

Abstract
Attention deficit hyperactivity disorder (ADHD) is a common neurodevelopmental disorder. Although genome-wide association studies (GWAS) identify the risk ADHD-associated variants and genes with significant P-values, they may neglect the combined effect of multiple variants with insignificant P-values. Here, we proposed a convolutional neural network (CNN) to classify 1033 individuals diagnosed with ADHD from 950 healthy controls according to their genomic data. The model takes the single nucleotide polymorphism (SNP) loci of P-values 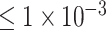 , i.e. 764 loci, as inputs, and achieved an accuracy of 0.9018, AUC of 0.9570, sensitivity of 0.8980 and specificity of 0.9055. By incorporating the saliency analysis for the deep learning network, a total of 96 candidate genes were found, of which 14 genes have been reported in previous ADHD-related studies. Furthermore, joint Gene Ontology enrichment and expression Quantitative Trait Loci analysis identified a potential risk gene for ADHD, EPHA5 with a variant of rs4860671. Overall, our CNN deep learning model exhibited a high accuracy for ADHD classification and demonstrated that the deep learning model could capture variants’ combining effect with insignificant P-value, while GWAS fails. To our best knowledge, our model is the first deep learning method for the classification of ADHD with SNPs data.
, i.e. 764 loci, as inputs, and achieved an accuracy of 0.9018, AUC of 0.9570, sensitivity of 0.8980 and specificity of 0.9055. By incorporating the saliency analysis for the deep learning network, a total of 96 candidate genes were found, of which 14 genes have been reported in previous ADHD-related studies. Furthermore, joint Gene Ontology enrichment and expression Quantitative Trait Loci analysis identified a potential risk gene for ADHD, EPHA5 with a variant of rs4860671. Overall, our CNN deep learning model exhibited a high accuracy for ADHD classification and demonstrated that the deep learning model could capture variants’ combining effect with insignificant P-value, while GWAS fails. To our best knowledge, our model is the first deep learning method for the classification of ADHD with SNPs data.
Keywords: ADHD identification, deep learning, saliency map, GWAS
Introduction
Attention deficit hyperactivity disorder (ADHD) is one of the most common mental disorders among children and adults with significant influence on attention, which causes the patient to appear with inattention, impulsiveness and hyperactivity [1, 2]. At least 5% of children have substantial difficulties with overactivity, inattention and impulsivity [3]. Family, twin and adoption studies indicate that ADHD has high heritability of 74% [4]. Identifying risk variants of ADHD and classification of ADHD cases from normal controls according to genomic data are essential for clinical diagnosis and treatment for ADHD.
Genome-wide association study (GWAS) has been applied to reveal genome-wide significant variants associated with ADHD [5–8]. Using thousands of ADHD cases and controls, GWAS examines thousands to millions of single nucleotide polymorphisms (SNPs) within the whole genome to detect significant variants. These variants with a P-value lower than the threshold in 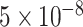 have been usually acknowledged as robust variants associated with traits [9]. The study of Neale et al. [10] investigated 2064 trios, 896 ADHD patients and 2455 controls and found a group of ADHD candidate genes. The study of Demontis et al. [8] discovered 12 robust variants that are significantly associated with ADHD using 20 183 ADHD cases, and 35 191 controls. These studies provided evidence that GWAS is an effective approach for identifying the risk variants or genes associated with ADHD. However, GWAS merely cares about the SNPs with significant P-values and may fail to capture the cumulative effect of insignificant SNPs and their overall contribution to ADHD.
have been usually acknowledged as robust variants associated with traits [9]. The study of Neale et al. [10] investigated 2064 trios, 896 ADHD patients and 2455 controls and found a group of ADHD candidate genes. The study of Demontis et al. [8] discovered 12 robust variants that are significantly associated with ADHD using 20 183 ADHD cases, and 35 191 controls. These studies provided evidence that GWAS is an effective approach for identifying the risk variants or genes associated with ADHD. However, GWAS merely cares about the SNPs with significant P-values and may fail to capture the cumulative effect of insignificant SNPs and their overall contribution to ADHD.
Deep learning techniques, especially Convolutional Neural Networks (CNNs), as a powerful tool for classification problems, have been widely applied to the classification of many diseases, such as skin cancers [11], interstitial lung diseases [12] and Alzheimer’s disease [13]. Several studies have also been reported for the classification of ADHD using deep learning model [14, 15] on the functional magnetic resonance imaging or structural magnetic resonance imaging data. These deep learning models achieved an accuracy of up to 69.15% for the classification of ADHD samples [15], which is insufficient for clinical diagnosis of ADHD. Meanwhile, there is a lack of deep learning models that use SNP data to identify ADHD.
In this study, we proposed a CNN-based deep learning model for the classification of ADHD with the SNPs data on a real dataset with 1033 individuals diagnosed with ADHD and 950 healthy controls. We test three single nucleotide polymorphism (SNP) locus sets as the features: loci of P-values 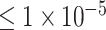 (10 SNPs),
(10 SNPs), 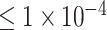 (109 SNPs) and
(109 SNPs) and 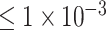 (764 SNPs). Our model achieved the classification performance with accuracy of 0.9018, AUC of 0.9570, sensitivity of 0.8980 and specificity of 0.9055 when using the SNP set with P-values
(764 SNPs). Our model achieved the classification performance with accuracy of 0.9018, AUC of 0.9570, sensitivity of 0.8980 and specificity of 0.9055 when using the SNP set with P-values 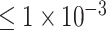 . Furthermore, we found a novel gene EPHA5 associated with ADHD, by incorporating the saliency analysis for our CNN-based deep learning model.
. Furthermore, we found a novel gene EPHA5 associated with ADHD, by incorporating the saliency analysis for our CNN-based deep learning model.
Materials and methods
Data collection
We used the data set from our previous study [16]. A total of 1033 ADHD patients (870 males, 84.2%) and 950 healthy controls (601 males, 63.3%) were included in the study (Figure 1A). This study was approved by the Ethics Committee of Peking University Sixth Hospital. The signed informed consent was obtained from all cases or from the parents of the children.
Figure 1 .
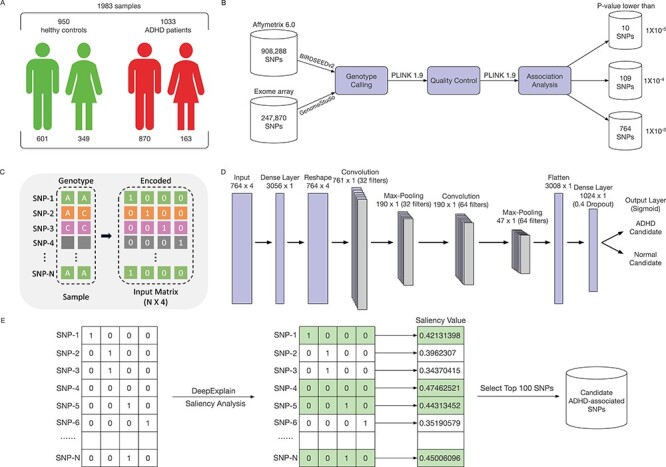
The overview of our ADHD classification pipeline. (A) The sample information of the our project. A total of 1033 ADHD patients (870 males, 84.2%) and 950 healthy controls (601 males, 63.3%) were included in this study. (B) The feature selection process for our deep leanring model. (C) The encoding schema for the genotype data. An input genotype data of one sample are first encoded to a 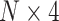 input matrix where N is the total SNP number. The gray cells means the genotype missing for SNPs. (D) The CNN architecture of our deep learning model. (E) The saliency analysis to find the candidate ADHD-associated SNPs.
input matrix where N is the total SNP number. The gray cells means the genotype missing for SNPs. (D) The CNN architecture of our deep learning model. (E) The saliency analysis to find the candidate ADHD-associated SNPs.
SNP genotyping
The collected DNA samples were genotyped by both the Affymetrix Genome-Wide Human SNP Array 6.0 [17] and the Illumina Infinium HumanExome-12v1 BeadChip [18]. The SNP genotypes were called by BIRDSEEDv2 and GenomeStudio v2011.1. In summary, each sample genotyped in the Affymetrix 6.0 and the Exome array contains 908 288 SNPs and 247 870 SNPs, respectively.
Quality control and association analysis
To conduct association analysis, samples and SNPs that meet the standard quality criteria were retained to reduce the potential bias due to genotyping technology and population size. Samples were removed if they did not satisfy the SNP heterozygosity rate (<50%) and SNP call rate (>90%) in either of the Affymetrix6.0 or the Exome array. Low-quality SNPs were eliminated if they had a low call rate (<0.95), low minor allele frequency (<0.05% for the Exome array and 1% for the Affymetrix6.0) or unexpected P-value(<1E-4) for Hardy–Weinberg equilibrium. The quality control process was performed by the whole genome data analysis toolset PLINK 1.9 [19]. After QC, all 1983 samples passed the criteria and 677 860 SNPs in the Affymetrix6.0 and 45 485 SNPs in the Exome array were retained, respectively. Association analysis between SNP genotypes and ADHD traits was performed by PLINK 1.9 with the command ‘–assoc’.
Feature selection
After quality control (QC), our dataset contains 1471 male samples (1471/1983, 74.2%) and male samples have one copy of the X chromosome. To eliminate the impact of the imbalance between males and females on the accuracy of binary classification of ADHD, only SNPs in the autosome were kept for further deep learning model. The SNPs in the Affymetrix6.0 and the Exome array were merged. For binary classification of ADHD, three subsets of all SNPs after QC and association analysis were selected: P-values lower than 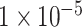 (10 SNPs),
(10 SNPs), 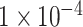 (109 SNPs) and
(109 SNPs) and 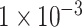 (764 SNPs). These three subsets were not mutually exclusive relationships, but include relationships. The SNPs in these subsets were regards as the features of each sample (Figure 1B).
(764 SNPs). These three subsets were not mutually exclusive relationships, but include relationships. The SNPs in these subsets were regards as the features of each sample (Figure 1B).
Deep learning model
Our model was designed to identify ADHD cases for a given sample. The first step occurs to encode an input SNPs data (Figure 1C). Each SNP was encoded to an 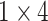 vector based on its genotype: AA to 1000, Aa to 0100, aa to 0010 and NA (missing) to 0001, where we assume A is the major allele, and a is the minor allele. Thus, the input SNPs data for each sample were encoded to an
vector based on its genotype: AA to 1000, Aa to 0100, aa to 0010 and NA (missing) to 0001, where we assume A is the major allele, and a is the minor allele. Thus, the input SNPs data for each sample were encoded to an 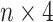 matrix. This encoded matrix will be passed into the input layer of our deep learning model.
matrix. This encoded matrix will be passed into the input layer of our deep learning model.
When using SNP sets with P-values 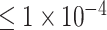 and
and 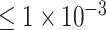 as input features, we built a CNN-based deep neural network to achieve the binary classification of ADHD samples, shown in Figure 1D. In detail, a new input sample is first passed to a fully connected layer and is then reshaped to the original input shape. The reshaped layer is accompanied by two convolutional layers, followed by a rectified linear unit (ReLU). After each convolutional layer, a maximum pooling layer is applied. This is then connected by a fully connected layer with dropout (0.4) and a sigmoid layer to indicate whether the input sample is an ADHD candidate. While when using the SNP set with P-values lower than
as input features, we built a CNN-based deep neural network to achieve the binary classification of ADHD samples, shown in Figure 1D. In detail, a new input sample is first passed to a fully connected layer and is then reshaped to the original input shape. The reshaped layer is accompanied by two convolutional layers, followed by a rectified linear unit (ReLU). After each convolutional layer, a maximum pooling layer is applied. This is then connected by a fully connected layer with dropout (0.4) and a sigmoid layer to indicate whether the input sample is an ADHD candidate. While when using the SNP set with P-values lower than 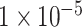 which contained only 10 SNPs, we developed a basic deep neural network with three fully connected layers followed by a sigmoid layer.
which contained only 10 SNPs, we developed a basic deep neural network with three fully connected layers followed by a sigmoid layer.
Training and evaluation
In order to train the model, the dataset (1983 samples) was randomly split into a training dataset (1486 samples, 75%), a validation dataset (100 samples, 5%) and a testing dataset (387, 20%). The deep learning model was constructed using Keras package [20] of TensorFlow [21]. First, the model was trained using the random weight initialization. The Adam method [22] was applied to the optimization process with an initial learning rate of 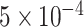 and a binary cross-entropy loss function. Then, the model was trained for 100 epochs with a batch size 512. We adopted the Dropout strategy to prevent the overfitting.
and a binary cross-entropy loss function. Then, the model was trained for 100 epochs with a batch size 512. We adopted the Dropout strategy to prevent the overfitting.
Benchmarking settings
To compare the performance of our model with other transitional machine learning models, we also performed the Random Forest model and the Support Vector Machine (SVM) model on our data. The ‘RandomForestClassifier’ model and the ‘svm‘ model of sklearn package [23] were used to perform the Random Forest model and SVM model, respectively. The default parameters were used for these two model.
Evaluation metrics
We used the accuracy (the percentage of cases where our model identified the correct class) to evaluate the performance of our model. Also, we measured the area under the receiver operating characteristic (ROC) curve (AUC), the sensitivity (the percentage of correctly predicted positive cohort cases from all positive cohort cases) and specificity (the percentage of correctly predicted negative cohort cases from all negative cohort cases) of our model.
Finding significant SNPs using saliency maps
Here, we performed the DeepExplain [24] using its ‘saliency’ method to determine a real saliency value for each input SNP. This saliency map value indicates the positive contribution of each SNP feature for ADHD binary classification. For each sample in the test dataset with an input dimension of 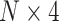 , a saliency value matrix with the same dimension can be calculated using DeepExplain. Since each SNP was encoded to a
, a saliency value matrix with the same dimension can be calculated using DeepExplain. Since each SNP was encoded to a 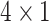 vector in the input matrix, the average saliency value of these four values of each SNP was selected to represent the final saliency value for this SNP. Thus, a saliency value matrix with the dimension of
vector in the input matrix, the average saliency value of these four values of each SNP was selected to represent the final saliency value for this SNP. Thus, a saliency value matrix with the dimension of 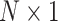 can be generated for each sample in the test dataset. To find the significant SNPs for ADHD binary classification, we took the average saliency value of all correctly predicted samples in the test dataset for each SNP as the measurement for SNP significance (Figure 1E). All samples in the test dataset with mispredicted labels were excluded from this process.
can be generated for each sample in the test dataset. To find the significant SNPs for ADHD binary classification, we took the average saliency value of all correctly predicted samples in the test dataset for each SNP as the measurement for SNP significance (Figure 1E). All samples in the test dataset with mispredicted labels were excluded from this process.
Gene Ontology enrichment and expression Quantitative Trait Loci analysis
To evaluate whether the SNPs with high saliency value are associated with ADHD, we performed the Gene Ontology (GO) terms for biological functions and expression Quantitative Trait Loci (eQTL) analysis for top 100 SNPs sorted by saliency value. The GO terms analysis was performed using FUMA [25] and Benjamini–Hochberg correction (FDR) was used for multiple test correction methods in gene-set enrichment testing. The maximum adjusted P-value for gene set association was 0.05. The Protein–Protein Interaction Networks analysis was performed using STRING online tool (https://string-db.org/). The gene list of the top 100 SNPs was uploaded to STRING website as the input gene set and the default parameters were used for all analysis in STRING. The eQTL analysis was conducted using BRAINEAC [26], the Brain eQTL Almanac, to investigate the genes and SNPs associated with neurological disorders.
Quantitative analyses for genes of interested
For the genes of interest identified from the above analyses, we further conducted quantitative analyses to explore the association of the genetic variants with ADHD core symptoms and cognitive functions which commonly showed deficits in ADHD including inhibition, working memory, shifting and processing speed [27]. The inhibition function was measured by Stroop color and word test, with color interference (IC) and word interference (IW) for analyses. Rey-Osterrieth complex figure test (RCFT) and Digit Span test were conducted to assess for visual and verbal working memory, respectively. For RCFT, structure and detail forgotten scores were generated and analyzed. For the Digit Span test, the forward, backward span number and total score were used for analyses. Trail making test was used for assessment of Shifting function with shift time for analyses. For processing speed, scores in the Coding test in the Chinese-Wechsler Intelligence Scale for Children (C-WISC) were used for analyses. These quantitative analyses were only conducted in ADHD samples using analysis of covariance (ANCOVA) with age and gender as covariant. Considering the multiple analyses, the corrected P-value was set as 0.05/8/18 = 3E-04 (8 represents the cognitive features for analyses; 18 presents the number of SNPs for quantitative analyses).
Results
Methodological development of deep learning model
Given an input SNPs data of one sample, the first step was encoding it to numbers-coding format (Figure 1C). Each SNP was encoded to a 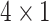 vector based on its genotype (see methods). Thus, the input SNPs data for each sample was encoded to an
vector based on its genotype (see methods). Thus, the input SNPs data for each sample was encoded to an 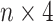 matrix. After encoding, the input matrix was fed into a CNN-based deep learning model to obtain the final ADHD candidates’ probability (Figure 1D). The model was initially trained using the training and validation dataset and then examined using a pre-excluded testing dataset (see methods). The performance of our model was evaluated by measuring the accuracy (the percentage of cases where our model identified the correct class). The accuracy emphasized the potential of our model as a reference toolkit in clinical ADHD detection. Also, we reported the area under the receiver operating characteristic (ROC) curve (AUC), the sensitivity and the specificity.
matrix. After encoding, the input matrix was fed into a CNN-based deep learning model to obtain the final ADHD candidates’ probability (Figure 1D). The model was initially trained using the training and validation dataset and then examined using a pre-excluded testing dataset (see methods). The performance of our model was evaluated by measuring the accuracy (the percentage of cases where our model identified the correct class). The accuracy emphasized the potential of our model as a reference toolkit in clinical ADHD detection. Also, we reported the area under the receiver operating characteristic (ROC) curve (AUC), the sensitivity and the specificity.
Binary classification of ADHD
For binary classification of ADHD, three subsets of all SNPs after QC and association analysis were selected: P-values lower than 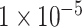 (10 SNPs),
(10 SNPs), 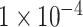 (109 SNPs) and
(109 SNPs) and 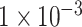 (764 SNPs). These three subsets of all SNPs are regarded as three different feature sets to train the model. The model was trained using 837 samples as a positive cohort and 749 other samples as a negative cohort. The test dataset contains 196 samples of ADHD and 201 non-ADHD samples. Figure 2 demonstrated the performance of our deep learning model with three different SNP sets as input features. As illustrated in Figure 2A–C, the model using SNP sets with P-values lower than
(764 SNPs). These three subsets of all SNPs are regarded as three different feature sets to train the model. The model was trained using 837 samples as a positive cohort and 749 other samples as a negative cohort. The test dataset contains 196 samples of ADHD and 201 non-ADHD samples. Figure 2 demonstrated the performance of our deep learning model with three different SNP sets as input features. As illustrated in Figure 2A–C, the model using SNP sets with P-values lower than 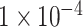 and
and 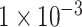 achieved over-fitting on the training dataset after around 20 epochs, while the model using SNP set with P-value lower than
achieved over-fitting on the training dataset after around 20 epochs, while the model using SNP set with P-value lower than 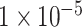 displayed a low degree of accuracy (lower than 0.7) after 100 epochs. On the validation dataset, the model using the SNP set with a P-value lower than
displayed a low degree of accuracy (lower than 0.7) after 100 epochs. On the validation dataset, the model using the SNP set with a P-value lower than 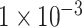 exhibited the highest accuracy with the value of about 0.85 among these three experiments (see Figure 2A–C). Figure 2D–F demonstrated the performance ROC curve of trained models for the testing dataset using these three SNP sets. Consistent with the performance on validation dataset, our model achieved the best performance using SNP set with P-value lower than
exhibited the highest accuracy with the value of about 0.85 among these three experiments (see Figure 2A–C). Figure 2D–F demonstrated the performance ROC curve of trained models for the testing dataset using these three SNP sets. Consistent with the performance on validation dataset, our model achieved the best performance using SNP set with P-value lower than 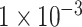 on testing dataset with the highest accuracy of 0.9018, AUC of 0.9570, sensitivity of 0.8980 and specificity of 0.9055 (see Table 1). We also compared the performance of our model with two traditional machine learning models, Random Forest and Support Vector Machine (SVM). The result of evaluation metrics are displayed in Table 1. Similar to our model, these two models also achieved the best classification performance on the testing dataset when using the SNP set with P-value lower than
on testing dataset with the highest accuracy of 0.9018, AUC of 0.9570, sensitivity of 0.8980 and specificity of 0.9055 (see Table 1). We also compared the performance of our model with two traditional machine learning models, Random Forest and Support Vector Machine (SVM). The result of evaluation metrics are displayed in Table 1. Similar to our model, these two models also achieved the best classification performance on the testing dataset when using the SNP set with P-value lower than 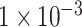 . For the results on the dataset with the P-value lower than
. For the results on the dataset with the P-value lower than 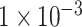 , the Random Forest model achieved the lowest classification performance. Compared with the other two models, its AUC and accuracy were more than 10% lower than the values of the other two models. Compared with the SVM model, our model still achieved the best classification performance, which was around 3% higher on all four evaluation metrics.
, the Random Forest model achieved the lowest classification performance. Compared with the other two models, its AUC and accuracy were more than 10% lower than the values of the other two models. Compared with the SVM model, our model still achieved the best classification performance, which was around 3% higher on all four evaluation metrics.
Figure 2 .
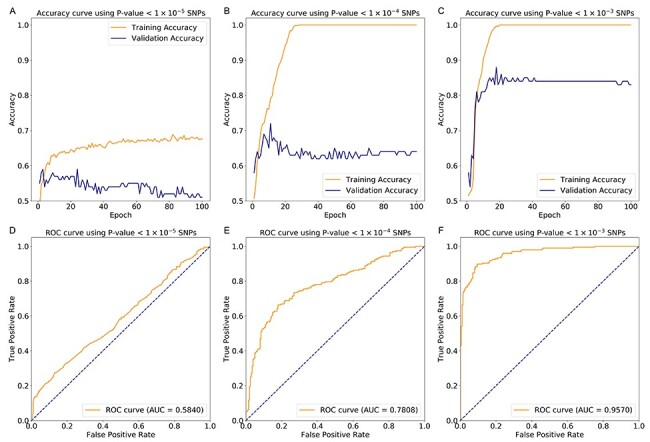
Model performance with three different number of SNPs. (A–C) The accuracy curve for training and validation datasets using three different SNP sets. (D–F) The performance ROC curve of trained models for testing dataset using three different SNP sets.
Table 1.
Model performance using three different SNP sets.
Dataset of P-value 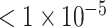
|
Dataset of P-value 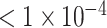
|
Dataset of P-value 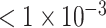
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Metrics | Random Forest | SVM | Our Model | Random Forest | SVM | Our Model | Random Forest | SVM | Our Model |
| AUC | 0.5898 | 0.5792 | 0.5840 | 0.7152 | 0.7878 | 0.7808 | 0.8027 | 0.9374 | 0.9570 |
| Accuracy | 0.5491 | 0.5840 | 0.5390 | 0.6751 | 0.7204 | 0.7229 | 0.7103 | 0.8640 | 0.9018 |
| Sensitivity | 0.4826 | 0.3930 | 0.5918 | 0.6119 | 0.7164 | 0.7449 | 0.5970 | 0.8607 | 0.8980 |
| Specificity | 0.6173 | 0.6888 | 0.4876 | 0.7398 | 0.7245 | 0.7015 | 0.8265 | 0.8673 | 0.9055 |
Common genome-wide association studies (GWAS) reveal significant SNPs with a genome-wide significant threshold of 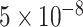 , which has been accepted as a standard for strong association [9]. The different classification performance for using above three SNP sets indicated that GWAS analysis focusing on the SNPs with significant P-values may fail to capture the cumulative effect of insignificant SNPs and their overall contribution to the ADHD binary classification, which could be captured using a deep learning model.
, which has been accepted as a standard for strong association [9]. The different classification performance for using above three SNP sets indicated that GWAS analysis focusing on the SNPs with significant P-values may fail to capture the cumulative effect of insignificant SNPs and their overall contribution to the ADHD binary classification, which could be captured using a deep learning model.
Finding significant SNPs using saliency map
Deep learning researchers have suggested that saliency map can be employed to find the real attribution for deep neural networks [24, 28, 29]. According to the result above, the model using SNP set with P-value lower than 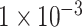 exhibited the best performance for ADHD binary classification. Therefore, this trained model was applied to saliency analysis. As described in the methods part, we calculated the saliency value for each SNP. To examine whether these SNPs with high saliency value are associated with ADHD, we performed Gene Ontology (GO) enrichment (biological processes) (http://amigo.geneontology.org) and Protein–Protein Interaction Network (protein interaction) (https://version11.string-db.org) analysis for top 100 SNPs out of 764 SNPs (see Supplementary Table S1).
exhibited the best performance for ADHD binary classification. Therefore, this trained model was applied to saliency analysis. As described in the methods part, we calculated the saliency value for each SNP. To examine whether these SNPs with high saliency value are associated with ADHD, we performed Gene Ontology (GO) enrichment (biological processes) (http://amigo.geneontology.org) and Protein–Protein Interaction Network (protein interaction) (https://version11.string-db.org) analysis for top 100 SNPs out of 764 SNPs (see Supplementary Table S1).
As illustrated in Figure 3, two significantly enriched GO terms were selected with a significant threshold of 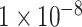 , including ‘GO central nervous system development’ with P-value =
, including ‘GO central nervous system development’ with P-value = 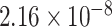 (adjusted P-value =
(adjusted P-value = 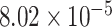 ) and ‘GO neurogenesis’ with P-value =
) and ‘GO neurogenesis’ with P-value = 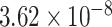 (adjusted P-value =
(adjusted P-value = 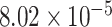 ), which indicated these SNPs with high saliency value may be associated with ADHD. Those genes both involved in the above two pathways included NRG3, TENM4, LIG4, MDGA2, BMP2, EPHA5, EPHA7, LPAR1 and TLR4.
), which indicated these SNPs with high saliency value may be associated with ADHD. Those genes both involved in the above two pathways included NRG3, TENM4, LIG4, MDGA2, BMP2, EPHA5, EPHA7, LPAR1 and TLR4.
Figure 3 .
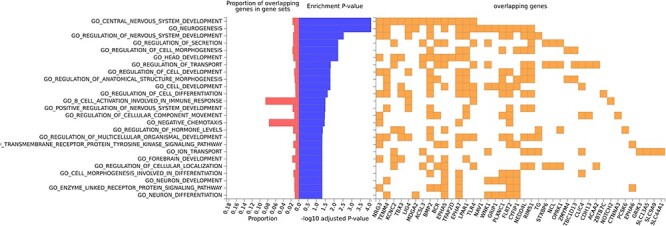
Gene Ontology enrichment for top 100 SNPs.
Protein interaction analyses based on STRING Interaction Network indicated significantly associated domains containing Ephrin receptor based on PFAM, INTERPRO or SMART database (FDR-corrected P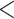 0.01). The involved genes included EPHA5, EHPA6, EPHA7 and EPHA10 (see Supplementary Table S2).
0.01). The involved genes included EPHA5, EHPA6, EPHA7 and EPHA10 (see Supplementary Table S2).
Moreover, we queried all genes of the top 100 SNPs from ‘GWAS Catalog’ database [30] and found that 14 genes (out of 100 genes) have been reported in previous ADHD-related studies (see Table 2), which proved that our model has the potential to discover ADHD-related genes using saliency analysis. It should be noticed that most of these ADHD-related genes were also involved in the above two significantly enriched GO terms. We further conducted quantitative analyses for the index variants of these genes with ADHD core symptoms and cognition functions. The NCL - rs16828074 was associated with ADHD core symptoms, however, the risk G-allele carriers were with lower hyperactive/impulsive [(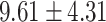 ) versus (
) versus (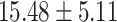 ), P = 2.85E-07] and total symptoms [(
), P = 2.85E-07] and total symptoms [(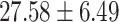 ) versus (
) versus (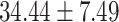 ), P = 1.27 E-10] than CC carriers. For the MDGA2 - rs12232114, the A allele was associated with severer inattentive symptoms (P = 0.004), indicating higher scores in AA carriers than that in AG [(
), P = 1.27 E-10] than CC carriers. For the MDGA2 - rs12232114, the A allele was associated with severer inattentive symptoms (P = 0.004), indicating higher scores in AA carriers than that in AG [(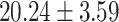 ) versus (
) versus (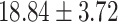 ), P = 0.002]or GG carriers [(
), P = 0.002]or GG carriers [(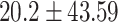 ) versus (
) versus (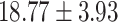 ), P = 0.001)]. However, the A allele was with lower frequency in ADHD than controls. For CDH13 - rs8055161, the risk A-allele was nominally associated with ADHD core symptoms indicating the higher hyperactive/impulsive [(
), P = 0.001)]. However, the A allele was with lower frequency in ADHD than controls. For CDH13 - rs8055161, the risk A-allele was nominally associated with ADHD core symptoms indicating the higher hyperactive/impulsive [(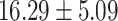 ) versus (
) versus ( ), P = 0.007] and total symptoms [(
), P = 0.007] and total symptoms [(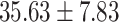 ) versus (
) versus (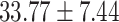 ), P = 0.007] than others.
), P = 0.007] than others.
Table 2.
Reported genes found in top 100 SNPs sorted by saliency value
| Chr | BP | Index variant | Gene | Region | P-value | Saliency value | Literatures |
|---|---|---|---|---|---|---|---|
| 2 | 232318754 | rs16828074 | NCL | UTR-3 |
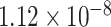
|
0.4497 | [31] |
| 3 | 143545596 | rs7621206 | SLC9A9 | intron |
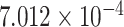
|
0.3306 | [33] |
| 4 | 66367589 | rs4860671 | EPHA5 | intron |
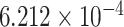
|
0.3432 | [32] |
| 6 | 72470319 | rs4707940 | RIMS1 | upstream |
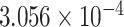
|
0.3679 | [34] |
| 8 | 6104273 | rs6559123 | CSMD1 | upstream |
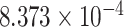
|
0.3457 | [35] |
| 10 | 85643777 | rs12244269 | NRG3 | downstream |
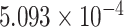
|
0.4221 | [36] |
| 11 | 79466812 | rs1944959 | TENM4 | upstream |
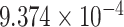
|
0.3327 | [37] |
| 13 | 108684837 | rs9514807 | LIG4 | downstream |
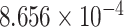
|
0.3703 | [38] |
| 14 | 25425964 | rs17200947 | STXBP6 | intron |
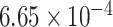
|
0.331 | [39] |
| 14 | 48219014 | rs12232114 | MDGA2 | upstream |
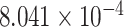
|
0.3311 | [40] |
| 16 | 6188504 | rs9935453 | RBFOX1 | intron |
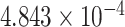
|
0.3501 | [41], [38] |
| 16 | 82443459 | rs8055161 | CDH13 | upstream |
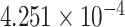
|
0.3507 | [42], [43] |
| 18 | 55771747 | rs1620068 | NEDD4L | intron |
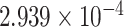
|
0.3383 | [44], [43] |
| 20 | 6970831 | rs952793 | BMP2 | downstream |
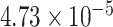
|
0.3281 | [43] |
When reviewing the above analyses, genes involved in the Ephrin receptor-related pathway attracted our attention, especially EPHA5 which have been revealed in Gene Ontology (GO) enrichment, STRING Interaction Network analyses, and previously reported ADHD-related genes. Then, we further explored the genetic influence of EPHA5 - rs4860671 on ADHD core symptoms and cognitive functions including inhibition, working memory, processing speed and shifting. A significant association was found for EPHA5 - rs4860671 with working memory measured by Digit Span Total score (P = 4.73 E-06) and processing speed measured by Coding score (P = 3.72 E-05), indicating the worse performance in the risk allele carriers (see Table 3).
Table 3.
Significant association of rs4860671-EPHA5 with ADHD diagnosis and ADHD-related cognitive functions.
| Digit Span Total Score | Coding Score | ||||||
|---|---|---|---|---|---|---|---|
| Genotype | ADHD | TDC | P-value | Mean 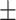 SE SE |
P-value | Mean 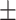 SE SE |
P-value |
| AA | 25 (2.5) | 46 (5.0) | 0.001 | 15.39 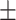 0.71 0.71 |
4.73 E-06 | 86.80 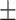 8.91 8.91 |
3.72E-05 |
| AG | 296 (29.4) | 307 (33.2) | 11.73 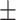 0.21 0.21 |
49.04 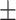 2.60 2.60 |
|||
| GG | 685(68.1) | 573 (61.9) | 11.94  0.14 0.14 |
45.89  1.70 1.70 |
|||
Note: TDC, typically developed controls.
To explore the potential neurological mechanism, we further conducted eQTL analyses (http://www.braineac.org/), indicating that the risk G-allele was associated with lower expression of EPHA5 in CRBL (P = 0.0029)(Figure 4). For the other gene, EPHA7 - rs16870710, no significant association with quantitative traits was found.
Figure 4 .
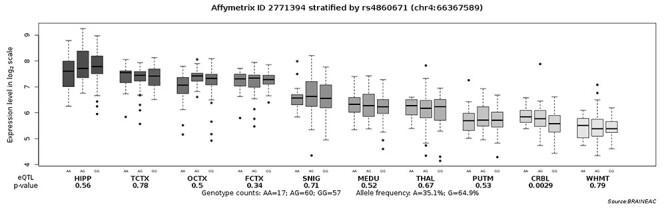
Analysis of the eQTL of EPHA5 - rs4860671 based on the data download from the UKBEC (http://www.braineac.org/). The significant association was found for rs4860671 with CRBL (P = 0.0029). Note. UKBEC = UK Brain Expression Cohort; eQTL = expression quantitative trait loci; THAL = thalamus; MEDU = medulla; FCTX = frontal cortex; TCTX = temporal cortex; WHMT = intralobular white matter; HIPP = hippocampus; SNIG = substantia nigra; OCTX = occipital cortex; PUTM = putamen; CRBL = cerebellar cortex.
Through the Brain eQTL analysis, we found a novel variant, rs6958168, showed significant association with the expression of gene CCM2 in temporal cortex (TCTX) with p-value = 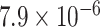 and intralobular white matter (WHMT) with P-value =
and intralobular white matter (WHMT) with P-value = 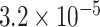 (see Figure 5).
(see Figure 5).
Figure 5 .
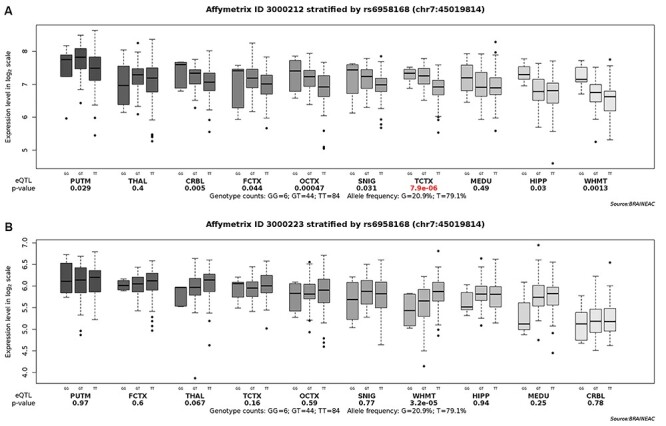
Brain eQTL analysis of gene CCM2 with rs6958168. The Significant association was found for rs6958168 with TCTX with P-value = 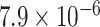 (A) and WHMT with P-value =
(A) and WHMT with P-value = 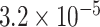 (B). Note. UKBEC = UK Brain Expression Cohort; eQTL = expression quantitative trait loci; THAL = thalamus; MEDU = medulla; FCTX = frontal cortex; TCTX = temporal cortex; WHMT = intralobular white matter; HIPP = hippocampus; SNIG = substantia nigra; OCTX = occipital cortex; PUTM = putamen; CRBL = cerebellar cortex.
(B). Note. UKBEC = UK Brain Expression Cohort; eQTL = expression quantitative trait loci; THAL = thalamus; MEDU = medulla; FCTX = frontal cortex; TCTX = temporal cortex; WHMT = intralobular white matter; HIPP = hippocampus; SNIG = substantia nigra; OCTX = occipital cortex; PUTM = putamen; CRBL = cerebellar cortex.
DISCUSSION
GWAS has been proved to be an effective approach for identifying the risk variants and genes associated with ADHD in the last decade. However, GWAS only cares about these SNPs with significant P-values and may fail to capture the cumulative effect of insignificant SNPs and their overall contribution to ADHD. Therefore, in this study, we proposed a CNN-based deep learning model for the classification of ADHD. We selected three SNP sets as the features of each sample: P-values lower than 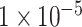 (10 SNPs),
(10 SNPs), 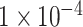 (109 SNPs) and
(109 SNPs) and 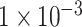 (764 SNPs). As demonstrated in Figure 2, the model using SNP sets with P-values lower than
(764 SNPs). As demonstrated in Figure 2, the model using SNP sets with P-values lower than 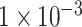 achieved the best classification performance, compared with the other two models. This result indicated that the deep learning model could capture the relationship between SNPs with insignificant P-values, while GWAS failed. Also, compared with reported deep learning models for the classification of ADHD [14, 15], which used the fMRI or sMRI data as the input data, our deep learning model achieved higher accuracy of 90.18%, compared with the accuracy of up to 69.15% of these models. The higher classification accuracy of our model proved the potential power of the deep learning model using SNPs data as input features for the classification of ADHD and this kind of model may be applied to the clinical diagnosis of ADHD.
achieved the best classification performance, compared with the other two models. This result indicated that the deep learning model could capture the relationship between SNPs with insignificant P-values, while GWAS failed. Also, compared with reported deep learning models for the classification of ADHD [14, 15], which used the fMRI or sMRI data as the input data, our deep learning model achieved higher accuracy of 90.18%, compared with the accuracy of up to 69.15% of these models. The higher classification accuracy of our model proved the potential power of the deep learning model using SNPs data as input features for the classification of ADHD and this kind of model may be applied to the clinical diagnosis of ADHD.
Deep learning researchers have suggested that a saliency map can be employed to find the real attribution for deep neural networks. Therefore, we conducted saliency analysis for our deep learning model to detect novel variants associated with ADHD. For the top 100 SNPs with high saliency values for ADHD classification, we further conducted Gene Ontology (GO) enrichment and STRING Interaction Network analyses. Then, the Ephrin receptor genes attracted our attention, especially EPHA5. Our further quantitative analyses of the SNP EPHA5 - rs4860671 with a saliency value of 0.343 indicated a significant association with working memory and processing speed performance in children with ADHD. When checking in GWAS Catalog, EPHA5 has been found to be associated with several ADHD-related deficits recently based really huge sample size, including mathematical ability (rs60178806, rs7659227) [45], cognitive function measurement (rs74944857) [45], educational attainment (rs10019169, rs74944857, rs4458506, rs13145146) [45], risk-taking behavior (rs28455852) [46] and intelligence (rs7655988, rs62300402, rs13145146) [47, 48]. After carefully check, the SNP rs4860671 found in our present study were independent locus from these above loci, because of the weak linkage disequilibrium (LD) with the above SNPs, showing the strongest LD with r2 of 0.448, 0.34 in CEU and CHB database, respectively. This suggested that the ADHD-associated SNP rs4860671 found in our present study was independent of these above loci (see Figure 6).
Figure 6 .
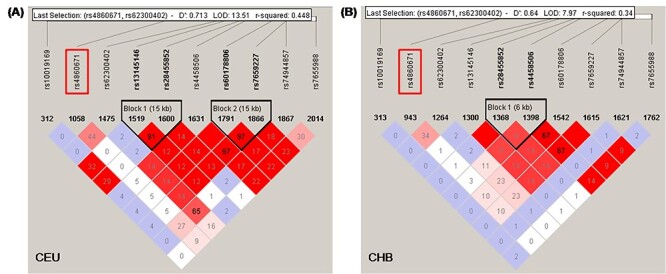
The linkage disequilibrium (LD) of rs4860671 with the previously reported SNPs of EPHA5 which have been suggested to be associated with ADHD-related deficits. The LD plots were determined by Haploview software, displaying LD values in r-squared.(i.e. 91 indicates r-squared of 0.91. Squares with no number indicate a r-squared of 1).
EPHA5 is located at 4q13, while duplication due to insertional translocation in this region has been found in two siblings with ADHD [32]. Besides, a genomic rearrangement that involved or near the EHPA5 gene was found in patients with autism spectrum disorder [49–51], which is often in co-occurrence with ADHD. Based on previous reports of the structural genetic variation in EPHA5, our present study should be the first one reporting the SNP related to ADHD. Further efforts are needed to explore the relationship between SNP and structural genetic variation, and their combined effects on the genetic etiology of ADHD. The EphA5 receptor, encoded by the EPHA5 gene, is involved in brain development, synaptic remodeling, plays a role in synaptic plasticity in the adult brain through regulation of synaptogenesis together with Ephrin A5 (EFNA5). For the SNP rs4860671, its precise function has not been reported. The eQTL analyses showed that the risk G-allele was associated with lower expression of EPHA5 in the cerebellar cortex. Notably, a recent genome-wide association analysis indicated that variants in EPHA5 might influence the volume of cerebellar vermal lobules [52]. The important role of the cerebellum in information processing speed has been supported in the existing literature [53, 54]. In the future, imaging genetic studies will be worth exploring the potential gene (EPHA5) -brain (structural/functional alteration in the cerebellum) -cognition (processing speed) relationship which might help us to illustrate a novel mechanism underlying the etiology of ADHD. Also, EphA5 has been indicated to be closely related to monoaminergic system [51, 55]; that gene–gene interactions between EPHA5 and monoaminergic genes might also be worth exploring.
In addition to EPHA5, consistent evidence from categorical and quantitative analyses was indicated for CDH13 - rs8055161. Several studies have supported the association of CDH13 with ADHD, especially the hyperactive-impulsive symptoms which was consistent with our present finding (rs6565113 [56]; rs11150556 [57]). Another novel finding is from the eQTL analysis of MYO1G - rs6958168 on the CCM2 gene. The CCM2 gene is associated with Cavernous Malformation diseases and related- pathways with Cavernous Malformation diseases are ‘Development HGF signaling pathway’ and ‘Development Endothelin-1/EDNRA signaling’ [58]. The top 1 of affiliating genes of these two pathways is ITGB1 gene [58]. ITGB1 gene can upregulate expression of CDC42 gene in ADHD neurodevelopmental signaling network [43] and CDC42 plays a requisite role in dopamine transporter endocytic trafficking [59]. These pieces of evidence indicate that the variant, rs6958168, is a susceptibility variant associated with ADHD. However, a more direct relationship of this SNP rs6958168, CCM2, and related pathways with ADHD needs to be investigated. The SNP rs6958168 is located upstream of CCM2 and is close to a long-noncoding RNA gene SNHG15. Future work could be conducted to explore whether long-noncoding RNAs participate in the relationship of rs6958168, CCM2 and ADHD.
Although our deep learning model achieved high accuracy of 0.9018, AUC of 0.9570 on our testing dataset, there are still several issues with our model. Firstly, the model performance only was evaluated by our testing data and has not been examined using other independent datasets, which weakens the credibility and the application to clinical diagnosis of our model. Secondly, the risk variants discovered using saliency analysis of our deep learning model still require further verification which can be done in our further studies.
Key Points
Common genome-wide association studies (GWAS) neglect the combined effect of multiple variants with insignificant P-values.
We proposed a convolutional neural network (CNN) to classify 1033 individuals diagnosed with ADHD from 950 healthy controls by using SNPs with insignificant P-values and achieved an accuracy of 0.9018, AUC of 0.9570, sensitivity of 0.8980 and specificity of 0.9055.
We applied saliency map analysis to the deep learning model and found potential ADHD-associated SNPs and genes.
To our best knowledge, our model is the first deep learning method for the classification of ADHD with SNPs data.
Supplementary Material
Acknowledgments
This study was funded by the National Natural Science Foundation of China (81873802, 81641163, 81571340, 81761148026), Beijing Natural Science Foundation (7172245) and the National Basic Research Program of China (2014CB846104, 2015CB856405).
Lu Liu, PhD, is currently an associate professor in the department of Child Psychiatry, Peking University Sixth Hospital/Institute of Mental Health, NHC Key Laboratory of Mental Health (Peking University), National Clinical Research Center for Mental Disorders (Peking University Sixth Hospital). She received her PhD in psychiatry in 2012. Her major interests are in the neuropsychology, neuroimaging and genetics of ADHD.
Xikang Feng, PhD, is an assistant professor at the School of Software, Northwestern Polytechnical University. He is good at the bioinformatics, deep learning and algorithm development.
Haimei Li, PhD, is a researcher at the Department of Child Psychiatry, Peking University Sixth Hospital/Institute of Mental Health, NHC Key Laboratory of Mental Health (Peking University), National Clinical Research Center for Mental Disorders (Peking University Sixth Hospital). She is good at the collection, quality control and analyses of experimental data.
Shuai Cheng Li, PhD, is an associate professor at the Department of Computer Science, City University of Hong Kong. He is good at the bioinformatics, deep learning and algorithm design.
Yufeng Wang, MD, PhD, is a professor in Peking University Sixth Hospital/Institute of Mental Health, NHC Key Laboratory of Mental Health (Peking University), National Clinical Research Center for Mental Disorders (Peking University Sixth Hospital). Her major interests are in therapeutics, neuropsychology, neurophysiology and genetics of ADHD.
Qiujin Qian, MD, PhD, is a professor and also the director in chief of the department of Child Psychiatry, Peking University Sixth Hospital/Institute of Mental Health, NHC Key Laboratory of Mental Health (Peking University), National Clinical Research Center for Mental Disorders (Peking University Sixth Hospital). She has got her PhD in 2002. Her major interests are in neuropsychology and neurophysiology of ADHD.
Contributor Information
Lu Liu, Peking University Sixth Hospital/Institute of Mental Health, National Clinical Research Center for Mental Disorders (Peking University Sixth Hospital) & the Key Laboratory of Mental Health, Ministry of Health (Peking University), 100191, Beijing, China.
Xikang Feng, School of Software, Northwestern Polytechnical University, Xi’an, 710072, Shaanxi, China.
Haimei Li, Peking University Sixth Hospital/Institute of Mental Health, National Clinical Research Center for Mental Disorders (Peking University Sixth Hospital) & the Key Laboratory of Mental Health, Ministry of Health (Peking University), 100191, Beijing, China.
Shuai Cheng Li, Department of Computer Science, City University of Hong Kong, Kowloon Tong, Hong Kong, China.
Qiujin Qian, Peking University Sixth Hospital/Institute of Mental Health, National Clinical Research Center for Mental Disorders (Peking University Sixth Hospital) & the Key Laboratory of Mental Health, Ministry of Health (Peking University), 100191, Beijing, China.
Yufeng Wang, Peking University Sixth Hospital/Institute of Mental Health, National Clinical Research Center for Mental Disorders (Peking University Sixth Hospital) & the Key Laboratory of Mental Health, Ministry of Health (Peking University), 100191, Beijing, China.
Data Availability
The annotation file for the Affymetrix Genome-Wide Human SNP Array 6.0 data was downloaded from the Affymetrix support website at http://www.affymetrix.com/support/technical/byproduct.affx?\product=genomewidesnp_6. The annotation file for the Illumina Infinium HumanExome-12v1 BeadChip data was downloaded from the Illumina support website at https://support.illumina.com/downloads/humanexome-12v1-2_product_support_files.html. The top 100 ADHD-related SNPs found in this study can be accessed in the Supplementary Table S1. The source code of our CNN model and the snps of three datasets used in this study are available at https://github.com/xikanfeng2/DeepADHD. For access to the full summary statistics with the results, interested researchers should contact the lead PI (QJ. Qian). Genotype data were not allowed to share, to protect the privacy of participants according to the commitment in the signed informed consent.
Author contributions statement
S.C.L., Q.Q. and Y.W. co-supervised the work. S.C.L. and X.F. designed and implemented the deep learning model and the saliency analysis. L.L., X.F. and H.L. conducted the data preprocessing and the downstream analysis. L.L., X.F. and S.C.L. revised the manuscript. All authors read and approved the final manuscript.
References
- 1. Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult adhd in the united states: results from the national comorbidity survey replication. Am J Psychiatry 2006;163(4):716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barkley RA, Poillion MJ. Attention-deficit hyperactivity disorder: A handbook for diagnosis and treatment. Behavioral Disorders, 1994;19(2):150–152. [Google Scholar]
- 3. Sayal K, Prasad V, Daley D, et al. Adhd in children and young people: prevalence, care pathways, and service provision. Lancet Psychiatry 2018;5(2):175–86. [DOI] [PubMed] [Google Scholar]
- 4. Faraone SV, Larsson H. Genetics of attention deficit hyperactivity disorder. Mol Psychiatry 2019;24(4):562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neale BM, Medland S, Ripke S, et al. Case-control genome-wide association study of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010;49(9):906–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fliers EA, Vasquez AA, Poelmans G, et al. Genome-wide association study of motor coordination problems in adhd identifies genes for brain and muscle function. World J Biol Psychiatry 2012;13(3):211–22. [DOI] [PubMed] [Google Scholar]
- 7. Sánchez-Mora C, Ramos-Quiroga JA, Bosch R, et al. Case–control genome-wide association study of persistent attention-deficit hyperactivity disorder identifies fbxo33 as a novel susceptibility gene for the disorder. Neuropsychopharmacology 2015;40(4):915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Demontis D, Walters RK, Martin J, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 2019;51(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clarke GM, Anderson CA, Pettersson FH, et al. Basic statistical analysis in genetic case-control studies. Nat Protoc 2011;6(2):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neale BM, Medland SE, Ripke S, et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010;49(9):884–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Esteva A, Kuprel B, Novoa RA, et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature 2017;542(7639):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anthimopoulos M, Christodoulidis S, Ebner L, et al. Lung pattern classification for interstitial lung diseases using a deep convolutional neural network. IEEE Trans Med Imaging 2016;35(5):1207–16. [DOI] [PubMed] [Google Scholar]
- 13. Liu S, Liu S, Cai W, et al. Early diagnosis of alzheimer’s disease with deep learning. In: 2014 IEEE 11th international symposium on biomedical imaging (ISBI). IEEE, 2014, 1015–8. [Google Scholar]
- 14. Kuang D, He L. Classification on adhd with deep learning. In: 2014 International Conference on Cloud Computing and Big Data. IEEE, 2014, 27–32. [Google Scholar]
- 15. Zou L, Zheng J, Miao C, et al. 3d cnn based automatic diagnosis of attention deficit hyperactivity disorder using functional and structural mri. IEEE Access 2017;5:23626–36. [Google Scholar]
- 16. Liu L, Zhang L, Li HM, et al. The snp-set based association study identifies itga1 as a susceptibility gene of attention-deficit/hyperactivity disorder in han chinese. Transl Psychiatry 2017;7(8):e1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edenberg HJ, Liu Y. Laboratory methods for high-throughput genotyping. Cold Spring Harb Protoc 2009;2009(11):pdb–top62. [DOI] [PubMed] [Google Scholar]
- 18. Grove ML, Bing Y, Cochran BJ, et al. Best practices and joint calling of the humanexome beadchip: the charge consortium. PloS one 2013;8(7):e68095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Purcell S, Neale B, Todd-Brown K, et al. Plink: a tool set for whole-genome association and population-based linkage analyses. The American journal of human genetics 2007;81(3):559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chollet F, et al. Keras, 2015.
- 21. Abadi M, Agarwal A, Barham P, et al. Tensorflow: Large-scale machine learning on heterogeneous distributed systems arXiv preprint arXiv:1603.04467. 2016.
- 22. Kingma DP, Ba J. Adam: A method for stochastic optimization arXiv preprint arXiv:1412.6980. 2014.
- 23. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: Machine learning in python. the Journal of machine Learning research 2011;12:2825–30. [Google Scholar]
- 24. Ancona M, Ceolini E, Öztireli C, et al. Towards better understanding of gradient-based attribution methods for deep neural networks arXiv preprint arXiv:1711.06104. 2017.
- 25. Watanabe K, Taskesen E, Van Bochoven A, et al. Functional mapping and annotation of genetic associations with fuma. Nat Commun 2017;8(1):1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramasamy A, Trabzuni D, Guelfi S, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci 2014;17(10):1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin J, Liu L, Gao Q, et al. The divergent impact of comt val158met on executive function in children with and without attention-deficit/hyperactivity disorder. Genes Brain Behav 2016;15(2):271–9. [DOI] [PubMed] [Google Scholar]
- 28. Simonyan K, Vedaldi A, Zisserman A. Deep inside convolutional networks: Visualising image classification models and saliency maps arXiv preprint arXiv:1312.6034. 2013.
- 29. Zeiler MD, Fergus R. Visualizing and understanding convolutional networks. In: European conference on computer vision. Salmon Tower Building New York City, United States: Springer, 2014, 818–33. [Google Scholar]
- 30. Buniello A, MacArthur JAL, Cerezo M, et al. The nhgri-ebi gwas catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2018;47(D1):D1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang L, Neale BM, Liu L, et al. Polygenic transmission and complex neuro developmental network for attention deficit hyperactivity disorder: Genome-wide association study of both common and rare variants. Am J Med Genet B Neuropsychiatr Genet 2013;162(5):419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matoso E, Melo JB, Ferreira SI, et al. Insertional translocation leading to a 4q13 duplication including the epha5 gene in two siblings with attention-deficit hyperactivity disorder. Am J Med Genet A 2013;161(8):1923–8. [DOI] [PubMed] [Google Scholar]
- 33. Mick E, Todorov A, Smalley S, et al. Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010;49(9):898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ekholm JM, Ogdie MN, Dang J, et al. Association analysis of candidate genes for adhd on chromosomes 5p13, 6q12, 16p and 17p. Open Psychiatry Journal 2007;1:34–42. [Google Scholar]
- 35. Brevik EJ, Donkelaar MMJ, Weber H, et al. Genome-wide analyses of aggressiveness in attention-deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 2016;171(5):733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meier S, Strohmaier J, Breuer R, et al. Neuregulin 3 is associated with attention deficits in schizophrenia and bipolar disorder. Int J Neuropsychopharmacol 2013;16(3):549–56. [DOI] [PubMed] [Google Scholar]
- 37. Cross-Disorder Group of the Psychiatric Genomics Consortium et al . Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. The Lancet 2013;381(9875):1371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Anney RJL, Lasky-Su J, Ó’Dúshláine C, et al. Conduct disorder and adhd: evaluation of conduct problems as a categorical and quantitative trait in the international multicentre adhd genetics study. Am J Med Genet B Neuropsychiatr Genet 2008;147(8):1369–78. [DOI] [PubMed] [Google Scholar]
- 39. Romanos M, Freitag C, Jacob C, et al. Genome-wide linkage analysis of adhd using high-density snp arrays: novel loci at 5q13. 1 and 14q12. Mol Psychiatry 2008;13(5):522. [DOI] [PubMed] [Google Scholar]
- 40. Lesca G, Rudolf G, Labalme A, et al. Epileptic encephalopathies of the landau-kleffner and continuous spike and waves during slow-wave sleep types: Genomic dissection makes the link with autism. Epilepsia 2012;53(9):1526–38. [DOI] [PubMed] [Google Scholar]
- 41. Klein M, Walters RK, Demontis D, et al. Genetic markers of adhd-related variations in intracranial volume. Am J Psychiatry 2019;176(3):228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lesch K-P, Timmesfeld N, Renner TJ, et al. Molecular genetics of adult adhd: converging evidence from genome-wide association and extended pedigree linkage studies. J Neural Transm 2008;115(11):1573–85. [DOI] [PubMed] [Google Scholar]
- 43. Poelmans G, Pauls DL, Buitelaar JK, et al. Integrated genome-wide association study findings: identification of a neurodevelopmental network for attention deficit hyperactivity disorder. Am J Psychiatry 2011;168(4):365–77. [DOI] [PubMed] [Google Scholar]
- 44. Ceccarini C, Sinibaldi L, Bernardini L, et al. Duplication 18q21. 31-q22. 2. Am J Med Genet A 2007;143(4):343–8. [DOI] [PubMed] [Google Scholar]
- 45. Lee JJ, Wedow R, Okbay A, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 2018;50(8):1112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Linnér RK, Biroli P, Kong E, et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet 2019;51(2):245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davies Gail, Lam Max, Harris Sarah E, Trampush Joey W, Luciano Michelle, Hill W David, Hagenaars Saskia P, Ritchie Stuart J, Marioni Riccardo E, Fawns-Ritchie Chloe, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun, 9(1):1–16, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lam M, David Hill W, Trampush JW, et al. Pleiotropic meta-analysis of cognition, education, and schizophrenia differentiates roles of early neurodevelopmental and adult synaptic pathways. The American Journal of Human Genetics 2019;105(2):334–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Girirajan S, Dennis MY, Baker C, et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. The American Journal of Human Genetics 2013;92(2):221–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shimada S, Okamoto N, Nomura S, et al. Microdeletions of 5.5 mb (4q13. 2–q13. 3) and 4.1 mb (7p15. 3–p21. 1) associated with a saethre–chotzen-like phenotype, severe intellectual disability, and autism. Am J Med Genet A 2013;161(8):2078–83. [DOI] [PubMed] [Google Scholar]
- 51. Pascolini G, Majore S, Valiante M, et al. Autism spectrum disorder in a patient with a genomic rearrangement that only involves the epha5 gene. Psychiatr Genet 2019;29(3):86–90. [DOI] [PubMed] [Google Scholar]
- 52. Zhao B, Luo T, Li T, et al. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat Genet 2019;51(11):1637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moroso A, Ruet A, Lamargue-Hamel D, et al. Posterior lobules of the cerebellum and information processing speed at various stages of multiple sclerosis. J Neurol Neurosurg Psychiatry 2017;88(2):146–51. [DOI] [PubMed] [Google Scholar]
- 54. Thakur B, Riches S, Costello A, et al. Calcifying pseudoneoplasm of the neuraxis, cerebellum and cognition: a rare opportunity to learn more. Cureus 2019;11(1):e3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Teng T, Gaillard A, Muzerelle A, et al. Ephrina5 signaling is required for the distinctive targeting of raphe serotonin neurons in the forebrain. eneuro 2017;4(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lasky-Su J, Neale BM, Franke B, et al. Genome-wide association scan of quantitative traits for attention deficit hyperactivity disorder identifies novel associations and confirms candidate gene associations. Am J Med Genet B Neuropsychiatr Genet 2008;147(8):1345–54. [DOI] [PubMed] [Google Scholar]
- 57. Salatino-Oliveira A, Genro JP, Polanczyk G, et al. Cadherin-13 gene is associated with hyperactive/impulsive symptoms in attention/deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 2015;168(3):162–9. [DOI] [PubMed] [Google Scholar]
- 58. Rappaport N, Twik M, Nativ N, et al. Malacards: A comprehensive automatically-mined database of human diseases. Curr Protoc Bioinformatics 2014;47(1):1–24. [DOI] [PubMed] [Google Scholar]
- 59. Sijia W, Melikian H. Cdc42 plays a requisite role in dopamine transporter endocytic trafficking (896.6). FASEB J 2014;28(1_supplement):896–6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The annotation file for the Affymetrix Genome-Wide Human SNP Array 6.0 data was downloaded from the Affymetrix support website at http://www.affymetrix.com/support/technical/byproduct.affx?\product=genomewidesnp_6. The annotation file for the Illumina Infinium HumanExome-12v1 BeadChip data was downloaded from the Illumina support website at https://support.illumina.com/downloads/humanexome-12v1-2_product_support_files.html. The top 100 ADHD-related SNPs found in this study can be accessed in the Supplementary Table S1. The source code of our CNN model and the snps of three datasets used in this study are available at https://github.com/xikanfeng2/DeepADHD. For access to the full summary statistics with the results, interested researchers should contact the lead PI (QJ. Qian). Genotype data were not allowed to share, to protect the privacy of participants according to the commitment in the signed informed consent.