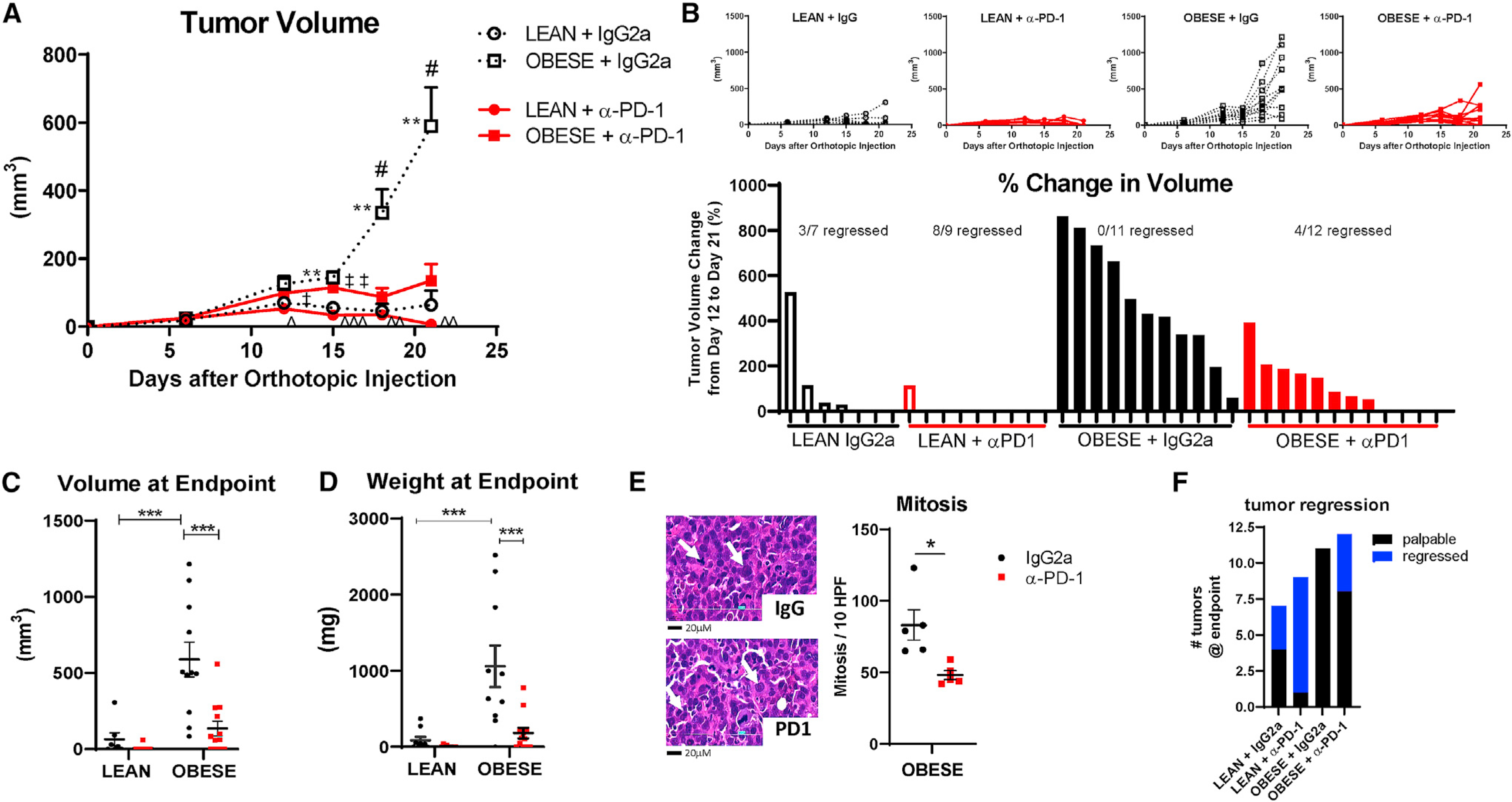
(A) E0771 tumor progression was measured by digital caliper over three weeks after orthotopic injection. Two-way ANOVA repeated measures with Tukey post hoc test was calculated in GraphPad Prism. #p < 0.05, obese IgG2a versus anti-PD-1; **p < 0.01, lean versus obese IgG2a; ˆp < 0.05, ˆˆp < 0.01, and ˆˆˆp < 0.001, lean anti-PD-1 versus obese IgG2a; and ‡p < 0.05 and ‡‡p < 0.01, lean versus obese anti-PD-1. n = 9–12.
(B) Tumor progression in individual mice in each diet and treatment group from (A). The percentage of change in tumor volume is shown below each rank-sorted treatment group. The number of mice with regressed tumors is shown out of the total.
(C) Tumor volume at the endpoint.
(D) Tumor weight at the endpoint. ***p < 0.001 by two-way ANOVA with Tukey post hoc test. n = 9–12.
(E) Number of mitosis per 10 high-power fields (HPFs) in obese tumors was quantified. *p = 0.013 by Student’s t test, n = 3–4. Representative H&E images of mitotic nuclei are indicated by the white arrow. Scale bar is 20 μM.
(F) Number of regressed (undetectable) tumors compared with total palpable tumors at the endpoint.
Data are shown as mean ± SEM for all images, except for (B) and (F). See also Figure S1.
Anti-PD-1 increased tumor regression in lean mice
In lean mice, there was an overall lack of tumor growth regardless of therapeutic intervention (Figures 1A–1D). Despite lack of progression in lean mice, anti-PD-1 treatment reduced tumor weight more than 12-fold in lean mice compared with IgG2a controls (Figure 1D). In addition, greater tumor regression was evident in lean mice after immunotherapy compared with obese mice. Of the tumors in obese mice treated with control IgG2a, none of the 11 tumors regressed (0% regression). In the obese mice treated with anti-PD-1, 4 of the 12 tumors regressed to undetectable by day 18 or 21 (33.3% regression, Figure 1F). However, in lean mice, 3 tumors regressed of the 7 detectable tumors with measurable volumes at day 6 or later in the IgG2a group (42.9% regression). Strikingly, of the 9 detectable tumors in the anti-PD-1 group, 8 regressed to undetectable (88.9% regression, Figure 1F).
Anti-PD-1 reversed immunosuppression in the tumor microenvironment of obese mice
We next examined the TME for the composition of immune cells that are established to mediate antitumor immune response. Dendritic cells (DCs) are of myeloid origin and are important in sampling the TME and antigen presentation for induction of an effective antitumor immunity (Chaib et al., 2020). In tumors from obese mice, anti-PD-1 therapy significantly elevated tumor DC (tDC) content (F4/80−, Ly6C−, major histocompatibility complex class II [MHCII]+, and CD11c+, Figure 2A) compared with isotype-treated obese tumors. Tumor-associated Macs (TAMs) and their precursors account for a large fraction of the myeloid infiltrate in most solid tumors. In tumors of obese mice, anti-PD-1 therapy did not change TAM frequencies (CD11b+, F4/80+, Ly6G−, and Ly6C-low, Figure 2B). However, treatment with anti-PD-1 significantly increased proinflammatory M1 TAMs (TAM-MHCII-high, Figure 2C) while significantly decreasing immunosuppressive protumoral M2 Macs (TAM-MHCII-lo, Figure 2C). Overall, the M1/M2 ratio in obese tumors indicated significant reprogramming toward a more potent antitumor phenotype with reduced immunosuppressive innate immune cells with PD-1 treatment (Figure 2D). Treatment with anti-PD-1 increased total T cells (CD45+ and CD3+) in the tumors of obese mice by flow cytometry and immunofluorescent quantification, respectively (Figures 2E and 2F), with a skewing of T cell subsets that included a significant increase in CD8+ T cells (Figure 2G).
Figure 2. Anti-PD-1 increased antitumor immunity in the tumor microenvironment.
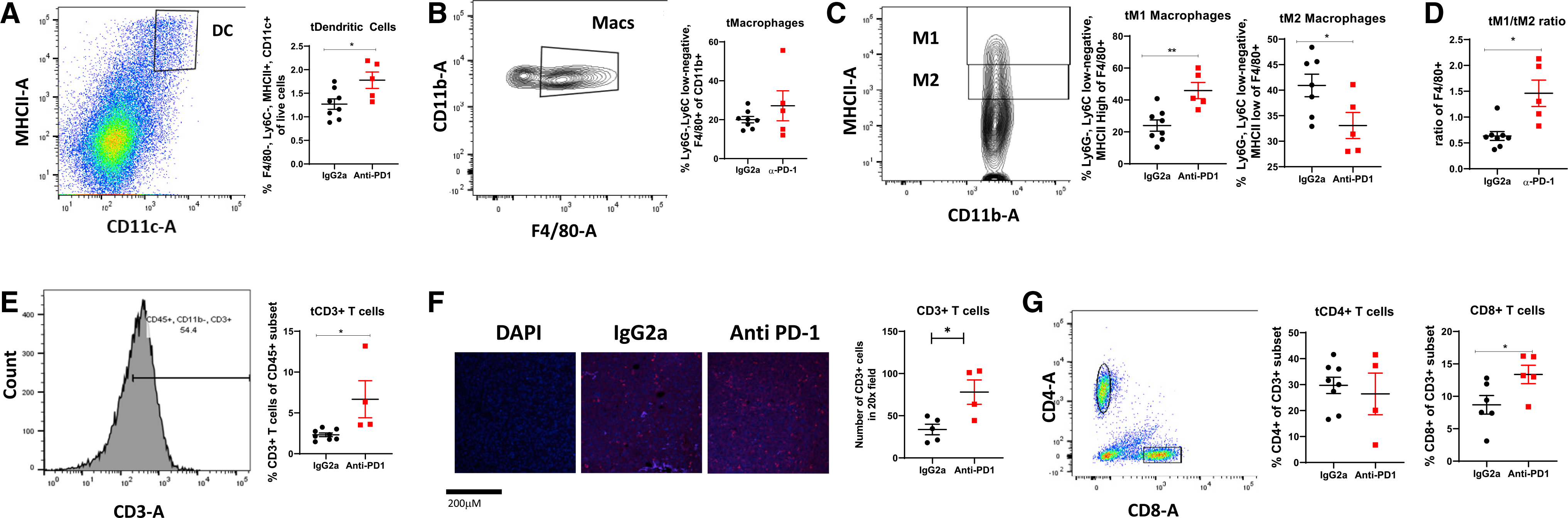
Flow cytometric analysis of tumors from obese mice treated with IgG2a or anti-PD-1 was conducted to quantify frequencies of immune cell infiltrates.
(A) Tumor dendritic cells (tDCs).
(B) Tumor macrophages (tMacs).
(C) M1-like and M2-like tMac subtypes.
(D) M1-like/M2-like ratio of tMacs.
(E) CD3+ T cells.
(F) CD3+ T cell staining by immunofluorescence and quantification in 6 20× fields. Representative 20× images are shown. Scale bar is 200 μM. DAPI nuclear stain shows negative control without primary antibody.
(G) CD8+ and CD4+ T cells.
Data are shown as mean ± SEM. *p < 0.05 and **p < 0.01 by two-way ANOVA. n = 5–8. See also Figures S1 and S2.
Heatmaps showing gene expression differences in tumors from obese mice treated with IgG2a compared with anti-PD-1 indicated significant ICB-induced alterations (Figure 3A; Figures S3A and S3C). Network analysis of genes upregulated by anti-PD-1 shows glycolysis, carbohydrate catabolism, and zinc ion homeostasis were enriched (Figure S3B). Genes downregulated by anti-PD-1 indicate that functions associated with integrin binding, response to hormone stimulus, transmembrane receptor protein serine threonine kinase signaling pathways, and bone morphogenic protein (BMP) signaling were enriched (Figure S3D). Congruent with flow cytometry analyses, treatment with anti-PD-1 increased tumor immune infiltration of CD8+ T cells and Macs, with elevations of several cell types not measured by flow cytometry, including CD4+ T helper 1 (Th1) and CD4+ memory T cells (Figure 3B). The immune score (Yoshihara et al., 2013) predicts immune cell infiltration in bulk RNA sequencing (RNA-seq) samples and was elevated by anti-PD-1 (Figure 3B). Because of the elevation of immune score and flow cytometry findings in response to anti-PD-1 therapy in obese tumors, we next examined the immunologic constant of rejection (ICR) (Roelands et al., 2020; Galon et al., 2013) as a prognostic signature for survival and response to immunotherapy. With anti-PD-1 treatment, tumors displayed a greater Th1, cytotoxic, and chemokine response compared with IgG2a-treated mice. Markers for adhesion were variably changed (Figure S4A). We also evaluated the gene’s tumor inflammation signature (TIS). Tumors with a high TIS score respond well to anti-PD-1 (Danaher et al., 2018; Cristescu et al., 2018). In tumors from obese mice, anti-PD-1 elevated 3 of the 5 genes for antigen-presenting cell (APC) abundance, all 3 genes for T and natural killer (NK) cells, half of the interferon gamma (IFNγ) signature, and every gene in the T cell exhaustion signature. In tumor-adjacent MFP, we note relatively high ICR and TIS in lean mice treated with IgG2a that mostly persists after anti-PD-1. A profound lack of APC and T/NK cell abundance is evident in IgG2a-treated obese tumor-adjacent MFP compared with lean IgG2a with greater T cell exhaustion, some chemokines, and markers for adhesion. The cytotoxic and adhesion signatures in the MFP were potently downregulated by anti-PD-1 in both lean and obese mice. Surprisingly, in contrast to the TME, anti-PD-1 dramatically downregulated the ICR and TIS to the lowest levels among the groups (Figure S4B). Lastly, Database for Annotation, Visualization and Integrated Discovery (DAVID) pathway analysis of tumors revealed key pathways significantly upregulated by anti-PD-1 compared with IgG2a controls, including cytokine activity, positive regulation of the apoptotic process, negative regulation of the mitotic cell cycle, tumor necrosis factor (TNF) signaling, cell chemotaxis, CCR2 chemokine receptor binding, immune response, cytokines and inflammatory response, and prostaglandin biosynthetic process (Figure S4C). Downregulated pathways included negative regulation of the calcium ion concentration, the lipid metabolic process, actin binding, thyroid hormone signaling, adhesion molecules, steroid hormone receptor activity, bile secretion, and cell adhesion (Figure S4D).
Figure 3. Anti-PD-1 alteration of immune cell infiltrates in murine tumors is reflected by up- and downregulated genes and immune cell content that predicts survival in ER− BC patients.
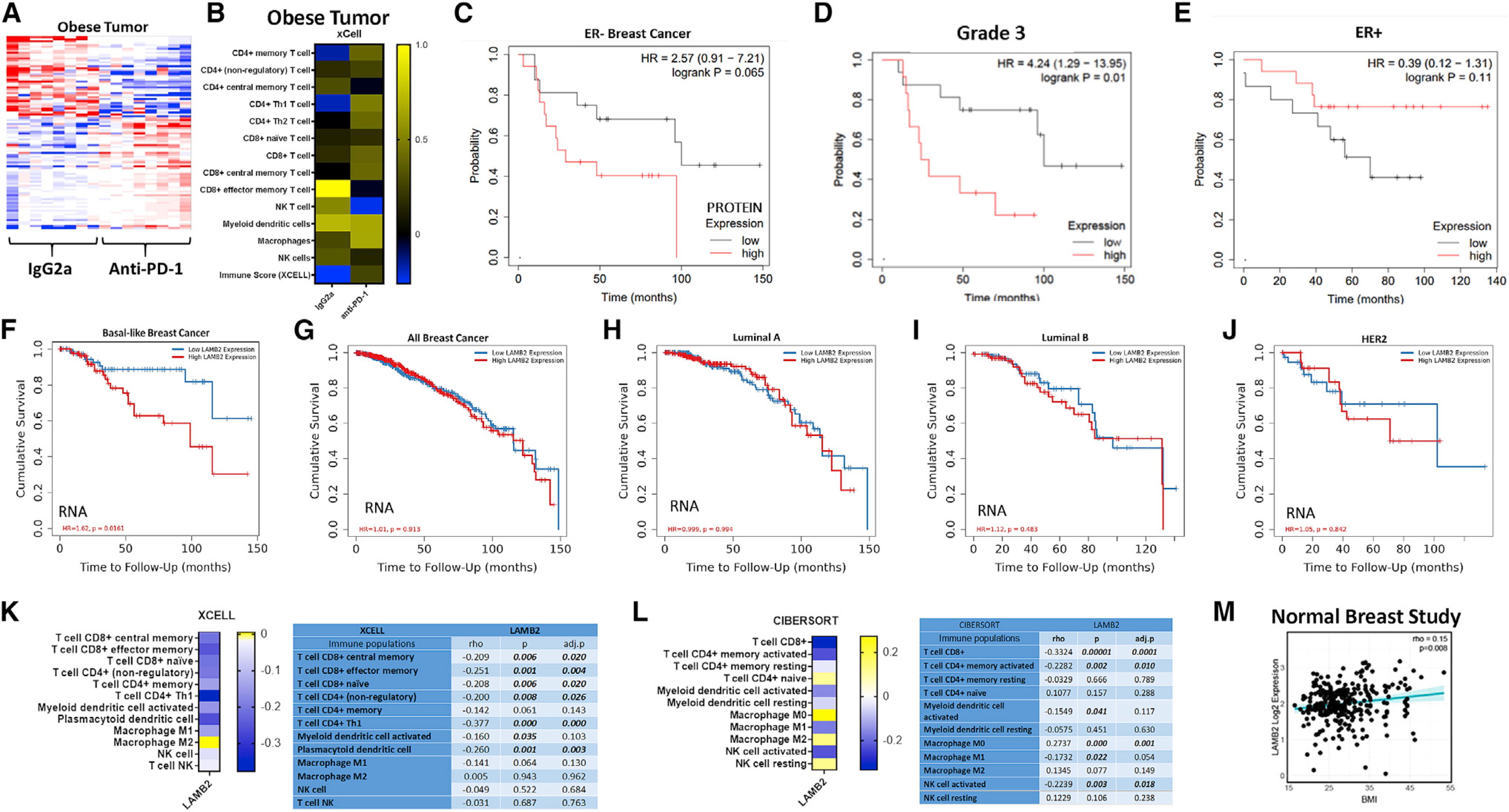
RNA-seq analysis of tumors from obese mice treated with IgG2a or anti-PD-1 was conducted, followed by bioinformatic analyses.
(A) Heatmap showing RNA-seq-normalized, median-centered gene expression (n = 100 genes) of mice treated with IgG2a (n = 8) and anti-PD-1 antibody (n = 8).
(B) Analysis of immune infiltration in tumors from obese mice treated with IgG2a or anti-PD-1 generated using the xCell algorithm in TIMER2.0.
(C–E) Overall survival in patients with low (gray line) or high (red line) protein expression for LAMB2 generated in KmPlotter using the Tang 2018 dataset in ER− tumors (C, HR = 2.57, log rank p = 0.65, n = 54 patients), grade 3 tumors (D, HR = 4.24, log rank p = 0.01, n = 48 patients), or ER+ tumors (E, HR = 0.39, log rank p = 0.11, n = 112).
(F–J) Cumulative survival in high (red line) and low (blue line) mRNA LAMB2-expressing samples using a Cox proportional hazard model in TIMER2.0 for basal-like BC patients from The Cancer Genome Atlas (TCGA) (F, n = 189 patients, HR = 1.62, p = 0.0161), all subtypes of BC combined (G, n = 1,100 patients), luminal A (H, n = 568), luminal B (I, n = 219), or HER2+ (J, n = 82).
(K and L) Prediction of LAMB2-associated immune cell infiltration of basal-like BC patient tumors from TCGA using (K) xCell and (L) CIBERSORT in the TIMER2.0 Immune Estimation module. Statistics are shown to the right of each heatmap.
(M) Association of LAMB2 and BMI in normal breast tissue from the Normal Breast Study (rho = 0.15, p = 0.008, n = 178 subjects).
See also Figures S2–S5.
Anti-PD-1-regulated genes predict the survival in estrogen receptor-negative (ER−) BC patients from The Cancer Genome Atlas (TCGA)
One gene downregulated by anti-PD-1 in obese tumors was laminin subunit beta-2 (Lamb2) (Figure S3C). In line with downregulation of Lamb2 with anti-PD-1 in obese murine tumors, low LAMB2 protein expression was associated with greater overall survival in estrogen receptor (ER)-negative (ER−) tumors (Ősz et al, 2020). In ER− tumors, median survival was 29 months in high expressers versus more than 3-fold longer at 100 months in low LAMB2 protein expressers (Figure 3C, HR = 2.57, and log rank p = 0.65, n = 54 patients). In addition, patients with aggressive tumors (grade 3) displayed improved survival when tumors had low LAMB2 protein expression (Figure 3D, HR = 4.24, log rank p = 0.01, n = 48). In contrast, low LAMB2 protein expression in ER+ patients was not protective but reduced survival compared with high expressers, although not significantly (Figure 3E, HR = 0.39, log rank p = 0.11, n = 112). Low LAMB2 mRNA expression is associated with significantly improved overall survival (OS) in basal-like BC patients (Figure 3F, HR = 1.62, p = 0.0161, n = 191 patients). Interestingly, LAMB2 expression did not affect OS upon examination of all BC patient subtypes (Figure 3G, n = 1,100 patients) or other subtypes, including luminal A (n = 568), luminal B (n = 219), or HER2+ (n = 82) (Figures 3H–3J, respectively). The immune landscape in patient samples was next evaluated in correlation with LAMB2 gene expression. Antitumor immune cells such as CD8+ T cells (central memory, effector memory, and naive), CD4+ T cells (non-regulatory and Th1), and plasmacytoid DCs were significantly negatively associated with LAMB2 expression in TCGA samples from basal-like BC patients (Figure 3K). Patient LAMB2 expression positively correlated with immunosuppressive M2 Macs (Figure 3K). LAMB2 negatively correlated with CD8+ T cells, CD4+ memory activated cells, and activated NK cells, whereas LAMB2 positively correlated with M0 (significantly), M2 Macs, resting NK cells, and naive CD4+ T cells (Figure 3L, cell-type identification estimating relative subsets of RNA transcripts [CIBERSORT]). Lastly, to determine whether obesity modulated LAMB2 expression, we examined patient data from the Normal Breast Study, which includes a collection of patient normal mammary glands (typically isolated from reduction mammoplasty) and tumor-adjacent breast tissue defined as pathologically normal (Sandhu et al., 2016; Sun et al., 2018a, 2018b), as well as BMI. Compared with normal-weight subjects, obesity increased LAMB2 (Figure 3M, p = 0.008, n = 178 subjects). Furthermore, RNA-seq analysis of tumor-adjacent MFP adipose tissue showed slight obesity-mediated elevations in Lamb2 compared with lean mice (data not shown).
Obesity and anti-PD-1 affected pathways and immune infiltrates associated with cancer progression in tumor-adjacent adipose tissue of the mammary fat pad
The MFP is primarily composed of adipose tissue, which includes not only lipid-laden adipocytes but also immune and other stromal cells that greatly affect BC risk, progression, and response to therapy (Cozzo et al., 2017; Lengyel et al., 2018; Sipe et al., 2020). Therefore, we next examined gene expression in tumor-adjacent MFP from both lean and obese mice. We first compared tumor-adjacent MFP from lean and obese controls treated with IgG2a to determine whether typical obesity-induced pathways were prevalent in MFP. RNA-seq analysis of tumor-adjacent MFP of obese versus lean mice treated with IgG2 revealed differential regulation of genes shown in a heatmap (Figure S5A). Increased immune infiltration of CD8+ T cells, DCs, monocytes, and NK cells, along with decreased B cells, was evident in obese compared with lean IgG2a-treated mice (Figure S5B). Pathway analysis identified upregulation of typical pathways associated with obesity (Cozzo et al., 2017) including inflammatory response, cytokine activity, chemotaxis, wound healing, and TNF receptor signaling, whereas many pathways were downregulated, including lipid metabolic processes, the tricarboxylic acid cycle (TCA) cycle, and insulin signaling pathways (Figures S5E and S5F, respectively).
Lastly, we examined the impact of ICB on tumor-adjacent MFP. A heatmap of RNA-seq analysis showing gene expression differences in tumor-adjacent MFP from obese mice treated with IgG2a or anti-PD-1 indicated significant variation of gene expression in response to anti-PD-1 in the MFP (Figure S5C). Anti-PD-1 not only increased CD8+ and CD4+ T cells in tumor-adjacent MFP but also increased Macs and DCs compared with IgG2a-treated obese mice (Figure S5D). Treatment with anti-PD-1 decreased B cells and NK cells (Figure S5D). Few pathways were found to be upregulated in DAVID analysis by anti-PD-1 immunotherapy in obese tumor-adjacent MFP compared with IgG2a-treated obese controls, including pleckstrin homology-like domain, potassium channel, and tetraspanin (Figure S5G). In contrast, many metabolic and signaling pathways were downregulated, including pathways in DNA repair and replication, unfolded protein response, and mitotic spindle assembly (subset shown in Figure S5H).
Obesity expanded protumor immunosuppressive splenic MDSCs and M2 macrophages that were reduced by anti-PD-1
To examine the impact of obesity on systemic antitumor immunity, we next queried the impact of obesity and ICB by comparing the splenic immune landscape of lean and obese tumor-free mice versus tumor-bearing mice. The spleen acts as a reservoir for Macs and myeloid progenitor cells such as MDSCs to mobilize in fighting cancer (Cortez-Retamozo et al., 2012; Franklin et al., 2014). MDSCs are a poorly understood immunosuppressive innate cell population that comprises two major subtypes: monocytic MDSCs (M-MDSCs, quantified as CD11b+ Ly6C-hi, Ly6G−) and polymorphonuclear MDSCs (PMN-MDSCs, quantified as CD11b+ Ly6C-lo, Ly6G+) (Tcyganov et al., 2018). Both MDSC subtypes suppress innate and adaptive immunity in the TME via production of immune suppressive factors (Groth et al., 2019). In spleens from lean and obese tumor-free mice, total splenic MDSCs (sMDSCs), sMDSCs expressing MHCII or PD-L1, and the PMN-sMDSC and M-sMDSC subsets were not significantly regulated by obesity (Figures 4A, 4C, 4E, and 4G). However, in tumor-bearing mice, obesity primed the systemic immune milieu to skew to a potent immunosuppressive phenotype. In tumor-bearing mice treated with IgG2a controls, obesity increased total sMDSC content by 6-fold (Figure 4B), including increases in MHCII+ sMDSCs and PD-L1+ sMDSCs (Figures 4D and 4F). Likewise, although obesity did not alter M-sMDSC or PMN-sMDSC subtypes in the absence of tumors, the presence of a tumor created a highly responsive microenvironment wherein both M-sMDSC and PMN-sMDSC subtypes were significantly increased by obesity by more than 3-fold and 5-fold, respectively (Figure 4H). Anti-PD-1 therapy decreased total sMDSCs, as well as every MDSC subtype examined, including MHCII+ sMDSC, PD-L1+ sMDSC, M-sMDSC, and PMN-sMDSC subtypes (Figures 4B, 4D, 4F, and 4H). In contrast, splenic DCs (sDCs) were slightly elevated in the spleen of obese tumor-free and tumor-bearing mice (Figures 4I and 4J, respectively), whereas anti-PD-1 therapy did not affect sDC frequencies (Figure 4J). Lastly, total splenic Macs (sMacs), as well as M1- and M2-like sMacs, were examined in tumor-free mice, with slight yet non-significant obesity-mediated increases in cell populations evident (Figures 4K and 4M). However, PD-L1+ sMacs significantly increased (Figure 4O). In contrast to the tumor-free state, sMacs in tumor-bearing mice, like sMDSCs, displayed dynamic responsiveness to obesity. In IgG2a controls, obesity significantly increased total sMacs (Figure 4L) primarily the M2-like subtype (Figure 4N). Importantly, similar to the potent efficacy of ICB in sMDSCs, anti-PD-1 significantly decreased total sMacs (Figure 4L). Although M1-like sMacs were not regulated by obesity or anti-PD-1, M2-like sMacs displayed a significant reversal of obesity-mediated upregulation after treatment with anti-PD-1 (Figure 4N). Lastly, PD-L1+ sMacs were increased by 3-fold in tumor-bearing mice (Figure 4P). Critically, after treatment with anti-PD-1, the percentage of PD-L1+ sMacs was significantly reduced to a concentration matching those detected in lean mice (Figure 4P).
Figure 4. Immune checkpoint blockade reprograms immunosuppressive splenic immune cells induced by obesity.
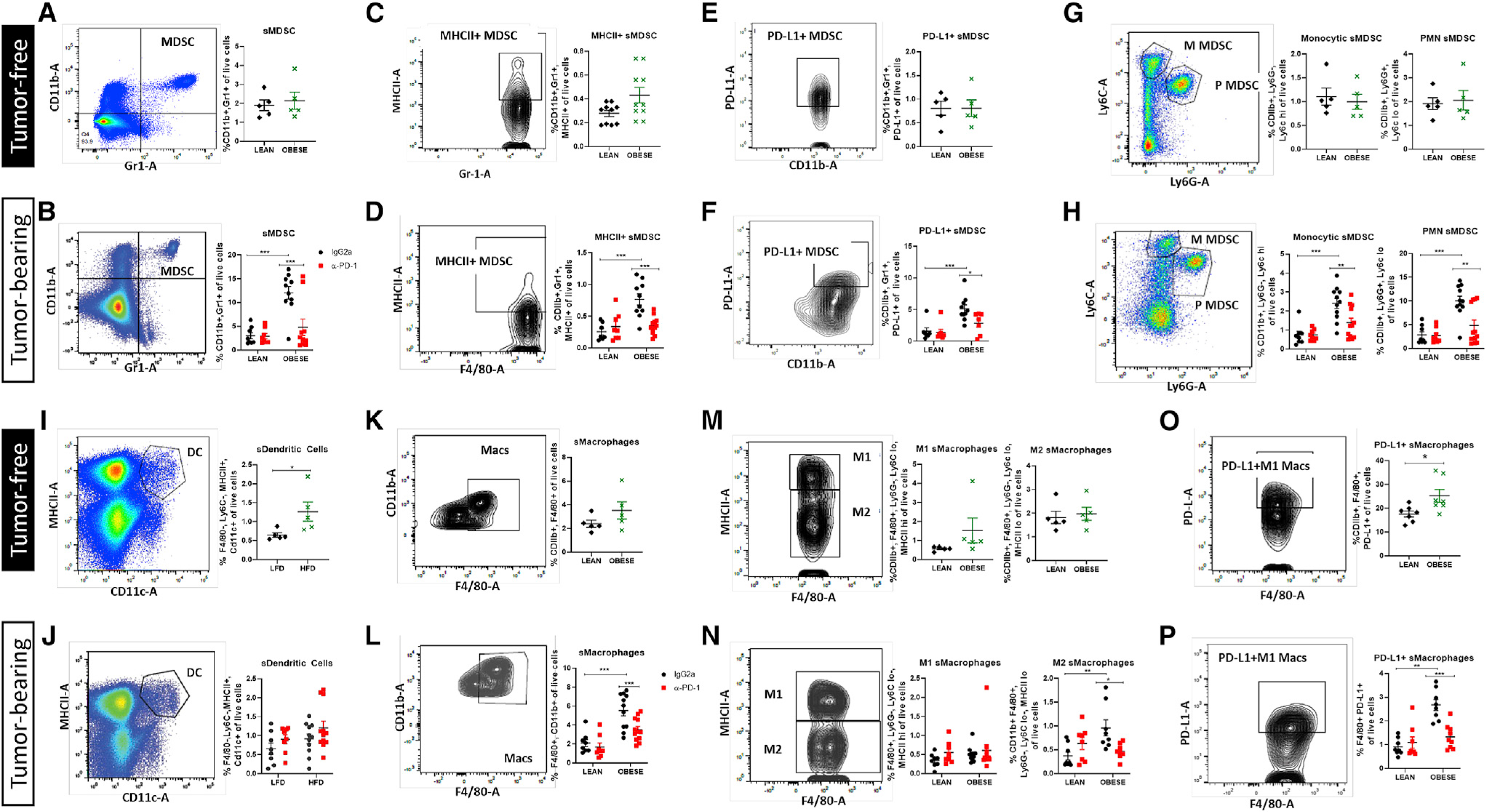
Flow cytometric analysis of spleens from lean and obese tumor-free mice (black diamond and green X) and lean and obese tumor-bearing mice treated with IgG2a or a-PD-1 (black circle and red square) is presented.
(A and B) Splenic myeloid-derived suppressor cells (sMDSCs).
(C–F) sMDSCs positive for MHCII (C and D) or immune checkpoint PD-L1 (E and F).
(G and H) Monocytic (M) and polymorphonuclear (PMN, P) subtypes of sMDSCs.
(I and J) Splenic DCs (sDCs).
(K and L) Splenic Macs (sMacs).
(M and N) M1-like and M2-like sMac subtypes.
(O and P) Frequency of PD-L1+ sMacs.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 by two-way ANOVA. n = 8–10. See also Figure S2B.
Obesity induced systemic changes in bone marrow myeloid cell composition that were reversed with anti-PD-1
Obesity dramatically altered the TME and systemic antitumor immune response; therefore, we next sought to investigate the obesity-mediated effect on the nascent immune system. We first examined bone marrow from tumor-free mice that were lean or obese. Bone marrow DCs (bDCs), bone marrow Macs (bMacs), and M1- and M2-like bMacs were increased in the bone marrow of obese mice compared with lean mice, but this upregulation was absent from tumor-bearing mice and was not altered by anti-PD-1 (data not shown). Obesity led to a significant increase in total bone marrow MDSCs (bMDSCs), primarily through a significant increase in M-bMDSCs, not PMN-bMDSCs, in the absence of tumors (Figures 5A and 5C). Total bMDSC content doubled in tumor-bearing mice (~45%) compared with tumor-free mice (~23%) (Figures 5A and 5B). In tumor-bearing mice, bMDSCs were significantly elevated by obesity (Figure 5B); however, the increase results from a significant increase in PMN-bMDSC frequencies, with no increase in M-bMDSCs (Figure 5D). Anti-PD-1 therapy significantly reduced total bMDSCs and PMN-bMDSCs in obese tumor-bearing mice (Figures 5B and 5D).
Figure 5. Immunosuppressive bone marrow immune cell changes induced by obesity are reprogrammed by immune checkpoint blockade.
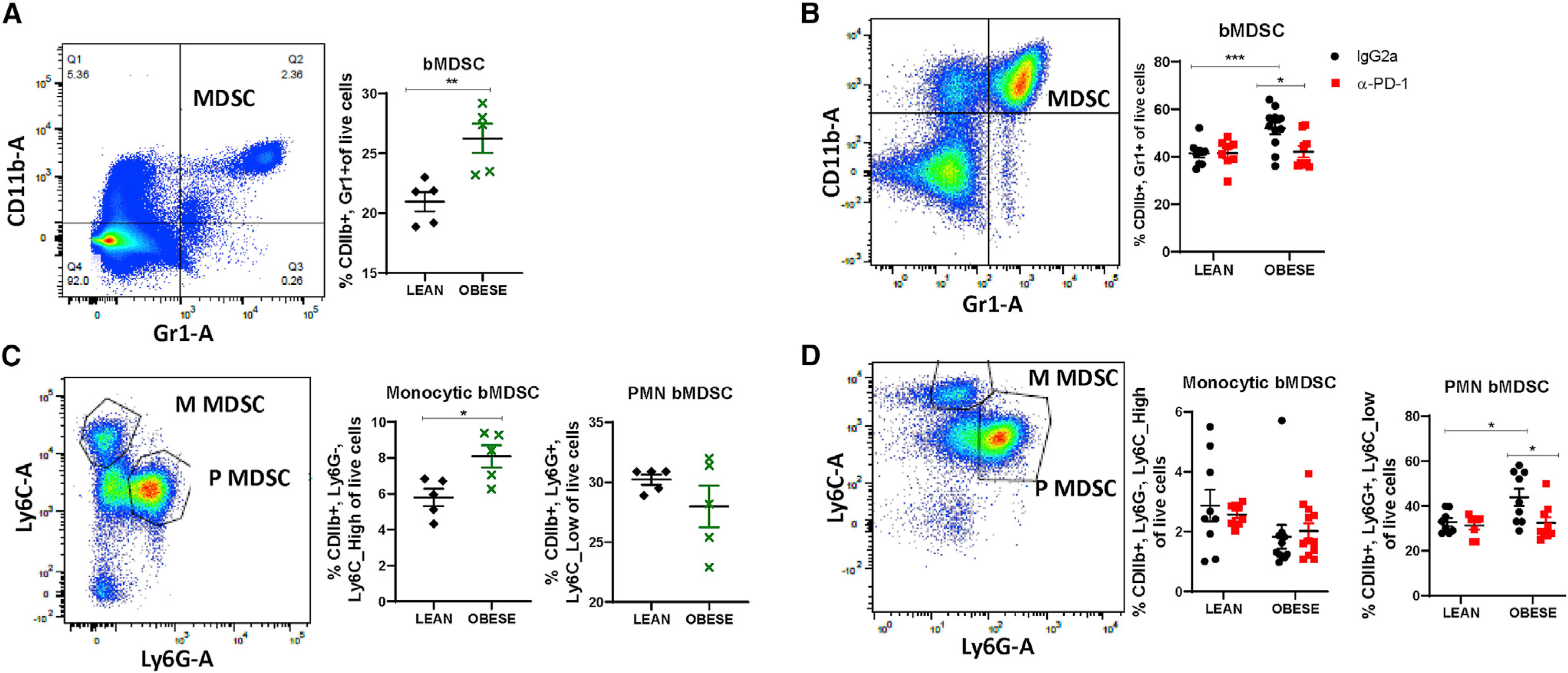
Flow cytometric analysis of bone marrow from lean and obese tumor-free mice (black and green) and lean and obese tumor-bearing mice treated with IgG2a or anti-PD-1 (black and red) is presented.
(A and B) Bone marrow-derived myeloid-derived suppressor cells (bMDSCs).
(C and D) M and P (PMN) subtypes of bMDSCs.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 by two-way ANOVA. n = 8–10. See alsoFigures S3 Figure S2.
Obesity and anti-PD-1 immunotherapy shape the gut microbiome
The gut microbiome can potently influence response to ICB in cancer (Frankel et al., 2017; Gopalakrishnan et al., 2018; Langan et al., 2018; Matson et al., 2018; Routy et al., 2018; Vétizou et al., 2015). Thus, to determine whether obesity, BC, and ICB affect the gut microbiome and how these changes associate with tumor progression, luminal content was isolated from the jejunum (JE, small intestine) and cecum (CE, large intestine) of obese tumor-bearing IgG2a and anti-PD-1-treated mice against untreated tumor-free obese controls to detect tumor-specific and ICB-specific effects (Figure 6; Figure S6). Consistent with prior microbiome obesity research work (Martinez-Guryn et al., 2018), HFD-induced obesity expanded the relative abundance of Clostridia and Bacilli at the phyla/class level in JE, partly through increased levels of the genera Enterococcus and Lactococcus, while concomitantly decreasing Bifidobacterium and Allobaculum (Figures S6A–S6G). Within the more taxonomically diverse CE, obesity-increased Clostridia and Gammaproteobacteria were evident at the phyla/class level, as driven by shifts in many taxa at the genus level (Figures S6H–S6N). Despite similar alpha diversity between diets, statistically significant shifts in beta diversity were diet dependent in CE, suggesting diet remains the dominant driver of community composition.
Figure 6. The gut microbiome is affected by tumor and anti-PD-L1 therapy.
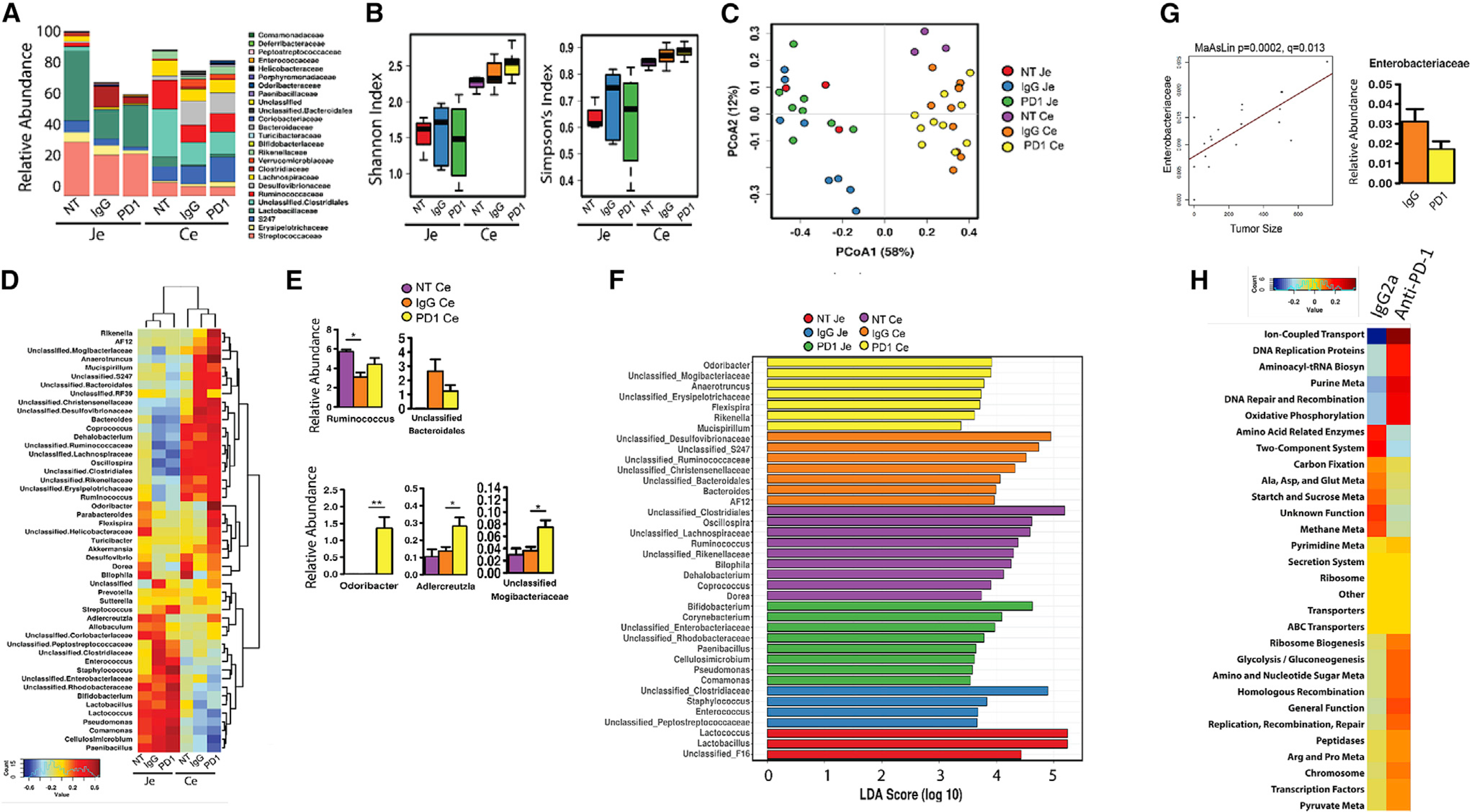
Fecal contents were isolated from the jejunum (JE) or cecum (CE) at sacrifice from obese tumor-free (no tumor [NT]) mice or obese mice with E0771 tumors treated with IgG2a control or anti-PD-1 as above.
(A) Relative abundance of taxonomic composition at the family level.
(B) Alpha diversity by Shannon and Simpson indices.
(C) Beta diversity calculated by Bray-Curtis analysis and displayed as principal coordinate analysis (PCoA). Permutational multivariate analysis of variance (PERMANOVA), R2 = 0.614, p = 0.0003; analysis of similarity (Anosim), R = 0.631, p = 0.001; analysis of multivariate homogeneity of group dispersions (PERMDISP2), p = 0.0026.
(D) Heatmap of the 50 most abundant microbial taxa as calculated by Spearman’s rank correlation coefficient. Dendrograms display group and taxa clustering.
(E) Select species shown for CE taxa altered by IgG2a and anti-PD-1 therapy compared with tumor-free mice (NT). Mean ± SEM of relative abundance is shown. *p < 0.05 and **p < 0.01 by two-way ANOVA.
(F) Linear discriminant analysis (LDA) effect size (LEfSe) by genus for JE and CE.
(G) Tumor size (volume) at the endpoint was correlated with cecal microbial community relative abundance using MaAsLin. Enterobacteriaceae is shown (p = 0.0002, q = 0.013). The graph on right is the relative abundance in CE.
(H) Heatmap of the 30 most abundant phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) predicted metabolic pathways calculated by Spearman’s rank correlation coefficient in CE. *p < 0.05, **p < 0.01, and ***p < 0.001.
n = 8–10. See also Figure S6.
The presence of a tumor reduced abundance in the CE and JE communities (Figure 6A). Alpha diversity determined by the Shannon and Simpson indices demonstrated greater CE diversity compared with JE overall (Figure 6B). Administration of PD-1 therapy was associated with a reduction in alpha diversity within the JE; the reciprocal trend was observed in CE, with immunotherapy correlating with a nominal increase in alpha diversity (Figure 6B). Beta diversity calculated by Bray-Curtis analysis showed significantly differential clustering between intestinal regions (Figure 6C, permutational multivariate analysis of variance [PERMANOVA], R2 = 0.614, p < 0.0003). Within JE, little divergence was observed, whereas the separation of tumor-free from tumor-bearing microbial beta diversity was more evident in the CE (Figure 6C). Spearman’s rank correlation coefficient clustering of the 50 most abundant microbial genera is represented by a heatmap, demonstrating that JE and CE have vastly different communities represented. Tumor-bearing mice clustered more similarly within sites, and tumor-free mice remained more distinct (Figure 6D). Clusters were evident in CE, in which several microbes were high in tumor-bearing compared with tumor-free mice, some were uniquely upregulated by anti-PD-1 compared with IgG2a or tumor-free mice, and others were dramatically downregulated by anti-PD-1 therapy (Figures 6D and 6E), with select significantly regulated cecal microbes highlighting tumor- and ICB-specific effects on abundance (Figure 6E). In particular, Akkermansia, Anaerotruncus, Rikenella, and AF12 increased in tumor-bearing IgG2a-treated obese mice compared with tumor-free mice. These 4 taxa were again elevated with anti-PD-1 in obese CE, whereas Enterobacteriaceae, Clostridiaceae, Sutterella, unclassified RF39, Mucispirillum, and Bacteroidales were increased by the presence of a tumor but reversed by anti-PD-1 ICB in obese murine CE. In contrast, a few microbes were reduced in tumor-bearing mice that were elevated by anti-PD-1, including Lactobacillus, Bilophila, Dorea, Ruminococcus, and Rikenellaceae. Several taxa were uniquely high in obese anti-PD-1-treated mice, including Adlercreutzia, Tuberibacter, Helicobacteraceae, Flexispira, Parabacterioides, and Odoribacter (Figures 6D and 6E).
Focusing on cecal correlations with ICB therapy, anti-PD-1 elevated the cecal content in lean mice of Akkermansia, Bacteroides, and Desulfovibrio (Figure S6K), with the greatest elevations in Coriobacteriaceae, Bifidobacterium, Sutterella, and Allobaculum (Figure S6N). Many microbes displayed reduced proportional abundance in lean anti-PD-1-treated murine CE, including Anaerotruncus, Coprococcus, Enterococcus, Ruminococcus, Rikenella, Mogibacteriaceae, Adlercreutzia, Mucispirillum, Clostridiaceae, Lactococcus, Rikenellaceae, and Dehalobacterium (Figures S6K and S6N). In obese mice, anti-PD-1 elevated cecal contents of Lactobacillus, Bifidobacterium, Adlercreutzia, Anaerotruncus, and Mogibacteriaceae, and some of the most increased microbes included Odoribacter, Parabacterioides, Flexispira, Helicobacteraceae, Turicibacter, Akkermansia, Dehalobacterium, and Rikenella (Figures S6K and S6N). Reductions by anti-PD-1 in obesity included lower Bacteroides, Paenibacillus, Cellulosimicrobium, and Enterobacteriaceae. Anti-PD-1 upregulated Akkermansia and Bifidobacterium in both lean and obese mice. In contrast, there were no common microbes downregulated by anti-PD-1 in lean and obese mice. However, dichotomous regulation was evident with several microbes, including Adlercreutzia, Anaerotruncus, Ruminococcus, Rikenella, Mogibacteriaceae, and Dehalobacterium, suggesting that there is no standard anti-PD-1 effect associated with microbiome but that lean versus obese state differentially affects the crosstalk between ICB and microbiome. To determine which taxa were enriched by the site, presence of tumors, and anti-PD-1 therapy, we assessed linear discriminant analysis (LDA) effect size (LEfSe) (Figure 6F). JE contained an abundance of Lactococcus and Lactobacillus in the absence of tumors, likely reflecting the bulk of the microbes from the diet (data not shown). Anti-PD-1 therapy induced enrichment of the genera Bifidobacterium, Corynebacterium, Paenibacillus, Cellulosimicrobium, Peudomonas, and Comamonas, as well as the families Enterobacteriaceae and Rhodobacteraceae, in the JE (Figure 6F). In the CE, the genera Odoribacter, Anaerotruncus, Flexispira, Rikenella, and Mucispirillum and families Mogibacteriaceae and Erysipelotrichaceae were enriched following anti-PD-1 therapy (Figure 6F).
Microbes correlate with tumor size
Because tumor volumes varied at the experimental endpoint, we employed multivariate analysis by linear models (MaAsLin) to determine whether tumor volume correlated with key taxa in tumor-bearing obese mice treated with IgG2a or anti-PD-1. No microbes correlated significantly with tumor size using obese jejunal contents (data not shown). However, Enterobacteriaceae abundance from the obese tumor-bearing CE significantly positively correlated with tumor size, which agreed with anti-PD-1 reducing the relative abundance of this family (Figures 6D and 6G). When cecal microbes isolated from lean mice treated with IgG2a or anti-PD-1 were examined in concert with obese treated mice, MaAsLin revealed microbes that significantly negatively correlated with tumor size, including Coriobacteriaceae, Bifidobacterium, and Allobaculum (Figure S6O). These 3 microbes were greatly elevated in lean PD-1-treated CE compared with IgG2a-treated mice yet were undetectable in either IgG2a or anti-PD-1 obese cecal contents (Figures S6K and S6N).
Predicted metabolic pathways correlate with anti-PD-1
Finally, to determine whether metabolic alterations in the microbial community were altered by anti-PD-1 that could potentially affect tumor outcomes and the immune milieu, we employed Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt). JE and CE contents markedly differ in predicted metabolic pathways, in line with observed differences in microbial abundance (Figures S6F and S6M). Within the CE, in which the most significant correlations with tumor size were detected, functional pathways were altered by anti-PD-1 compared with IgG2a controls, including upregulation of ion-coupled transport, DNA replication pathways, and oxidative phosphorylation, glycolysis, and pyruvate, arginine, and proline metabolism. In contrast, metabolic pathways predicted to be downregulated by anti-PD-1 included amino acid, methane, and starch and sucrose metabolism (Figure 6H).
DISCUSSION
This study uniquely highlights the impact of ICB on the peripheral innate and adaptive immune milieu in lean and obese mice, as well as in the local TME in obesity-associated BC. Obesity leads to sustained low-grade inflammation and elevated immunosuppression, which can increase the risk of cancer and negatively affect therapy (Cozzo et al., 2017; Lengyel et al., 2018). To be able to determine effects of ICB in obesity, we used the C57BL/6J strain, a murine model well established to become obese on a HFD (Engber, 2018). We demonstrate that young female mice on obesogenic diets gained weight through large increases in adiposity without the need for ovariectomy. Importantly, anti-PD-1 therapy had no impact on weight or body composition throughout the study, which rules out confounding effects of therapy-induced weight loss. This study emphasizes the complexity of examining immunotherapy in immune-competent mice with consideration of the natural variance in weight gain of the C57BL/6 model (Engber, 2018), tumor growth, innate regression, and response to ICB (Gao et al., 2020; Kasikara et al., 2019; Crosby et al., 2018; Wu et al., 2018). A caveat of this study is that in the comparison of ICB efficacy in tumors between obese and lean states, one must consider that the size and immune content of the tumors vary inherently as tumors progress. Future studies matched on tumor size may enable more controlled comparisons, potentially by allowing lean tumors to progress longer. Likewise, the impact on obesity per se compared with diet composition needs to be compared to fully understand the response to ICB. Another limitation of our study is that we did not examine additional outcomes, such as survival, metastasis, or recurrence after removal of therapy, which are critical factors that would affect translation to future patient care.
Obesity leads to increased immune escape by the tumor through several pathways, including immune cell dysfunction and exhaustion (Woodall et al., 2020). In other cancer or non-cancer models, PD-1 and PD-L1 expression or frequency of PD-L1-positive immune cells are reported to be increased by obesity, leading to increased immunosuppression (Yang et al., 2016; Del Cornò et al., 2016; Shirakawa et al., 2015, Circulation, abstract). The obese microenvironment regulates PD-1 and PD-L1 transcription or protein stabilization through leptin and other inflammatory cytokines (Lim et al., 2016; Wang et al., 2019). In melanoma, CD4+ and CD8+ T cells from obese patients, mice, and primates displayed elevated exhaustion compared with lean counterparts with greater PD-1 expression and reduced proliferative and activation capacity (Wang et al., 2019). Furthermore, adipocytes express high levels of PD-L1 that affect ICB efficacy. Pharmacologically blunting adipocyte differentiation reduced PD-L1 and led to increased ICB antitumor response (Wu et al., 2018). Transgenic mice lacking adipocyte-specific PD-L1 have reduced tumor progression, indicating reduced immunosuppression mediated by adipocyte PD-L1 (Wu et al., 2020a). Because tumors were exceedingly small or regressed in lean mice in our study, we were unable to examine lean compared with obese impacts in the TME. Interestingly, obesity-induced elevations in PD-L1 or PD-1 on some systemic cells, such as splenic MDSCs, were not detected in the absence of tumors. However, in the presence of a tumor, cells were primed for potent obesity-mediated increases in PD-L1 on splenic MDSCs and Macs.
Transcriptomic and flow analysis demonstrated elevations of CD8+ T cells, CD4+ Th1 cells, and overall immune score in obese TME after ICB. CD8+ T cells also increased in obese tumor-adjacent mammary fat pad compared with lean controls. ICB therapy reinvigorates DC function to generate potent anticancer T cell immunity (Chaib et al., 2020), which was evident in tumors from obese mice. In the periphery, DCs were elevated by obesity in the spleen in both tumor-free and tumor-bearing mice showing a system-wide impact on these antigen-presenting cells. Surprisingly, although anti-PD-1 increased tumor-localized DCs, treatment with anti-PD-1 did not alter DC populations in lean or obese spleens from tumor-bearing mice, which suggests a TME-specific impact of ICB on DC content. Future studies must quantify obesity’s impacts on DC subtypes and function, including cDC1, cDC2, or plasmacytoid DC, which affect T cell function differentially (Chaib et al., 2020).
Based on stimuli provided by chemokines, cytokines, and other factors secreted by tumor and immune cells, monocytes are recruited to the TME, where they can differentiate to TAMs, whereas tissue-resident Macs have the capacity to proliferate (Cozzo et al., 2017; Franklin et al., 2014). In the TME, Macs typically lose antitumor function because local cytokines and metabolites promote an immunosuppressive TAM (M2-like) polarization state. TAMs affect every aspect of tumorigenesis and metastasis and negatively influence response to anticancer therapies. However, Macs can also play an important role in tumor immunosurveillance as M1-like classically activated Macs, which are antitumor via the production of proinflammatory cytokines and activation of cytotoxic T cells. More M1-like TAMs are associated with better prognosis and outcomes. In the obese TME herein, treatment with anti-PD-1 elevated the M1/M2 ratio in the tumors of obese mice, demonstrating that anti-PD-1 was successful in reversing Mac polarization in the TME despite continued intake of HFD and persistence of obesity. Systemically, M2 Macs were dramatically upregulated by obesity in the IgG2a controls, but only when tumors were present. Critically, the obesity-driven increase in M2s was significantly blunted by anti-PD-1 to frequencies of lean control IgG2a-treated mice, as was quantified in the TME. Immune checkpoint ligand expression on splenic Macs was only modestly increased with obesity in the absence of tumors yet highly increased in tumor-bearing mice. ICB therapy blunted this PD-L1+ splenic Mac population in obese tumor-bearing mice. In vitro studies showed that when PD-L1 of Macs is inhibited, antitumor potential is activated, with increased proliferation and upregulation of multiple Mac inflammatory pathways (Hartley et al., 2018). Altogether, findings provide evidence of the potent systemic effects of the tumor on the splenic myeloid compartment in the obese-permissive environment.
Another innate population, albeit immature and poorly defined, consists of MDSCs, which are generated in the bone marrow (Veglia et al., 2021). In pathologic conditions such as cancer, MDSCs dramatically expand. The spleen acts as a reservoir for MDSCs (Wu et al., 2020b), and patients who have high levels of circulating MDSCs respond poorly to ICB (Chaib et al., 2020; Weber et al., 2018). Our study demonstrated that total sMDSCs and subsets were not significantly affected by obesity in tumor-free mice. This important finding suggests that immunosuppression associated with obesity is not primarily mediated by sMDSCs in the absence of tumors. In contrast, in tumor-bearing mice, both M- and PMN-sMDSC subsets were dramatically upregulated by obesity in the IgG2a controls. Anti-PD-1 significantly reduced total, monocytic, and granulocytic sMDSCs in obese mice to frequencies comparable to those in lean control mice, demonstrating highly effective abrogation of this immunosuppressive population. The percentage of PD-L1+ sMDSCs was not changed by obesity when mice were tumor free; however, this population was highly upregulated with obesity in mice with BC. Thus, obesity in the absence of tumors does not vastly affect splenic myeloid cells. In contrast, the presence of a tumor greatly increases immunosuppressive MDSCs and M2 sMacs, with greater PD-L1-positive cells in obese mice. PD-L1+ MDSCs regulate T cell function in a cell-contact-dependent manner, and blockade of PD-L1 impairs MDSC-mediated T cell suppression (Ballbach et al., 2017).
Lastly, bone marrow is the center for myelopoiesis and is the location of MDSC production in cancer (Tcyganov et al., 2018). Our findings support previous work demonstrating that PMN-bMDSCs are increased with the presence of tumors from melanoma, colon, and lung cancer models (Bian et al., 2018). It appears that bone marrow is more permissive to obesity-mediated impacts compared with the spleen, because total bMDSCs were increased by obesity in both tumor-free and tumor-bearing mice. Like in spleen, anti-PD-1 significantly reduced total bMDSCs, as well as the most prevalent subtype, PMN-bMDSCs, compared with IgG2a treatment in obese mice. As highly prevalent and potently immunosuppressive myeloid cells, ICB-mediated downregulation of PMN-MDSCs in both spleen and bone marrow may mediate observed reductions on obesity-driven tumor progression.
Transcriptomics of the TME and tumor-adjacent MFP suggest that anti-PD-1 has potent effects on both the tumor and the surrounding mammary gland, which supports the role for tumor-adjacent adipose crosstalk to influence tumor progression and response to therapy (Lengyel et al., 2018; Cozzo et al., 2017). We were not able to assess how ICB affected pathways in lean tumors because of tumor regression in this non-immunocompromised model; however, several targets arose from transcriptomic analysis of obese tumors treated with ICB that had relevance to human pathways. For example, anti-PD-1 therapy downregulated several genes in obese tumors, including Lamb2. LAMB2 is part of a complex glycoprotein laminin, consisting of three polypeptide chains (alpha, beta, and gamma). Laminins are important in the basal lamina, with effects on cell differentiation, migration, and adhesion (Rowe and Weiss, 2008). Survival data demonstrate that lower LAMB2 in tumors predicts greater survival uniquely in patients with more aggressive basal-like or ER− BC tumors. Reduced LAMB2 expression positively associates with antitumor immune cells and negatively associates with immunosuppressive cells in patients with BC, which together support the concept that downregulated or lower expression of LAMB2 is protective. We have previously published that tumor-adjacent breast can predict tumor subtype in patients (Casbas-Hernandez et al., 2015), which suggests that elevated LAMB2 measured in normal breast of patients with higher BMI may be a biomarker of high risk and is modifiable by interventions to reduce obesity. Because LAMB2 is increased by obesity and is important in cell adhesion and migration, it could be critical in metastasis, a point to be examined in future studies.
A major concern of ICB or combination therapy is toxicity with treatment-limiting effects, including immune-related adverse events (irAEs) (Larkin et al., 2015). Ideally, finding biomarkers to predict a high rate of response to ICB therapy with few toxicities would enable better personalization of medicine. Hence, we explored associations with obesity-mediated factors that could influence antitumor immunosurveillance to identify putative biomarkers of anti-PD-1 efficacy, namely, the gut microbiome. Obesity affects the gut microbiome (Pierre et al., 2016; Aron-Wisnewsky et al., 2021; Newman et al., 2021). Microbiota, including bacteria and fungi, are increasingly tied to response to ICB or chemotherapy through unknown mechanisms, but recent work suggests mechanisms including microbially derived metabolite signaling on immune cells (Mager et al., 2020; Sipe et al., 2020; Simpson et al., 2021; Langan et al., 2018; Gopalakrishnan et al., 2018; Frankel et al., 2017; Vétizou et al., 2015; Matson et al., 2018; Routy et al., 2018; Iida et al., 2013; Viaud et al., 2013; Sepich-Poore et al., 2021; Sivan et al., 2015). Some specific microbes have arisen as interesting mediators of ICB in preclinical and clinical studies, such as Akkermansia (Roshanravan et al., 2021). Administering Akkermansia increased tumor-infiltrating lymphocytes (TILs) and improved ICB efficacy in germ-free mice following microbial transplant (Routy et al., 2018). Clinical data also support microbial involvement with ICB responders harboring a higher abundance of Akkermansia and Alistipes (Routy et al., 2018; Roshanravan et al., 2021). We found that Akkermansia was upregulated in both lean and obese mice by anti-PD-1 (along with Bifidobacterium), which supports patient findings. Patients who responded to anti-PD-1 therapy displayed greater microbial diversity and specific enrichment of Clostridiales, Ruminococcaceae, and Faecalibacterium (Gopalakrishnan et al., 2018; Matson et al., 2018), whereas poor responders had lower gut microbial diversity and elevated Bacteroidales (Gopalakrishnan et al., 2018). In CE, but not JE, we found that anti-PD-1 increased diversity, consistent with patient data. In other work, ICB responders displayed greater Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium in fecal samples. Fecal microbial transplant of such ICB responder’s fauna reduced melanoma and other cancer progression in mice through the microbially derived metabolite inosine (Matson et al., 2018; Mager et al., 2020; Peng et al., 2020). Fecal microbial transplant also appears to potentiate ICB in patients (Davar et al., 2021; Baruch et al., 2021). In accordance with our data, elevated gut Lactobacillus was associated with ICB response in gastrointestinal cancer (Peng et al., 2020). Three bacterial species—Bifidobacterium pseudolongum, Lactobacillus johnsonii, and Olsenella spp.—significantly enhanced efficacy of immune checkpoint inhibitors (Mager et al., 2020). Consistently, we report that anti-PD-1 elevated Bifidobacterium, Lactobacillus, and Adlercreutzia, the latter being closely related to Olsenella within the family Coriobacteriaceae. Adlercreutzia, Odoribacter, and Mogibacteriaceae were uniquely upregulated by anti-PD-1 in obese mice CE. Within the CE of obese mice, we identified decreased relative abundance of Ruminococcus but greatly elevated Bacteroidales in IgG2a-treated mice, which was reversed with anti-PD-1 therapy, consistent with clinical data. Thus, it appears that a higher ratio of Ruminococcus/Bacteroidales is critical to ICB efficacy in both preclinical and patient studies. In addition, greater cecal Enterobacteriaceae content significantly correlated with increased tumor size but decreased with anti-PD-1. Although the data presented herein is exploratory and correlative, ongoing studies investigating causality will aid in interpretation of obesity-mediated impacts of the gut microbiome on ICB. BMI failed to correlate with microbial diversity in the tumor microbiome of pancreatic cancer (Riquelme et al., 2019), but new studies report that obesity modifies breast tumor microbiota populations. Patient obesity increased the proportional abundance of breast tumor Enterobacteriaceae and reduced the abundance of Ruminococcaceae compared with intratumoral microbiota from non-obese BC patients (Chiba et al., 2020). Collectively, our results support the growing body of literature that demonstrates specific microbial changes in the presence of tumors and potential roles for microbes affecting ICB efficacy (Simpson et al., 2021). We posit that the cecal microbes, including low frequencies of Bacteroidales and Enterobacteriaceae plus high frequencies of Ruminococcus, Odoribacter, Lactobacillus, Adlercreutzia, Mogibacteriaceae, Bifidobacterium, and Akkermansia, represent an ICB-associated signature that is either causal or a putative biomarker of response to be investigated in further preclinical and clinical BC studies.
Altogether, the permissive immunosuppressive milieu of obesity in tumor-free mice was greatly exacerbated in mice bearing tumors, which likely contributes to rapid tumor progression and overall elevated tumor burden in obese mice. ICB studies in mice that are tumor free cannot be extrapolated to mice with tumors, because the immune milieu is incomparable. BC primes the systemic immune system to be highly responsive to obesity, which could have subsequently enabled efficacy of anti-PD-1 to reinvigorate antitumor immunity systemically and in TME (Video S1). Surprisingly, anti-PD-1 increased antitumor immunity even in the immunosuppressive obese state, demonstrating that obesity-induced alterations to the immune milieu are not immutable. Although anti-PD-1 potently reduced obesity-induced tumor progression, anti-PD-1 also led to tumor regression in lean mice. The elevated regression observed in lean mice could result from a greater proportion of antitumor immune cells, such as CD8 T cells or NK cells as detected in tumor-adjacent adipose, and the reduction in inflammatory signals, emphasizing the importance of tumor-stroma crosstalk. Thus, a critical finding is that anti-PD-1 immunotherapy was effective in restoring antitumor immunity, a lean-like immunophenotype, despite persistent obesity. Importantly, these preclinical results corroborate recent clinical findings demonstrating significantly improved outcomes in obese cancer patients treated with PD-1/PD-L1 inhibitors (Woodall et al., 2020).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Liza Makowski liza.makowski@uthsc.edu.
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA-seq expression datasets for tumor and tumor adjacent adipose MFP generated during this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO: GSE174055 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174055) specifically for tumor (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174053) or mammary fat pad (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174044), as described in greater detail on our STAR Methods website. Human Normal Breast Study (NBS) is publicly available at NCBI GEO: GSE50939 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50939.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Eight-week-old female C57BL/6J (Jackson stock number: 000664) mice were fed with either obesogenic high fat diet (HFD, D12492i – 60% kcal derived from fat) or low fat diet (LFD, D12450Ji- 10% kcal derived from fat, Research Diets Inc., New Brunswick, NJ) for seventeen weeks at Jackson labs (Figure S1A). Mice were shipped to the University of Tennessee Health Science Center at 25 weeks of age. Animal studies were performed with approval and in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at the University of Tennessee Health Science Center and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animals were housed in a temperature-controlled facility with a 12-h light/dark cycle and ad libitium access to food and water. Weights are provided in Figure S1. Mice were never singly housed. Mice were randomized to IgG2a or anti-PD-1 interventions.
Cell lines
E0771-luciferase (luc), a kind gift from Dr. Hasan Korkaya, Augusta University) murine adenocarcinoma breast cancer cell line was originally isolated from a spontaneous tumor from C57BL/6 mouse (Kasikara et al., 2019). Cells were cultured as described previously (Sirotnak et al., 1984; Sugiura and Stock, 1952). Briefly, cells were cultured in RPMI containing 10% FBS, 100 UI/mL of penicillin, and (100 μg/ml) streptomycin in a humidified chamber at 37°C under 5% CO2.
METHOD DETAILS
Reagents
All reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Fetal bovine serum (FBS) (GIBCO, Waltham, MA) RPMI 1640 (Corning, Tewksbury, MA), 100X L-glutamine, 100X penicillin/streptomycin HyClone (Pittsburgh, PA), and GIBCO 100X antibiotic mix were obtained from Thermo Fisher (Waltham, MA). Matrigel (Corning, Tewksbury, MA). Antibodies, compensation beads, and reagents are described in the Key Resources Table (Tonbo, (San Diego, CA), Thermo Fisher (Waltham, MA) and Biolegend (San Diego, CA)).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| RedFluor 710 anti-Human/Mouse CD11b (M1/70) Antibody | Tonbo Biosciences | 80-0112 RRID: AB_2621979 |
| VioletFluor 450 anti-Mouse CD45 (30-F11) | Tonbo Biosciences | 75-0451 RRID:AB_2621947 |
| FITC anti-Mouse CD8a (53-6.7) antibody | Tonbo Biosciences | 35-0081 RRID:AB_2621671 |
| PE anti-Mouse F4/80 Antigen (BM8.1) antibody | Tonbo Biosciences | 50-4801 RRID:AB_2621795 |
| PerCp Cy5.5 anti-Mouse Ly-6G (1A8) antibody | Tonbo Biosciences | 651276: RRID:AB_2621899 |
| APC anti-mouse Ly-6C antibody | Biolegend | Cat# 128015, RRID:AB_1732087 |
| Brilliant Violet 711 anti-mouse CD274 (B7-H1, PD-L1) antibody | Biolegend | 124319, RRID:AB_2563619 |
| Brilliant Violet 605 anti-mouse CD4 antibody | Biolegend | 100548, RRID:AB_2563054 |
| Brilliant Violet 421 anti-mouse CD279 (PD-1) antibody | Biolegend | Cat# 109121, RRID:AB_2687080 |
| Brilliant Violet 650 anti-mouse CD11c antibody | Biolegend | Cat# 117339, RRID:AB_2562414 |
| Brilliant Violet 785 anti-mouse CD3epsilon antibody | Biolegend | Cat# 100355, RRID:AB_2565969 |
| PE/Cy 7 anti-mouse I-A/I-E antibody | Biolegend | Cat# 107629, RRID:AB_2290801 |
| Anti-Mouse CD16 / CD32 (2.4G2) antibody | Tonbo Biosciences | Cat# 70-0161, RRID:AB_2621487 |
| Alexa Fluor 488 anti-mouse Gr-1 antibody | Biolegend | Cat# 108419, RRID:AB_493480 |
| Anti-PD-1 antibody | Bio X Cell | BE0146, RRID:AB_10949053 |
| rat IgG2a isotype control antibody | Bio X Cell | Cat# BE0089, RRID:AB_1107769 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Corning Matrigel Membrane Matrix HC: | Corning | 354263 |
| UltraComp eBeads | Thermo Fisher | 01-2222-41 |
| ArC Amine Reactive Compensation Bead Kit | Thermo Fisher | A10628 |
| Flow Staining Buffer (1X) | Tonbo biosciences | TNB-4222-L500 |
| Foxp3 / Transcription Factor Fix/Perm Concentrate (4X) | Tonbo biosciences | TNB-1020-L050 |
|
| ||
| Critical commercial assays | ||
|
| ||
| NCBI Geo (GSE) - GSE174055 SuperSeries | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174055 | Immune checkpoint blockade reprograms systemic immune landscape and tumor microenvironment in obesity-associated breast cancer |
| NCBI Geo (GSE) - Tumor GSE174053 SubSeries | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174053: | Immune checkpoint blockade reprograms systemic immune landscape and tumor microenvironment in obesity-associated breast cancer [tumor] |
| NCBI Geo (GSE) - Tumor Adjacent Adipose in Mammary Fat Pad GSE174044 SubSeries | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174044: | Immune checkpoint blockade reprograms systemic immune landscape and tumor microenvironment in obesity-associated breast cancer [Mammary fat pad, MFP] |
| Human Normal Breast Study (NBS) NCBI Geo GSE50939 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50939. | Tumor Intrinsic Subtype is Reflected in Cancer-Adjacent Benign Tissue |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| E0771-luciferase (derived from ATCC CRL-3461. A gift from Dr. Hassan Korkaya, August University, Augusta GA) | ATCC | CRL-3461 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratories | stock number: 000664 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| FlowJo Software | FlowJo | https://www.flowjo.com/ |
| Timer 2.0 | Li et al., 2020 | http://timer.cistrome.org/ |
| CIBERSORT | Newman et al., 2021 | https://cibersort.stanford.edu/ |
| xCell | Aran et al., 2017 | https://xcell.ucsf.edu/ |
|
| ||
| Other - diets | ||
|
| ||
| High fat diet (60% kcal from fat) | Research Diets Inc | D12492i |
| Low fat diet (10% kcal from fat) | Research Diets Inc | D12450Ji |
Mice and body composition
After lean or obese female mice were received from Jackson labs, mice were acclimated for one week before the start of experiments and were maintained on the same HFD or LFD until the endpoint. Bodyweight was measured at the start of 25 weeks of age and weekly until sacrifice. Body composition including lean mass, fat mass, free water content, and total water content of non-anesthetized mice was measured using EchoMRI-100 quantitative magnetic resonance whole-body composition analyzer (Echo Medical Systems, Houston, TX) in the same time course. Fat mass is presented as percent fat mass over total body weight.
Tumor cell implantation and ICB
E0771-luc cells were injected in the left fourth mammary fat pad (MFP) of 26-week-old C57BL/6J females fed LFD or HFD at 250,000 cells in 100 μL of 25% Matrigel. Injected cells that did not take at time of injection were not counted as tumors (data not shown). When tumors became palpable (typically on day 6 after implantation), tumor growth was monitored every third day by measuring the length and width of the tumor using digital calipers. Tumor volume was calculated using the following formula: Volume = (width)2 × (length)/2 (Faustino-Rocha et al., 2013). Number of mice with regressed (undetectable) tumors compared to total palpable tumors at endpoint was recorded. At the end of the experiments (day 21), excised tumor mass was determined. Anti-PD-1 antibody (BE0146) and IgG2a control (BE0089) were purchased from BioXcell (West Lebanon, NH). Anti PD-1 antibody administration by intraperitoneal (i.p.) injection began three days after E0771-luc cell injection. Mice were injected every third day for three weeks until sacrifice (200 μg/mouse in 50μl) (Rigo et al., 2017).
Flow cytometric analysis
Tumors, adjacent mammary adipose tissue, spleen, and bone marrow were analyzed by flow. Excised tumors (~500 mg) were minced using scissors in RPMI media containing enzyme cocktail mix from Miltenyi Biotec mouse tumor dissociation kit (Miltenyi Biotec, Auburn, CA). Tumor pieces were further digested as per manufacturer’s instructions and digested tissue was filtered through 70 mm strainer to obtain a single cell suspension. Spleen single cell suspensions were obtained by grinding spleens against a 70 μm filter using a syringe plunger. Bone marrow cells were obtained by flushing the femurs of the mice by spinning down at 12,000 g for 10 s. Following red blood cell (RBC) lysis (Millipore Sigma, St. Louis, MO), viability was determined by staining with Ghost dye (Tonbo Biosciences Inc.) followed by FcR-blocking (Tonbo Biosciences Inc). Antibodies in the Key Resources Table were titrated, and the separation index was calculated using FlowJo v. 10 software. Cells were stained with fluorescently labeled antibodies as previously described (Schmid et al., 2018a) and fixed in Perm/fix buffer (Tonbo Biosciences). Stained cells were analyzed using Bio-Rad ZE5 flow cytometer. Fluorescence minus one (FMO) stained cells and single color Ultracomp Beads (Invitrogen, Carlsbad CA) were used as negative and positive controls, respectively. Data were analyzed using FlowJo v 10 software (Treestar, Woodburn, OR). Lean tumors were too small for flow cytometric analysis. Total immune cells in the TME were gated by plotting forward scatter area versus CD45+, live cells by plotting forward scatter area versus Ghost viability dye, single cells by plotting side scatter height versus side scatter area, and CD11b+ by plotting count versus CD11b area. Immune cells were gated as follows in the tumor Macs: (CD11b+ F4/80+Ly6G− Ly6C-low/-); M1-like TAM (CD11b+ F4/80+ Ly6G− Ly6C-low/- MHCII-high); M2-like TAM (CD11b+ F4/80+ Ly6G− Ly6C-low/- MHCII-low); dendritic cells (F4/80− Ly6C− CD11c+ MHCII+). CD3+ T cells (CD11b− CD3+), CD4+ T cells (CD11b− CD3+ CD4+, CD8−), and CD8+ T cells (CD11b− CD3+ CD4−, CD8+). Gating schema is shown in Figure S2A. The following gating scheme was adopted for spleen and bone marrow: Total MDSC (CD11b+ Gr-1+); M-MDSC (CD11b+ Ly6C-high Ly6G−); PMN-MDSC (CD11b+ Ly6C-low Ly6G+); Macs: (CD11b+ Gr-1− F4/80+); M1-like TAM (CD11b+ Gr-1− F4/80+ MHCII-high); M2-like TAM (CD11b+ Gr-1− F4/80+ MHCII-low/-); dendritic cells (F4/80− Ly6C− CD11c+ MHCII+). Gating schema is shown in Figure S2B.
RNA sequencing (RNA-seq)
RNA was extracted from tumor tissue using RNeasy mini kit (Invitrogen) and tumor adjacent MFP using a kit specific for lipid rich tissue (Norgen Biotek, Ontario, Canada). The integrity of RNA was assessed using Agilent Bioanalyzer and samples with RIN > 5.0 were used. Libraries were constructed using NEBNext® Ultra RNA Library Prep Kits (non-directional) for Illumina, following manufacturer protocols. mRNA was enriched using oligo-dT beads. Libraries were sequenced on NovaSeq 6000 using paired-end 150 bp reads. There was no PhiX spike-in. Data was analyzed as described previously (Law et al., 2014; Patro et al., 2017; Soneson et al., 2015). RNA degradation was examined by SCISSOR (shape changes in selecting sample outliers in RNA-seq) which uses unsupervised screening of a range of structural alterations in RNA-seq data (Feng et al., 2015; Wang et al., 2016; Choi et al., 2021). RNA-seq statistical differences between experimental groups were determined as described previously (Ritchie et al., 2015). Benjamini-Hochberg procedure was used to control false discovery rate (FDR) for adjusted P value. RNA-seq data has been uploaded (pending link). Transcript-level abundance was imported into gene-level abundance with the R package tximport (Soneson et al., 2015). Genes with low expression were identified and filtered out from further analysis using filterByExpr function of the edgeR package in R software (Robinson et al., 2010). Voom transformation function was applied to normalize log2-cpm values using mean-variance trend in the limma software package (Law et al., 2014). ClaNC was used to create classifier genes that characterize the groups of interest for heatmap and tables (Dabney, 2006). Summary spreadsheet of all gene expression for protein coding genes is reported with Voom transformation and log2 presented from tumor and tumor adjacent MFP adipose tissue (Table S1). Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 was used for pathway analysis (Huang et al., 2009). Immune infiltration estimations based on bulk gene expression data from RNA-seq was plotted using TIMER2.0, an easy-to-use web interface resource for systematic analysis of immune cell infiltrates across many cancers. In TIMER2.0 several immune deconvolution methods are available to integrate tumor immunological, clinical and genomic features comprehensively. (Li et al., 2020). In TIMER2.0, xCell or CIBERSORT algorithms were used to examine human homologs and immune cell content in basal-like breast cancer or other BC subtype patient expression data from TCGA. xCell is a gene signature-based algorithm developed in 2017 to infer dozens of immune and stromal cells based on human cell expression and a curve fitting approach for linear comparison with compensation techniques to identify cells (Aran et al., 2017). Cell-type identification estimating relative subsets of RNA transcripts (CIBERSORT) is a method developed in 2015 to identify cell types in complex bulk sequencing from tissue (Newman et al., 2015). TIMER2.0 was used to predict patient survival in the Gene Outcome module using candidate genes identified on the “Cancer Exploration” tab (Aran et al., 2017; Li et al., 2020; Racle et al., 2017; Newman et al., 2015). Partial Spearman’s correlation was used and p value of less than 0.05 was determined to be significant. For example, N = 191 patients were included in the analysis for the BRCA-Basal cohort. In Timer2.0, the Cox Proportional Hazard Model examined overall survival and gene expression “Surv(OS, EVENT) ~ LAMB2 + Stage.” We did not stratify by other optional clinical parameters due to small N. We used a 45% Split expression percentage of patients to maximize the difference in expression for LAMB2. Network analysis was completed on up- and downregulated genes in obese tumor treated with PD-1 antibody or its isotype control using GeneMANIA (Franz et al., 2018).
Human samples-normal breast study
Normal breast samples from the Normal Breast Study (NBS) were examined for candidate genes identified by murine RNA-seq findings. Study design details and participant recruitment, demographics, and medical history were previously reported, and expression data from histologically normal breast samples is publicly available (Sandhu et al., 2016; Sun et al., 2018a, 2018b). Herein, key genes of interest were correlated with patient body mass index (BMI) by Spearman correlations from N = 178 subjects.
Histology and quantification
Tumors and mammary glands (in obese tumor adjacent and in lean normal 4th gland contralateral to the injected tumor-bearing mammary glands) were isolated at the time of sacrifice and fixed in 10% formalin. Then stored in 70% ethanol at room temperature before embedding. Formalin-fixed paraffin embedded (FFPE) sections from tumors and adipose were cut at 5 μm thickness. FFPE sections were stained with Hematoxylin and Eosin, for mitosis and adipocyte size, and scanned by Thermo Fisher digital scanner (Panoramic 250 Flash III, Thermo Fisher, Tewksbury, MA) and quantified using software (Case Viewer). Mitotic nuclei were scored by pathologist (R. Sekhri) by examining 10–20 40X high power fields (HPF) for mitotic nuclei to calculate the mitotic index (number of mitosis/10 HPF). Tumor sections from obese were deparaffinized and rehydrated. Sections were then incubated with the primary antibody against anti-CD3 antibody (Rabbit anti-mice monoclonal ab16999 Abcam) and Alexa fluor 568 donkey anti-rabbit secondary antibody (Invitrogen) was used to detect the primary antibody. Images were captured using Zeiss 710 with 20 × numerical aperture objective lens. Total number of CD3+ cells were quantified using ImageJ 1.8 (Rueden et al., 2017).
Microbiota analysis
Cecal and jejunum content were collected at the time of sacrifice and stored at −80°C. To assess bacterial community structure, we used primers specific for the 16S rRNA V4–V5 region (forward, 338F: 5′-GTGCCAGCMGCCGCGGTAA-3′; reverse, 806R: 5′-GGACTACHVGGGTWTCTAAT −3′) that contained Illumina 3′ adaptor sequences, and a 12-bp barcode. Sequences were generated by an Illumina MiSeq DNA platform at Argonne National Laboratory and analyzed by the program Quantitative Insights Into Microbial Ecology (QIIME) (Kuczynski et al., 2011). Operational Taxonomic Units (OTUs) were picked at 97% sequence identity using open reference OTU picking against the GreenGenes database (v13.8). Surgical instruments and reagents were surveyed as sequencing controls, which failed to produce amplification. LFD and HFD pellets resulted entirely in Lactococcus, a genus involved in food manufacturing (data not shown). Statistical analyses were executed using the Calypso software (v8.84) (Zakrzewski et al., 2017). with a minimum of 5,000 sequences per sample. Alpha diversity was analyzed by Shannon and Simpson’s indexes. Beta diversity was calculated by Bray Curtis and is displayed as principal Coordinate analysis (PCoA). Permutational multivariate analysis of variance (PERMANOVA), Analysis of similarity (Anosim), and analysis of multivariate homogeneity of group dispersions (PERMDISP2) were determined for beta diversity. The unbiased analysis was used to generate a heatmap of the 50 most abundant microbial taxa, as calculated by Spearman’s rank correlation coefficients. Linear discriminant analysis (LDA) effect size (LEfSe) was calculated for significantly enriched taxa. Using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt), we predicted metabolic pathways calculated by Spearman’s rank correlation coefficient, which is presented in a heatmap of the 50 most abundant pathways. Regression analyses of key operational taxonomic unit (OTUs) with significant correlation with tumor volume at endpoint (day 21) were performed with Multivariate Analysis by Linear Models (MaAsLin) with 0.05 false discovery rate significance thresholding and 0.0001 minimum feature relative abundance filtering (Morgan et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical differences between experimental groups were determined by unpaired Student’s t tests or two-way ANOVA with Tukey correction for multiple comparisons using statistical software within GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). For body weight data, to account for the repeated-measures over time within animals, data analyzed by two-way ANOVA (Sundaram et al., 2014). All data are shown as mean ± standard error of the mean (SEM). P values less than 0.05 were considered statistically significant as noted in legends. Statistics are reported in figure legends with N indicated.
Supplementary Material
Highlights.
Anti-PD-1 reverses obesity-exacerbated immunosuppression and breast cancer burden
Anti-PD-1 reinvigorates antitumor immunity despite persistent obesity
Anti-PD-1 affects clinically relevant tumor and mammary fat pad gene signatures
Potential microbial signature biomarker of ICB efficacy in obesity is identified
ACKNOWLEDGMENTS
All flow cytometry and flow sorting data were generated in the Flow Cytometry and Cell Sorting (FCCS) core at The University of Tennessee Health Science Center (UTHSC). RNA-seq data generation was prepared and analyzed by the Translational Sciences Shared Resource in the UTHSC Center for Cancer Research. L.M. was supported by the UTHSC Center for Cancer Research, NIH NCI R37CA226969, NIH NCI R01CA253329, V Foundation, and Mary Kay Foundation. L.M.S. was supported by the Obesity Society/Susan G. Komen Cancer Challenge award, Transdisciplinary Research on Energetics and Cancer (TREC) R25CA203650, and the 2020 American Association for Cancer Research (AACR) Triple Negative Breast Cancer Foundation Research Fellowship. J.F.P. was supported by NIH NCI R01CA253329, NIH NIDDK R01DK127209, Tennessee Governor Pediatric Recruitment Grant, and Tennessee Clinical and Translational Science Institute. D.N.H. was supported by the UTHSC Center for Cancer Research, NIH NCI U24CA210988, NIH NCI UG1CA233333, NIH NCI R37CA226969, and V Foundation. M.A.T. was supported by NIH U01ES019472 and the University of North Carolina University Cancer Research Fund. A.M.H. was supported by F31CA257388. R.N. was supported by NIH NCI R01CA229164 and R01CA253329. S.A. was supported by NIH NCI R01CA253329. K.L.C. was supported by NIH NCI R01CA253329 and DOD BCRP W81XWH-20-1-0014.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109285.
A video abstract is available at https://doi.org/10.1016/j.celrep.2021.109285/mmc4.
REFERENCES
- Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, et al. (2019a). Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Ann. Oncol 30, 405–411. [DOI] [PubMed] [Google Scholar]
- Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, Cescon DW, Iwata H, Campone M, Nanda R, et al. (2019b). Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Ann. Oncol 30, 397–404. [DOI] [PubMed] [Google Scholar]
- Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron-Wisnewsky J, Warmbrunn MV, Nieuwdorp M, and Clément K (2021). Metabolism and Metabolic Disorders and the Microbiome: The Intestinal Microbiota Associated With Obesity, Lipid Metabolism, and Metabolic Health-Pathophysiology and Therapeutic Strategies. Gastroenterology 160, 573–599. [DOI] [PubMed] [Google Scholar]
- Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, Rieber N, and Hartl D (2017). Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol. Lett 192, 1–6. [DOI] [PubMed] [Google Scholar]
- Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S, Bloch N, et al. (2021). Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371, 602–609. [DOI] [PubMed] [Google Scholar]
- Bian Z, Shi L, Venkataramani M, Abdelaal AM, Culpepper C, Kidder K, Liang H, Zen K, and Liu Y (2018). Tumor conditions induce bone marrow expansion of granulocytic, but not monocytic, immunosuppressive leukocytes with increased CXCR2 expression in mice. Eur. J. Immunol 48, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, and Hines WC (2011). Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med 17, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casbas-Hernandez P, Sun X, Roman-Perez E, D’Arcy M, Sandhu R, Hishida A, McNaughton KK, Yang XR, Makowski L, Sherman ME, et al. (2015). Tumor intrinsic subtype is reflected in cancer-adjacent tissue. Cancer Epidemiol. Biomarkers Prev 24, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaib M, Chauhan SC, and Makowski L (2020). Friend or Foe? Recent Strategies to Target Myeloid Cells in Cancer. Front. Cell Dev. Biol 8, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WY, Wu CY, and Yu J (2020). The role of gut microbiota in cancer treatment: friend or foe? Gut 69, 1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba A, Bawaneh A, Velazquez C, Clear KYJ, Wilson AS, Howard-McNatt M, Levine EA, Levi-Polyachenko N, Yates-Alston SA, Diggle SP, et al. (2020). Neoadjuvant Chemotherapy Shifts Breast Tumor Microbiota Populations to Regulate Drug Responsiveness and the Development of Metastasis. Mol. Cancer Res 18, 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HY, Jo H, Zhao X, Hoadley KA, Newman S, Holt J, Hayward MC, Love MI, Marron JS, and Hayes DN (2021). SCISSOR: a framework for identifying structural changes in RNA transcripts. Nat. Commun 12, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia AL, and Bissell MJ (2012). The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat 15, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, Ryan RJ, Iwamoto Y, Marinelli B, Gorbatov R, et al. (2012). Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 109, 2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzo AJ, Fuller AM, and Makowski L (2017). Contribution of Adipose Tissue to Development of Cancer. Compr. Physiol 8, 237–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby EJ, Wei J, Yang XY, Lei G, Wang T, Liu CX, Agarwal P, Korman AJ, Morse MA, Gouin K, et al. (2018). Complimentary mechanisms of dual checkpoint blockade expand unique T-cell repertoires and activate adaptive anti-tumor immunity in triple-negative breast tumors. OncoImmunology 7, e1421891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney AR (2006). ClaNC: point-and-click software for classifying microarrays to nearest centroids. Bioinformatics 22, 122–123. [DOI] [PubMed] [Google Scholar]
- Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I, Wallden B, Marincola FM, and Cesano A (2018). Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, Deblasio RN, Menna C, Ding Q, Pagliano O, et al. (2021). Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Cornò M, D’Archivio M, Conti L, Scazzocchio B, Varì R, Donninelli G, Varano B, Giammarioli S, De Meo S, Silecchia G, et al. (2016). Visceral fat adipocytes from obese and colorectal cancer subjects exhibit distinct secretory and ω6 polyunsaturated fatty acid profiles and deliver immunosuppressive signals to innate immunity cells. Oncotarget 7, 63093–63105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engber D (2018). What models eat. Nat. Med 24, 692–695. [DOI] [PubMed] [Google Scholar]
- Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG, Colaço B, Pires MJ, Colaço J, Ferreira R, and Ginja M (2013). Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. (NY) 42, 217–224. [DOI] [PubMed] [Google Scholar]
- Feng H, Zhang X, and Zhang C (2015). mRIN for direct assessment of genome-wide and gene-specific mRNA integrity from large-scale RNA-sequencing data. Nat. Commun 6, 7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, and Koh AY (2017). Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 19, 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, and Li MO (2014). The cellular and molecular origin of tumor-associated macrophages. Science 344, 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz M, Rodriguez H, Lopes C, Zuberi K, Montojo J, Bader GD, and Morris Q (2018). GeneMANIA update 2018. Nucleic Acids Res. 46 (W1), W60–W64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoi MA, Romano E, and Piccart M (2021). Immunotherapy for early breast cancer: too soon, too superficial, or just right? Ann. Oncol 32, 323–336. [DOI] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D, and Marincola FM (2013). The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 39, 11–26. [DOI] [PubMed] [Google Scholar]
- Gao M, Wang T, Ji L, Bai S, Tian L, and Song H (2020). Therapy With Carboplatin and Anti-PD-1 Antibodies Before Surgery Demonstrates Sustainable Anti-Tumor Effects for Secondary Cancers in Mice With Triple-Negative Breast Cancer. Front. Immunol 11, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, and Umansky V (2019). Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 120, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hartley GP, Chow L, Ammons DT, Wheat WH, and Dow SW (2018). Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol. Res 6, 1260–1273. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. (2013). Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar NM, Zhou XK, Gucalp A, Morris PG, Howe LR, Giri DD, Morrow M, Wang H, Pollak M, Jones LW, et al. (2016). Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin. Cancer Res 22, 2283–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaora S, Nagler A, Nejman D, Alon M, Barbolin C, Barnea E, Ketelaars SLC, Cheng K, Vervier K, Shental N, et al. (2021). Identification of bacteria-derived HLA-bound peptides in melanoma. Nature 592, 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasikara C, Davra V, Calianese D, Geng K, Spires TE, Quigley M, Wichroski M, Sriram G, Suarez-Lopez L, Yaffe MB, et al. (2019). Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 79, 2669–2683. [DOI] [PubMed] [Google Scholar]
- Kuczynski J, Stombaugh J, Walters WA, González A, Caporaso JG, and Knight R (2011). Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinformatics 36, 10.7.1–10.7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langan EA, Grätz V, Billmann F, Zillikens D, and Terheyden P (2018). Does the gastrointestinal microbiome contribute to the ‘obesity paradox’ in melanoma survival? Br. J. Dermatol 179, 225–226. [DOI] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Naour A, Rossary A, and Vasson MP (2020). EO771, is it a well-characterized cell line for mouse mammary cancer model? Limit and uncertainty. Cancer Med. 9, 8074–8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel E, Makowski L, DiGiovanni J, and Kolonin MG (2018). Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 4, 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, and Liu XS (2020). TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48 (W1), W509–W514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al. (2016). Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, Paik S, Stagg J, Groves RA, Gallo M, et al. (2020). Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489. [DOI] [PubMed] [Google Scholar]
- Makowski L, Chaib M, and Rathmell JC (2020). Immunometabolism: From basic mechanisms to translation. Immunol. Rev 295, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, Pierre JF, Miyoshi J, Sontag TJ, Cham CM, et al. (2018). Small Intestine Microbiota Regulate Host Digestive and Absorptive Adaptive Responses to Dietary Lipids. Cell Host Microbe 23, 458–469.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, and Gajewski TF (2018). The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik A, Monjazeb AM, and Decock J (2019). The Obesity Paradox in Cancer, Tumor Immunology, and Immunotherapy: Potential Therapeutic Implications in Triple Negative Breast Cancer. Front. Immunol 10, 1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, et al. (2016). Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol 34, 2460–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman TM, Shively CA, Register TC, Appt SE, Yadav H, Colwell RR, Fanelli B, Dadlani M, Graubics K, Nguyen UT, et al. (2021). Diet, obesity, and the gut microbiome as determinants modulating metabolic outcomes in a non-human primate model. Microbiome 9, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ősz A, Lánczky A, and Győrffy B (2020). Survival analysis in breast cancer using proteomic data from four independent datasets. medRxiv. 10.1101/2020.12.03.20242065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parida S, and Sharma D (2020). Microbial Alterations and Risk Factors of Breast Cancer: Connections and Mechanistic Insights. Cells 9, 1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Cheng S, Kou Y, Wang Z, Jin R, Hu H, Zhang X, Gong JF, Li J, Lu M, et al. (2020). The Gut Microbiome Is Associated with Clinical Response to Anti-PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol. Res 8, 1251–1261. [DOI] [PubMed] [Google Scholar]
- Pierre JF, Martinez KB, Ye H, Nadimpalli A, Morton TC, Yang J, Wang Q, Patno N, Chang EB, and Yin DP (2016). Activation of bile acid signaling improves metabolic phenotypes in high-fat diet-induced obese mice. Am. J. Physiol. Gastrointest. Liver Physiol 311, G286–G304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racle J, de Jonge K, Baumgaertner P, Speiser DE, and Gfeller D (2017). Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 6, e26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC (2021). Obesity, Immunity, and Cancer. N. Engl. J. Med 384, 1160–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo V, Emionite L, Daga A, Astigiano S, Corrias MV, Quintarelli C, Locatelli F, Ferrini S, and Croce M (2017). Combined immunotherapy with anti-PDL-1/PD-1 and anti-CD4 antibodies cures syngeneic disseminated neuroblastoma. Sci. Rep 7, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et al. (2019). Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 178, 795–806.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelands J, Hendrickx W, Zoppoli G, Mall R, Saad M, Halliwill K, Curigliano G, Rinchai D, Decock J, Delogu LG, et al. (2020). Oncogenic states dictate the prognostic and predictive connotations of intratumoral immune response. J. Immunother. Cancer 8, e000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshanravan N, Bastani S, Tutunchi H, Kafil B, Nikpayam O, Mesri Alamdari N, Hadi A, Sotoudeh S, Ghaffari S, and Ostadrahimi A (2021). A comprehensive systematic review of the effectiveness of Akkermansia muciniphila, a member of the gut microbiome, for the management of obesity and associated metabolic disorders. Arch. Physiol. Biochem Published online January 15, 2021. 10.1080/13813455.2021.1871760. [DOI] [PubMed] [Google Scholar]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97. [DOI] [PubMed] [Google Scholar]
- Rowe RG, and Weiss SJ (2008). Breaching the basement membrane: who, when and how? Trends Cell Biol. 18, 560–574. [DOI] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu R, Chollet-Hinton L, Kirk EL, Midkiff B, and Troester MA (2016). Digital histologic analysis reveals morphometric patterns of age-related involution in breast epithelium and stroma. Hum. Pathol 48, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MC, Khan SQ, Kaneda MM, Pathria P, Shepard R, Louis TL, Anand S, Woo G, Leem C, Faridi MH, et al. (2018a). Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun 9, 5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, et al. ; IMpassion130 Trial Investigators (2018b). Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med 379, 2108–2121. [DOI] [PubMed] [Google Scholar]
- Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N, et al. ; KEYNOTE-522 Investigators (2020). Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med 382, 810–821. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, and Knight R (2021). The microbiome and human cancer. Science 371, eabc4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shively CA, Register TC, Appt SE, Clarkson TB, Uberseder B, Clear KYJ, Wilson AS, Chiba A, Tooze JA, and Cook KL (2018). Consumption of Mediterranean versus Western Diet Leads to Distinct Mammary Gland Microbiome Populations. Cell Rep. 25, 47–56.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RC, Shanahan E, Scolyer RA, and Long GV (2021). Targeting the Microbiome to Overcome Resistance. Cancer Cell 39, 151–153. [DOI] [PubMed] [Google Scholar]
- Sipe LM, Chaib M, Pingili AK, Pierre JF, and Makowski L (2020). Microbiome, bile acids, and obesity: How microbially modified metabolites shape anti-tumor immunity. Immunol. Rev 295, 220–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirotnak FM, DeGraw JI, Schmid FA, Goutas LJ, and Moccio DM (1984). New folate analogs of the 10-deaza-aminopterin series. Further evidence for markedly increased antitumor efficacy compared with methotrexate in ascitic and solid murine tumor models. Cancer Chemother. Pharmacol 12, 26–30. [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura K, and Stock CC (1952). Studies in a tumor spectrum. I. Comparison of the action of methylbis (2-chloroethyl)amine and 3-bis(2-chloroethyl)aminomethyl-4-methoxymethyl −5-hydroxy-6-methylpyridine on the growth of a variety of mouse and rat tumors. Cancer 5, 382–402. [DOI] [PubMed] [Google Scholar]
- Sun X, Shan Y, Li Q, Chollet-Hinton L, Kirk EL, Gierach GL, and Troester MA (2018a). Intra-individual Gene Expression Variability of Histologically Normal Breast Tissue. Sci. Rep 8, 9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Stewart DA, Sandhu R, Kirk EL, Pathmasiri WW, McRitchie SL, Clark RF, Troester MA, and Sumner SJ (2018b). Correlated metabolomic, genomic, and histologic phenotypes in histologically normal breast tissue. PLoS ONE 13, e0193792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram S, Le TL, Essaid L, Freemerman AJ, Huang MJ, Galanko JA, McNaughton KK, Bendt KM, Darr DB, Troester MA, and Makowski L (2014). Weight Loss Reversed Obesity-Induced HGF/c-Met Pathway and Basal-Like Breast Cancer Progression. Front. Oncol 4, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcyganov E, Mastio J, Chen E, and Gabrilovich DI (2018). Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol 51, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Sanseviero E, and Gabrilovich DI (2021). Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol Published online February 1, 2021. 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. (2015). Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, et al. (2013). The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Nie J, Sicotte H, Li Y, Eckel-Passow JE, Dasari S, Vedell PT, Barman P, Wang L, Weinshiboum R, et al. (2016). Measure transcript integrity using RNA-seq data. BMC Bioinformatics 17, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, Mirsoian A, Minnar CM, Stoffel KM, Sturgill IR, et al. (2019). Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med 25, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, Utikal J, and Umansky V (2018). Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol 9, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodall MJ, Neumann S, Campbell K, Pattison ST, and Young SL (2020). The Effects of Obesity on Anti-Cancer Immunity and Cancer Immunotherapy. Cancers (Basel) 12, 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Sun X, Gupta HB, Yuan B, Li J, Ge F, Chiang HC, Zhang X, Zhang C, Zhang D, et al. (2018). Adipose PD-L1 Modulates PD-1/PD-L1 Checkpoint Blockade Immunotherapy Efficacy in Breast Cancer. OncoImmunology 7, e1500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chiang HC, Sun X, Yuan B, Mitra P, Hu Y, Curiel TJ, and Li R (2020a). Genetic ablation of adipocyte PD-L1 reduces tumor growth but accentuates obesity-associated inflammation. J. Immunother. Cancer 8, e000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Hua Q, and Zheng L (2020b). Generation of Myeloid Cells in Cancer: The Spleen Matters. Front. Immunol 11, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Zhang Q, Liu S, Wang AR, and You Z (2016). PD-1, PD-L1 and PD-L2 expression in mouse prostate cancer. Am. J. Clin. Exp. Urol 4, 1–8. [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewski M, Proietti C, Ellis JJ, Hasan S, Brion MJ, Berger B, and Krause L (2017). Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 33, 782–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq expression datasets for tumor and tumor adjacent adipose MFP generated during this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO: GSE174055 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174055) specifically for tumor (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174053) or mammary fat pad (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174044), as described in greater detail on our STAR Methods website. Human Normal Breast Study (NBS) is publicly available at NCBI GEO: GSE50939 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50939.