

Abstract
Epigenomics and transcriptomics data from high-throughput sequencing techniques such as RNA-seq and ChIP-seq have been successfully applied in predicting gene transcript expression. However, the locations of chromatin loops in the genome identified by techniques such as Chromatin Interaction Analysis with Paired End Tag sequencing (ChIA-PET) have never been used for prediction tasks. Here, we developed machine learning models to investigate if ChIA-PET could contribute to transcript and exon usage prediction. In doing so, we used a large set of transcription factors as well as ChIA-PET data. We developed different Gradient Boosting Trees models according to the different tasks with the integrated datasets from three cell lines, including GM12878, HeLaS3 and K562. We validated the models via 10-fold cross validation, chromosome-split validation and cross-cell validation. Our results show that both transcript and splicing-derived exon usage can be effectively predicted with at least 0.7512 and 0.7459 of accuracy, respectively, on all cell lines from all kinds of validations. Examining the predictive features, we found that RNA Polymerase II ChIA-PET was one of the most important features in both transcript and exon usage prediction, suggesting that chromatin loop anchors are predictive of both transcript and exon usage.
Keywords: gene expression, transcript, exon usage, machine learning, ChIA-PET, chromatin loop anchors, histone modifications, alternative splicing
Introduction
Transcription is the synthesis of RNA from DNA. An exon is any part of a gene that will encode a part of the final mature RNA produced by that gene after introns have been removed during pre-messenger RNA splicing (Figure 1A). Differential exon usage via alternative splicing can add greatly to the diversity of gene products encoded by the genome [1]. More specifically, the usage of exons has the potential to affect the stability, localization or translation of RNAs, as well as the specificity, efficiency, localization or life cycle of proteins [2]. Dysregulation in gene transcription and exon usage is related to disease [3–6]. For example, certain isoforms of PKM2 and MAP2 gene have higher expression in later stage of neuroblastoma compared with other isoforms [5]. This preference of certain isoforms may be a result of tumorigenesis, and therefore, exon usage in cancer can reveal more information about cancer pathways.
Figure 1 .
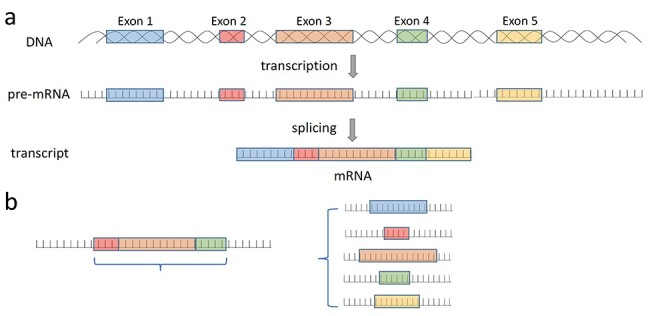
(A) The relationship between transcript and exon. (B) An illustration of the difference between our method and other prediction methods. Other methods mainly predict the splicing level with more than two exons (left), our method focuses on one exon to predict its usage regardless of the splicing event it included (right).
RNA sequencing (RNA-seq) plays an important role in revealing the expression levels of genetic features such as genes, transcripts or exons between samples [7]. RNA-Seq can be used to identify exon usage, meaning the number of reads that fall within a particular exon. Additionally, RNA-seq data can be used to quantify alternative splicing events by using the junction reads of RNA-Seq data. For example, Kakaradov et al. [8] presented several probabilistic models of position-specific read counts with increasing complexity to estimate the percent inclusion of alternatively spliced junctions from RNA-seq data.
Depending on the nature of the question studied, exon usage or percent inclusion of alternatively spliced junctions can be used to interrogate the question of alternative splicing in RNA-Seq. Papers that used percent inclusion included Goldstein et al. and Zhang et al. Goldstein et al. [9] assembled the splice junctions and exons in terms of mapped reads into a genome-wide splice graph and identify the splice events from the graph. Zhang et al. [10] developed a bayesian hypothesis testing statistical model to infer the differential alternative splicing by integrating empirical evidence in a specific RNA-seq dataset with prior probability of differential alternative splicing. In contrast, Lee et al. used exon usage. Specifically, Lee et al. [11] measured the splicing events according to exonic expression level represented by fragment per kilobase per million reads mapped FPKM from RNA-seq.
Chromatin Immunoprecipitation Sequencing (ChIP-Seq) facilitates the identification of whole-genome localization of protein–DNA binding sites [12]. ChIP-Seq produces data for transcription factors (TFs) on gene expression. Histone modification (HM) is one set of critical chemical reactions at the chromatin that plays a crucial role in regulating gene expression by altering chromatin structure or recruiting histone modifiers [13]. ChIP-seq peaks for TFs and transcriptome data are used individually or in a combinatorial manner for gene regulation and expression analysis or prediction. For example, BETA combined ChIP-seq and transcriptome data to unravel the regulation of gene expression [14], and DeepDiff interprets how dependencies among HMs control the differential patterns of gene regulation [13]. Different kinds of TF synthetic indexes extracted from ChIP-seq data are used to predict the gene expression level [15] and predict TF binding [16]. Moreover, some studies use the combination of the ChIP-Seq and RNA-Seq data to reveal the association between specific HMs and various aspects of differential splicing. For example, Hu et al. [17] developed computational approaches to model the association between alternative splicing and histone posttranslational modifications in mammalian brain.
Recently, machine learning has gained widespread attention and has been successfully applied to predict splicing using RNA-seq data, ChIP-seq data or DNA sequences. Leung et al. [18] presented a model inferred from mouse RNA-Seq data to predict splicing events in individual tissues and differences across tissues, Jha et al. [19] proposed a modeling framework that leverages transfer learning to incorporate CLIP-Seq, knockdown and over expression experiments in mouse tissues and a computational framework named DARTS was developed to infer differential alternative splicing between biological samples by integrating deep learning-based predictions with empirical RNA-seq evidence [10].
Studies such as Epigenome-based Splicing Prediction using a Recurrent Neural Network (ESPRNN) also considered other types of data such as DNase-seq, eCLIP, methylation and MNase-seq [11]. However, ESPRNN only used adjacent epigenetic signals around the splice sites, but not across the entire gene. Besides, none of these methods considered any chromatin interaction related data, i.e. Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET) data.
ChIA-PET is a technique that incorporates sonication-based chromatin fragmentation, chromatin ChIP-based enrichment, chromatin proximity ligation and Paired-End Tag high-throughput sequencing to determine genome-wide de novo long-range chromatin interactions [20]. Chromatin interactions can regulate gene transcription [21]. Oncogenes and remote regulatory elements can be brought into close spatial proximity through chromatin interactions [22], which may serve structural basis for protein binding and thus lead to transcription regulation. Chromatin organization has been shown to be correlated with alternative splicing [23]. A recent study showed that the degradation of RNA polymerases can affect local chromatin architectures and these local regions include high RNA polymerases binding sites and active promoters [24]. However, no other studies report on whether chromatin interactions have any ability to predict exon usage.
Based on the evidence discussed above, we hypothesized that in addition to ChIP-seq and RNA-seq data, ChIA-PET data indicating chromatin loop anchors might also be of predictive value, but it has not been tested in transcript and exon usage prediction. Hence, here, we asked the question: can we apply ChIA-PET data to transcript and exon usage prediction and will such data contribute to the prediction? What are the chromatin factors that can predict transcript and exon usage, if any? Therefore, in this work, we aimed to use machine learning method, i.e. gradient boosting trees (GB), to predict the transcript and exon usage using the ChIA-PET data as well as TF data.
As opposed to previous splicing event prediction methods which use the level of splicing calculated from the exonic expression level involved in that splicing event, we predicted exon usage independently (Figure 1B) and quantified transcripts directly. Previous prediction methods mainly predict alternate usages of cassette exons (inclusion or exclusion of exons), which is the most common type of alternative splicing; hence, they only detect one type of alternative splicing event. But we should note that there are other events (for example stand-alone expression of a single exon) that will affect exon usage. Our exon usage measurements include cassette exons, 5′ and 3′ terminal exons, mutually exclusive exons and even single-exon (intronless) transcripts. According to the best of our knowledge, no previous methods had used machine learning model to predict the usage level for single exon transcripts.
Another difference between previous methods and ours is that previous methods such as ESPRNN use DNA sequences and epigenomic signals adjacent to splice sites, whereas we are use epigenomic signals present throughout the gene, not just at splice sites. A comparison between our method and other prediction methods was listed as Supplementary Table S1, see Supplementary Data available online at http://bib.oxfordjournals.org/. We collected and integrated the RNA-seq, ChIP-seq, ChIA-PET and DNase-seq data of GM12878, HeLaS3 and K562 cell lines from different sources. Then, we developed GB models to see if we can apply these data successfully to different prediction tasks, including predicting the verified transcripts and transcript abundance, and predicting exon usage. The workflow to conduct this study is illustrated in Figure 2.
Figure 2 .
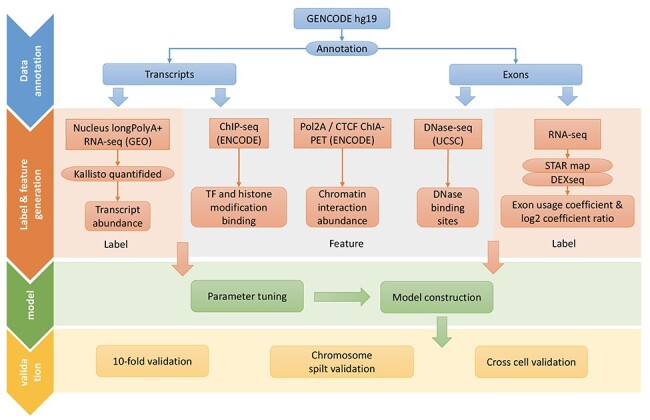
The workflow to conduct the study.
We evaluated our models via 10-fold cross validation, as well as the chromosome split and cross cell validations, which could break the dependencies between training and test data. Results show that our model realizes accuracy larger than 0.7707 (average), 0.7755 and 0.7512 for verified transcripts prediction and 0.7700 (average), 0.7459 and 0.7463 for exon usage prediction, respectively, regarding to the above three validation methods in different cell lines.
The validation results here illustrate the model’s effectiveness and robustness, as well as the plausibility to predict the transcript and exon usage. Interestingly, we noticed that the ChIA-PET and chromatin-related TF binding motif related features could play more significant roles than some of the most frequently used ChIP-seq features in both the transcription and exon usage prediction. Our results suggested that there may be a connection between chromatin factors and exon usage.
Material and methods
Data collection and definition
We collected all the publicly available datasets from cell lines from the ENCODE consortium [25], which have both high-quality HM ChIP-Seq as well as high-quality ChIA-PET information. GENCODE hg19 GTF file was used as the reference annotation for transcripts and exons. The scrambled GTF was generated by randomly shuffling the coordinates and accession ID in the reference GTF file. This scrambled GTF was used to generate a negative dataset for transcript and exon matrices.
After that, we generated the feature matrix of transcript and exon for both positive and negative datasets.
Transcript prediction feature matrix
Transcript abundance. Nucleus longPolyA RNA-seq data of K562, GM12878 and HeLaS3 cells were downloaded from GEO (GSM765387) and quantified using kallisto (0.45.1). We built two kallisto indexes using positive and negative GTF file. This resulted in a correctly quantified transcript abundance and a scrambled negative transcript abundance. We further quantiled the transcript abundance into four categories: not_transcribed, lowly_transcribed, transcribed and highly_transcribed according to the TPM (Transcripts Per Million). TPM is a normalization method for RNA-seq, it is calculated following the steps of: (i) divide the read counts by the length of each gene in kilobases, (ii) count up all the reads per kilobase (RPK) values in a sample and divide this number by 1 000 000 and (iii) divide the RPK values by the ‘per million’ scaling factor. In each cell line, we sorted the TPM values from smallest to largest; then, we found the first quartile (Q1), the second quartile (Q2) (median) and the third quartile (Q3) correspondingly. We defined the transcription with TPM value range from smallest to Q1 as not_transcribed, Q1 to Q2 as lowly_transcribed, Q2 to Q3 as transcribed and Q3 to largest as highly_transcribed. The value of Q1, Q2 and Q3 for three cell lines was recorded in Supplementary Table S2, see Supplementary Data available online at http://bib.oxfordjournals.org/.
TF and HM binding. ChIP-seq peaks of different TFs and HMs were downloaded from UCSC and overlapped with the transcript in the matrix. ChIP-seq peaks are the genomic regions with significantly more enrichment for proteins or HMs compared with genomic background, which are normally several hundreds to several kb long.
DNase binding sites. DNase binding sites were downloaded from UCSC and overlapped with the transcript in the matrix.
Chromatin interaction abundance. RNA Polymerase II (Pol2A) (GM12878 and K562) or CTCF (GM12878 and HeLaS3) ChIA-PET data were downloaded from ENCODE and overlapped with transcripts in the matrix. The number of ChIA-PET interactions that have either end overlapping with the transcript was used as the feature of chromatin interaction abundance.
We performed two prediction tasks using the data collected in this part. The first one is to predict verified transcripts as a two classes classification problem, and the other one is to predict the transcript abundance as a four classes classification problem. The corresponding number of positive and negative samples in each class for three cell lines was recorded in Supplementary Table S3, see Supplementary Data available online at http://bib.oxfordjournals.org/.
Exon usage prediction feature matrix
Similarly, exon feature matrices were prepared following the process of transcript feature matrices.
Exon usage coefficient. We first used STAR (2.7.3a) to map the RNA-seq reads using the true hg19 fasta and GTF file. The alignment results of the RNA-seq data were further prepared and counted for the number of reads mapped to different exons using DEXseq helping scripts. Then, DEXseq (1.28.0) was used to infer coefficient of the exon usage in K562, GM12878 and HeLaS3 cells, where the relative usage of an exon is defined as number of transcripts from the gene that contain this exon divided by number of all transcripts from the gene.
TF and HM binding. ChIP-seq peaks of different TFs and HMs were downloaded from UCSC and overlapped with the transcript in the matrix.
DNase binding sites. DNase binding sites were downloaded from UCSC and overlapped with the exon in the matrix.
Chromatin interaction abundance. Pol2A or CTCF ChIA-PET data were downloaded from ENCODE and overlapped with transcripts in the matrix. The number of ChIA-PET interactions that have either end overlapping with the transcript was used as the feature of chromatin interaction abundance.
The corresponding data amounts in each class for three cell lines in predicting the exon usage were recorded in Supplementary Table S4, see Supplementary Data available online at http://bib.oxfordjournals.org/, and the features used in each cell line for both transcription and exon usage prediction were listed in Supplementary Table S5, see Supplementary Data available online at http://bib.oxfordjournals.org/.
Model construction
We used GB trees to build the prediction model. GB is an ensemble method that combining weak learners such as Decision Trees to a strong learner. GB fits new trees to minimize the loss or error, and it would assign more weights to observations that are hard to classify. Therefore, GB works great with categorical and numerical values and hence is suitable to be used on our datasets. We trained the model using ChIP-seq, DNase-seq and ChIA-PET data on three cell lines for transcription and exon usage prediction, respectively, including GM12878, K562 and HeLaS3. The model parameters for each prediction task were determined using Python function ‘sklearn.model_selection.GridSearchCV’ with cv = 5 and score = ‘accuracy’ on the corresponding datasets. The candidate parameters and the parameters chosen to train the model for transcription and exon usage prediction were recorded in Supplementary Tables S6 and S7 separately, see Supplementary Data available online at http://bib.oxfordjournals.org/.
Validation methods
We implemented three types of experiments for each cell line to validate the effectiveness of the model. Firstly, we adopted the stratified 10-fold cross validation. The usage of stratified cross validation could help to keep the same proportions of class labels in each fold. In each validation, one-fold would be used as validation data and the reaming folds would be used as training data. Such practice generally reflects the robustness of the model.
The second type of validation method is the chromosome split validation. We adopted the same chromosome-split strategy from work of [26] to split samples. Under such strategy, all samples on the same chromosome were either all in the training or all in the test set. The chromosome used in training and test dataset was listed in Supplementary Table S8, see Supplementary Data available online at http://bib.oxfordjournals.org/.
We also adopted the cross cell line validation to evaluate the model. We used all samples from one cell line to train the model but validate the model on all samples from different cell lines. It should be noted that we used the shared features when implementing cross cell line validation. Both the chromosome split validation and the cross cell validation enabled us to exclude the data similarity between samples in training and test datasets, hence could give us a fair implication about the robustness and effectiveness of the model.
Evaluation metrics
We used the criteria of accuracy, sensitivity, specificity, Area Under Receiver Operating Characteristic curve (AUROC) and Area Under Precision-Recall Curves (AUPRC) to evaluate the model performances on different datasets in each classification task. The accuracy, sensitivity and specificity were calculated as
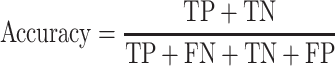 |
(1) |
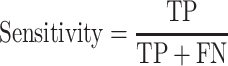 |
(2) |
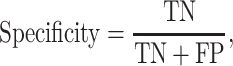 |
(3) |
where TP, FN, TN and FP denote the numbers of true positive, false negative, true negative and false positive, respectively.
Due to the imbalanced datasets in exon usage prediction, we also calculated the random guess values for fair comparisons. The random guess values for AUROC and AUPRC were obtained as the average results by shuffling the true prediction scores 10 times, and the random guess value for accuracy was obtained as the average results by shuffling the true label 10 times.
Besides, we used the XGBoost to obtain the feature importance, where the importance score for each feature was obtained as the average across all the decision trees within the GB model according to the amount that each attribute split point improves the performance measure. We applied function ‘xgboost.XGBClassifier’ in Python with parameters determined in section of Material and Methods–Model construction using all data for each task to build the trees; then, the feature importance scores were extracted using function of ‘feature_importances_’ from the XGBoost model for each dataset.
Results
Verified transcripts can be predicted from TFs and ChIA-PET data
We first investigate if the verified transcripts can be predicted from TFs as well as ChIA-PET data. A verified transcript indicates that there is gene expression located at a bona fide gene locus as indicated by GENCODE hg19 gene annotation. We treat verified transcription as positive data while treating the unannotated transcripts as the negative data. Different validation methods are evaluated on all three cell lines using the corresponding optimal parameters determined in the section of Material and Methods–Model construction. The results are demonstrated in Figure 3.
Figure 3 .
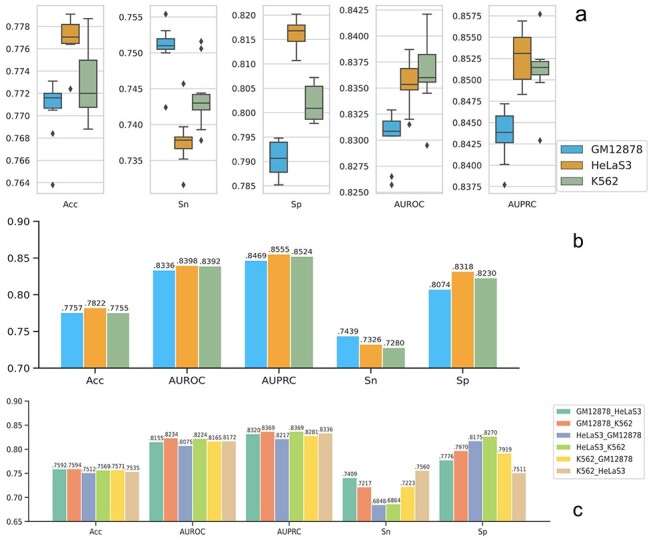
The prediction results in verified transcript prediction. Sn (sensitivity) represents the positive prediction accuracy and Sp (specificity) represents the negative prediction accuracy; y-axis represents the corresponding values for each evaluation criteria; (A) 10-fold validation. The box and whisker plot here shows the spread and centers of the 10-fold validation results. The five horizon lines from bottom to top represent minimum (the smallest number in the data set), the first quartile, the median, the third quartile and the maximum (the largest number in the data set), respectively. (B) Cross chromatin split validation. (C) Cross cell line validation.
The accuracies of 10-fold validation range from 0.7638 to 0.7731, 0.7724 to 0.7791 and 0.7688 to 0.7787 for GM12878, HeLaS3 and K562 cell, respectively (Figure 3A). This high accuracy of the model confirms that transcription could be predicted from transcriptomics data with TFs and ChIA-PET data. The prediction performance on GM12878 cells is slightly lower than that of the other two cell lines. The average AUROC and AUPRC values for GM12878 cell of 10-fold validation is 0.8303 and 0.8436 separately; however, the average AUROC and AUPRC values for HeLaS3 and K562 cells are 0.8354 and 0.8527 and 0.8364 and 0.8511, respectively. Notably, the small variations from 10-fold validation for all evaluation matrices on all cell lines, i.e. less than 0.0148, indicate the model robustness.
Using the data from each cell line, we are able to identify some important features such as H3K4me3, H3K9ac and H3K36me3 to distinguish the verified transcriptions from those not verified (Supplementary Figure S1, see Supplementary Data available online at http://bib.oxfordjournals.org/) in all three cell lines. The top identified important features show strong prediction powers. According to the average results of the 10-fold validation, training with the top 10 important features only slightly reduces 0.0047–0.0094 on the prediction accuracies and AUROC values from training with all features; training with the top 5 important features reduces 0.0083–0.0308 on the prediction accuracies and AUROC values from training with all features (Supplementary Figure S2, see Supplementary Data available online at http://bib.oxfordjournals.org/). Our findings about the important features are supported by several studies. First, H3K4me3 is a near-universal chromatin modification at the transcription start site of active genes in eukaryotes. As reviewed in Howe et al. [27], H3K4me3 may not be the initiator but the result of transcription, thus influencing processes such as splicing, transcription termination, memory of previous states and transcriptional consistency. Second, H3K9ac leads to increased transcription elongation via functioning in transcription to recruit the Super-Elongation Complex to chromatin and facilitate subsequent pol II pause release [28]. Third, H3K36me3 is thought to be a mark that is catalyzed by the SETD2 enzyme concomitantly with RNA polymerase II transcriptional elongation [29]. Interestingly, we noticed that H3K4me2 plays a more significant role in GM12878 and K562 cells than in HeLaS3 cell. Moreover, ChIA-PET Pol2A is also an important feature in K562 cells, and Pol2 is dominant in HeLaS3 but not very important in the other two cell lines.
We then evaluate our models on independent test datasets. The prediction results on chromosome split validation (Figure 3B) are similar to that of the 10-fold validation. The best accuracy is achieved on HeLaS3 data and the AUROC and AUPRC values for GM12878 cell are slightly worse than the other two cell lines. Interestingly, although the values of accuracy, AUROC and AUPRC are comparable on all cross cell line validations (Figure 3C), the model trained on HeLaS3 data leads to much lower sensitivity but much higher specificity values when testing on both GM12878 and K562 data. Conversely, the models trained on GM12878 and K562 data lead to higher sensitivity but lower specificity values when testing on HeLaS3 data. Such phenomenon further indicates the similarity of data characteristics between GM12878 and K562 cell line and their diversity from HeLaS3 cell as what we observed from the feature importance, which might be explained as the fact that the lymphoblastoid and myeloid cell lines are both suspension cells, while hela are adherent (monolayer) cells.
We also compare our GB model with other baseline models such as Adaptive Boosting (AdaBoost), Random Forest (RF), Convolutional Neural Network (CNN) and Forward Neural Network (NN). The results of the average values for 10-fold validation and chromosome split validation are shown in Supplementary Figures S3 and S4 separately, see Supplementary Data available online at http://bib.oxfordjournals.org/. Although the average performance of RF model on 10-fold validation is the highest in all three cell lines, the RF model only achieves slightly better performances than AdaBoost model in the chromosome split validation while performs worse than other models. GB model performs slightly worse than RF model in 10-fold validation but it is the best model in chromosome split validation, which shows the ability to predict independent data.
Transcription abundance prediction
TPM is used to measure gene or transcript expression level in RNA-seq. We aim to investigate if epigenomics can be used to predict expression level, and leverage on transcriptomics to measure and evaluate the accuracy of the predictions. For this purpose, we classify the data to four classes according to their TPM values: not_transcribed, lowly_transcribed, transcribed and highly_transcribed. More details can be found in the section of Material and Methods–Data collection and definition. We train and test the GB model on 10-fold cross validation. During the training process, we down-sample the classes of the majority class to the same data amount as the minority class to avoid prediction bias caused by the imbalanced dataset.
The results for 10-fold validation are shown in Supplementary Figure S5, see Supplementary Data available online at http://bib.oxfordjournals.org/. The average accuracies of the training results are 0.425, 0.426 and 0.444 for GM12878, HeLaS3 and K562 cells, respectively. These values are higher when compared with random guess value of 0.25 in the balanced dataset, indicating that transcription abundance can be predicted using epigenomic data and transcriptomics data. Several HMs previously related to transcription are identified as the most important features in these models (Supplementary Figure S6, see Supplementary Data available online at http://bib.oxfordjournals.org/). H3K36me3, an HM related to RNA Polymerase II passage and transcription fidelity maintenance [30], is found to be the most predictive across all three cell lines. This suggests that the levels of H3K36me3 are closely related to transcription activity. In addition, H3K79me2 is ranked the second most important feature except in K526 cells. H3K79me2 is a histone mark is involved in alternative splicing regulation [31], indicating that the transcription machinery is closely connected to exon usage. Another transcription elongation-related HM, H3K9ac, is also identified as one of the most important features.
Interestingly, DNase Hypersensitive sites (a measure of open chromatin which is associated with increased ability of the transcription machinery to access the region) and ChIA-PET loop anchors are also important in predicting transcription levels. We think that DNase may be an indicator of the open regions so that the transcription machinery can access the gene to produce transcripts. We reason that the presence of ChIA-PET loop anchors may reflect that the gene is interacting with distal regulatory elements and these regulatory elements may harbor TF binding motifs that can recruit TFs or mediators, which together orchestrate a stable transcription structure for RNA Polymerase II [32].
The importance of H3K36me3, H3K79me2 and H3K9ac in predicting gene expression has also been reported in other studies. Rosa et al. reported that H3K9ac is one of the most important HMs in predicting the expression of promoter with different CpG content in human T-cells [33]. In addition, Dong et al. [34] showed that H3K36me3, H3K79me3 and H3K9ac are predictive of gene expression from either CAGE or RNA-seq data. Our results are consistent with the reported correlation between gene expression and HMs. Taken together, we demonstrate that specific HMs, chromatin loop anchors and open chromatin are predictive of transcription abundance.
Again, the top identified important features show strong prediction powers in transcription abundance prediction (Supplementary Figure S7, see Supplementary Data available online at http://bib.oxfordjournals.org/). But the RF performs best on this classification problem (Supplementary Figures S8 and S9, see Supplementary Data available online at http://bib.oxfordjournals.org/).
Exon usage prediction
We perform exon usage prediction using exon usage coefficient as target. The exon usage coefficient indicates the number of reads at a particular exon present in a particular cell. We treat the samples whose exon usage coefficient values are non-Nan (the exon is included in one or more transcripts) as positive while treating those with Nan (the exon is not included in any transcripts) value as negative. Because we consider all exons, and many exons do not have reads, hence, there are much more negative samples and the dataset is imbalanced. Therefore, we evaluate the model performances mainly based on the criteria of accuracy, AUROC and AUPRC. Because with a large size of negative dataset, using AUPRC which is based on precision (TP/(TP + FP)) and recall (TP/(TP + FN)) makes it possible to assess the performance of a classifier on the minority class by excluding the influence of TN (where TP, FN, TN and FP denote the numbers of true positive, false negative, true negative and false positive, respectively).
In the 10-fold validation prediction (Figure 4A), the average accuracy values for GM12878, HeLaS3 and K562 cells are as high as 0.7700, 0.7893 and 0.8014, respectively. The same tendencies can also be found from the corresponding AUROC values for GM12878, HeLaS3 and K562 cells, whose average AUROC values are 0.6825, 0.6979 and 0.7001, respectively.
Figure 4 .
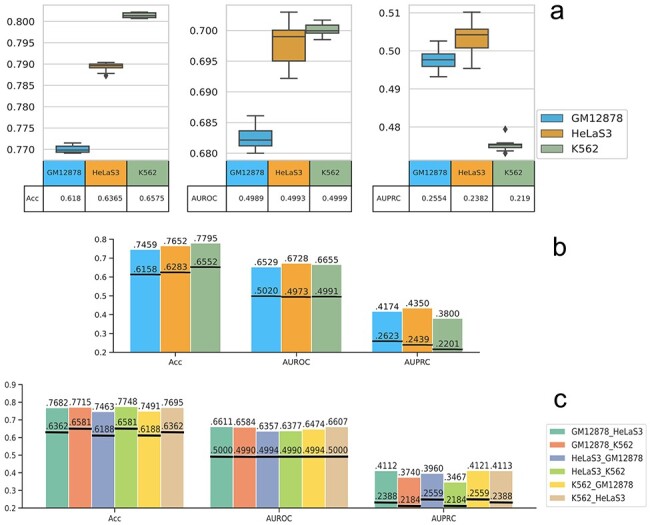
The prediction results in exon usage prediction. Sn (sensitivity) represents the positive prediction accuracy and Sp (specificity) represents the negative prediction accuracy; y-axis represents the corresponding values for each evaluation criteria; (A) 10-fold validation. The box and whisker plot here shows the spread and centers of the 10-fold validation results. The five horizon lines from bottom to top represent minimum (the smallest number in the data set), the first quartile, the median, the third quartile and the maximum (the largest number in the data set), respectively. The bottom table represents the random values. (B) Cross chromatin split validation. The black horizon lines represent the random results. (C) Cross cell line validation. The black horizon lines represent the random results.
H3K36me3 is found to be important in distinguishing exon usage level in all three cell lines (Supplementary Figure S10, see Supplementary Data available online at http://bib.oxfordjournals.org/), which are consistent to the verified transcription classification. H3K27ac is another important feature for the exon usage prediction, but it is not a very important feature in transcription prediction. The observation that H3K27ac is associated with gene exon usage has not been made before, and we suggest that perhaps different enhancer usage (since enhancers associated with H3K27ac) may play a role in specifying which exons are to be transcribed and included in the mRNA by splicing. Also, RNA Polymerase II ChIA-PET chromatin interaction anchors and H3K4me2 are important features for exon prediction in GM12878 and K562 cells and H3K9ac is an important feature for exon prediction in GM12878 and HeLaS3 cells. Similarly, the top identified important features show strong prediction powers in exon usage prediction. According to the average results of the 10-fold validation, training with the first 10 important features only reduces 0.0126–0.0569 on the prediction accuracies, AUROC and AUPRC values from training with all features; training with the first 5 important features reduces 0.0169–0.0929 on the prediction accuracies, AUROC and AUPRC values from training with all features (Supplementary Figure S11, see Supplementary Data available online at http://bib.oxfordjournals.org/).
The performances of chromosome split validation (Figure 4B) and cross cell validation (Figure 4C) are similar to that of the 10-fold validation. The prediction results on K562 cell data have higher accuracy values but lower AUPRC values considering the data amount ratio. However, compared with the random prediction results, the models work effectively and robustly in predicting the exon usage on independent test datasets. We show some exons that have predicted exon usage in GM12878 but not HelaS3 cell (Supplementary Figure S12, see Supplementary Data available online at http://bib.oxfordjournals.org/) and in HeLaS3 but not GM12878 cell (Supplementary Figure S13, see Supplementary Data available online at http://bib.oxfordjournals.org/) trained with K562 cell. The RNA-seq signal of H1hESC, K562, GM12878 and HeLaS3 cells is shown in RPKM (Reads Per Kilobase of transcript, per Million mapped reads) to represent the normalized transcript expression. The results here illustrate that our prediction is in agreement with the real detected exon usage.
We also compare our GB model with other baseline models (AdaBoost, RF, CNN and NN) on the average values for 10-fold validation and chromosome split validation (Supplementary Figures S14 and S15, see Supplementary Data available online at http://bib.oxfordjournals.org/). Results show that GB model is the second best model in 10-fold validation and the best model on independent test datasets (chromosome split validation).
Prediction time complexity analysis
We report the time complexity for each prediction task by providing the average training and test time for 10-fold validation (Supplementary Tables S9–S11, see Supplementary Data available online at http://bib.oxfordjournals.org/). The operation system is Linux with CPU type ‘Intel(R) Xeon® CPU E5-2698v4 @ 2.2GHz’. The training process takes an average of 64–81 s on three cell lines in verified transcript prediction with around 220 000 entries. The training and test times for exon usage prediction are much longer than that of the transcription prediction, i.e. more than 2000s, because the dataset size of exon is about three times of the dataset size of transcript and the number of estimators in the exon usage prediction model is five times of the transcription prediction model. And also, the training time for four classes (transcription abundance prediction) is longer than that of two classes (verified transcript prediction) even with relatively less training data.
Discussion
Exon usage is important in revealing the abundance of different transcript variants of the same gene. Pre-mRNA alternative splicing can result from different transcription kinetics, often changing transcript abundance and biological function, which can be implicated in diseases [35]. Here, we include chromatin loop data (i.e. ChIA-PET data) in the transcript and exon usage prediction. Our prediction results on 10-fold validation as well as the chromatin split and cross cell line validation, which ensure the independence of the data illustrated the effectiveness and robustness of our approaches. The different validation methods together with the large datasets on three cell lines also indicate that our model is scalable.
Specifically, in the transcription prediction part, the machine learning framework revealed close relationships between transcription abundance and transcription-related HMs, including H3K36me3, H3K79me3 and H3K9ac. These important features again re-emphasized the importance of epigenetic marks in maintaining a permissive and high-fidelity environment for gene transcription. Moreover, our GB tree model was able to recapture these features, as they were also reported to be important in predicting gene expression using conventional regression models [33, 34].
HMs have been reported to be involved in regulating differential exon usage in human cells [36]. For example, H3K9ac hyperacetylation at a splice site of NCAM gene can result in skipping of an exon in neuron cells [37]. H3K36me3 histone marks can be recognized and bound by splicing regulators (such as MRG15 and Psip1) and thus regulate the splicing of the transcript [36]. Importantly, our model also identified H3K36me3 and H3K9ac as important features in predicting exon usage. The link between splicing and chromatin interactions is unclear. While it has been shown previously that chromatin loops can bring exons into close spatial proximity with promoters and enhancers [23], it was unclear how important chromatin loops were in predicting splicing occurrences. Here, we observed that RNA Pol2A ChIA-PET anchors are important in predicting transcription and exon usage.
Splicing mainly occurs while transcription is ongoing, and in vivo studies have shown that the spliceosomes are physically close to RNA Polymerase II [38, 39]. Splicing will give rise to different exon usage because different exons may be included in the final transcripts. Our model identified H3K36me3, H3K9ac and RNA Pol2A ChIA-PET anchors as important for transcription prediction as well as exon usage prediction, which highlights the tight connection between splicing and transcriptional machinery. Based on our results, we extended this ‘splicing-transcription’ model whereby we proposed that the splicing regulators, RNA polymerase II and chromatin loop anchors bound by RNA polymerase II all come together in close proximity. This may be facilitated in part by key HMs such as H3K36me3 and H3K9ac helping to recruit splicing regulatory proteins (Figure 5).
Figure 5 .
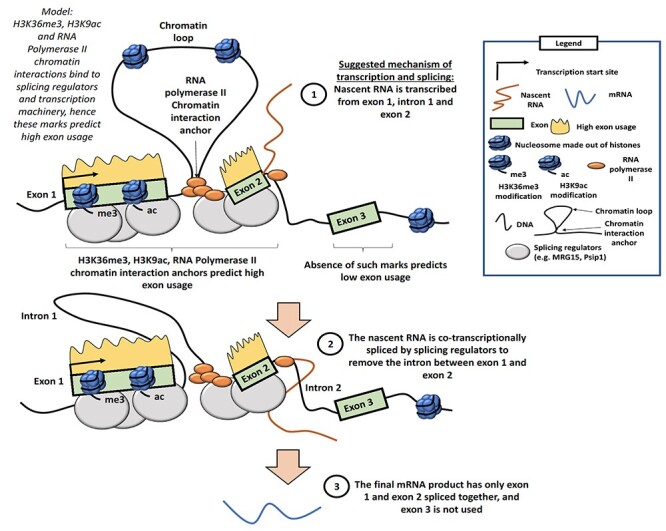
Schematic of our proposed model of alternative splicing, which gives rise to differential exon usage, chromatin loops and transcription machinery. In this model, we speculate that HMs, such as H3K36me3, H3K9ac and chromatin interactions, can bind to splicing regulators and transcription machinery. The transcriptional machinery which includes RNA Polymerase II produces a nascent RNA transcript. Splicing of the nascent RNA takes place in a co-transcriptional manner, to remove introns. The final mRNA product contains the exons that are used and does not include exons that are not used nor introns. This would explain our findings that HMs and chromatin interactions can predict high exon usage.
We speculate that perhaps, chromatin interactions between the exon being transcribed and distal regulatory elements may help to stabilize the transcription machinery [32]. When the splicing and/or transcription machineries are assembled at the transcript, it creates structural scaffolds for DNA or RNA to fold and interact with each other. Such interactions between DNA/RNA and distal regulatory elements can be captured by chromatin interaction profiling techniques such as ChIA-PET. Therefore, this interaction information was learnt by our model when predicting transcript and exon usage.
Taken together, our results show that transcription-related HMs are important in predicting transcript abundance and exon usage in human cells. More importantly, we show that chromatin interaction data are also important in predicting transcript and exon usage, suggesting a close relationship between transcription, splicing and chromatin structural organization.
Key Points
We did not just confine ourselves to splice sites, in contrast to previous splicing event prediction methods which use level of splicing calculated from the exonic expression level involved in that splicing event. We predicted the exon usage independently and quantified the transcript directly.
We successfully applied a set of transcription factors and ChIA-PET data which indicates locations of chromatin loops in the genome in transcript and exon usage prediction.
Our model performed well on independent test datasets, which indicates its robustness.
Examining the predictive features, we found that ChIA-PET Pol2A data were one of the most important features in both transcript and exon usage prediction, suggesting that chromatin loop anchors are predictive of both transcription and exon usage.
Supplementary Material
Acknowledgements
We thank Mr Ngiam Jia Jun for his contribution to the idea of verified transcription analysis.
Yu Zhang received her BEng degree from Shandong University, China, and the MSc degree (distinction degree) from Imperial College London, UK, in 2017 and 2018, respectively. She is currently a PhD candidate in Nanyang Technological University, Singapore. Her research interests include bioinformatics and deep learning.
Yichao Cai received the bachelor’s degree in bioinformatics from the Huazhong University of Science and Technology, and the PhD degree in bioinformatics from the National University of Singapore. He is currently a postdoctoral research fellow in the Cancer Science Institute of Singapore. His research interests include bioinformatics, chromatin interactions and cancer genomics.
Xavier Roca received his bachelor in biology in the University of Barcelona and PhD degree in the Autonomous University of Barcelona Spain in 2000. Upon completing his post-doctoral studies under supervision of Prof Adrian Krainer at Cold Spring Harbor Laboratory (NY, USA), he joined the School of Biological Sciences at Nanyang Technological University, Singapore, in 2011, as Assistant Professor, and in 2019, he became an Associate Professor. For most of his career, he used molecular biology to research on splicing mechanisms in human cells with implications in human genetic diseases, with a recent focus on immune cells and cancer.
Chee Keong Kwoh received the bachelor’s degree in electrical engineering (first class) and the master’s degree in industrial system engineering from the National University of Singapore, Singapore, in 1987 and 1991, respectively. He received the PhD degree from the Imperial College of Science, Technology and Medicine, University of London, in 1995. He has been with the School of Computer Engineering, Nanyang Technological University (NTU), since 1993. He is the program director of the MSc in Bioinformatics program at NTU. His research interests include data mining, soft computing and graph-based inference; applications areas include bioinformatics and biomedical engineering.
Melissa Jane Fullwood received the BSc degree (Hons.) from Stanford University, USA, and the PhD degree from the National University of Singapore, Singapore, in 2005 and 2009, respectively. She is a principal Investigator in the Cancer Science Institute of Singapore, National University of Singapore, an Assistant Professor in the School of Biological Sciences, Nanyang Technological University and an adjunct principal investigator in the Institute of Molecular and Cell Biology, A*STAR Singapore. Her research interests include investigating 3D genome organization in cancer.
Contributor Information
Yu Zhang, School of Computer Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798, Singapore.
Yichao Cai, Cancer Science Institute of Singapore, National University of Singapore, 14 Medical Dr, Singapore 117599, Singapore.
Xavier Roca, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Dr, Singapore 637551, Singapore.
Chee Keong Kwoh, School of Computer Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798, Singapore.
Melissa Jane Fullwood, Cancer Science Institute of Singapore, National University of Singapore, 14 Medical Dr, Singapore 117599, Singapore; School of Biological Sciences, Nanyang Technological University, 637551, Singapore; Institute of Molecular and Cell Biology, Agency for Science, Technology and Research (A*STAR), 61 Biopolis Dr, Singapore 138673, Singapore.
Data Availability
The data used in this work is freely available at https://researchdata.ntu.edu.sg/dataverse/chrom_pred_exon. The code used in this work is freely available at https://github.com/mjflab/exon-prediction.
Funding
National Research Foundation (NRF) Singapore through an NRF Fellowship (NRF-NRFF2012-054 to M.J.F); Nanyang Technological University start-up funds to M.J.F.; RNA Biology Center at the Cancer Science Institute of Singapore, NUS, as part of funding under the Singapore Ministry of Education Academic Research Fund Tier 3 to D.T. as lead PI with M.J.F. as co-investigator (MOE2014-T3-1-006); National Research Foundation Competitive Research Programme grant to V.T. as lead PI and M.J.F. as co-PI (NRF-CRP17-2017-02); National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative; Ministry of Education Tier II grant to M.J.F. (T2EP30120-0020); Advancing Creativity and Excellence (ACE) grant awarded by Nanyang Technological University to M.J.F. as PI, C.K.K. as co-PI and X.R. as co-PI (NTU-ACE2019-03).
References
- 1. Bjørklund SS, Panda A, Kumar S, et al. Widespread alternative exon usage in clinically distinct subtypes of invasive ductal carcinoma. Sci Rep 2017; 7: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reyes A, Anders S, Weatheritt RJ, et al. Drift and conservation of differential exon usage across tissues in primate species. Proc Natl Acad Sci 2013; 110: 15377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qu X, Alsager S, Zhuo Y, et al. Hox transcript antisense rna (hotair) in cancer. Cancer Lett 2019; 454: 90–7. [DOI] [PubMed] [Google Scholar]
- 4. Eiholzer RA, Mehta S, Kazantseva M, et al. Intronic tp53 polymorphisms are associated with increased d133tp53 transcript, immune infiltration and cancer risk. Cancer 2020; 12: 2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo X, Chen QR, Song YK, et al. Exon array analysis reveals neuroblastoma tumors have distinct alternative splicing patterns according to stage and mycn amplification status. BMC Med Genomics 2011; 4: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegeneration 2008; 3: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soneson C, Love MI, Patro R, et al. A junction coverage compatibility score to quantify the reliability of transcript abundance estimates and annotation catalogs. Life Science Alliance 2019; 2: e201800175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kakaradov B, Xiong HY, Lee LJ, et al. Challenges in estimating percent inclusion of alternatively spliced junctions from RNA-seq data. InBMC Bioinformatics 2012; 13 (6):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldstein LD, Cao Y, Pau G, et al. Prediction and quantification of splice events from rna-seq data. PLoS One 2016; 11: e0156132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Z, Pan Z, Ying Y, et al. Deep-learning augmented rna-seq analysis of transcript splicing. Nat Methods 2019; 16: 307–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee D, Zhang J, Liu J, et al. Epigenome-based splicing prediction using a recurrent neural network. PLoS Comput Biol 2020; 16: e1008006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson DS, Mortazavi A, Myers RM, et al. Genome-wide mapping of in vivo protein-dna interactions. Science 2007; 316: 1497–502. [DOI] [PubMed] [Google Scholar]
- 13. Sekhon A, Singh R, Qi Y. Deepdiff: deep-learning for predicting differential gene expression from histone modifications. Bioinformatics 2018; 34: i891–900. [DOI] [PubMed] [Google Scholar]
- 14. Wang S, Sun H, Ma J, et al. Target analysis by integration of transcriptome and chip-seq data with beta. Nat Protoc 2013; 8: 2502–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang LQ, Li QZ, Su WX, et al. Predicting gene expression level by the transcription factor binding signals in human embryonic stem cells. Biosystems 2016; 150: 92–8. [DOI] [PubMed] [Google Scholar]
- 16. Schmidt F, Gasparoni N, Gasparoni G, et al. Combining transcription factor binding affinities with open-chromatin data for accurate gene expression prediction. Nucleic Acids Res 2017; 45: 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu Q, Kim EJ, Feng J, et al. Histone posttranslational modifications predict specific alternative exon subtypes in mammalian brain. PLoS Comput Biol 2017; 13: e1005602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leung MK, Xiong HY, Lee LJ, et al. Deep learning of the tissue-regulated splicing code. Bioinformatics 2014; 30 (12):i121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jha A, Gazzara MR, Barash Y. Integrative deep models for alternative splicing. Bioinformatics 2017; 33 (14):i274–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fullwood MJ, Liu MH, Pan YF, et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature 2019; 462 (7269):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dekker J, Misteli T. Long-range chromatin interactions. Cold Spring Harb Perspect Biol 2015; 7: a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. See YX, Wang BZ, Fullwood MJ. Chromatin interactions and regulatory elements in cancer: from bench to bedside. Trends Genet 2019; 35: 145–58. [DOI] [PubMed] [Google Scholar]
- 23. Mercer TR, Edwards SL, Clark MB, et al. Dnase i–hypersensitive exons colocalize with promoters and distal regulatory elements. Nat Genet 2013; 45: 852–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang Y, Huang J, Lua K, et al. Genome-wide analyses of chromatin interactions after the loss of pol i, pol ii, and pol iii. Genome Biol 2020; 21: 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davis CA, Hitz BC, Sloan CA, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 2018; 46 (D1):D794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao F, Fullwood MJ. Inflated performance measures in enhancer–promoter interaction-prediction methods. Nat Genet 2019; 51: 1196–8. [DOI] [PubMed] [Google Scholar]
- 27. Howe FS, Fischl H, Murray SC, et al. Is h3k4me3 instructive for transcription activation? Bioessays 2017; 39: 1–12. [DOI] [PubMed] [Google Scholar]
- 28. Gates LA, Shi J, Rohira AD, et al. Acetylation on histone h3 lysine 9 mediates a switch from transcription initiation to elongation. J Biol Chem 2017; 292: 14456–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wagner EJ, Carpenter PB. Understanding the language of lys36 methylation at histone h3. Nat Rev Mol Cell Biol 2012; 13: 115–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang C, Zhu B. Roles of h3k36-specific histone methyltransferases in transcription: antagonizing silencing and safeguarding transcription fidelity. Biophys Reports 2018; 4: 170–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li T, Liu Q, Garza N, et al. Integrative analysis reveals functional and regulatory roles of h3k79me2 in mediating alternative splicing. Genome Med 2018; 10: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soutourina J. Transcription regulation by the mediator complex. Nat Rev Mol Cell Biol 2018; 19: 262. [DOI] [PubMed] [Google Scholar]
- 33. Karlić R, Chung HR, Lasserre J, et al. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci 2010; 107 (7):2926–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dong X, Greven MC, Kundaje A, et al. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol 2012; 13 (9):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kelemen O, Convertini P, Zhang Z, et al. Function of alternative splicing. Gene 2013; 514 (1):1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou HL, Luo G, Wise JA, et al. Regulation of alternative splicing by local histone modifications: potential roles for RNA-guided mechanisms. Nucleic Acids Res 2013; 42 (2): 701–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schor IE, Rascovan N, Pelisch F, et al. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci 2009; 106 (11):4325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Herzel L, Ottoz DS, Alpert T, et al. Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat Rev Mol Cell Biol 2017; 18 (10):637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang S, Aibara S, Vos SM, et al. Structure of a transcribing RNA polymerase II–U1 snRNP complex. Science 2021; 371 (6526):305–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this work is freely available at https://researchdata.ntu.edu.sg/dataverse/chrom_pred_exon. The code used in this work is freely available at https://github.com/mjflab/exon-prediction.