

Abstract
DNA N6-methyladenine is an important type of DNA modification that plays important roles in multiple biological processes. Despite the recent progress in developing DNA 6mA site prediction methods, several challenges remain to be addressed. For example, although the hand-crafted features are interpretable, they contain redundant information that may bias the model training and have a negative impact on the trained model. Furthermore, although deep learning (DL)-based models can perform feature extraction and classification automatically, they lack the interpretability of the crucial features learned by those models. As such, considerable research efforts have been focused on achieving the trade-off between the interpretability and straightforwardness of DL neural networks. In this study, we develop two new DL-based models for improving the prediction of N6-methyladenine sites, termed LA6mA and AL6mA, which use bidirectional long short-term memory to respectively capture the long-range information and self-attention mechanism to extract the key position information from DNA sequences. The performance of the two proposed methods is benchmarked and evaluated on the two model organisms Arabidopsis thaliana and Drosophila melanogaster. On the two benchmark datasets, LA6mA achieves an area under the receiver operating characteristic curve (AUROC) value of 0.962 and 0.966, whereas AL6mA achieves an AUROC value of 0.945 and 0.941, respectively. Moreover, an in-depth analysis of the attention matrix is conducted to interpret the important information, which is hidden in the sequence and relevant for 6mA site prediction. The two novel pipelines developed for DNA 6mA site prediction in this work will facilitate a better understanding of the underlying principle of DL-based DNA methylation site prediction and its future applications.
Keywords: DNA modification, 6mA, self-attention mechanism, deep learning, LSTM, attention interpretation
Introduction
Epigenetics is regarded as a critical component of genetics, which can be expressed at different levels including protein posttranslational modifications, RNA interference, DNA modifications, etc. Briefly, epigenetics results in heritable gene expression or cell phenotype changes through certain mechanisms without changing the sequence [1]. As a novel epigenetic regulation, DNA methylation is discovered across various species and is found to be closely correlated with a myriad of biological processes such as cell differentiation, neural development and cancer suppression [2, 3]. Different types of methylation can occur with respect to the different positions of modifications. For example, 4-methyl-cytosine (4mC) occurs at the 4th position of the pyrimidine ring of cytosine, 5-methyl-cytosine (5mC) appears at the 5th position of the pyrimidine ring, whereas 6-methyl-adenine (6mA) occurs on the 6th position of the purine ring of adenine [4]. Among all of those mentioned above, 4mC and 5mC have been extensively studied due to their widespread distributions.
For a long time, 6mA had been believed to occur only in bacteria, whereas the distribution and the function of 6mA in eukaryotes had remained largely unknown because it was not detectable in early-stage studies [5]. In recent years, benefiting from the advances and applications of high-throughput sequencing technology, 6mA has also been detected in eukaryotic species [6–12]. 6mA sites can be detected through a series of wet-lab experimental methods, which include but are not limited to methylated DNA immunoprecipitation sequencing [13], capillary electrophoresis and laser-induced fluorescence [14] and PacBio single-molecule real-time sequencing [15]. The experimental results provide a wealth of information and meanwhile also suffer from obvious shortcomings, such as high expense and low efficiency. 6mA sites are sparsely and unevenly distributed across the genome; our current understanding of the functional role of 6mA modification is still limited. Therefore, predicting 6mA sites at single-nucleotide resolution and exploring the key information surrounding the targeted methylation sites are of great significance to the characterization of their role in epigenetic regulation of gene expression and associations with human diseases.
Several computational methods have been developed for 6mA site prediction in eukaryotes. Early-stage methods focus on the representation and extraction of hand-crafted features and employ traditional machine learning algorithms for making the prediction. For example, Chen et al. [16] proposed the first Machine Learning (ML)-based method i6mA-Pred for 6mA site identification, which uses support vector machine (SVM) and utilizes nucleotide chemical properties and nucleotide frequency as the input features. i6mA-DNCP [17] employs dinucleotide composition and dinucleotide-based DNA properties to represent the input DNA sequence and uses a bagging classifier for the prediction. iDNA6mA-Rice [18] adopts the mononucleotide binary encoding for sequence representation and utilizes Random Forest for classification. SDM6A [19] uses five different encodings to identify the optimal feature sets as the input into SVM and extremely randomized tree classifiers. 6mA-Finder [20] uses the recursive feature elimination strategy to select the optimal feature group from seven types of sequence-derived features and three physicochemical-based features. Hand-crafted features such as accumulated nucleotide frequency, frequency of certain k-mer motifs, electronion interaction pseudopotential, position-specific triple-nucleotide propensity and pseudo nucleotide composition are often being used for sequence representation [21–24]. In addition, statistical models have also been employed. For example, MM-6mAPred [25] uses a 1st-order Markov model for identifying 6mA sites in the rice genome. However, the combination of hand-crafted features and machine learning classifiers is widely used to process the genomic sequence, which inevitably suffers from certain disadvantages. For example, manually crafted features have redundant information and are very subjective, although they are interpretable. Besides, the hidden information in the sequence is often neglected, making it difficult for the manually crafted features to be explored as an optimal option for training the classifiers. With the development of deep learning (DL), some sequence-based end-to-end algorithms have been employed for 6mA site identification. These methods include iDNA6mA (five-step rule) [26], SNNRice6mA [27], DeepM6A [28], Deep6mA [29] and i6mA-DNC [30] all of which are convolutional neural network (CNN)-based models by taking one-hot encoding of the sequence as the input. In particular, i6mA-DNC [30] splits the DNA sequences into dinucleotide components before being fed into the CNN model to detect the N6-methyladenine sites. Deep6mA [29] combines CNN with LSTM to predict 6mA sites and finds that similar patterns around 6mA sites are shared across different species.
Despite the promising predictive performance, DL-based models are often criticized as ‘blackbox’ and suffer from issues such as lack of interpretability. A typical workflow of DL is as follows: The encoded sequences are fed into a DL-based model, and then the hyperparameters or structures are further modified to achieve better performance. In this context, a natural question to ask is why the model could make accurate predictions? What important information did it learn from the input sequences and use for making the final prediction? From the end users’ perspective, understanding how and why it works are much more meaningful than just training an accurate model based on trial-and-error efforts. There have been some ongoing efforts in using CNN-based methods to learn characteristic motifs that correspond to the regulators associated with the mechanism of DNA methylation. Each filter of the first convolutional layer can be regarded as a motif detector, which is widely used to understand how the network responds to an input sequence [31–33]. Meanwhile, in the field of natural language processing, an increasing number of studies have also been recently conducted to leverage and interpret the attention mechanism in an effort to improve the model interpretability [34–37].
In light of the achievements already made in DNA 6mA site prediction, we are curious about which key information the model pays special attention to when making the prediction. In this study, two end-to-end methods are proposed for 6mA site prediction, termed LA6mA and AL6mA. These two sequence-based methods extract sequence features automatically and distinguish 6mA site from non-6mA site using the DNA sequence as the only input, thereby avoiding the trouble and overreliance on the extraction of hand-crafted features. In addition, bidirectional long short-term memory (Bi-LSTM) is used to capture the important short-range and long-range information from DNA sequences, and the self-attention mechanism is also adopted to capture the position information of the sequences. Throughout the benchmarking experiments, different combinations of LSTM and self-attention mechanism are adopted to examine the efficiency of the methods. Detailed analyses of the attention matrix are also conducted, including the key positions of input sequences, the variation of attention vectors when attending to these key positions and similarities and differences of the attention layers from the two models and two model organisms. The differences in the attention layer for both true positive (TP) examples and true negative (TN) examples are found to be beneficial for our understanding of why the models make correct predictions. Extensive experiments demonstrate the competitive performance of the proposed LA6mA and AL6mA methods in comparison with other existing state-of-the-art methods for 6mA prediction. An online web server of AL6mA and LA6mA are implemented and made publicly accessible at http://csbio.njust.edu.cn/bioinf/al6ma/.
Materials and methods
In this section, we will first provide a description of the curation of the benchmark datasets and then introduce the two proposed methods. Implementation of the methods and performance evaluation metrics will also be provided afterward.
Benchmark datasets
In this study, the DNA 6mA data of the two model organisms Arabidopsis thaliana and Drosophila melanogaster were taken from [28]. The raw data came from the PacBio public database [38]. Candidates were further filtered out by excluding those with the sequence variance located between 10 bp upstream and 5 bp downstream of the identified modification site and the variation ratio of the estimated methylation level of greater than 30%. After this filtering procedure, 19 632 and 10 653 6mA sites for A. thaliana and D. melanogaster were obtained, respectively. Non-6mA sites of the same number were used as negative samples. Each non-6mA site was at least 200 bp away from any neighboring 6mA site. For more detailed information on the data set construction, please refer to [28].
We further screened the sequences to weed out those sequences that contained sites with uncertain DNA bases. Finally, 19 616 positive samples and 10 653 positive samples were retained for A. thaliana and D. melanogaster, respectively. A statistical summary of the two datasets is provided in Table 1. For each organism, the samples were randomly divided with a ratio of 9:1 as the training and independent test datasets.
Table 1.
Statistical summary of the datasets curated in this study
| Dataset | Positive | Negative | Total |
|---|---|---|---|
| Arabidopsis thaliana | 19 616 | 19 616 | 39 232 |
| Drosophila melanogaster | 10 653 | 10 653 | 21 306 |
Feature representation
The proposed models take as the input a DNA sequence centered on 6mA site or non-6mA site. The binary one-hot encoding scheme is adopted to represent the input DNA sequences with the following rules: 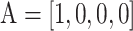 ,
, 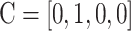 ,
, 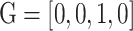 and
and 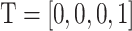 . Such encoding scheme makes the elements in the encoding matrix correspond to the bases in the input sequence, which is convenient for the analysis of the attention matrix/vector. Accordingly, each DNA sequence of length
. Such encoding scheme makes the elements in the encoding matrix correspond to the bases in the input sequence, which is convenient for the analysis of the attention matrix/vector. Accordingly, each DNA sequence of length 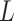 is converted to a 2D matrix of the size
is converted to a 2D matrix of the size 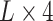 after the encoding. The length of each sequence is 41 bp, which is composed of 20 flanking nucleotides at each side and the centered adenine site (refer to the Supplementary Figure S1 for performance comparison of models trained with different lengths of flanking sequence, with experimental details in the Supplementary Material S1).
after the encoding. The length of each sequence is 41 bp, which is composed of 20 flanking nucleotides at each side and the centered adenine site (refer to the Supplementary Figure S1 for performance comparison of models trained with different lengths of flanking sequence, with experimental details in the Supplementary Material S1).
Network architecture
The input sequences are encoded and fed into the end-to-end networks directly. DNA data analysis is analogous to natural language processing [39], in which recurrent neural networks (RNNs) can be used to process the sequential data. As a popular and powerful RNN architecture, long short-term memory (LSTM) [40] has been widely used to address sequence analysis problems [41] and has achieved excellent performance. Here we employ Bi-LSTM to capture the short-range and long-range information of DNA sequences. The use of LSTM makes up the shortcoming for the lack of time information in the one-hot encoding. The architecture of each LSTM unit is shown in Figure 1E. The formulas of each LSTM unit can be expressed as:
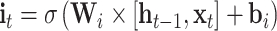 |
(1) |
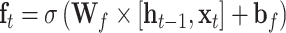 |
(2) |
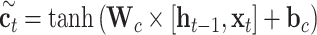 |
(3) |
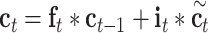 |
(4) |
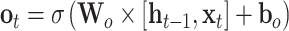 |
(5) |
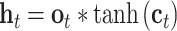 |
(6) |
where 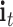 ,
, 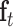 and
and 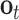 denote the input gate, forget gate and output gate, respectively.
denote the input gate, forget gate and output gate, respectively. 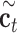 and
and 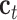 are new memory cell and final memory cell, respectively.
are new memory cell and final memory cell, respectively. 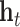 is the hidden state vector at the position
is the hidden state vector at the position 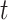 .
. 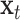 is the input vector at the position
is the input vector at the position 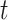 .
. 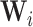 ,
, 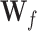 ,
, 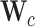 and
and 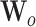 are weight matrices that need to be learned.
are weight matrices that need to be learned. 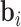 ,
, 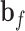 ,
, 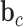 and
and 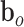 are bias vectors of the corresponding parts.
are bias vectors of the corresponding parts. 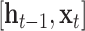 represent the concatenation of the vector
represent the concatenation of the vector 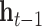 and vector
and vector 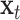 .
.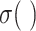 and
and 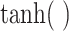 are the sigmoid function and hyperbolic tangent function, respectively.
are the sigmoid function and hyperbolic tangent function, respectively. 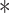 is the element-wise multiplication.
is the element-wise multiplication.
Figure 1.
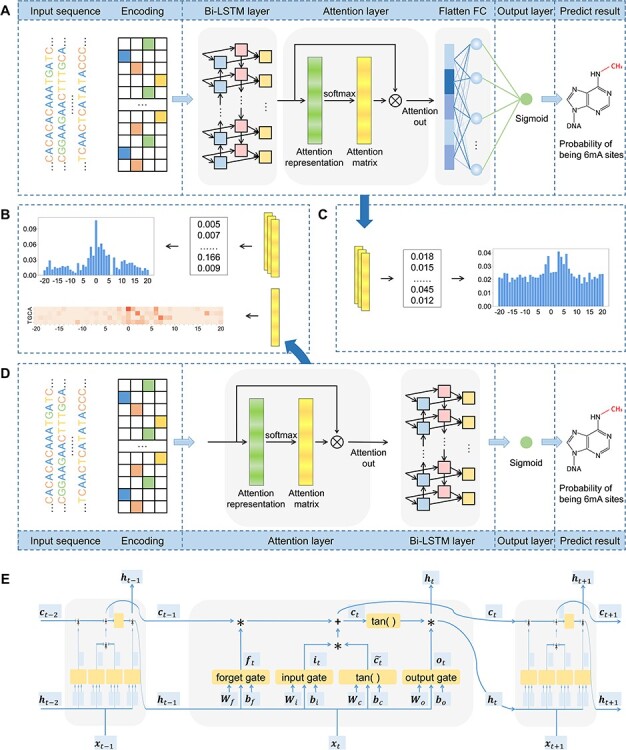
Network architectures of the proposed LA6mA and AL6mA methods for the prediction and analysis of DNA 6mA sites: (A) network architecture of LA6mA; (B) analysis of the attention matrix of AL6mA; (C) analysis of the attention matrix of LA6mA; (D) network architecture of AL6mA and (E) network architecture of an LSTM unit.
In addition to the Bi-LSTM architecture, the attention mechanism is also employed to capture the position information of the DNA sequence. It was originally proposed to solve machine translation tasks [42] and has proven to be capable of identifying the key information [43]. In recent years it has been applied to bioinformatics to address the problems faced by RNNs [44] and has been shown to achieve a competitive performance in a wide range of biological sequence analysis problems [45–47]. Hence, it is adopted in this study to investigate the key information that affects DNA methylation site prediction. The attention layer is able to compute the weight coefficients matrix 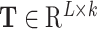 using the following formula:
using the following formula:
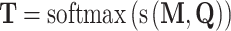 |
(7) |
where 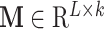 is the input matrix,
is the input matrix, 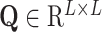 represents the weight matrix of attention and
represents the weight matrix of attention and 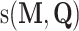 is the attention scoring function represent as
is the attention scoring function represent as 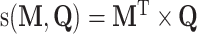 .
.
To address the problem of DNA N6-methyladenine site identification, we proposed two networks based on Bi-LSTM and the attention mechanism. The frameworks of the proposed AL6mA and LA6mA methods are illustrated in Figure 1. As can be seen, both networks take the encoded matrix as the input; however, there are some differences in the network structures of the two methods.
The framework of LA6mA is shown in Figure 1A. Its Bi-LSTM layer is connected to the encoding matrix firstly, in which two bidirectional LSTM layers with num_units set of 32 are employed. Then each time step of LSTM is used to connect the attention layer, and the parameter 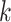 in the weight coefficient matrix
in the weight coefficient matrix 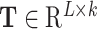 equals 32. Finally, the attention layer is flattened and connected to the output after the fully connected (FC) layer. The number of nodes in the FC layer is set to be 100.
equals 32. Finally, the attention layer is flattened and connected to the output after the fully connected (FC) layer. The number of nodes in the FC layer is set to be 100.
The structure of AL6mA is a little different from that of LA6mA, as shown in Figure 1D. The input sequence of length 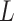 is encoded and then directly connected with the attention layer. After the attention layer is one bidirectional LSTM layer, the parameters of which are as follows: num_units set is at 128, whereas the time_steps is set at 41. Finally, the output of the last time step of Bi-LSTM is used as the final predict result.
is encoded and then directly connected with the attention layer. After the attention layer is one bidirectional LSTM layer, the parameters of which are as follows: num_units set is at 128, whereas the time_steps is set at 41. Finally, the output of the last time step of Bi-LSTM is used as the final predict result.
It is noteworthy that the two proposed methods are not merely used to predict potential methylation sites. They also enable us to perform an in-depth analysis of the hidden information that the model pays attention to and utilizes to make the prediction. Figure 1B and C depict how the attention matrices are analyzed and interpreted for this purpose.
Implementation
The models are implemented in Keras (version 2.3.1) and trained on one NVIDIA TITAN X GPU. The batch size is set to be 128. The Adam optimizer is employed with the default learning rate of 0.001, beta_1 = 0.9, beta_2 = 0.999, and a learning rate decay of 0.5 with patience of 7. 5-fold cross-validation is performed to determine the model structure and hyperparameters on the training data. After the model structure is determined, we take 8/9 (about 8/10 of the whole dataset) and 1/9 (about 1/10 of the whole dataset) of the training data to train and verify the trained model, respectively. Early stopping with patience of 7 is adopted on the validation set to avoid overfitting, which means the training process will terminate when the prediction performance does not improve on the validation set.
Performance measurement
Five performance measures are adopted to assess the performance of the proposed methods. Among these, four performance metrics for evaluating the binary prediction output can be derived from the confusion matrix. These include sensitivity (Sen), specificity (Spe), accuracy (Acc) and Matthew’s correlation coefficient (MCC), which are respectively defined as follows:
 |
(8) |
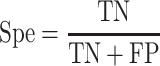 |
(9) |
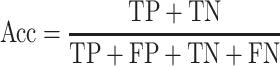 |
(10) |
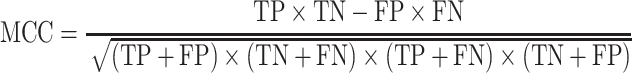 |
(11) |
where TP, false positive (FP), TN and false negative (FN) represent the numbers of TPs (i.e. number of correctly predicted samples as 6mA sites), FPs (i.e. number of incorrectly predicted samples as 6mA sites), TNs (i.e. number of correctly predicted samples as non-6mA sites) and FNs (i.e. number of incorrectly predicted samples as non-6mA sites), respectively. In addition, the area under the receiver operating characteristic (ROC) curve (AUC) is also used as a measure to comprehensively evaluate and compare the performance of different models.
Results and discussion
Rapid identification of key positions
As shown in Figure 1B and C, the attention matrix 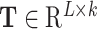 is extracted from each input sequence being fed into AL6mA or LA6mA. Then the rows of the attention matrix are averaged and the matrix is transformed into an attention vector with the same length as the input sequence. These vectors can be used to identify the key positions relevant for making the prediction by the model. Specifically, for an input DNA sequence articulated as
is extracted from each input sequence being fed into AL6mA or LA6mA. Then the rows of the attention matrix are averaged and the matrix is transformed into an attention vector with the same length as the input sequence. These vectors can be used to identify the key positions relevant for making the prediction by the model. Specifically, for an input DNA sequence articulated as 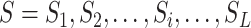 , where
, where 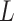 was the length of the sequence, it was converted into an attention vector
was the length of the sequence, it was converted into an attention vector 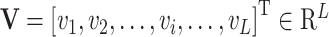 . A larger value of
. A larger value of 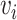 meaned
meaned 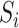 made a more important contribution to the prediction result.
made a more important contribution to the prediction result.
We selected all the TP samples (i.e. correctly predicted 6mA sites) in the test dataset to generate the final attention vectors and displayed them in an intuitive way. Figure 2 illustrates the experimental results on A. thaliana by AL6mA and LA6mA, including randomly initialized attention vectors, attention vectors of the final model and changes in the attention vectors.
Figure 2.
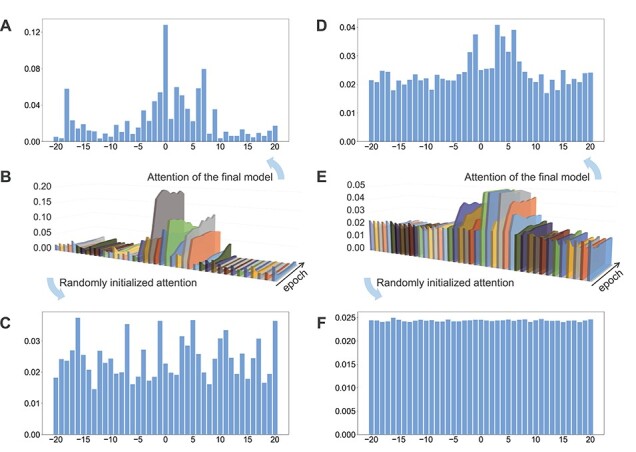
Characterization of the attention vectors of the AL6mA and LA6mA methods for predicting 6mA sites in A. thaliana: (A) attention vectors of well-trained AL6mA; (B) changes of the attention vectors of AL6mA for 10 epochs; (C) randomly initialized attention vectors of AL6mA; (D) attention vectors of well-trained LA6mA; (E) changes of the attention vectors of LA6mA for 10 epochs and (F) randomly initialized attention vectors of LA6mA. The x-axis of the panels B and E represents the position of the sequence; the y-axis denotes the increase of epochs, whereas the z-axis denotes the value of the attention vector, respectively.
Figure 2C and F display the randomly initialized attention vectors of the AL6mA and LA6mA methods, respectively. As can be seen, the initial attention weights of AL6mA appeared to be distributed randomly throughout the sequence (Figure 2C), which obviously differed from each other. In the case of LA6mA, its initialized attention weights were almost evenly distributed (Figure 2F). When an input sequence was fed into an initialized LA6mA model, the LSTM layer extracted the features of the sequence, which were then passed into the attention layer. That is the main reason why the values in the extracted initial attention vector of LA6mA were distributed evenly.
Figure 2A and D show the final attention vectors of AL6mA and LA6mA, respectively. Regardless of the distribution of the initial attention, it is apparent that the weights of the central region in the attention of the final model are larger than those of the marginal region. This suggests that the central region made more contributions to the prediction of the final result. Furthermore, we conjecture that the right flanking region contributed more to the prediction than the left flanking region. Specifically, the region of 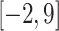 made the most significant contributions to the prediction in terms of the values of the attention weights. From a biological perspective, mutations in this region might affect the methylation possibility of the centered adenine site, which has been confirmed in [48]. In addition, the changes in methylation caused by such mutation may lead to abnormal biological processes.
made the most significant contributions to the prediction in terms of the values of the attention weights. From a biological perspective, mutations in this region might affect the methylation possibility of the centered adenine site, which has been confirmed in [48]. In addition, the changes in methylation caused by such mutation may lead to abnormal biological processes.
To observe the changes of the attention vectors, the attention weights were extracted during model optimization. The changes are then displayed as 3D graphics, shown in Figure 2B and E. It can be seen that, in accordance with the increase of the epochs, the values of the central regions increased, whereas the values of the marginal regions decreased, highlighting that the models could automatically focus on the key areas during the optimization. Surprisingly, the changes of the attention vectors that attended to the key positions appeared to occur after only a few epochs, and as the iteration progressed, the values of the attention vectors were being constantly fine-tuned and eventually reached the plateau. Figure 2B and E verified rapid identification of the key positions that are relevant for making the prediction by the model.
Similarities and differences of different attention layers
In this section, we performed a detailed analysis of the attention vectors on the two model organisms. The attention vectors of AL6mA and LA6mA on A. thaliana are displayed in Figure 2A and D. The final attention vectors of the well-trained AL6mA and LA6mA on D. melanogaster are displayed in Figure 3.
Figure 3.
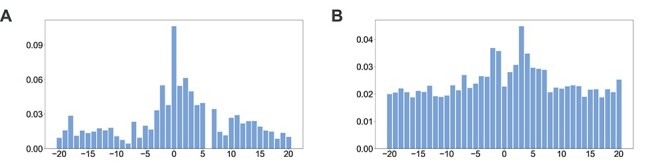
Distributions of the final attention vectors of AL6mA and LA6mA for predicting 6mA sites in D. melanogaster: (A) attention vectors of AL6mA and (B) attention vectors of LA6mA.
We made the following important observations:
(i) The right flanking region made more important contributions to the prediction than the left flanking region. In general, the values of the attention vectors within the region of
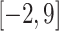 were comparably larger. It is also worth noting that LA6mA and AL6mA exhibited different characteristics with respect to the attention vectors. More specifically, the attention vectors calculated by the final LA6mA model of two model organisms seemed to be more similar (e.g. both values at the positions −2, −1, 3 and 4 were larger), whereas the attention vectors of the AL6mA model appeared to be significantly different (e.g. the values at the positions 0, 2, 6 and 7 were larger for A. thaliana, whereas the values at the positions −2, 0, 1, 2 and 3 were larger for D. melanogaster). These results suggest that the attention mechanism could indeed attend to the key differential features and was good at identifying key areas of universality. In contrast, the attention layers connected to the original sequence tended to find the key positions of individuality.
were comparably larger. It is also worth noting that LA6mA and AL6mA exhibited different characteristics with respect to the attention vectors. More specifically, the attention vectors calculated by the final LA6mA model of two model organisms seemed to be more similar (e.g. both values at the positions −2, −1, 3 and 4 were larger), whereas the attention vectors of the AL6mA model appeared to be significantly different (e.g. the values at the positions 0, 2, 6 and 7 were larger for A. thaliana, whereas the values at the positions −2, 0, 1, 2 and 3 were larger for D. melanogaster). These results suggest that the attention mechanism could indeed attend to the key differential features and was good at identifying key areas of universality. In contrast, the attention layers connected to the original sequence tended to find the key positions of individuality.(ii) Although for both AL6mA and LA6mA, the attention weights of the central region were larger than those of the marginal region, they differed from each other in terms of the distribution of values. Taking Figure 3A and B as an example, the attention weight of the central region for the AL6mA method was considerably larger than that of the marginal region (approximately 7–12 times larger). In contrast, the weight difference for the LA6mA method was only 2.5–4 times larger. This result was not due to the different initialization values, but due to the use of different structures. For the LA6mA method, the attention layer was placed following the feature extraction layer and thus resulted in the attention distraction.
(iii) For the AL6mA method, there existed an abnormal region whose weight increased at the end of the right flanking region (i.e. the position −18, 19 and 20 for A. thaliana and the positions −18 for D. melanogaster, respectively). In addition, it can be observed that they gradually increased with increasing iterations, as shown in Figure 2B. A possible explanation is that only the output of the last time step of LSTM was used for making the prediction. Therefore, after continuing the iteration for some time, the attention weight of the right end would become larger. But fortunately, this change did not affect the pattern of the central key region.
In summary, the LA6mA method placed the attention layer after the feature extraction layer prior to being connected to an FC layer. Thus, the attention layer of the LA6mA model paid attention to the extracted features instead of the original sequence. To a certain extent, the attention may be distracted and become more abstract. On the other hand, the AL6mA model directly connected the attention layer to the input matrix, which paid attention to the underlying information and facilitated the discovery of the key position information.
AL6mA revealed key nucleotides for 6mA prediction
As aforementioned, the attention vectors of LA6mA reveal key areas of universality, whereas AL6mA tends to find the key positions of individuality. The attention layer in AL6mA is directly connected to the encoding matrix, and the size is the same as that of the input 2D matrix. In this section, we further analyzed the detailed parameters in the attention layer of AL6mA.
To facilitate the analysis of the attention mechanism, we used pLogo [49] to generate the sequence logo representations for each position in the aligned groups of sequences. Specifically, the sequences surrounding the A bases at the center of A. thaliana and D. melanogaster were examined and motifs identified. The base heights were adjusted according to the statistical significance with P-value <0.05. As shown in Figure 4, in both datasets, the enriched and depleted nucleotides in the DNA sequences surrounding the 6mA and non-6mA sites were significantly different.
Figure 4.
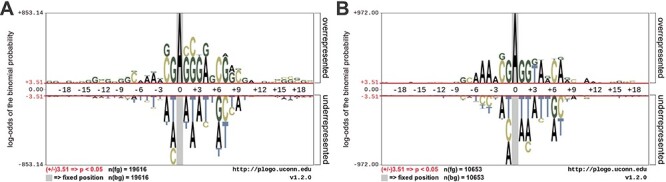
Sequence logo representations of the nucleotide preferences of the upstream and downstream positions surrounding 6mA sites and non-6mA sites: (A) A. thaliana and (B) D. melanogaster. The height of each base represents its over- or underrepresentation at the given positions, whereas the red line indicates significant enrichment.
For the well-trained model, an attention matrix with the size of 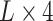 can be extracted from each input sequence. Different attention matrices were obtained from different sequence inputs, and the values in the attention matrix reflect which specific areas the model paid attention to when making the prediction. As shown in Figure 5A, the heatmap provides a visualization of the matrix, in which the values were highlighted by dark or light colors, with darker colors indicating larger values of the attention matrix, whereas lighter colors being the opposite.
can be extracted from each input sequence. Different attention matrices were obtained from different sequence inputs, and the values in the attention matrix reflect which specific areas the model paid attention to when making the prediction. As shown in Figure 5A, the heatmap provides a visualization of the matrix, in which the values were highlighted by dark or light colors, with darker colors indicating larger values of the attention matrix, whereas lighter colors being the opposite.
Figure 5.
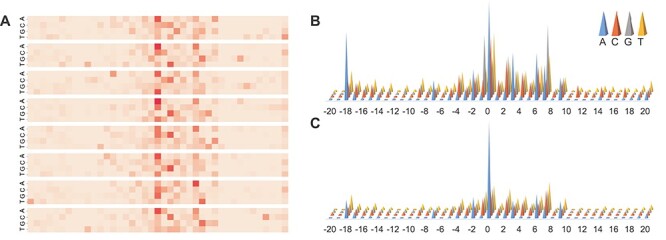
Illustration of the attention matrix of AL6mA for predicting 6mA sites of A. thaliana. It consists of (A) heatmap of key areas for a single input sequence; (B) attention matrix for TPs and (C) attention matrix for TNs. The darker colors in the heatmap indicate larger values in the attention matrix, whereas lighter colors denote the opposite. The height of the cones in B and C is proportional to the contribution to the prediction.
Widespread short nucleotides of DNA sequences that are conjectured to have a functional role are defined as DNA sequence motifs [50]. In this context, except for the key area with a single sequence, we also analyzed the results based on a set of samples. Specifically, all the test samples were fed into the well-trained AL6mA model and accordingly all the attention matrices were extracted. Subsequently, the attention matrices of the TP and TN samples were picked out to calculate the mean value of the attention matrices. The mean attention matrix could be further mapped to a sequence to indicate the amount of information provided by different positions in the sequence. Figure 5B and C and Figure 6 show the results on A. thaliana and D. melanogaster, respectively. Taking the middle site in Figure 5B as an example, which referred to an adenine encoded with 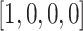 , it shows that the 1st position (i.e. adenine) provides more important information than the other three positions.
, it shows that the 1st position (i.e. adenine) provides more important information than the other three positions.
Figure 6.
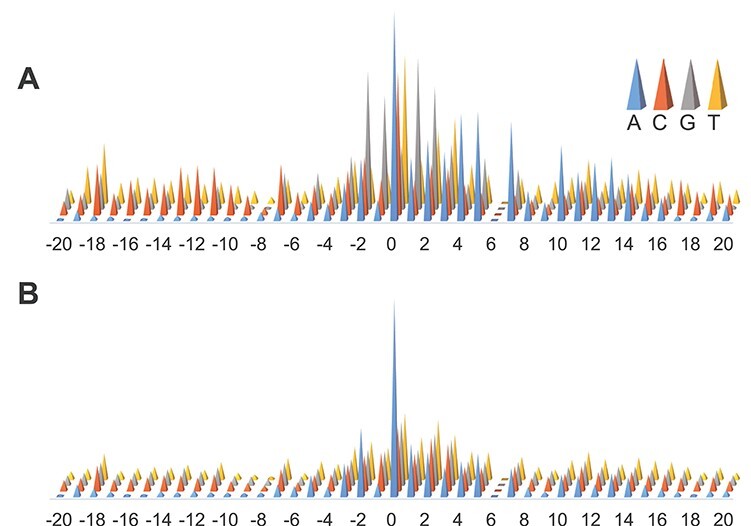
Illustration of the attention matrix of AL6mA for predicting 6mA sites of D. melanogaster. (A) Attention matrix for TPs. (B) Attention matrix for TNs.
We further revealed key nucleotides for 6mA prediction in lieu of the importance of each location in the key area through the analyses of the attention matrices of the TP examples and TN examples. With reference to the attention matrix in Figure 5B and C and Figure 6, we list the key nucleotides that collectively contributed to the correct prediction. The key nucleotides for A. thaliana and D. melanogaster are listed in Tables 2 and 3, respectively. We consider the number of statistically significant bases. Taking the position 1 in the Sequence logo of positive samples in Figure 4A as an example, both nucleotides C and G were found to be statistically significant. Thus, the two nucleotides with the highest height at the position 1 in Figure 5B are listed in the Table. Comparing the key nucleotides with the result of statistical significance one by one, we can find that for the TP samples, only several key nucleotides weight share consistent patterns with nucleotide sequence logos, whereas most key nucleotides are different from the nucleotide sequence logos. Specifically, for the TP samples of A. thaliana, only the positions −1, 1, 2 and 7 share the same pattern with the sequence logos (G, C/G G/C, G respectively). And for the TP samples of D. melanogaster, only the positions −1, 1, 2, 4, 7 shared the same motifs (G, G, G, A, A/G). In contrast, for the TN samples of the two model organisms, the nucleotides with the high attention weight are almost identical to the sequence logos.
Table 2.
Key nucleotides at specific positions for A. thaliana
| −2 | −1 | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TP | A/C | G | A | C/G | G/C | A | C/T | C | A | G | C/G | A |
| TN | A/T | T/C/G | A | C/T | T | A | T | C/G | A | C/T | C/G | A/T |
Table 3.
Key nucleotides at specific positions for D. melanogaster
| −3 | −2 | −1 | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TP | G | G | G | A | G | G | T/A | A | A/G | – | A/G |
| TN | A/T | A | C/G/T | A | A/T | T/C/G | C | A/T | A/T | – | A |
This is an interesting phenomenon. The correct prediction of negative samples is largely attributed to the attention to enriched nucleotides. For the type of binary classification problem, the number of negative samples is huge. The experiment randomly selects samples with the same number of positive samples as negative samples. If the algorithm needs to identify negative samples correctly, it should pay attention to the distribution of nucleotides. The correct prediction mechanism of the positive sample is different. The key nucleotides are not the same as significant distribution. It automatically pays more attention to the C site and G site.
Impact of the attention layer on the model performance
In this section, we investigated the potential impact of the attention layer on the model performance. Specifically, we removed the attention layers of LA6mA and AL6mA, respectively, and compared the performance of these methods. For convenience, the attention-layer-removed LA6mA is named as LA6mA-al, and the attention-layer-removed AL6mA is named as AL6mA-al. The performance results on the test datasets are shown in Table 4. It can be seen that nearly all the performance measures of the models without the attention layer decreased, compared with those of the corresponding models with the attention layer. Of the five performance measures in Table 4, AUROC can be regarded as a comprehensive prediction performance measure as it does not rely on the prediction cutoff threshold. LA6mA-al and AL6mA-al achieved an AUROC of 0.958 and 0.935 on A. thaliana, which were 0.004 and 0.01 lower than that of LA6mA and AL6mA, respectively. On the dataset of D. melanogaster, LA6mA-al and AL6mA-al achieved an AUROC of 0.962 and 0.937, which were decreased by 0.004 and 0.004, compared with that of LA6mA and AL6mA, respectively. On the other hand, the results highlight effectiveness of the attention layer on the performance of the two proposed methods. It can be seen from Table 4 that LA6mA performed better than AL6mA with an improvement of 0.017 and 0.025 in terms of AUROC on A. thaliana and D. melanogaster, respectively. The performance difference is due to by their different structures (Figure 1A and D). In addition, it is worth mentioning that with the attention layer removed, LA6mA-al still performed better than AL6mA. This is because LA6mA used the FC layers, but AL6mA did not.
Table 4.
Performance comparison of the proposed AL6mA and LA6mA methods with LA6mA-al and AL6mA-al with the removal of the attention layer on the test datasets
| Dataset | Method | Sen | Spe | Acc | MCC | AUROC |
|---|---|---|---|---|---|---|
| Arabidopsis thaliana | LA6mA | 0.899 | 0.917 | 0.909 | 0.817 | 0.962 |
| LA6mA-al | 0.904 | 0.891 | 0.897 | 0.794 | 0.958 | |
| AL6mA | 0.862 | 0.905 | 0.884 | 0.768 | 0.945 | |
| AL6mA-al | 0.854 | 0.888 | 0.871 | 0.743 | 0.935 | |
| Drosophila melanogaster | LA6mA | 0.909 | 0.915 | 0.912 | 0.824 | 0.966 |
| LA6mA-al | 0.896 | 0.907 | 0.902 | 0.803 | 0.962 | |
| AL6mA | 0.840 | 0.916 | 0.878 | 0.758 | 0.941 | |
| AL6mA-al | 0.866 | 0.872 | 0.869 | 0.738 | 0.937 |
Performance of the proposed models on 5-fold cross-validation
We evaluated the performance of the proposed models on the training datasets on 5-fold cross-validation. To do so, we randomly divided the training datasets into five nonoverlapping subsets. In each validation step, four subsets were used to train the model, whereas the remaining subset was used to test the performance of the trained model. The unweighted averages of the 5-fold cross-validation were calculated as the final results. To make a fair comparison, the division of the subsets was fixed for the methods on the same model organisms. The average performance results, along with the SD, are provided in Table 5. It can be seen that LA6mA achieved the best performance. The AUROC value was 0.960 and its SD was 0.002 on A. thaliana, whereas the AUROC value was 0.963 and its SD was 0.003 on D. melanogaster, respectively. The performance of AL6mA was slightly lower than that of LA6mA. The SD of the performance measures of LA6mA ranged from 0.002 to 0.016, whereas those of AL6mA ranged from 0.003 to 0.018, which reflects the robustness of the proposed models on the 5-fold cross-validation.
Table 5.
Performance of the proposed AL6mA and LA6mA methods using 5-fold cross-validation
| Dataset | Method | Sen | Spe | Acc | MCC | AUROC | |
|---|---|---|---|---|---|---|---|
| Arabidopsis thaliana | LA6mA | Average | 0.895 | 0.908 | 0.901 | 0.803 | 0.960 |
| SD | 0.014 | 0.002 | 0.003 | 0.006 | 0.002 | ||
| AL6mA | Average | 0.852 | 0.897 | 0.874 | 0.750 | 0.938 | |
| Standard | 0.015 | 0.018 | 0.005 | 0.010 | 0.003 | ||
| Drosophila melanogaster | LA6mA | Average | 0.911 | 0.902 | 0.906 | 0.812 | 0.963 |
| SD | 0.004 | 0.013 | 0.008 | 0.016 | 0.003 | ||
| AL6mA | Average | 0.859 | 0.895 | 0.877 | 0.755 | 0.942 | |
| SD | 0.013 | 0.015 | 0.009 | 0.017 | 0.006 | ||
Performance comparison with the existing methods
The categories of the methods mentioned in the introduction section can be generally categorized into three major groups, i.e. ML-based models, statistical models and DL-based models. To evaluate the performance of our two methods for predicting m6A sites, we further compared them with both DL-based methods and classical k-mer-based logistic regression (LR) method. The compared DL-based methods include DeepM6A [28], i6mA-DNC [30] and iDNA6mA [26]. All the methods used the same training dataset and test set to make a fair performance comparison. Table 6 summarizes the main characteristics of the compared 6mA site prediction methods, including the features employed, performance evaluation strategy, the corresponding species and the number of samples as well as the availability of code or web server.
Table 6.
A summary of the main characteristics of the compared methods for 6mA site prediction
| Method/Tool | Year | Features | Evaluation strategy | Speciesa | Code or web server |
|---|---|---|---|---|---|
| DeepM6A [28] | 2020 | One-hot encoding | 10-fold cross-validation |
Arabidopsis thaliana (19 632 + 19 632), Drosophila melanogaster (10 653 + 10 653), Escherichia coli (33 700 + 33 700) |
https://github.com/tanfei2007/DeepM6A/tree/master/Code |
| i6mA-DNC [30] | 2020 | Dinucleotide components | 10-fold cross-validation | Rice (880 + 880) | http://nsclbio.jbnu.ac.kr/tools/i6mA-DNC/ |
| iDNA6mA [26] | 2019 | One-hot encoding | Independent test | Rice (880 + 880) | http://nsclbio.jbnu.ac.kr/tools/iDNA6mA/ |
The source code of DeepM6A [28] was downloaded from https://github.com/tanfei2007/DeepM6A/tree/master/Code. To make a fair comparison, we maintained its structure and hyperparameters, adopted the same optimizer with an early stopping strategy and used the final well-trained models for the prediction. The length of the input sequence was adjusted to 41, consistent with the length used in the original DeepM6A work. For both A. thaliana and D. melanogaster, the models were trained on the training datasets independently without transfer learning. i6mA-DNC [30] and iDNA6mA [26] were reimplemented on the same dataset in this work. The k-mer encoding scheme, a commonly used method of DNA sequence encoding, was used to generate all the possible subsequence frequencies of the input DNA sequences. We set the length of the subsequences as 3, so that each sequence was encoded as a vector of length 64.
We used the scikit-learn library to train the LR model (https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.LogisticRegressionCV.html). Five-fold cross-validation was performed to obtain the optimal parameters for the 3-mer-LR method. Table 7 summarizes the performance results of the six different 6mA prediction methods on the two test datasets. In Table 7, the performance values of Sen, Spe, Acc and MCC were calculated based on the prediction cutoff threshold set as 0.5. To facilitate the performance comparison, the values of Sen were calculated by fixing the Specificity. In addition the ROC curves of these methods are displayed in Figure 7.
Table 7.
Performance comparison of the proposed AL6mA and LA6mA methods with other existing methods for predicting 6mA sites on the test datasets
| Dataset | Method | Sen1 | Spe1 | Acc1 | MCC1 | AUROC | Sen2 | Sen3 |
|---|---|---|---|---|---|---|---|---|
| Arabidopsis thaliana | DeepM6Aa | 0.894 | 0.931 | 0.913 | 0.826 | 0.966 | 0.920 | 0.956 |
| i6mA-DNCb | 0.846 | 0.909 | 0.878 | 0.757 | 0.944 | 0.853 | 0.912 | |
| iDNA6mAc | 0.843 | 0.889 | 0.866 | 0.733 | 0.932 | 0.833 | 0.902 | |
| 3-mer-LRd | 0.669 | 0.728 | 0.699 | 0.397 | 0.773 | 0.411 | 0.577 | |
| LA6mA | 0.899 | 0.917 | 0.909 | 0.817 | 0.962 | 0.912 | 0.948 | |
| AL6mA | 0.862 | 0.905 | 0.884 | 0.768 | 0.945 | 0.867 | 0.927 | |
| Drosophila melanogaster | DeepM6Aa | 0.901 | 0.939 | 0.920 | 0.841 | 0.969 | 0.930 | 0.959 |
| i6mA-DNCb | 0.869 | 0.917 | 0.893 | 0.787 | 0.947 | 0.878 | 0.916 | |
| iDNA6mAc | 0.883 | 0.843 | 0.863 | 0.727 | 0.937 | 0.846 | 0.904 | |
| 3-mer-LRd | 0.680 | 0.702 | 0.691 | 0.383 | 0.753 | 0.347 | 0.558 | |
| LA6mA | 0.909 | 0.915 | 0.912 | 0.824 | 0.966 | 0.921 | 0.955 | |
| AL6mA | 0.840 | 0.916 | 0.878 | 0.758 | 0.941 | 0.848 | 0.920 |
aResult obtained by retraining and retesting the source code of DeepM6A [28].
bResult obtained by the re-implementation of i6mA-DNC [30] on benchmark datasets.
cResult obtained by the re-implementation of iDNA6mA [26] on benchmark datasets.
dResult obtained by testing the well-trained 3-mer-based LR model.
1With prediction cutoff threshold value set as 0.5.
2With the fixed Specificity at 0.9.
3With the fixed Specificity at 0.8.
Figure 7.
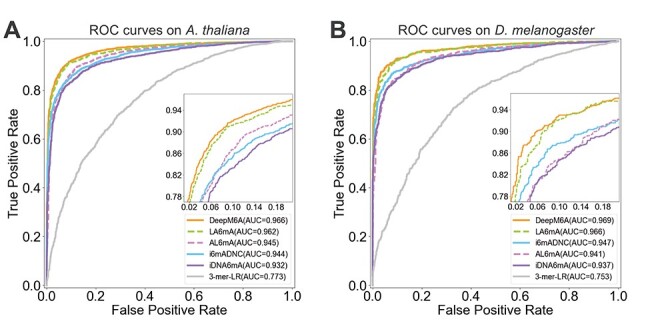
ROC curves of different methods for 6mA prediction on the two organisms: (A) ROC curves on A. thaliana and (B) ROC curves on D. melanogaster.
As can be seen, DeepM6A achieved the overall best performance for 6mA prediction with AUROC = 0.966 on A. thaliana and AUROC = 0.969 on D. melanogaster, respectively, both of which were higher than all the other compared methods. Not surprisingly, the 3-mer-LR method, which was developed based on conventional machine learning, achieved the lowest predictive performance in terms of all performance metrics. For 6mA prediction in A. thaliana, our proposed LA6mA method achieved an AUROC value of 0.962, which was the 2nd-best performance and slightly lower than that of DeepM6A, which achieved an AUROC of 0.966. While for 6mA prediction in D. melanogaster, the other three DL-based methods, including i6mA-DNC, AL6mA and iDNA6mA achieved a very similar performance with AUROC values ranging from 0.937 to 0.947 (Table 7). True positive rate is also named as Sen, and the sum of false positive rate (FPR) and Spe is 1, which means FPR = 1−Spe. In addition, Table 7 also provides performance comparison of different methods in terms of Sen under the fixed Specificity (i.e. 0.8 and 0.9). For both model organisms, it can be concluded that DeepM6A consistently performed best under fixed Specificity, followed by LA6mA. As shown in Figure 7, for A. thaliana, the Sen of AL6mA and i6mADNC reached the same value under the fixed Specificity of 0.93. The Sen of AL6mA achieved a larger value when the Specificity was smaller than 0.93; however, the situation was the opposite when the Specificity was larger than 0.93. In contrast, for D. melanogaster in Figure 7B, the threshold of the Specificity between AL6mA and i6mADNC was 0.82. It is worth mentioning that DeepM6A is a deep convolutional network with 315 481 parameters, whereas the proposed AL6mA and LA6mA only have 138 043 (43.76% of DeepM6A) and 159 235 (50.47% of DeepM6A) parameters, respectively. This suggests that LA6mA could achieve a competitive performance with that of DeepM6A by using only half of its parameters.
Conclusions
DL can automatically extract useful features from raw genome sequence data that are pertinent to the prediction task. Nevertheless, such capability can be a ‘double-edged sword’: on one hand, researchers are liberated from the complicated, tedious and manual feature extraction process, but on the other hand, researchers find it challenging to address the ‘blackbox’ issue and interpret the models. In this study, we have proposed two novel computational methods for DNA 6mA site identification, which are termed LA6mA and AL6mA, respectively. The networks of these two methods mainly consist of the LSTM layer and attention layer. The LSTM layer can automatically capture the short-range and long-range information from the encoded input sequences, whereas the attention layer provides biologically meaningful interpretations through introducing sequence context information and identifying the key positions surrounding the potential 6mA/non-6maA sites that contribute the most to the final prediction. Specifically, the attention layer after the LSTM layer is responsible for processing the extracted features and generating the abstract attentions to find the key areas of universality between the organisms (e.g. A. thaliana and D. melanogaster), whereas the attention layer preceding the LSTM layer pays attention to the individuality between organisms. Benchmarking experiments have demonstrated that the two methods could achieve a competitive performance for DNA 6mA site prediction. In future work, a potentially useful strategy based on the multi-head attention mechanism [51] can be employed, which has been successfully applied to address protein classification and generation tasks [52]. Analogous to the simultaneous use of multiple filters in CNNs, the multi-head attention allows the model to capture information from different representation subspaces, which might be helpful for extracting richer information from the input sequences. Interpreting the attention mechanism with complex RNN network architectures or the attention mechanism with RNN and CNN can be also investigated. The two proposed methods herein are promising and are generally applicable to address other sequence-based problems in the fields of bioinformatics and computational biology.
Key Points
Accurate prediction of DNA 6mA sites is important for the characterization of their functional roles in multiple biological processes.
Two novel methods, termed LA6mA and AL6mA, are developed to automatically capture the short-range and long-range information from DNA sequence using LSTM.
The self-attention mechanism is employed to effectively capture the position information from DNA sequence.
In-depth analyses of the changes in the attention weights, similarities and differences of the attention layer from the two 6mA prediction models for A. thaliana and D. melanogaster, and the differences of the attention layer for TP and TN examples are conducted to interpret what key information underpins the model prediction.
An online web server is implemented and publicly available at http://csbio.njust.edu.cn/bioinf/al6ma/, which can be exploited as useful tool for the prediction of DNA 6mA sites.
Supplementary Material
Ying Zhang received her MS degree in Control Science and Engineering from Yangzhou University in 2020. She is currently a PhD candidate in the School of Computer Science and Engineering at Nanjing University of Science and Technology. Her research interests include bioinformatics, machine learning and pattern recognition.
Yan Liu received his MS degree in Computer Science from Yangzhou University in 2019. He is currently a PhD candidate in the School of Computer Science and Engineering at Nanjing University of Science and Technology and a member of the Pattern Recognition and Bioinformatics Group. His research interests include bioinformatics, machine learning and pattern recognition.
Jian Xu received his PhD degree from Nanjing University of Science and Technology, on the subject of Data Mining in 2007. He is currently a full professor in the School of Computer Science and Engineering, Nanjing University of Science and Technology. His research interests include event mining, log mining and their applications to complex system management and machine learning. He is a member of both China Computer Federation (CCF) and IEEE.
Xiaoyu Wang received her MS degree in Information Technology from The University of Melbourne in 2020. She is currently a research assistant in the Biomedicine Discovery Institute, Monash University and a member of the Bioinformatics and Computational Biomedicine Lab. Her research interests include bioinformatics, computational biomedicine and machine learning.
Xinxin Peng received his MS degree in Physics Science from La Trobe University in 2020. He is currently a research assistant in the Biomedicine Discovery Institute, Monash University and a member of the Bioinformatics and Computational Biomedicine Lab. His research interests include bioinformatics, computational biomedicine, machine learning and medical imaging.
Jiangning Song is an associate professor and group leader in the Monash Biomedicine Discovery Institute and the Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia. He is also affiliated with the Monash Data Futures Institute, Monash University. His research interests include bioinformatics, computational biomedicine, machine learning and pattern recognition.
Dong-Jun Yu received the PhD degree from Nanjing University of Science and Technology in 2003. He is currently a full professor in the School of Computer Science and Engineering, Nanjing University of Science and Technology. His research interests include bioinformatics, machine learning and pattern recognition. He is a senior member of the China Computer Federation (CCF) and a senior member of the China Association of Artificial Intelligence (CAAI).
Contributor Information
Ying Zhang, School of Computer Science and Engineering at Nanjing University of Science and Technology, 200 Xiaolingwei, Nanjing 210094, China.
Yan Liu, School of Computer Science and Engineering at Nanjing University of Science and Technology, 200 Xiaolingwei, Nanjing 210094, China.
Jian Xu, School of Computer Science and Engineering, Nanjing University of Science and Technology, 200 Xiaolingwei, Nanjing 210094, China.
Xiaoyu Wang, Monash Biomedicine Discovery Institute and the Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia.
Xinxin Peng, Monash Biomedicine Discovery Institute and the Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia.
Jiangning Song, Monash Biomedicine Discovery Institute and the Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia.
Dong-Jun Yu, School of Computer Science and Engineering, Nanjing University of Science and Technology, 200 Xiaolingwei, Nanjing 210094, China.
Funding
National Natural Science Foundation of China (62072243, 61772273, 61872186); Natural Science Foundation of Jiangsu (BK20201304); Foundation of National Defense Key Laboratory of Science and Technology (JZX7Y202001SY000901); Fundamental Research Funds for the Central Universities (30918011104); National Health and Medical Research Council of Australia (NHMRC) (APP1127948, APP1144652); Australian Research Council (ARC) (LP110200333, DP120104460); National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965); Major Inter-Disciplinary Research (IDR) project, Monash University.
References
- 1. Bird A. Perceptions of epigenetics. Nature 2007;447:396–8. [DOI] [PubMed] [Google Scholar]
- 2. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology 2013;38:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol 2014;6:a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ye P, Luan Y, Chen K, et al. MethSMRT: an integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res 2017;45:D85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ratel D, Ravanat JL, Berger F, et al. N6-methyladenine: the other methylated base of DNA. BioEssays 2006;28:309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liang Z, Shen L, Cui X, et al. DNA N(6)-adenine methylation in Arabidopsis thaliana. Dev Cell 2018;45:406–16.e3. [DOI] [PubMed] [Google Scholar]
- 7. Liu J, Zhu Y, Luo GZ, et al. Abundant DNA 6mA methylation during early embryogenesis of zebrafish and pig. Nat Commun 2016;7:13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu TP, Wang T, Seetin MG, et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 2016;532:329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mondo SJ, Dannebaum RO, Kuo RC, et al. Widespread adenine N6-methylation of active genes in fungi. Nat Genet 2017;49:964–8. [DOI] [PubMed] [Google Scholar]
- 10. Fu Y, Luo GZ, Chen K, et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 2015;161:879–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greer EL, Blanco MA, Gu L, et al. DNA methylation on N6-adenine in C. elegans. Cell 2015;161:868–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang G, Huang H, Liu D, et al. N6-methyladenine DNA modification in Drosophila. Cell 2015;161:893–906. [DOI] [PubMed] [Google Scholar]
- 13. Pomraning KR, Smith KM, Freitag M. Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods 2009;47:142–50. [DOI] [PubMed] [Google Scholar]
- 14. Krais AM, Cornelius MG, Schmeiser HH. Genomic N(6)-methyladenine determination by MEKC with LIF. Electrophoresis 2010;31:3548–51. [DOI] [PubMed] [Google Scholar]
- 15. Flusberg BA, Webster DR, Lee JH, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 2010;7:461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen W, Lv H, Nie F, et al. i6mA-Pred: identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics 2019;35:2796–800. [DOI] [PubMed] [Google Scholar]
- 17. Kong L, Zhang L. i6mA-DNCP: computational identification of DNA N(6)-methyladenine sites in the rice genome using optimized dinucleotide-based features. Genes 2019;10:828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lv H, Dao FY, Guan ZX, et al. iDNA6mA-rice: a computational tool for detecting N6-methyladenine sites in rice. Front Genet 2019;10:793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Basith S, Manavalan B, Shin TH, et al. SDM6A: a web-based integrative machine-learning framework for predicting 6mA sites in the rice genome. Mol Ther Nucleic Acids 2019;18:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu H, Hu R, Jia P, et al. 6mA-finder: a novel online tool for predicting DNA N6-methyladenine sites in genomes. Bioinformatics 2020;36:3257–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brayet J, Zehraoui F, Jeanson-Leh L, et al. Towards a piRNA prediction using multiple kernel fusion and support vector machine. Bioinformatics 2014;30:i364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nair AS, Sreenadhan SP. A coding measure scheme employing electron-ion interaction pseudopotential (EIIP). Bioinformation 2006;1:197–202. [PMC free article] [PubMed] [Google Scholar]
- 23. He W, Jia C, Zou Q. 4mCPred: machine learning methods for DNA N4-methylcytosine sites prediction. Bioinformatics 2019;35:593–601. [DOI] [PubMed] [Google Scholar]
- 24. Chen W, Feng P, Ding H, et al. iRNA-methyl: identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal Biochem 2015;490:26–33. [DOI] [PubMed] [Google Scholar]
- 25. Pian C, Zhang G, Li F, et al. MM-6mAPred: identifying DNA N6-methyladenine sites based on Markov model. Bioinformatics 2020;36:388–92. [DOI] [PubMed] [Google Scholar]
- 26. Tahir M, Tayara H, Chong KT. iDNA6mA (5-step rule): identification of DNA N6-methyladenine sites in the rice genome by intelligent computational model via Chou's 5-step rule. Chemom Intel Lab Syst 2019;189:96–101. [Google Scholar]
- 27. Yu H, Dai Z. SNNRice6mA: a deep learning method for predicting DNA N6-methyladenine sites in rice genome. Front Genet 2019;10:1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tan F, Tian T, Hou X, et al. Elucidation of DNA methylation on N6-adenine with deep learning. Nat Mach Intell 2020;2:466–75. [Google Scholar]
- 29. Li Z, Jiang H, Kong L, et al. Deep6mA: a deep learning framework for exploring similar patterns in DNA N6-methyladenine sites across different species. PLoS Comput Biol 2021;17:e1008767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park S, Wahab A, Nazari I, et al. i6mA-DNC: prediction of DNA N6-methyladenosine sites in rice genome based on dinucleotide representation using deep learning. Chemom Intel Lab Syst 2020;204:104102. [Google Scholar]
- 31. Zeng H, Gifford DK. Predicting the impact of non-coding variants on DNA methylation. Nucleic Acids Res 2017;45:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 2016;26:990–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alipanahi B, Delong A, Weirauch MT, et al. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 2015;33:831–8. [DOI] [PubMed] [Google Scholar]
- 34. Zhong R, Shao S, Mckeown K. Fine-grained sentiment analysis with faithful attention. arXiv preprint arXiv:190806870. 20 August 2019, preprint: not peer reviewed.
- 35. Wiegreffe S, Pinter Y. Attention is not not Explanation. In: Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP). Hong Kong, China: Association for Computational Linguistics, 2019, 11–20. [Google Scholar]
- 36. Clark K, Khandelwal U, Levy O, et al. What does BERT look at? An analysis of BERT's attention. In: Proceedings of the 2019 ACL Workshop BlackboxNLP: Analyzing and Interpreting Neural Networks for NLP. Florence, Italy: Association for Computational Linguistics, 2019;276–86. [Google Scholar]
- 37. Htut PM, Phang J, Bordia S, et al. Do attention heads in BERT track syntactic dependencies? arXiv preprint arXiv:191112246v1. 28 November 2019, preprint: not peer reviewed.
- 38. Kim KE, Peluso P, Babayan P, et al. Long-read, whole-genome shotgun sequence data for five model organisms. Sci Data 2014;1:140045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hirschberg J, Manning CD. Advances in natural language processing. Science 2015;349:261–6. [DOI] [PubMed] [Google Scholar]
- 40. Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput 1997;9:1735–80. [DOI] [PubMed] [Google Scholar]
- 41. Lecun Y, Bottou L, Bengio Y, et al. Gradient-based learning applied to document recognition. Proc IEEE 1998;86:2278–324. [Google Scholar]
- 42. Vaswani A, Shazeer N, Parmar N, et al. Attention Is All You Need. Proceedings of the 31st International Conference on Neural Information Processing Systems. Long Beach, CA: Curran Associates Inc., 2017, 6000–10. [Google Scholar]
- 43. Lin Z, Feng M, Santos C, et al. A structured self-attentive sentence embedding. In: Conference paper in 5th International Conference on Learning Representations (ICLR). Toulon, France, 2017. [Google Scholar]
- 44. Li H, Tian S, Li Y, et al. Modern deep learning in bioinformatics. J Mol Cell Biol 2021;12:823–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park S, Koh Y, Jeon H, et al. Enhancing the interpretability of transcription factor binding site prediction using attention mechanism. Sci Rep 2020;10:13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zou Z, Tian S, Gao X, et al. mlDEEPre: multi-functional enzyme function prediction with hierarchical multi-label deep learning. Front Genet 2019;9:article 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hong Z, Zeng X, Wei L, et al. Identifying enhancer-promoter interactions with neural network based on pre-trained DNA vectors and attention mechanism. Bioinformatics 2020;36:1037–43. [DOI] [PubMed] [Google Scholar]
- 48. Wahab A, Mahmoudi O, Kim J, et al. DNC4mC-deep: identification and analysis of DNA N4-methylcytosine sites based on different encoding schemes by using deep learning. Cell 2020;9:1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. O'Shea JP, Chou MF, Quader SA, et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nat Methods 2013;10:1211–2. [DOI] [PubMed] [Google Scholar]
- 50. Xiao CL, Zhu S, He M, et al. N(6)-methyladenine DNA modification in the human genome. Mol Cell 2018;71:306–18.e7. [DOI] [PubMed] [Google Scholar]
- 51. Devlin J, Chang MW, Lee K, et al. BERT: pre-training of deep bidirectional transformers for language understanding. arXiv preprint arXiv:181004805. 28 May 2019, preprint: not peer reviewed.
- 52. Vig J, Madani A, Varshney LR, et al. BERTology meets biology: interpreting attention in protein language models. arXiv preprint arXiv:200615222v3. 30 March 2021, preprint: not peer reviewed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.