

Abstract
Different spatiotemporal abnormalities have been implicated in different neuropsychiatric disorders and anthropometric social traits, yet an investigation in the temporal network modularity with brain tissue transcriptomics has been lacking. We developed a supervised network approach to investigate the genome-wide association study (GWAS) results in the spatial and temporal contexts and demonstrated it in 20 brain disorders and anthropometric social traits. BrainSpan transcriptome profiles were used to discover significant modules enriched with trait susceptibility genes in a developmental stage-stratified manner. We investigated whether, and in which developmental stages, GWAS-implicated genes are coordinately expressed in brain transcriptome. We identified significant network modules for each disorder and trait at different developmental stages, providing a systematic view of network modularity at specific developmental stages for a myriad of brain disorders and traits. Specifically, we observed a strong pattern of the fetal origin for most psychiatric disorders and traits [such as schizophrenia (SCZ), bipolar disorder, obsessive–compulsive disorder and neuroticism], whereas increased co-expression activities of genes were more strongly associated with neurological diseases [such as Alzheimer’s disease (AD) and amyotrophic lateral sclerosis] and anthropometric traits (such as college completion, education and subjective well-being) in postnatal brains. Further analyses revealed enriched cell types and functional features that were supported and corroborated prior knowledge in specific brain disorders, such as clathrin-mediated endocytosis in AD, myelin sheath in multiple sclerosis and regulation of synaptic plasticity in both college completion and education. Our study provides a landscape view of the spatiotemporal features in a myriad of brain-related disorders and traits.
Keywords: BrainSpan, temporal gene expression, network modularity, brain-related disorders and traits, EW_dmGWAS
Introduction
The course of human brain development involves complex regulation of transcriptional gene expression with an orchestrated dynamic that has both temporal and spatial specificities. Neuropsychiatric disorders are postulated as neurodevelopmental or neurodegenerative in nature and thus may be pathophysiologically linked to an abnormal temporal and spatial gene expression in the brain. This notion raises the possibility that investigation of the dynamics of brain spatiotemporal transcription is an opportunity that can lead to a better understanding of the pathophysiology of these disorders. Indeed, the veracity of this approach has been corroborated by recent studies using spatiotemporal transcriptome data that have implied some specific brain regions and stages enriched with susceptibility genes [1–4] in autism [5] and SCZ [1, 6–9]. However, distinct neuropsychiatric disorders may have their origins in distinct spatiotemporal disturbances, yet, until now, no study has thoroughly systematically charted the dynamics of spatiotemporal gene expression, transcript variants and modularity in the brain across separate neuropsychiatric disorders and traits to ascertain their conceivable individual and common neurodevelopmental and neurodegenerative trajectories [10, 11] by unveiling features of transcription that are either unique or shared in different brain regions and developmental stages.
According to the polygenic model of the disease, many genes are contributive to the disease and they are co-regulated or correlated to confer risk to disease, including brain disorders [5, 12, 13]. Gene co-expression network is one of the major approaches to study convergence and divergence of disease susceptibility genes [14]. Importantly, gene expression profiles from various tissues, cell types and developmental stages will empower our investigation of gene functions in dynamic cellular systems. Weighted gene co-expression network analysis [15] represents the unsupervised approach for such purposes, where co-expression modules are constructed using normal samples and are subsequently tested for disease associations [16–19]. Another category of methods, including two from our group [20, 21], search for subnetwork modules in a disease-focused or context-focused way. These methods typically construct modules guided by scoring systems such as genetics evidence or gene co-expression regulations and, hence, represent the supervised group to prioritize disease-associated genes [22, 23]. We recently developed an algorithm that enables the node- and edge-weighted dense module search of genome-wide association study (GWAS) signals and that can integrate gene co-expression profiling (EW_dmGWAS) [21]. Because gene expression data can be dynamic and can be obtained in different biological contexts, such as tissues, brain regions, developmental stages and disease versus healthy samples, this new technique opened a new avenue to leverage the rich information in gene expression profile and to subsequently boost the discoveries from GWAS data. It has been applied and validated in various disorders including SCZ [24], multiple sclerosis (MS) [25] and cancer [26].
Here, we first propose a conditional gene expression correlation measurement, namely conditional Pearson correlation coefficient (PCC), by including the developmental stage information as covariant. In comparison with the canonical PCC, we found that there are brain disorder genes undergoing transcriptional changes over the developmental stages, sometimes with an opposite trend. We then modified our original EW_dmGWAS algorithm to identify subnetworks constrained by the temporal transcriptomic context. Finally, we collected GWAS data in 20 brain-related disorders and traits and conducted a comparative study to explore their distinct features during different developmental stages, in this case, prenatal and postnatal stages. The resultant modules also enabled us to investigate modular gene functions at a fine scale. Indeed, the module genes of several disorders and traits yielded functional enrichment that is consistent with prior knowledge, what corroborates and further validates our method.
Methods and Materials
Selection of multi-trait GWAS data
We collected GWAS summary statistics for 20 brain-related disorders or traits in three major groups: psychiatric disorders and psychiatric-related traits [attention deficit hyperactivity disorder (ADHD) [27], alcohol use disorder (AUD) [28], anxiety [29], autism spectrum disorder (ASD) [30], bipolar disorder (BD) [31], depressive symptoms (DS, ‘DSM-oriented depression subscale of the age-appropriate survey from the ASEBA taxonomy’ as defined in the original study) [32], internalizing problems (IP) [33], major depressive disorder (MDD) [34], neuroticism (NEU) and two of their subtypes, anxiety tension special factor of neuroticism (ANEU) and general factor of neuroticism (GNEU) [32], obsessive compulsive disorder (OCD) [35] and SCZ] [36]; neurological diseases [Alzheimer’s disease (ALZ) [37], amyotrophic lateral sclerosis (ALS) [38], multiple sclerosis (MS) [39] and Parkinson’s disease (PD)] [40] and anthropometric social traits [college completion (COL) [41], educational attainment (EDU) [42] and subjective well-being (SWB) [43]]. All participants were from European ancestry.
We used Multi-marker Analysis of GenoMic Annotation (MAGMA) V1.07 to calculate gene-based P-values [44]. MAGMA combines multi-markers that are mapped to a gene and takes into account the effects of the gene length, SNP density and local linkage disequilibrium structure. For each gene, we considered all single nucleotide polymorphisms (SNPs) located in the gene body or the flanking regions (50 kb upstream and 35 kb downstream). Then, we obtained the mean of the SNP P-values based on the χ2 statistic as the measurement of the gene-level P-value. We used the 1000 Genome Project Phase 3 European population as the reference panel. A summary of all GWAS datasets and the MAGMA results is available in Supplementary Table S1 available online at https://academic.oup.com/bib.
Brain gene expression data
We downloaded the BrainSpan developmental transcriptome data [accessed date: April 2020, currently part of the PsychENCODE data [45, 46]] for normal brains, which contains gene expression for multiple brain regions in multiple developmental stages [47]. We excluded lowly expressed genes, defined as those with RPKM (Reads Per Kilobase of transcript per Million mapped reads) value <1 in more than half of the samples. Gene expression was measured using log2 (RPKM+1). Five samples were excluded based on the principal component analysis (PCA) (Figure 1A). Finally, we obtained 229 samples for the prenatal and 344 for the postnatal stages. Mean age was 16.28 post-conception weeks (pcw) (range: 8–26 pcw) for the prenatal samples and to 14.52 years for the postnatal samples (range: 4 months to 40 years). Regarding sex, 22 were males and 19 females.
Figure 1 .
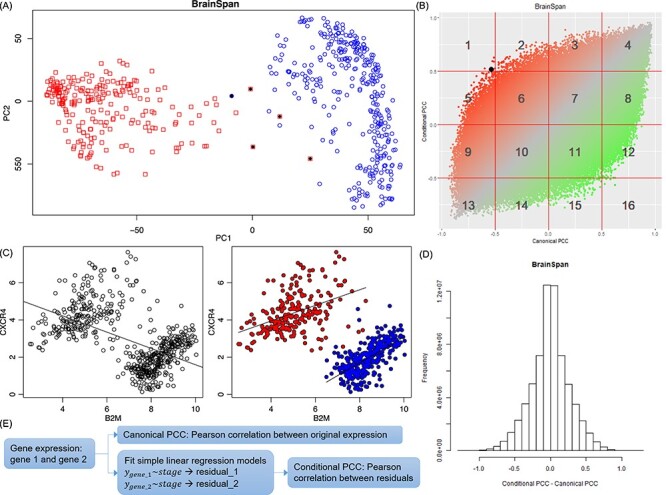
Distinct expression of selected genes at the prenatal and postnatal stages. (A) PCA of the BrainSpan data. Red: Prenatal brain samples. Blue: Postnatal brain samples. Outlier samples (black dots) were excluded in follow-up analyses. (B) Comparison of ConPCC (y-axis) with canonical PCC (x-axis) using the BrainSpan data. The black dot in area 1 is the example shown in (C). (C) An example of a pair of genes showing negative correlation among all BrainSpan samples but positive correlation in either prenatal (red dots) or postnatal (blue dots) subgroup. (D) Distribution of the difference between conditional and canonical PCC values using the BrainSpan data. (E) Summary of calculation of canonical PCC and ConPCC.
Background network data
We downloaded the gene–gene association data from PathwayCommons [48] (V12, download date: 12 December 2019) that were curated and integrated from public pathway and interaction databases (Supplementary Table S2 available online at https://academic.oup.com/bib). We excluded 2291 ribosomal genes and 351 genes located in the MHC region (chr6:26000000_34000000). The resultant network served as the background network and was subsequently assigned with edge weights in different conditions (see below).
Definition of correlation coefficient
In this study, we used PCC to measure gene co-expression. We defined a conditional correlation coefficient by regressing out the stage conditions and correlating variables using the residuals. Specifically, given two genes with expression values across N samples, we fit the linear regression model as below: Y1 ~ stage and Y2 ~ stage, where Y1 and Y2 are the gene expression (a vector of length N) for the two genes and stage is a categorical variable indicating the stage of the sample: prenatal or postnatal. ConPCC is thus defined as the PCC of residuals from the two models.
Temporal EW_dmGWAS
We modified our previously developed method EW_dmGWAS [21] for the integration of temporal gene expression data. EW_dmGWAS aims to identify groups of interacting genes (termed modules) whose joint effects from GWAS are significant, while the module genes are also significantly concordantly expressed. In temporal EW_dmGWAS (tpGWAS), we define a score for each module to measure the combined effect of the GWAS signal and gene expression correlation among multiple genes: 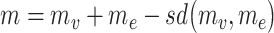 , where
, where 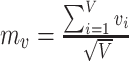 ,
, 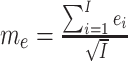 and
and 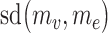 measures the deviation between
measures the deviation between 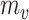 and
and 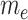 . V is the number of vertices in the module,
. V is the number of vertices in the module, 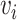 is the normalized node weight, I is the number of interactions and
is the normalized node weight, I is the number of interactions and 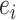 is the normalized edge weight. Node weight was transformed from gene-based P-values calculated by MAGMA. Edge weight was based on the PCC calculated using prenatal samples only or postnatal samples only and then was transformed to
is the normalized edge weight. Node weight was transformed from gene-based P-values calculated by MAGMA. Edge weight was based on the PCC calculated using prenatal samples only or postnatal samples only and then was transformed to 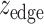 following the original work [21]. We then quantile normalized
following the original work [21]. We then quantile normalized 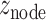 and
and 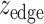 by taking the standard normal distribution as the reference, resulting in
by taking the standard normal distribution as the reference, resulting in 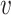 and
and 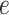 (i.e.
(i.e. 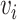 and
and 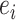 as the node and edge weight, respectively, for the ith node and the ith edge), both of which are approximate to the standard normal distribution. Modules were obtained by using nodes with a positive
as the node and edge weight, respectively, for the ith node and the ith edge), both of which are approximate to the standard normal distribution. Modules were obtained by using nodes with a positive  from GWAS as the seed gene and applying a greedy search algorithm [20], followed by node and edge trimming [49]. We used the default parameter for module search. More details of the algorithm can be found in our previous works [20, 21, 24, 49].
from GWAS as the seed gene and applying a greedy search algorithm [20], followed by node and edge trimming [49]. We used the default parameter for module search. More details of the algorithm can be found in our previous works [20, 21, 24, 49].
Significant modules
To evaluate the significance of the resultant modules, we generated random modules by switching the node and weight (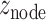 ) pairing relationships, as well as the edge and weight (
) pairing relationships, as well as the edge and weight (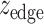 ) pairing relationships. The resultantly random modules were used to form the null distribution and to normalize the module scores for those genuine modules obtained using the actual GWAS and brain expression data, resulting in
) pairing relationships. The resultantly random modules were used to form the null distribution and to normalize the module scores for those genuine modules obtained using the actual GWAS and brain expression data, resulting in 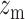 . The normalized module z-scores (
. The normalized module z-scores (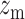 ) were then transformed to P-values, followed by the Benjamini and Hochberg (BH) procedure [50] for multiple testing correction. We defined significant modules as those with PBH < 0.05. We also applied a restriction to filter modules that showed extreme node/edge ratios, e.g.
) were then transformed to P-values, followed by the Benjamini and Hochberg (BH) procedure [50] for multiple testing correction. We defined significant modules as those with PBH < 0.05. We also applied a restriction to filter modules that showed extreme node/edge ratios, e.g. 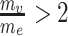 or
or 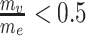 , as such modules were mainly driven by either GWAS or co-expression but not by a concordant effect.
, as such modules were mainly driven by either GWAS or co-expression but not by a concordant effect.
Cell type-specific enrichment analysis
We collected two single-cell RNA sequencing (scRNA-seq) datasets: DER-20 and DER-22 [51, 52]. The DER-20 dataset merged three studies: PsychENCODE (developmental) [45], Darmanis et al. [53] and Lake et al. [51]. The panel has 35 cell types identified from both fetal and adult brains, including 13 cell types from the fetal brain [astrocytes, endothelial, excitatory neuron (ExN), inhibitory neuron (InN), intermediate progenitor cells, microglia, neuroepithelial cells, oligodendrocytes, oligodendrocyte precursor cells (OPCs), pericytes, quiescent newly born neurons, replicating neuronal progenitors and transient cell type (trans)] and 22 cell types from the adult brain [astrocytes, endothelial, eight types of excitatory neurons (Ex1–Ex8), eight subtypes of InNs (In1–In8), microglia, neurons, oligodendrocytes and OPC] [45]. The DER-22 dataset merged two studies: PsychENCODE (adult) [45] and Lake et al. [52]. It has 25 cell types, including 9 types of excitatory neurons (Ex1, Ex2, Ex3e, Ex4, Ex5b, Ex6a, Ex6b, Ex8 and Ex9), 10 InNs (In1a, In1b, In1c, In3, In4a, In4b, In6a, In6b, In7 and In8), as well as astrocytes, endothelial, microglia, oligodendrocytes, OPC and pericytes. For each dataset, we fit linear regression models to assess the cell type specificity of each gene in each cell type, following the strategy we developed for measurement of tissue specificity [54]. As a result, we obtained a t-score for each gene in each cell type to measure its cell type specificity. Subsequently, in the cell type-specific enrichment analysis of module genes, we applied Fisher’s exact test to assess whether a list of query genes was associated with cell type marker genes (defined as those with a t-score within the top 10% quantile).
Results
Temporal gene expression patterns in normal human brain
After excluding lowly expressed genes and outlier samples (Figure 1A), we built a working matrix with 11 466 genes in 573 brain samples (229 prenatal and 344 postnatal samples). We calculated the canonical PCC and conditional PCC (ConPCC) for all gene pairs using all 573 samples. The former measured the overall trend of gene co-expression in samples across all stages, whereas the latter measured gene co-expression conditioned on stages. Figure 1B illustrated using one example: genes B2M and CXCR4 had an overall negative correlation across the whole time points (canonical PCC), but a positive correlation in both the prenatal and postnatal stages when stage was taken into account (ConPCC). The distribution of the difference between these two PCC values (ConPCC − PCC) was shown in Figure 1C. Large differences indicated that the gene pairs had different co-expression patterns in all stages compared to each individual stage. On the plot of the two PCC values for each gene pair, we defined 16 areas by using threshold values of 0.5 and −0.5 (1–16 as labeled in Figure 1B). Some gene pairs showed moderate changes of correlation, e.g. from strong to weak, while their correlation trend (positive or negative PCC values) remained the same, such as area 3 (PCC in the range of [0, 0.5] and ConPCC in the range of [0.5, 1.0]), area 8 (PCC in [0.5, 1.0] while ConPCC in [0, 0.5]) and, similarly, areas 9 and 14. Other gene pairs showed strong changes, where gene pairs changed from weak positive (negative) to strong negative (positive) correlations, such as area 15 (area 2), or strong positive (negative) to weak negative (positive) correlations, such as area 12 (area 5). To further explore the gene functions involved in these areas, we conducted functional enrichment analysis using ToppGene [55] for areas 2 and 15. As shown in Supplementary Tables S3 and S4 available online at https://academic.oup.com/bib, we found many genes from areas 2 and 5 (i.e. genes involved in edges that went through dramatic changes during brain development) enriched in brain- and neuron-related processes. In area 2, we found the genes enriched in GO terms for post-synapse (P = 1.02 × 10−25), dendritic tree (P = 1.99 × 10−21) and dendrite (P = 1.99 × 10−21). In area 15, we found dendritic tree (P = 6.00 × 10−19), dendrite (P = 6.00 × 10−19) and axon (P = 1.95 × 10−21). Overall, we noted that there were indeed gene pairs that underwent different co-expression in different developmental stages, implying a necessity to analyze disease susceptible genes in each stage separately.
Genes with temporal expression change enriched in brain-related disorders or traits
Next, we investigated whether GWAS-implied genes were involved in the gene pairs that showed strong changes in all samples compared to that in the samples of each stage. We transformed the normalized difference between the two types of PCC (ConPCC − PCC) to z-scores (Figure 1D) and focused on pairs with P-value <5 × 10−5, where the P-values were reversely transformed from the z-score. There were, totally, 877 genes involved in 4630 pairs, which showed positive correlation across the whole developmental stages, while a negative correlation was found in either the prenatal or postnatal stages. For these 877 genes, we obtained their gene-based P-values calculated based on the GWAS data for 20 brain-related disorders or traits. We found that these genes tend to have smaller gene-based P-values than other protein-coding genes in 7 of the 20 disorders or traits: AUD (P = 0.027), BD (P = 2.624 × 10−3), NEU (P = 0.042) and its two sub-phenotypes (P = 3.422 × 10−3 for anxiety tension factor of NEU and P = 0.05 for GNEU), MS (P = 6.112 × 10−3) and SCZ (P = 7.623 × 10−5) (Table 1). The opposite trend was observed for a total of 854 genes involved in 4686 pairs (negative correlation using all samples and positive correlation in prenatal and postnatal brains analyzed separately). These genes tended to have smaller gene-based P-values in five disorders/traits: AUD (P = 0.022), ANEU (P = 0.011), ADHD (P = 0.036), GNEU (P = 0.03), MS (P = 8.653 × 10−3) and SCZ (P = 6.214 × 10−3). These results suggested that GWAS-implied genes for brain-related disorders or traits were likely to be involved in co-expression correlations undergoing substantial changes during development. Accordingly, we separated the brain samples of the prenatal stage and samples of the postnatal stage to construct gene co-expression weighted networks for the following analyses.
Table 1.
Enrichment results of GWAS genes and genes presenting strong positive or negative co-expression in prenatal and postnatal brain transcriptomes
| Positive | Negative | Union | ||||
|---|---|---|---|---|---|---|
| Disorder or trait | P | P BH | P | P BH | P | P BH |
| AUD | 0.027 | 0.076 | 0.022 | 0.112 | 0.027 | 0.089 |
| ALZ | 0.205 | 0.273 | 0.293 | 0.417 | 0.136 | 0.226 |
| ALS | 0.183 | 0.262 | 0.286 | 0.417 | 0.147 | 0.226 |
| Anxiety | 0.645 | 0.645 | 0.611 | 0.611 | 0.751 | 0.751 |
| ANEU | 3.422 × 10 −3 | 0.023 | 0.011 | 0.074 | 0.027 | 0.089 |
| ADHD | 0.021 | 0.070 | 0.036 | 0.121 | 0.042 | 0.094 |
| ASD | 0.158 | 0.244 | 0.253 | 0.417 | 0.290 | 0.341 |
| BD | 2.624 × 10 −3 | 0.023 | 0.262 | 0.417 | 4.448 × 10 −3 | 0.044 |
| DS | 0.048 | 0.100 | 0.312 | 0.417 | 0.086 | 0.157 |
| EDU | 0.058 | 0.106 | 0.263 | 0.417 | 0.086 | 0.157 |
| GNEU | 0.050 | 0.100 | 0.030 | 0.118 | 0.021 | 0.089 |
| IP | 0.270 | 0.318 | 0.611 | 0.611 | 0.454 | 0.478 |
| MDD | 0.020 | 0.070 | 0.358 | 0.447 | 0.041 | 0.094 |
| MS | 6.112 × 10 −3 | 0.031 | 0.009 | 0.074 | 0.019 | 0.089 |
| Neuroticism | 0.042 | 0.100 | 0.067 | 0.191 | 0.031 | 0.089 |
| OCD | 0.307 | 0.341 | 0.392 | 0.461 | 0.326 | 0.363 |
| PD | 0.221 | 0.276 | 0.098 | 0.245 | 0.288 | 0.341 |
| SCZ | 7.623 × 10 −5 | 1.525 × 10 −3 | 6.214 × 10−3 | 0.074 | 5.124 × 10 −4 | 0.010 |
| COL | 0.094 | 0.157 | 0.516 | 0.573 | 0.245 | 0.327 |
| SWB | 0.403 | 0.424 | 0.128 | 0.285 | 0.162 | 0.231 |
Positive pairs: ConPCC > canonical PCC. Negative pairs: ConPCC < canonical PCC.
Bold values indicate nominal significance (unadjusted P < 0.05).
Distinct prenatal expression in psychiatric disorders and postnatal expression in neurological disorders
We conducted tpGWAS for each of the 20 GWAS datasets using co-expression weighted networks in two conditions: the weight calculated using the prenatal samples only or the postnatal samples only. The workflow is summarized in Figure 2. For each run of tpGWAS, we constructed the null distribution for module scores (see Methods) and defined significant modules as those with PBH < 0.05 (Table 2). Then, significant modules were merged to form the disorder/trait-associated network.
Figure 2 .
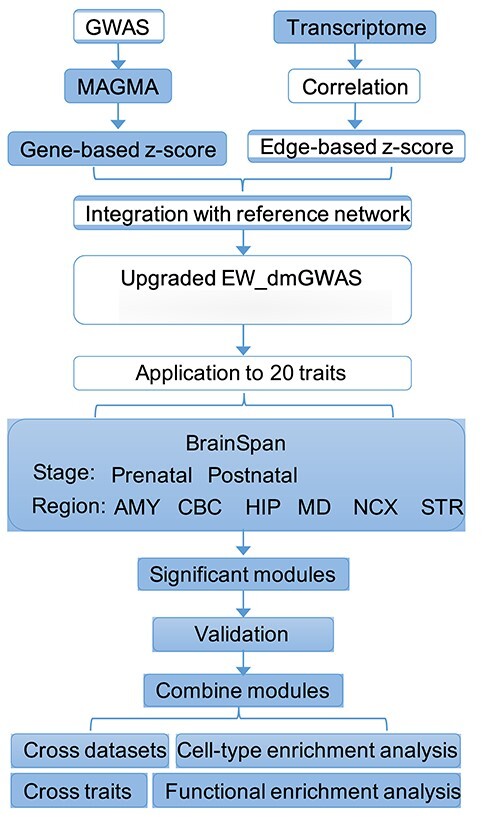
Analysis pipeline. Gene-based z-scores calculated from GWAS data and edge-based z-scores calculated based on transcriptomic data were integrated into the background reference gene–gene association network. We conducted the analysis by stratifying the transcriptome data into prenatal samples and postnatal samples as well as six brain regions: the NCX, HIP, amygdala (AMY), striatum (STR), mediodorsal nucleus of the thalamus (MD) and cerebellar cortex (CBC). After obtaining significant modules in different conditions, we next conducted cross-datasets, cross-trait and functional enrichment analysis to investigate the results.
Table 2.
Summary of the EW_dmGWAS results for each brain-related traits or disorders in the prenatal or postnatal stages
| Prenatal | Postnatal | |||||
|---|---|---|---|---|---|---|
| No. of modules (BS) | No. of module genes | No. of avg −log10(P) | No. of modules (BS) | No. of module genes | No. of avg −log10(P) | |
| AUD | 22 | 33 | 3.38 | 0 | 0 | − |
| ALZ | 3 | 10a | 57.22 | 69 | 94 | 7.12 |
| ALS | 10 | 18a | 2.27 | 35 | 54 | 2.17 |
| ANEU | 1 | 4 | 6.55 | 0 | 0 | − |
| Anxiety | 0 | 0 | − | 5 | 8 | 2.43 |
| ADHD | 0 | 0 | − | 7 | 13 | 3.44 |
| ASD | 1 | 3 | 2.67 | 0 | 0 | − |
| BD | 22 | 33 | 3.88 | 2 | 5 | 4.92 |
| DS | 5 | 13 | 4.17 | 22 | 43 | 4.28 |
| EDU | 0 | 0 | − | 36 | 54 | 5.47 |
| GNEU | 3 | 4 | 7.38 | 63 | 82 | 3.96 |
| Height | 0 | 0 | − | 0 | 0 | − |
| IP | 11 | 15 | 2.04 | 4 | 9 | 2.18 |
| MDD | 0 | 0 | − | 0 | 0 | − |
| MS | 4 | 12a | 5.71 | 83 | 102 | 3.70 |
| NEU | 4 | 10 | 4.74 | 39 | 65 | 4.09 |
| OCD | 28 | 48 | 1.99 | 0 | 0 | − |
| PD | 0 | 0 | − | 29 | 48 | 1.95 |
| SCZ | 44 | 77 | 6.97 | 16 | 24 | 7.79 |
| COL | 2 | 4 | 4.25 | 20 | 38 | 3.02 |
| SWB | 0 | 0 | − | 8 | 16 | 2.92 |
BS, BrainSpan.
−log10(P): average gene-based P-value from GWAS for module genes.
Bold cases indicate the conditions that were considered significant (see main text).
aCell growth and DNA replication-related pathway genes.
As summarized in Table 2 and Figure 3, we found substantially different patterns of gene activities. Psychiatric disorders (such as AUD, BD, IP, OCD and SCZ) tended to have significant modules in the prenatal stage, whereas neurological diseases (such as ALZ, ALS, MS and PD) tended to have more significant modules in the postnatal stage. These results are consistent with previous reports that psychiatric disorder genes were found to be more active in prenatal brain transcriptome, while genes associated with neurological diseases become more active during the life course of an affected individual, and towards aging stages [56]. However, by integrating GWAS data with gene co-expression modules, we provided an additional line of evidence to support the genetic basis of these two different categories of brain disorders, and at specific temporal levels. In addition, EDU, COL, SWB and two subtypes of neuroticism yielded more enriched modules in postnatal transcriptome, which may suggest that these traits are partially influenced by the environment. Importantly, for SCZ, we found significant modules in both prenatal and postnatal transcriptomes, although relatively more modules in prenatal than in postnatal. This is in line with the current understanding of the pathophysiology of SCZ that preaches that SCZ has its roots in abnormalities in both neurodevelopmental and neurodegenerative processes. However, we did not find any alteration for MDD, neither in the prenatal nor in the postnatal stages. This later finding might reflect the fact that MDD is a rather heterogeneous condition that can be better conceptualized as a syndrome as opposed to a unitary disorder.
Figure 3 .
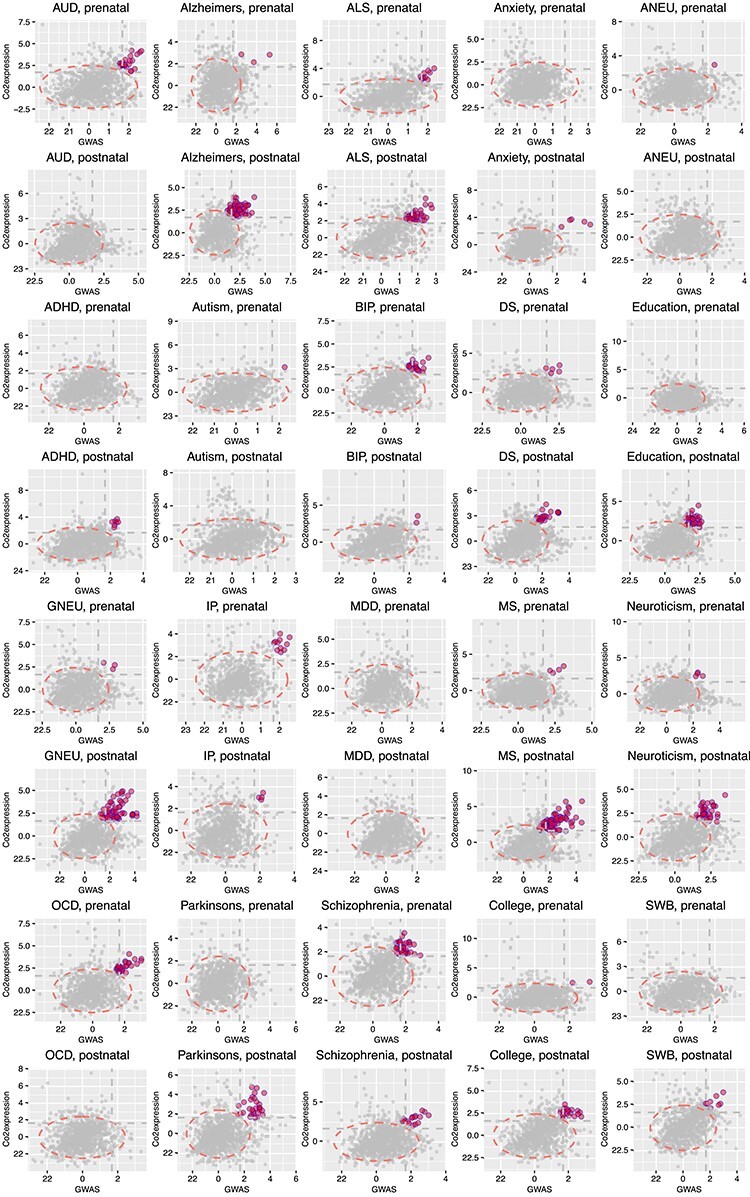
tpGWAS revealed different patterns of module distribution in 20 brain-related disorders or traits. In each panel, a dot represents a module. The x-axis is the normalized node score from 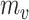 , which is calculated using gene-based P-value from GWAS. The y-axis is the normalized edge score from
, which is calculated using gene-based P-value from GWAS. The y-axis is the normalized edge score from 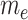 , which is calculated using gene co-expression. Normalization was conducted using random modules, which are not shown in the figure, but the 95% confidence interval estimated by using random modules is shown as the red eclipse. Color dots indicate significant modules selected to define module genes. The horizontal and vertical gray dash lines indicate where x = 1.68 or y = 1.68 (95% of modules having the scores below the line). ANX, anxiety; COL, college.
, which is calculated using gene co-expression. Normalization was conducted using random modules, which are not shown in the figure, but the 95% confidence interval estimated by using random modules is shown as the red eclipse. Color dots indicate significant modules selected to define module genes. The horizontal and vertical gray dash lines indicate where x = 1.68 or y = 1.68 (95% of modules having the scores below the line). ANX, anxiety; COL, college.
The BrainCloud (GEO ID: GSE30272) dataset [57] was used to validate the modules. This dataset profiled 269 human prefrontal cortex samples ranging from fetal development through aging (80 years) using the microarray platform. We implemented two methods for the validation, one using a one-sided t-test and the other estimating the random expectation to test if the module edges had significantly higher edge weight than chance. Details of both methods were presented in the Supplementary Material available online at https://academic.oup.com/bib. In short, more than half of the modules discovered using BrainSpan were validated by both methods (Supplementary Table S5 and Supplementary Figure S1 available online at https://academic.oup.com/bib). When future data with more samples are available, we will do further validations.
As a result, we focused on modules and genes for 5 disorders and traits in the prenatal stage (AUD, BD, IP, OCD and SCZ) and 12 disorders and traits in the postnatal stage (ALZ, ALS, ADHD, DS, EDU, GNEU, MS, NEU, PD, SCZ, COL and SWB). Specifically, we excluded those ‘disorder/trait – stage’ pairs where 10 or less genes were identified, as these were likely random co-occurrence, or the resultant networks would be too small (Table 2). Particularly, for the few modules found in the prenatal stage for ALZ, ALS, NEU and MS (Table 2), we found that these modules were all from the cell growth and DNA replication pathways. We have noticed that cell cycle regulation-related pathways were particularly more active during fetal brain development than in adult brains (Supplementary Figure S2 available online at https://academic.oup.com/bib). Such pattern was also validated using the BrainCloud data. Thus, we discarded such modules since they were not related to diseases but rather to general cellular processes.
Distinct temporal expression patterns of brain disorders stratified by brain regions
The original BrainSpan project categorized the brain samples into 16 brain regions, which can be further combined as six major regions including the neocortex areas (NCX), hippocampus (HIP), amygdala, striatum, mediodorsal nucleus of the thalamus, and cerebellar cortex (CBC) (Supplementary Table S6 available online at https://academic.oup.com/bib). By stratifying samples based on both the region and the stage annotations, we conducted tpGWAS for each of the 20 GWAS datasets (6 regions × 2 stages × 20 traits = 240 runs). As shown in Figure 4, the enrichment patterns that we observed when combining all regions (Figure 3) were largely observed when we stratified samples by regions: psychiatric disorders (ASD, AUD, IP and OCD) were mainly enriched in prenatal, neurological diseases (ALS, ALZ, MS and PD) and most other traits (COL, DS, EDU, GNEU, NEU and SWB) were mainly enriched in postnatal, and some disorders, notably BD and SCZ, were found enriched in both stages. As for region specificity, we found the neuropsychiatric traits were mainly enriched in two brain regions, NCX and HIP, in the prenatal stage. In the postnatal stage, several traits (COL, DS, EDU, GNEU, NEU and SWB) were enriched in NCX and CBC. On the other hand, neurological diseases (ALS, MS and PD) were found enriched in nearly all six regions. Interestingly, we did not find significant modules for ASD in any stages when combining all regions (Figure 3), likely due to the limited power of the original GWAS study [30]. However, in the region-stratified analysis, we found significant modules in the HIP, the brain region that is mainly responsible for learning and memory. Abnormality of HIP has been linked to ASD in many studies [58, 59]. In our results, we only found enrichment of ASD genes in prenatal HIP, further underscoring the importance of temporally and spatially stratified analyses. However, considering that the sample sizes were small in some temporal and spatial conditions, some previously reported ‘disorder/trait – region’ associations were missing and future validations with large sample sizes are required.
Figure 4 .
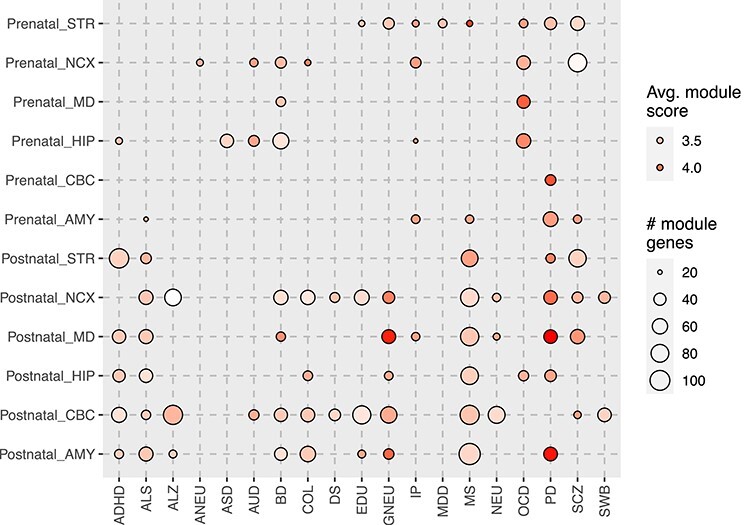
Module discovery using region and developmental stage transcriptome profiles. Full names of the brain regions and traits can be found in the main text and in the legend of Figure 3.
Cell type-specific expression of module genes
We next examined the cell type-specific features of the module genes for each brain-related disorder/trait using two brain scRNA-seq panels (DER-20 and DER-22). We found that the module genes of each trait were generally consistent with their discovering stages: genes found in prenatal samples were mainly enriched in cell types identified in fetal brain and genes found in postnatal samples were mainly enriched in cell types from adult brain (Figure 5A). For example, all five trait-associated networks in prenatal stage were significantly enriched in fetal ExN (AUD: PBH = 0.12; BD: PBH = 2.87 × 10−3; IP: PBH = 0.059; OCD, PBH = 0.059; SCZ: PBH = 0.059) and fetal transient cells (AUD: PBH = 0.049; BD: PBH = 0.012; IP: PBH = 0.012; OCD, PBH = 0.027; SCZ: PBH = 0.10). Note that we considered PBH < 0.2 as statistically significant in these analyses. In the postnatal stage, all traits showed enrichment in one or more excitatory neurons, as observed in both the DER-20 and DER-22 panels. EDU, NEU and its subtype GNEU were also enriched in the InNs.
Figure 5 .
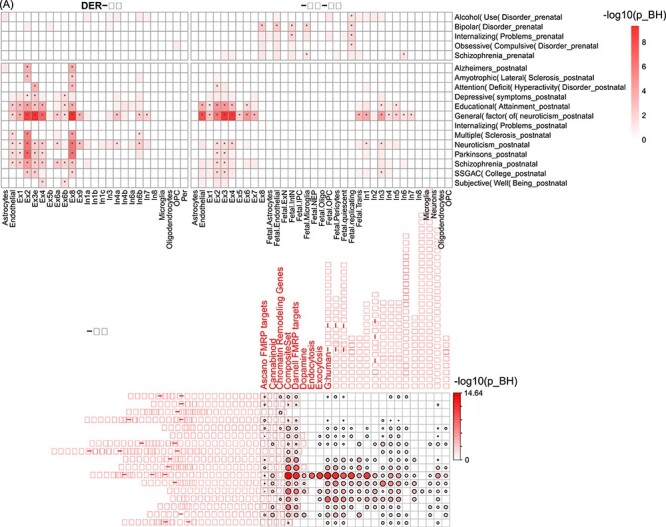
Enrichment analysis of module genes using cell type expression and functional annotations. (A) Cell type-specific enrichment analysis using two panels of brain scRNA-seq data (DER-22 and DER-20). (B) Functional enrichment of module genes (rows) using curated gene sets (columns) previously shown to be involved in brain disorders. The color of each cell is proportional to −log10(PBH).
There was some notable difference in enrichment types between the DER-20 and DER-22 datasets. In addition to the batch differences between Lake et al. [51] and Lake et al. [52], the different cell types in the two panels are, probably, the main reason for such differences. In each panel, the t-score was calculated by comparing one cell type to the remaining cells (considered as the reference group in the regression model). Thus, the same cell type (e.g. Ex1) in DER-20 was compared to different reference cells as analyzed in DER-22, especially because fetal cells are relatively more distinguishable from adult cells.
We conducted a gene set enrichment analysis to investigate the functions of module genes using 40 custom gene sets curated for brain-related functions in our previous study [60]. These gene sets included neurotransmitter signaling (e.g. dopamine, neurotrophin, serotonin, and glutamate), postsynaptic density proteins (PSD), synaptic genes and ion balance, among others [1, 60]. Figure 5B shows that the module genes found in postnatal were enriched in PSD-related genes except ADHD genes. Module genes of neuroticism were enriched in many gene sets, especially PSD-related sets and neurotransmitter signaling sets. However, module genes of traits found in prenatal brains were only marginally enriched in a few functional sets.
tpGWAS prioritized distinct genes in prenatal and postnatal stages
We next examined the module genes and their co-expression patterns in each brain-related disorder or trait. We explored the functional enrichment results of subnetworks, the interactions in both stages and the trajectory expression of the major principal components of the module genes. We demonstrated tpGWAS by using the following representative disorders and traits:
Alcohol use disorder
The significant modules found in the prenatal stage for AUD GWAS were merged as one disease network, consisting of 33 genes (Figure 6A). To examine the expression changes over stages, we conducted PCA of these genes to identify the major trajectory patterns. The first three PCs explained 78.73% of the total variance (Figure 6C). Nine genes showed a pattern similar to PC1 (absolute PCC > 0.7), including seven genes (ABL1, CAD, CHD3, CTNND1, DCAF7, EP300 and GATAD2B) highly expressed in the prenatal stage with decreasing expression along the developmental stages (PCC < −0.7 with PC1; opposite trend with PC1) and two genes (ALDOA and FMNL1, PCC > 0.7) showing an opposite trend (lowly expressed in the prenatal stage followed by increasing expression). Seven genes showed a pattern similar to PC2 and two genes with PC3 (GTF3A and PSMA3). In summary, most of the module genes had high expression in the prenatal stage when compared to the postnatal stage. Functional enrichment analysis showed that the top Gene Ontology (GO) Biological Process term for the AUD network was ‘regulation of cell-matrix adhesion’ (PBH = 1.04 × 10−3, including ABL1, DAPK3, LIMCH1, PRKCZ and RHOA). This confirmed the previous report that cell matrix likely changed the behavioral response to drug abuse [61], such as alcohol abuse [62].
Figure 6 .
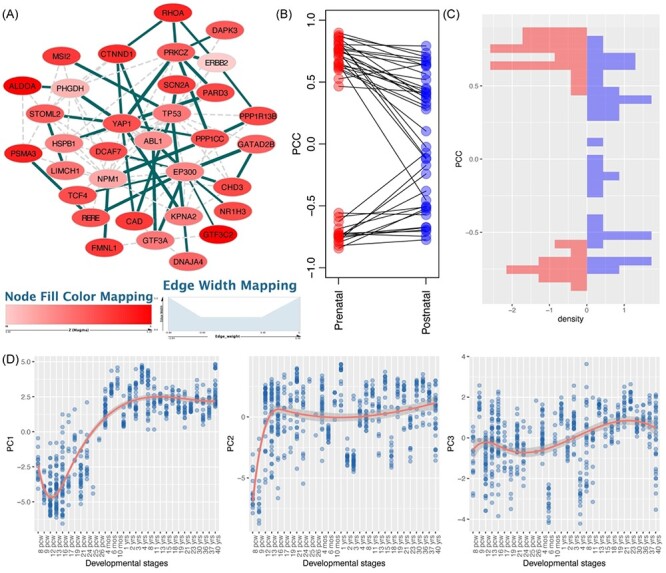
AUD-associated network and features of module genes. (A) Network visualization of AUD module genes identified in prenatal transcriptome. Blue edges indicate those involved in the identified modules, while gray edges are only connected using the reference interactome. Node color is proportional to the gene-based P-value. Edge width is proportional to the absolute PCC value in the prenatal stage. Gray edges are not involved in the discovery of modules. (B-C) Distribution of module gene interactions (edge scores) in prenatal (left panel) and postnatal (right panel) data. These interactions show consistent high co-expression in prenatal stage in both the discovery and validation datasets, but such a pattern is not observed in the postnatal stage in either the discovery or the validation dataset. (D) The first three principal components (PCs) of the module genes and their trajectory expression across the whole developmental stage.
Schizophrenia
The SCZ GWAS was found enriched in both the prenatal and postnatal brain transcriptomes, which was the only trait that showed such a pattern among all the 20 disorders and traits examined. There are, thus, two distinctive networks from either stage, and they shared five genes (CACNA1C, CACNB1, CACNB2, NFATC3 and XRCC3) (Figure 7). These findings support the hypothesis that SCZ is both neurodevelopmental and neurodegenerative in nature. The prenatal network (consisting of 77 module genes) was enriched with biological processes such as protein C-terminus binding (PBH = 3.19 × 10−4), several processes of transcription factor binding regulation and several regulation processes of cell cycle. The postnatal network (24 genes), although with fewer genes, was found highly enriched in synapse and central nervous system (CNS)-related regulations. Three of the shared genes were involved in voltage-gated calcium ion channels, which have been extensively studied in SCZ [63, 64]. We also find enriched pathways in cardiac functions. This is not surprising, as investigators have shown that the risk genes of SCZ may be promoting pleiotropic effects of cardiovascular dysfunction [65].
Figure 7 .
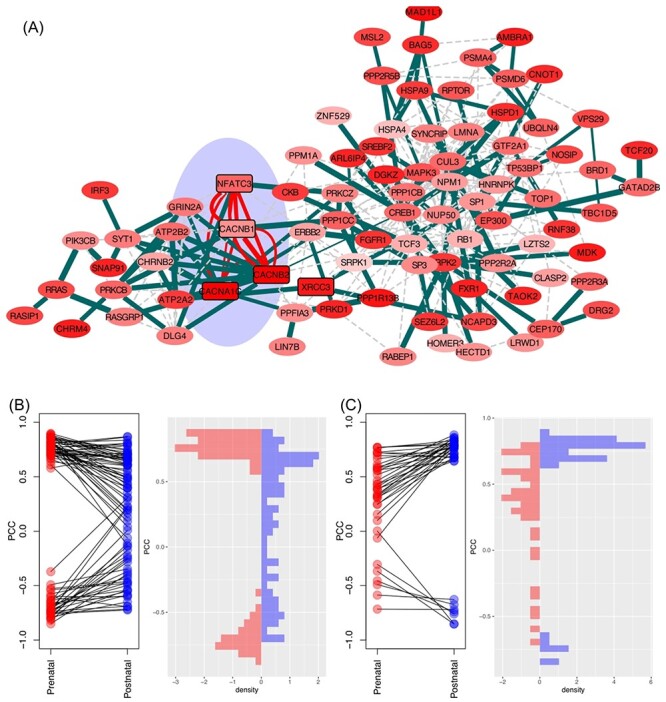
SCZ-associated network and features of module genes. (A) Network visualization of SCZ module genes identified in prenatal and postnatal transcriptomes. The two networks shared five genes (shaded cyan area). Distribution of module gene interactions (edge scores) in prenatal (B) and postnatal stages (C).
Alzheimer’s disease
ALZ is associated with both cognitive and motor deficits in late life. In our results, we found 69 modules for the ALZ GWAS in the postnatal stage (Figure 3). After merging these modules, we found that the resultant network was centered on the well-studied ALZ gene, APP (Figure 8A). This network included 94 genes, 31 of which were positively associated with PC1, where 22 genes showed higher expression in the postnatal stage than the prenatal stage and 9 genes showed an opposite trend. Seventeen genes presented a negative correlation with PC2, which means that these genes were more highly expressed in the postnatal stage than in the prenatal stage, and 14 genes presented a positive correlation with PC2, which means that they were more highly expressed in the prenatal stage than in the postnatal stage. Functional enrichment analysis of the module genes revealed that they were enriched in protein kinase binding (PBH = 8.19 × 10−4), lipid binding (PBH = 0.01), regulation of vesicle-mediated transport (PBH = 2.26 × 10−3), regulation of synaptic plasticity (PBH = 2.26 × 10−3), clathrin-coated vesicle membrane (PBH = 3.08 × 10−4) and synaptic vesicle (PBH = 3.08 × 10−4). Among them, clathrin-coated vesicles are well studied and highly involved in the pathology of ALZ [66–68]. The clathrin chains of proteins make up the coating of vesicles, which are essential to neuronal functions [66]. Abnormal clathrin-mediated endocytosis has shown to contribute to pathological processes of some neurological diseases [66, 67]. Furthermore, amyloid-β, which has been largely implicated in the pathophysiology of ALZ, is produced from amyloid precursor protein primarily after amyloid precursor protein is internalized by clathrin-mediated or clathrin-independent endocytosis [66, 69]. Interestingly, three genes (BACE1, APOE and APP) were involved in amyloidosis-related regulation, such as amyloid-beta metabolic process (PBH = 0.025) and amyloid fibril formation (PBH = 0.030).
Figure 8 .
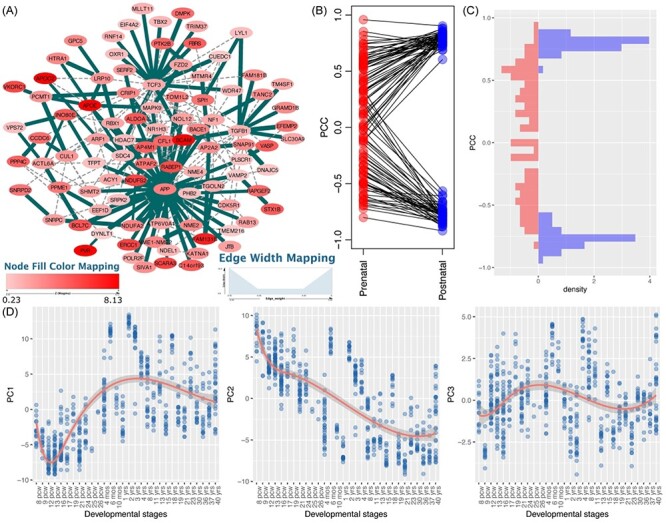
Alzheimer’s disease (ALZ)-associated network and features of module genes. (A) Network visualization of ALZ module genes identified in the postnatal stage. (B-C) Distribution of module gene interactions (edge scores) in postnatal (left panel) and prenatal (right panel) stages. These interactions showed consistent high co-expression in both the discovery and validation datasets using samples of the postnatal stage; however, such a pattern was not observed in the prenatal stage in either discovery or the validation dataset. (D) The first three principal components (PCs) of the module genes and their trajectory expression across the whole developmental stage. pcw, post-conception week.
Multiple sclerosis
The MS GWAS was also more aligned with adult gene expression, resulting in a subnetwork with 102 genes. The top enriched GO Cellular Component terms for MS included cellular response to lipid (PBH = 5.94 × 10−6), myelin sheath (PBH = 4.42 × 10−3, component genes: ATP1B1, CLTC, GNB1, NSF, NDUFS3, PHGDH and SLC25A12), among others. In MS, the autoimmune attacks are directed toward myelin sheath of the CNS [25, 70], and our findings support this previous knowledge.
Educational attainment
EDU (an anthropometric social trait that might be used as a proxy for cognition) GWAS was better aligned with the adult gene expression data (Figure 3). The top enriched GO Biological Process terms for the 54 component genes included synapse organization (PBH = 9.19 × 10−5, component genes ACTB, ACTR2, APP, BSN, GABRB2, MEF2C, MTMR2, PAK1, PTPRF, RAB3A and STAU1) and learning (PBH = 6.73 × 10−3, ACTR2, APP, CLN3, HTT and PAK1). Regulation of synaptic plasticity plays an essential role in learning and memory, both of which are very important processes to EDU [71].
College completion
We found significant modules for the COL GWAS in the adult gene expression data, resulting in a network with 38 genes. One of the top 10 GO Biological Process terms enriched for this network was regulation of synaptic plasticity (PBH = 0.02) with component genes ARF1, HTT, PRNP, SNAP25 and YWHAG. Notably, the genes HTT and YWHAG were present in the disease networks for both EDU and COL. HTT encodes the protein huntingtin. This protein is essential in brain development, although it has a poorly understood function [72]. The gene YWHAG encodes for a signal transduction protein that has been implicated in the function of smooth muscle [73]. Our results suggest that both HTT and YWHAG might play essential roles in synaptic plasticity, which is crucial to learning and memory.
Discussion
In this work, we conducted temporal gene co-expression network analyses of GWAS data for 20 brain-related disorders or traits. Our analytical strategy falls in the area of supervised analyses and our results complement insights from previous studies. We demonstrated that the integration analysis of GWAS and co-expression data can reveal biologically insightful patterns when conducted in a developmental stage-stratified manner. Our results revealed different stages in which the trait-associated genes were more coordinately expressed. Also, our tpGWAS method identified smaller modules that are more accessible for biological interpretation and, thus, more relevant for translation and future implementation into actionable knowledge. Further functional enrichment analyses revealed that the module genes had significant relevance to the disorders and traits examined.
Our results capitalized and confirmed previous reports that disorders traditionally classified as psychiatric tend to have a neurodevelopmental origin while neurological diseases tend to show high gene activity in the postnatal stage. For instance, BD, OCD and NEU showed marked alterations in the prenatal stage, and AD, ALS and PD, together with the anthropometric traits, such as college completion, education and well-being, were mostly altered in the postnatal brains. Of note, SCZ was the only condition that was found altered at both the prenatal and postnatal stages. This helps to conceptualize and further supports the traditional division of psychiatric and neurological disorders. It also supports the notion that SCZ is a more distinct phenotype with some differentiation from BD, a psychiatric disorder that shares some of SCZ biological correlates. In addition, the reported module genes supported biological functions that are related to disorders or traits, such as cell–matrix adhesion in AUD, clathrin chain cellular components in AD, myelin sheath in MS and synaptic plasticity in learning (EDU and COL). Some of these discoveries provide insights for maximizing results when searching future optimal molecular therapeutic targets. For example, the inhibition of clathrin-mediated endocytosis has been studied as a therapeutic option to prevent neuronal damage in AD [68]. We prioritized genes enriched for clathrin-coated vesicle membrane as potentially contributing to AD development, as well as possible therapeutic targets: AP2A2, APOE, DNAJC5, FZD2, SNAP91, TGOLN2 and VAMP2. Taken together, our work not only revealed the critical developmental stages in which each trait was actively expressed but also prioritized genes and functions that, much probably, constitute more promising targets that might be prioritized in future investigations.
Our work has some limitations. First, we only defined the prenatal and postnatal stages for temporal analysis. Brain development involves multiple stages with precise molecular cascades. With increased sample size and data from more representative stages, in the future we will integrate in our network analysis more refined time points that are critical to brain development such as adolescence. Second, it is important to notice that we cannot infer causality of the found altered genes in the 20 conditions considered, or their alterations per se that are sufficient condition for the development of the disease/trait analyzed. Most likely, there is a fine interplay between genes and environment. For example, exposure to environmental insults, such as tobacco and alcohol, can, at least in some instances, be a necessary trigger for the development of the disorders. In future, results from epigenome-wide association studies, as well as transcriptional or post-transcriptional regulatory mechanisms (e.g. DNA and RNA methylation, histone modification and chromosome remodeling), may be included in such network modularity-based analysis to identify epigenetic regulations during brain development. Third, although we stratified samples based on regions, the sample size in each spatiotemporal condition was small. Future work with more samples is warranted to enable the identification of modules in different brain regions and provide more detailed insights into how disease genes are coordinately expressed in different contexts.
In conclusion, we conducted a network-assisted analysis of GWAS-implied genes using temporal transcriptome data. Our results not only revealed the brain developmental stages in which GWAS genes were enriched but also how these genes were related. Our study not only shed light on the still relatively obscure pathophysiology of these disorders by illuminating implicated biological pathways, it also will aid the prioritization of promising molecular targets in future endeavors dedicated to drug development for these serious and often debilitant disorders.
Key Points
We conducted a systematic investigation of the temporal expression patterns of a myriad of 20 brain-related disorders, providing a systematic view of network modularity at specific developmental stages for these disorders and traits.
Our analyses unveiled a strong pattern of a fetal origin for most psychiatric disorders, whereas increased co-expression activities of genes associated with neurological diseases and anthropometric traits were more strongly altered in postnatal brains.
Further analyses revealed enriched cell types and functional features that were supported and corroborated prior knowledge in specific brain disorders.
Supplementary Material
Acknowledgements
We thank Dr Chunyu Liu and Dr Daifeng Wang for valuable discussions on the usage of PsychENCODE data and the PsychENCODE Consortium for making the data publicly available. The authors would like to thank members of the Bioinformatics and Systems Medicine Laboratory for valuable discussion.
Peilin Jia is an assistant professor of bioinformatics in the Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston. Her research interest includes bioinformatics, machine learning, methodology development and integrative genomics.
Astrid M. Manuel is a PhD candidate of bioinformatics in the Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston. Her research interest includes human genetics, bioinformatics, and methodology development.
Brisa S. Fernandes is a postdoctoral fellow in the Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston. Her research interest includes precision medicine in psychiatric disorders, biomarkers, bioinformatics and integrative analysis.
Yulin Dai is a research assistant professor of bioinformatics in the Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston. His research interest includes bioinformatics, genetics and integrative genomics
Zhongming Zhao is the chair professor for precision health and the founding director of the Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston. He directs the Bioinformatics and Systems Medicine Laboratory and UTHealth Cancer Genomics Core. His research interest includes bioinformatics, integrative genomics and methodology development.
Contributor Information
Peilin Jia, Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston, 7000 Fannin St. Suite 600, Houston, TX 77030, USA.
Astrid M Manuel, Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston, 7000 Fannin St. Suite 600, Houston, TX 77030, USA.
Brisa S Fernandes, Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston, 7000 Fannin St. Suite 600, Houston, TX 77030, USA.
Yulin Dai, Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston, 7000 Fannin St. Suite 600, Houston, TX 77030, USA.
Zhongming Zhao, Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston, 7000 Fannin St. Suite 600, Houston, TX 77030, USA.
Data availability
All data are publicly available. Computational codes are available upon request.
Funding
National Institutes of Health (NIH) grants [R01LM012806 and R01DE030122 to Z.Z.]; Cancer Prevention and Research Institute of Texas [CPRIT RP180734 to Z.Z.]. A.M.M. was supported by a training fellowship from the Gulf Coast Consortia, on the NIH National Library of Medicine (NLM) Training Program in Biomedical Informatics & Data Science (T15LM007093). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1. Jia P, Chen X, Xie W, et al. Mega-analysis of odds ratio: a convergent method for a deep understanding of the genetic evidence in schizophrenia. Schizophr Bull 2019;45:698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011;474:380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parikshak NN, Luo R, Zhang A, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013;155:1008–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gupta S, Ellis SE, Ashar FN, et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 2014;5:5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Willsey AJ, Sanders SJ, Li M, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013;155:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilman SR, Chang J, Xu B, et al. Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat Neurosci 2012;15:1723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin GN, Corominas R, Lemmens I, et al. Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron 2015;85:742–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jia P, Chen X, Fanous AH, et al. Convergent roles of de novo mutations and common variants in schizophrenia in tissue-specific and spatiotemporal co-expression network. Transl Psychiatry 2018;8:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gulsuner S, Walsh T, Watts AC, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 2013;154:518–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shohat S, Ben-David E, Shifman S. Varying intolerance of gene pathways to mutational classes explain genetic convergence across neuropsychiatric disorders. Cell Rep 2017;18:2217–27. [DOI] [PubMed] [Google Scholar]
- 11. Tebbenkamp AT, Willsey AJ, State MW, et al. The developmental transcriptome of the human brain: implications for neurodevelopmental disorders. Curr Opin Neurol 2014;27:149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J, Hu S, Zhang K, et al. A comparative study of the genetic components of three subcategories of autism spectrum disorder. Mol Psychiatry 2019;24:1720–31. [DOI] [PubMed] [Google Scholar]
- 13. Lee PH, Feng YA, Smoller JW. Pleiotropy and cross-disorder genetics among psychiatric disorders. Biol Psychiatry 2021;89:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shohat S, Amelan A, Shifman S. Convergence and divergence in the genetics of psychiatric disorders from pathways to developmental stages. Biol Psychiatry 2021;89:32–40. [DOI] [PubMed] [Google Scholar]
- 15. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 2005;4:Article 17. [DOI] [PubMed] [Google Scholar]
- 16. Dam S, Vosa U, Graaf A, et al. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform 2018;19:575–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller JA, Ding SL, Sunkin SM, et al. Transcriptional landscape of the prenatal human brain. Nature 2014;508:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jensen M, Girirajan S. An interaction-based model for neuropsychiatric features of copy-number variants. PLoS Genet 2019;15:e1007879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J, Cai T, Jiang Y, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry 2016;21:290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jia P, Zheng S, Long J, et al. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics 2011;27:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Q, Yu H, Zhao Z, et al. EW_dmGWAS: edge-weighted dense module search for genome-wide association studies and gene expression profiles. Bioinformatics 2015;31:2591–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jia P, Zhao Z. Network-assisted analysis to prioritize GWAS results: principles, methods and perspectives. Hum Genet 2014;133:125–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Creixell P, Reimand J, Haider S, et al. Pathway and network analysis of cancer genomes. Nat Methods 2015;12:615–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jia P, Wang L, Fanous AH, et al. Network-assisted investigation of combined causal signals from genome-wide association studies in schizophrenia. PLoS Comput Biol 2012;8:e1002587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manuel AM, Dai Y, Freeman LA, et al. Dense module searching for gene networks associated with multiple sclerosis. BMC Med Genet 2020;13:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reyna MA, Leiserson MDM, Raphael BJ. Hierarchical HotNet: identifying hierarchies of altered subnetworks. Bioinformatics 2018;34:i972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Demontis D, Walters RK, Martin J, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 2019;51:63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kranzler HR, Zhou H, Kember RL, et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun 2019;10:1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Otowa T, Hek K, Lee M, et al. Meta-analysis of genome-wide association studies of anxiety disorders. Mol Psychiatry 2016;21:1391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium . Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism 2017;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stahl EA, Breen G, Forstner AJ, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet 2019;51:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baselmans BML, Jansen R, Ip HF, et al. Multivariate genome-wide analyses of the well-being spectrum. Nat Genet 2019;51:445–51. [DOI] [PubMed] [Google Scholar]
- 33. Benke KS, Nivard MG, Velders FP, et al. A genome-wide association meta-analysis of preschool internalizing problems. J Am Acad Child Adolesc Psychiatry 2014;53:667, e667–76. [DOI] [PubMed] [Google Scholar]
- 34. Wray NR, Ripke S, Mattheisen M, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 2018;50:668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. International Obsessive Compulsive Disorder Foundation Genetics Collaborative and OCDCGA studies . Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Mol Psychiatry 2018;23:1181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pardinas AF, Holmans P, Pocklington AJ, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 2018;50:381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 2019;51:404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rheenen W, Shatunov A, Dekker AM, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 2016;48:1043–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. International Multiple Sclerosis Genetics Consortium . Low-frequency and rare-coding variation contributes to multiple sclerosis risk. Cell 2018;175:1679, e1677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pankratz N, Beecham GW, DeStefano AL, et al. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann Neurol 2012;71:370–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rietveld CA, Medland SE, Derringer J, et al. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science 2013;340:1467–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okbay A, Beauchamp JP, Fontana MA, et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 2016;533:539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okbay A, Baselmans BM, De Neve JE, et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat Genet 2016;48:624–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leeuw CA, Mooij JM, Heskes T, et al. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 2015;11:e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang D, Liu S, Warrell J, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018;362:eaat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li M, Santpere G, Imamura Kawasawa Y, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018;362:eaat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brain Span Atlas . http://www.brainspan.org/.
- 48. Rodchenkov I, Babur O, Luna A, et al. Pathway commons 2019 update: integration, analysis and exploration of pathway data. Nucleic Acids Res 2020;48:D489–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jia P, Pei G, Zhao Z. CNet: a multi-omics approach to detecting clinically associated, combinatory genomic signatures. Bioinformatics 2019;35:5207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995;57:289–300. [Google Scholar]
- 51. Lake BB, Ai R, Kaeser GE, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016;352:1586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lake BB, Chen S, Sos BC, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 2018;36:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Darmanis S, Sloan SA, Zhang Y, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A 2015;112:7285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pei G, Dai Y, Zhao Z, et al. deTS: tissue-specific enrichment analysis to decode tissue specificity. Bioinformatics 2019;35:3842–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen J, Bardes EE, Aronow BJ, et al. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009;37:W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sey NYA, Hu B, Mah W, et al. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat Neurosci 2020;23:583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Colantuoni C, Lipska BK, Ye T, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 2011;478:519–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeLong GR. Autism, amnesia, hippocampus, and learning. Neurosci Biobehav Rev 1992;16:63–70. [DOI] [PubMed] [Google Scholar]
- 59. Codagnone MG, Podesta MF, Uccelli NA, et al. Differential local connectivity and neuroinflammation profiles in the medial prefrontal cortex and hippocampus in the Valproic acid rat model of autism. Dev Neurosci 2015;37:215–31. [DOI] [PubMed] [Google Scholar]
- 60. Jia P, Han G, Zhao J, et al. SZGR 2.0: a one-stop shop of schizophrenia candidate genes. Nucleic Acids Res 2017;45:D915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lasek AW. Effects of ethanol on brain extracellular matrix: implications for alcohol use disorder. Alcohol Clin Exp Res 2016;40:2030–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith AC, Scofield MD, Kalivas PW. The tetrapartite synapse: extracellular matrix remodeling contributes to corticoaccumbens plasticity underlying drug addiction. Brain Res 2015;1628:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li W, Fan CC, Maki-Marttunen T, et al. A molecule-based genetic association approach implicates a range of voltage-gated calcium channels associated with schizophrenia. Am J Med Genet B Neuropsychiatr Genet 2018;177:454–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang T, Zhu L, Ni T, et al. Voltage-gated calcium channel activity and complex related genes and schizophrenia: a systematic investigation based on Han Chinese population. J Psychiatr Res 2018;106:99–105. [DOI] [PubMed] [Google Scholar]
- 65. Chen X, Lee G, Maher BS, et al. GWA study data mining and independent replication identify cardiomyopathy-associated 5 (CMYA5) as a risk gene for schizophrenia. Mol Psychiatry 2011;16:1117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu F, Yao PJ. Clathrin-mediated endocytosis and Alzheimer's disease: an update. Ageing Res Rev 2009;8:147–9. [DOI] [PubMed] [Google Scholar]
- 67. Nakamura Y, Takeda M, Yoshimi K, et al. Involvement of clathrin light chains in the pathology of Alzheimer's disease. Acta Neuropathol 1994;87:23–31. [DOI] [PubMed] [Google Scholar]
- 68. Kuboyama T, Lee YA, Nishiko H, et al. Inhibition of clathrin-mediated endocytosis prevents amyloid beta-induced axonal damage. Neurobiol Aging 2015;36:1808–19. [DOI] [PubMed] [Google Scholar]
- 69. Alsaqati M, Thomas RS, Kidd EJ. Proteins involved in endocytosis are upregulated by ageing in the normal human brain: implications for the development of Alzheimer's disease. J Gerontol A Biol Sci Med Sci 2018;73:289–98. [DOI] [PubMed] [Google Scholar]
- 70. Lemus HN, Warrington AE, Rodriguez. Multiple sclerosis: mechanisms of disease and strategies for myelin and axonal repair. Neurol Clin 2018;36:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Humeau Y, Choquet D. The next generation of approaches to investigate the link between synaptic plasticity and learning. Nat Neurosci 2019;22:1536–43. [DOI] [PubMed] [Google Scholar]
- 72. Young AB. Huntingtin in health and disease. J Clin Invest 2003;111:299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jin Y, Dai MS, Lu SZ, et al. 14-3-3gamma binds to MDMX that is phosphorylated by UV-activated Chk1, resulting in p53 activation. EMBO J 2006;25:1207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are publicly available. Computational codes are available upon request.