

Abstract
Intratumoral heterogeneity is a well-documented feature of human cancers and is associated with outcome and treatment resistance. However, a heterogeneous tumor transcriptome contributes an unknown level of variability to analyses of differentially expressed genes (DEGs) that may contribute to phenotypes of interest, including treatment response. Although current clinical practice and the vast majority of research studies use a single sample from each patient, decreasing costs of sequencing technologies and computing power have made repeated-measures analyses increasingly economical. Repeatedly sampling the same tumor increases the statistical power of DEG analysis, which is indispensable toward downstream analysis and also increases one’s understanding of within-tumor variance, which may affect conclusions. Here, we compared five different methods for analyzing gene expression profiles derived from repeated sampling of human prostate tumors in two separate cohorts of patients. We also benchmarked the sensitivity of generalized linear models to linear mixed models for identifying DEGs contributing to relevant prostate cancer pathways based on a ground-truth model.
Keywords: linear mixed model, prostate cancer, transcriptomics, RNA-seq, multiple sampling, variance
Introduction
Large-scale pan-cancer genomic analyses, such as the Pan-Cancer Atlas [1] and the Pan-Cancer Analysis of Whole Genomes [2], have firmly established the heterogeneous and diverse nature of many primary tumors. Indeed, deep sequencing experiments have revealed that most cancers harbor complex phylogenies with distinct driver and passenger alterations such that one region of a cancer may demonstrate a different phenotype than another part of the same tumor [3]. This spatial tumoral heterogeneity not only creates challenges for clinical diagnostics but also adversely impacts clinical outcome [4]. While the clinical convention of biopsying tumors to confirm diagnoses has historically been based on a single sample of tissue for most cancers, the regional histologic heterogeneity of prostate cancer (PCa), in particular, lends itself to multiple sampling for a final diagnosis [5–8]. Paradoxically, PCa molecular diagnostic tests, like many others, still rely on a single biopsy sample; the delicate balance between expensive and comprehensive testing is justified by treating the most aggressive-appearing tumor when given a choice [9]. Given the extent of tumor heterogeneity, however, sampling for research purposes has recapitulated diverse genomic and genotypic results across multiple samples of the same human tumor, which can similarly be reflected in discordant within-patient diagnostic scores [5–8].
For studies of the tumor transcriptome, measures of differentially expressed genes (DEGs) are used to discover relationships between biological activity and phenotypes of interest, such as response to drug treatment or the impact of genomic alterations. With multiple-sampling strategies increasingly becoming common due to lower costs of sequencing and computational power, key concerns linger pertaining to the consolidation or aggregation of multiple sources of (valuable) information from the same biological sample (i.e. a patient) with an outcome that is ascribed to the whole patient and thus all samples as well [4, 10–12]. For example, how much variance is acceptable to lose in the process of reconciling an evolutionarily and phenotypically diverse tumor to a discrete outcome? Perhaps, and more importantly, what measures related to outcome harbor intrinsically more variance deemed by repeated measures of the tumor?
For ribonucleic acid (RNA) sequencing (RNA-seq), there is an underlying assumption that biological variability is often much greater between samples of different individuals than from within the same individual. Numerous bioinformatic solutions have been developed to assess the relationship between gene expression and outcome, often within the context of well-defined biological model systems with negligible within-individual variance and highly reproducible analytical measures such as RNA-seq. The most common tools include EdgeR, Limma/Voom and DESeq2, and these tools are frequently used to compare phenotypes with a defined number of biological replicates for each phenotype such that each replicate is given equal weighting using generalized linear models [13–15]. If applied to multiple samples of the same individual, options thus would include aggregating all within-individual data to individual-level values, or weighting each sample equally, which would skew observations. For example, an approach using DESeq2 would collapse pseudo-replicates by summing RNA-seq counts, which reduces the information available to discern fixed effects (variables that are constant across individuals, like treatment outcome) from random effects (categorical variables like somatic mutation status). Although Limma/Voom can accommodate the modeling of a single random effect, this would assume there are no other sources of variance to model, which is rarely the case when working with human biospecimens, especially if batch effects or purity are considered. To wit, when extended to translational cancer research, modeling the transcriptomes of patient samples that are acquired or compared based on innumerable salient phenotypes requires the inclusion of both fixed and random effects, which can be accommodated in linear mixed models (LMMs). The R package for LMMs, lme4 [16], is used by the Dream package [17] for repeated-sampling analysis by RNA-seq. However, its performance with real-world tumor data benchmarked against conventional analyses is unknown.
Here, we consider three potential genotypes based on somatic mutations, which are relevant to PCa (TMPRSS2-ERG fusion, PTEN loss and 18q deletion), the potential interaction between TMPRSS2-ERG fusion and PTEN loss and the sampling of tumors from patients with those genotypes. The TMPRSS2-ERG fusion occurs very early in the natural history of approximately 50% of PCa and frequently co-occurs with losses to PTEN, which manifest subclonally [18]. We explore the impact of five different analytical methods on the differential expression analysis from two PCa cohorts in which repeated-sampling design is a consideration and in which four of the methods collapse the specimens from the same lesion into a single sample (collectively termed aggregation), while the fifth method employs a LMM. By comparing between-sample individuals of each cohort with known heterogeneous cancer somatic phenotypes to robust ‘gold standard’ PCa transcriptomes from The Cancer Genome Atlas (TCGA), we show that LMMs demonstrate superior sensitivity for identifying DEGs when controlling for heterogeneous confounders such as PTEN loss. Aggregation approaches show similar sensitivity to LMMs when working with fewer samples per individual or when working with phenotypes that result in little within-patient variability. As such, comparing the sensitivity of these methods further reveals sources of variance depending on target phenotypes and is a practical exercise for identifying occult causes of heterogeneity within a cohort. We thus provide this computational code as a resource for translational cancer researchers exploring heterogeneity within their own datasets.
Materials and methods
Patient cohorts
Cohort 1 consists of tumor samples from 37 patients diagnosed with intermediate- to high-risk PCa and subsequently treated with neoadjuvant hormone therapy and radical prostatectomy [8]. These tumor samples were acquired from magnetic resonance imaging (MRI)-targeted biopsies prior to the onset of therapy and were guided by immunohistochemistry prior to laser capture microdissection. The manuscript by Wilkinson et al. [8] describes this cohort and each laser capture microdissected (LCM) tumor focus.
Cohort 2 consists of tumor samples from 39 patients diagnosed with intermediate- to high-risk PCa and subsequently treated by radical prostatectomy [19]. These tumor samples were acquired from tissue punches of radical prostatectomy tissue, guided by a pathologist’s determination of low-grade or high-grade cancer regions. The manuscript by Charmpi et al. [19] describes this cohort and each tumor region.
Raw sequence data processing
Paired-end RNA-seq FASTQ files for 130 tumor samples (cohort 1), 66 tumor samples (cohort 2) and 494 tumor cases (TCGA-PRAD) were downloaded from dbGaP (cohort 1), SRA (cohort 2) and the NCI Genomic Data Commons (TCGA-PRAD) and were trimmed using Trimmomatic [20] version 0.36 using Illumina adaptor sequences with the parameter 3:50:10 MINLEN:36. Gene-level counts were estimated from paired-end reads aligned to version hg19 of the human genome using RSEM [21] version 1.3.2 as a wrapper around STAR [22] version 2.7.0f. Candidate gene fusions were detected using deFuse [23] version 0.8.1.
Paired-end BAM files corresponding to exome capture libraries for the matched samples in cohort 1 and cohort 2 were downloaded and converted to FASTQ files using GATK SamToFastq. FASTQ files were then re-aligned with the Burrows Wheeler Aligner [24] BWA-MEM version 0.7.17 to version hg19 of the human genome (b37 with decoy chromosomes). The SAM alignment files were coordinate-sorted and duplicate-marked using PICARD version 2.18.27 SortSam and MarkDuplicates and then quality score-recalibrated using version 4.1.3.0 of the Genome Analysis Toolkit (GATK) BaseRecalibrator and ApplyBQSR. Mutation calling for ascertaining PTEN status was performed using MuTect2 (part of the GATK4 package), first by running MuTect2 in tumor-only mode on all of the normal tissue BAM files individually, with the parameters disable-read-filter set to MateOnSameContigOrNoMappedMateReadFilter and max-mnp-distance set to 0. The ‘padded’ bait BED file (Agilent Human All Exon V7 for cohort 1 and Agilent Human All Exon V5 + UTR for cohort 2) was used as the interval file of covered regions for all MuTect2 and filtering steps. Each of the output VCF files from this analysis of normal BAMs was merged into a database using GATK GenomicsDBImport with the setting merge-input-intervals set to true and then by generating a panel of normal from the database using GATK CreateSomaticPanelOfNormals. MuTect2 was run in somatic mode on each tumor–normal BAM pair and the panel of normals with af-of-alleles-not-in-resource set to 0.0000025 to exclude sites present in gnomAD and with disable-read-filter set to MateOnSameContigOrNoMappedMateReadFilter. GetPileupSummaries and CalculateContamination were used on each tumor BAM file, and the resultant contamination table was used to filter somatic mutations using FilterMutectCalls. CollectSequencingArtifactMetrics and FilterByOrientationBias were used to further filter mutations for 8-oxoG artifacts using the settings -AM G/T -AM C/T. These pass-filter mutations were then functionally annotated using Oncotator version 1.9.70 (database version April052016).
PTEN copy number changes were called following whole-genome assessment of somatic copy number alterations (SCNAs) across genomic intervals specified by the Agilent library design BED file, with variable resolution depending on bait spacing. The design BED file was preprocessed with GATK PreprocessIntervals, with bin-length set to 0 and interval-merging-rule set to OVERLAPPING_ONLY. The BED file was also annotated with GC content using AnnotateIntervals. These interval files were used in all copy number calling steps. Read counts were first obtained from all tumor and normal BAM files using GATK CollectReadCounts. The normal read count files were compiled into a panel of normals using GATK CreateReadCountPanelOfNormals, excluding cases that had read depth coverage outside the 95% confidence interval (CI) for the dataset. The panel of normals was used to smooth read counts across all samples using GATK DenoiseReadCounts. Normal BAM files were then processed with GATK CollectAllelicCounts to identify regions of potential LOH. GATK CollectAllelicCounts was also applied to each tumor BAM file. GATK ModelSegments used smoothed read counts from each tumor BAM along with the paired normal/tumor allelic counts for generating copy number estimates.
Calls of PTEN somatic copy number changes were performed using GISTIC 2.0.23 on the GenePattern platform (module version 6.15.28). The SEG file from GATK ModelSegments was used as input with the following parameters: focal length cutoff set to 0.50, gene GISTIC set to yes, confidence level set to 0.90, cap value set to infinite, broad analysis set to on (to call arm-level events), max sample segments set to 10 000, arm peel set to no and gene collapse method set to extreme. Post-GISTIC log2-copy number ratio threshold values were used for calling discrete gene-level calls as follows: >1.3 = 2-copy gain; 0.1–1.3 = 1-copy gain; −1.3 to −0.1 = 1-copy loss; <−1.3 = 2-copy loss. Chromosome 18q changes followed the same threshold scheme per tumor focus by using the convention that at least 50% of the chromosome arm (noncontiguously) must be affected.
Genotype/phenotype determination
For all TCGA cases, the reported status of TMPRSS2-ERG, PTEN and 18q were used as deposited on cBioPortal for the PanCancer Atlas analysis (comprising 489 tumors). Fusions involving TMPRSS2 or ERG and other genes (such as SLC45A3-ERG) were excluded. Cases with undocumented 18q status, or gains to PTEN or 18q were also excluded. For cohort 1, PTEN status was determined jointly based on the status of anti-PTEN immunohistochemistry reported for each focus [8] and the somatic copy number or mutation calls. For cohort 2, PTEN status was determined based on somatic copy number or mutation calls. For cohort 1, TMPRSS2-ERG status was determined jointly based on the status of nuclear anti-ERG immunohistochemistry reported for each focus [8] and RNA-seq fusions determined by deFuse. For cohort 2, TMPRSS2-ERG status was determined based on RNA-seq fusions determined by deFuse supplemented with any observed interstitial deletions between ERG and TMPRSS2 on chromosome 21 detected by somatic copy number analysis.
Aggregation of ground-truth phenotypes from foci to lesions and/or patients was as follows: a pre-determined threshold was set for a minimum number of foci to harbor that alteration to call a particular copy number event for the entire lesion. For lesions sampled by 7 or 8 foci, the threshold was 3; for 3, 4, 5 or 6 foci, the threshold was 2 and for 1 or 2 foci, the threshold was 1. For each focus, PTEN or 18q somatic copy number calls from GISTIC (i.e. −1, −2, 0, 1 or 2) were averaged across all foci from each lesion. If the absolute value of that average was greater than the pre-determined threshold, the averaged value was rounded to the nearest integer. For PTEN point mutations or the TMPRSS2-ERG, the lesion (cohort 1) or patient (cohort 2) was considered to harbor the mutation if any number of foci harbored it. Similarly, if any focus harbored PTEN loss or ERG expression by immunohistochemistry, the entire lesion (cohort 1) or patient (cohort 2) was considered to have harbored it. Lesions marked as the index lesion were used as patient-level data for cohort 1.
Sample counts aggregation
The input to the five analysis methods were count data with >3 million reads. Five (cohort 1) or four (cohort 2) different methods of transcriptomic analysis of multiple-sampled tumors were investigated as depicted in Figure 1. In the first method, concatenation, paired-end FASTQ files derived from samples of the same lesion or tumor were concatenated into a single pair of FASTQ files representing the entire lesion or tumor. These FASTQ pairs were then processed as above with RSEM and STAR, arriving at a single counts file per lesion or tumor. The remaining analyses were performed with sample counts obtained after alignment and count estimation as stated above. In the second method, averaging, counts from the same lesion were averaged across genes, creating a representative lesion-level transcriptome profile for each lesion or patient. In method three, RNA weighting, gene counts were normalized based on the weighted average of the specimen RNA input amounts into library preparation as seen in Equation (1).
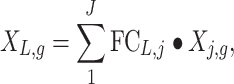 |
(1) |
where XL,g is the count of gene g, FCL,j is defined as the fractional concentration of specimen j obtained from lesion L, which is calculated as Equation (2).
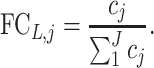 |
(2) |
Figure 1.

Schematic illustration of the work flow for cohort 1. Biopsies were obtained from one or more lesions per patient. For each lesion, one or more foci were laser capture microdissected (LCM) from biopsied materials. RNA sequencing was performed on each focus and the associated ERG and PTEN protein status were determined by immunohistochemistry of adjacent slides, while PTEN and chromosome 18q status were obtained from exome sequencing data. The gene expression data were analyzed by five different methods: counts averaging, RNA weighting, FASTQ concatenation, counts summation and LMM.
Due to missing data, RNA weighting was only performed for cohort 1. In method four, summation, counts of the same lesion/patient were added across genes to create a high coverage profile of the tumor’s transcriptome. Finally, in method five, the LMM was used with fixed and random effects to account for inter-patient and inter-lesion variability.
Purity calculation
Determination of tumor sample purity was performed using ESTIMATE version 1.0.13 for R [25], which uses the single sample gene set enrichment algorithm to infer the transcriptomic signal from tumor gene expression datasets. Transcripts per million (TPM) values from RSEM-STAR alignments were filtered for common genes with the ESTIMATE algorithm and the tumor purity score was outputted as a projection interpolated against a priori ground-truth purity percentages. For cohort 1, mean purity was 96.6% (95% CI: 96.1–97.1%) and was thus not factored into any gene expression model. For cohort 2, mean purity was 87.4% (95% CI: 85.2–89.7%) and was included as a covariate.
Differential gene expression analysis
Differential gene expression analysis for aggregated samples in cohorts 1 and 2 was performed using the DESeq2 package available in R [15]. Gene count matrices were prepared as described above using RSEM/STAR. Genes with zero counts in more than half of the samples were removed. Next, counts were normalized by the median ratio method from DESeq followed by differential expression analysis by using the following 5 phenotypic classifications as fixed effect parameters: (1) TMPRSS2-ERG fusion-positive versus TMPRSS2-ERG fusion-negative, (2) PTEN loss versus PTEN intact, (3) TMPRSS2-ERG fusion-positive and PTEN intact versus TMPRSS2-ERG fusion-negative and PTEN intact, (4) TMPRSS2-ERG fusion-positive and PTEN loss versus TMPRSS2-ERG fusion-negative and PTEN intact and (5) chr18q loss versus chr18q intact. Genes with false discovery rate (Padj) <0.05 and log2 fold-change <−1 or >1 were considered to be differentially expressed.
LMM analysis
Differential gene expression analyses using a LMM were performed using the variancePartition package available in R [17]. First, gene counts were normalized by the trimmed mean method in edgeR. Next, the genomic alterations of interest and sample purity (for cohort 2) were modeled as fixed effects, while patients and their lesions were modeled as nested random effects in the LMM model using the formula: ~Fixed_effect + (1|Patient:Lesion) for cohort 1. For cohort 2, the genomic alterations of interest were modeled as fixed effects while patients were modeled as a random effect variable using the formula: ~Fixed_effect + purity + (1|Patient). For the aggregation methods (averaged, concatenation and summation), the average purity of the specimens were used as an adjustment to the linear model.
Derivation of PTEN loss, TMPRSS2-ERG and 18q-loss gene sets
Gene count matrices from TCGA-PRAD were annotated with PTEN, TMPRSS2-ERG and 18q status, and differential expression analysis based on each genotype was performed using DESeq2. After removing genes with zero counts in at least half of the samples, genes were normalized by the median of ratios method as described in DESeq2. Gene signatures were generated by performing differential expression analysis using DESeq2 by using each genotype as an input parameter and by accepting DEGs with Padj < 0.05 by Wald test.
Statistics
Similarities between groups of samples were determined using unsupervised hierarchical clustering (Euclidean distance) of all genes. Null hypothesis tests of associations between affected and unaffected samples were performed using two-sided Fisher’s exact tests. Generalized linear models using the likelihood ratio test were used to compare gene expression between groups of singly sampled tumors. Unless noted otherwise, alpha of 0.05 was used as a significance threshold for all statistical analyses.
Software
All mathematical analyses were performed using R version 4.0.2 and RStudio 1.3.
Results
Transcriptomic consequences of genomic heterogeneity in prostate tumors
Despite its common genetic background, within-patient tumor heterogeneity manifests itself through variable histologies and genomic alterations, even by geographically proximal clusters of tumor cells. As a framework for assessing salient gene expression as a function of histologic or genomic heterogeneity, we selected three somatic alterations common to PCa: genomic deletion or deleterious alterations to the tumor suppressor PTEN, deletion of at least half of chromosome 18q and the fusion of TMPRSS2 and ERG on chromosome 21. Alterations to PTEN and 18q were determined by analysis of whole-exome sequencing data from matched samples (compared to benign controls), while TMPRSS2-ERG fusion was determined by the expression of nuclear ERG by immunohistochemistry.
We assessed the frequency of these alterations in two distinct cohorts. As depicted in Figure 1, cohort 1 was comprised of 130 LCM foci, sampled from 51 different biopsies targeting 50 distinct imaging-visible lesions from 39 patients, with 1–3 lesions per patient and 1–8 foci per patient [8]. Foci in cohort 1 were microdissected based on distinct histologic features, such as gland architecture, proximity to other tumor cells and immunochemistry against common PCa markers, such as anti-ERG and anti-PTEN. Cohort 2 was comprised of 66 tumor punches from either high-grade or low-grade tumors from prostatectomy samples assessed without imaging, with 1–2 foci per patient [19]. Regions sampled in cohort 2 were determined solely on tumor aggressivity.
We first performed unsupervised clustering on gene expression data from each focus individually, in both cohorts, annotating each sample for PTEN, ERG and 18q status. Of these three factors, clustering revealed that foci clustered most strongly based on ERG fusion status and then by PTEN status, both in cohort 1 (Fisher’s exact test, adjusted P = 4.29 × 10−24; 5.39 × 10−11 and 1, respectively, k = 3; Figure 2A and B) and in cohort 2 (Fisher’s exact test, adjusted P = 2.10−5; 0.041 and 1, respectively, k = 2; Figure 2C and D). As ERG fusion and PTEN loss showed significant per-focus co-occurrence in cohort 1 but not cohort 2 (Fisher’s exact test, P = 6.30 × 10−9 and 0.6, respectively), we assessed the inter- and intra-lesion variations of ERG, PTEN and chr18q alterations of cohort 1 and the intra-patient variation in cohort 2. PTEN loss had the highest inter- and intra-lesion variabilities of the three alterations in cohort 1 (Figure 2A and B) and the highest intra-patient variability in cohort 2 (Figure 2C and D), suggesting that the subclonal nature of major driver alterations, such as PTEN loss, might affect tumor phenotypes within a patient.
Figure 2.

Summary of gene expression profile of cohort 1 and 2. (A) Unsupervised clustering of focus level gene expression data from cohort 1 with TMPRSS2-ERG, PTEN and chr18q loss annotation. The results revealed a strong tendency for focus to group based on TMPRSS2-ERG and PTEN status (Fisher’s exact test, adjusted P = 4.29 × 10−24 and 5.39 × 10−11, respectively) but not chr18q loss (P = 1). Corresponding patient identifier and lesion identifier were mapped below the dendrogram. Black lines corresponding to foci or lesions without TMPRSS2-ERG fusion were drawn connecting to lesions and patients, while red lines correspond to foci or lesions with TMPRSS2-ERG fusion foci. (B) Assessment of TMPRSS2-ERG, PTEN and chr18q inter and intra lesion heterogeneity. PTEN loss remains the most variable feature within and between lesions in cohort 1. (C) Unsupervised clustering of focus level gene expression data from cohort 2 with TMPRSS2-ERG, PTEN and chr18q status annotation. The results revealed a strong tendency for foci to group based on TMPRSS2-ERG and PTEN status (Fisher’s exact test, adjusted P = 2.10−5 and 0.041, respectively) but not chr18q loss (P = 1). (D) Assessment of TMPRSS2-ERG, PTEN and chr18q intra lesion heterogeneity. PTEN loss remains the most variable feature within lesions in cohort 2. Clustering was performed on the basis of total gene expression.
We thus further subdivided each cohort based on the dual status of TMPRSS2-ERG and PTEN to examine whether the ‘double-hit’ frequent co-occurrence of TMPRSS2-ERG fusion and PTEN loss was significantly different than TMPRSS2-ERG fusion on a wild-type (intact) PTEN background. For both cohorts, the TMPRSS2-ERG fusion-positive samples separated distinctly from TMPRSS2-ERG fusion-negative samples by the first two principal components (Supplementary Figure S1 available online at http://bib.oxfordjournals.org/), with PTEN intact versus loss samples showing adjacent grouping with the TMPRSS2-ERG-positive structure. In contrast, TMPRSS2-ERG-negative cases with PTEN loss versus intact are admixed on the first two principal components, suggesting that PTEN loss alone is a weak effect on global transcription. These high-level views of these data further suggest that the effect of PTEN may be challenging to dissect from the dominant signal arising from TMPRSS2-ERG fusion.
Transcriptomic consequences of somatic genomic alterations
Patient response to cancer therapies are often distilled to a binary value: remission or recurrence, response or failure and survival or mortality. Precision medicine workflows to predict such clinical responses most often rely upon a single sample from which various measures are taken. However, assessments of tumor heterogeneity (i.e. multiple region sampling) are exploratory in nature and are for research purposes; given the geometric increase in cost for clinical assays and the challenges associated with interpreting multiple and potentially conflicting results from a single patient, repeated clinical assays of the same sample per patient are not feasible. Thus, the translatability of human oncology research depends upon a definitive outcome wherein the evidence supporting that result takes into account phenotypic variability due to heterogeneity.
We thus considered five different methods for deriving DEGs from our two cohorts when working with tumors that were sampled multiple times: (1) averaging of counts within each case (averaged), (2) weighted averaging of counts based on RNA input amounts into library preparation (RNA weighting), (3) concatenation of FASTQ files prior to a single analysis per case (concatenation), (4) summation of counts (summation) and (5) use of a LMM. DEGs were derived for each cohort and method based on the status of ERG (TMPRSS2-ERG fusion-positive versus -negative), and PTEN (reduced or lost versus intact). Due to the strong potential of interaction between TMPRSS2-ERG fusion and PTEN in driving changes in gene expression, we modeled two additional subsets: (1) samples with only TMPRSS2-ERG fusion-positive and (PTEN being intact) versus TMPRSS2-ERG fusion-negative (and PTEN intact); and (2) combined TMPRSS2-ERG fusion-positive and PTEN loss versus combined TMPRSS2-ERG fusion-negative and PTEN intact. Finally, as a negative control, we modeled a highly subclonal event that was predicted to have minimal effects on gene expression: chromosome 18q (lost versus intact). For aggregation methods 1–4, if a single sample was considered altered, the entire case was considered altered. For the LMM (method 5), TMPRSS2-ERG, PTEN and 18q status were specified as fixed effects, while cases were modeled as random effect. Sample purity, which was highly variable for cohort 2 only, was modeled as an adjustment to the linear model for aggregation methods 1, 3 and 4 and as a fixed effect for the LMM.
Table 1 shows the number of statistically significant DEGs (Padj < 0.05). Generally, the number of DEGs was dependent on the genomic alteration and the method of aggregation. However, we noted significant differences for a subset of the DEGs based, in part, on sample segregation observed in the PCA plot (Supplementary Figure S1 available online at http://bib.oxfordjournals.org/). TMPRSS2-ERG fusion, a categorically early event in the natural history of prostate tumors, yielded among the largest number of DEGs for all three comparisons (all fusion-positive cases, only fusion-positive cases with PTEN loss and only fusion-positive cases with intact PTEN). Notably, for cohort 1, the greatest number of DEGs was observed when comparing ‘double-hit’ TMPRSS2-ERG fusion-positive and PTEN loss versus ‘double-neutral’ TMPRSS2-ERG fusion-negative and PTEN intact cases on account of the majority of individual samples being concordant (positive or negative) for alterations to ERG and PTEN. TMPRSS2-ERG fusion accounting for a majority of transcriptomic variance in cohort 1 (with PTEN loss contributing subtly to the variance) explains the ability of LMM to be sensitive to tracking subclonal ERG and PTEN mutations better than aggregate methods, resulting in the greatest number of DEGs for cohort 1.
Table 1.
The total number of DEGs for each aggregation method per cohort, per genotype
| Method | Cohort 1 | Cohort 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ERG | PTEN | ERG fus /PTEN wt | ERG fus /PTEN loss | 18q | ERG | PTEN | ERG fus /PTEN wt | ERG fus /PTEN loss | 18q | |
| Count averaging | 804 | 551 | 69 | 971 | 6 | 869 | 542 | 618 | 1038 | 2 |
| RNA input weighting | 807 | 580 | 101 | 943 | 4 | N/A | N/A | N/A | N/A | N/A |
| Concatenation | 785 | 557 | 103 | 942 | 6 | 880 | 554 | 625 | 1056 | 2 |
| Count summation | 840 | 464 | 105 | 949 | 10 | 881 | 562 | 634 | 1061 | 2 |
| LMM | 894 | 17 | 510 | 1012 | 0 | 415 | 309 | 634 | 616 | 1 |
ERG, TMPRSS2-ERG fusion-positive versus TMPRSS2-ERG fusion-negative; PTEN, PTEN loss versus PTEN intact; ERGfus/PTENwt, TMPRSS2-ERG fusion-positive and PTEN intact versus TMPRSS2-ERG fusion-negative and PTEN intact; ERGfus/PTENloss, TMPRSS2-ERG fusion-positive and PTEN loss versus TMPRSS2-ERG fusion-negative and PTEN intact; 18q, chr18q loss versus chr18q intact.
In both cohorts, PTEN loss alone contributes very little to global transcriptomic variance by PCA and thus is modeled more accurately by LMM, with the fewest number of genes for both cohorts. In cohort 2, the frequency of co-occurring TMPRSS2-ERG and PTEN mutations is less than that of cohort 1, which accounts for all ERG and double-hit DEGs being fewer than TMPRSS-ERG only (with intact PTEN) dominating the DEG list. Indeed, when visualized using an UpSet plot (Supplementary Figure S2 available online at http://bib.oxfordjournals.org/), we observed the greatest number of shared DEGs within method were between the TMPRSS2-ERG fusion/PTEN loss and the TMPRSS2-ERG fusion genotypes, further suggesting that the greater number of DEGs among aggregation methods versus LMM (and especially for PTEN loss) represents ‘spillover’ from the TMPRSS2-ERG fusion genotype. The complete lists of DEGs for each genotype are given in Supplementary Tables S1–S5 (cohort 1) and Supplementary Tables S6–S10 (cohort 2) available online at http://bib.oxfordjournals.org/.
We next performed set analysis to compare each aggregation or LMM method directly. 467, 7, 24, 616 and 0 DEGs were shared between the five methods for TMPRSS2-ERG fusion, PTEN loss, TMPRSS2-ERG fusion/PTEN intact, TMPRSS2-ERG fusion/PTEN loss and chr18q loss, respectively in cohort 1 (Figure 3A). Similarly, a total of 331, 182, 316, 482 and 0 genes were common for cohort 2 (Figure 3B). Within both cohorts, a large number of genes were common to all methods except LMM, given the common generalized lineal models used to identify those DEGs. Interestingly, however, despite the similarities between concatenation and summation (pre-alignment versus post-alignment addition), there were very few DEGs exclusive to just these two aggregation methods.
Figure 3.

Set analysis of genes related to TMPRSS2-ERG fusion, PTEN loss, TMPRSS2-ERG fusion/PTEN intact, TMPRSS2-ERG fusion/PTEN loss and chr18q loss in cohorts 1 and 2. (A) Venn diagram depicting the shared DEGs between each analysis method in cohort 1. The number of DEGs common between all five methods are 467, 7, 24, 616 and 0 for TMPRSS2-ERG fusion, PTEN loss, TMPRSS2-ERG fusion/PTEN intact, TMPRSS2-ERG fusion/PTEN loss, and chr18q loss, respectively, in cohort 1. (B) Venn diagram depicting the shared DEGs between each analysis method in cohort 2. The number of DEGs common between the four methods are 331, 182, 316, 482 and 0 for TMPRSS2-ERG fusion, PTEN loss, TMPRSS2-ERG fusion/PTEN intact, TMPRSS2-ERG fusion/PTEN loss and chr18q loss, respectively, in cohort 2.
To determine if the number of DEGs produced by different methods was sensitive to an alpha threshold, the number of unique DEGs obtained from all five methods were pooled and measured as a function of increasing alpha for all five genomic features in cohorts 1 (Figure 4, top) and 2 (Figure 4, bottom). When increasing alpha from 0.05 to 0.2, the number of unique DEGs increased only in cohort 1 for the TMPRSS2-ERG fusion and TMPRSS2-ERG fusion/PTEN loss genotypes, in part, due to the strong effect of TMPRSS2-ERG fusion on global transcription and the concordance of TMPRSS2-ERG and PTEN mutations in these samples (Figure 2). The lack of convergence for the PTEN and TMPRSS2-ERG fusion/PTEN intact genotypes in cohort 1 likely represents the sensitivity of LMM for handling PTEN heterogeneity as a blocking factor. Because cohort 2 was derived from tissue punches and not microdissected based on ERG and PTEN status like cohort 1, the lack of convergence represents hidden heterogeneity that is incompletely addressed by any model.
Figure 4.

Sensitivity analysis of DEGs to alpha. For each genomic alteration analysis, the total number of unique DEGs from each method were pooled. The filtering threshold were set to |log2 fold-change |>1 followed by increasing alpha to determine the percentage of significant DEGs. The increase in the alpha from 0.05 to 0.20 did not lead to a convergence >75% of unique DEGs across genotype in cohort 1. Sensitivity curves were grouped the closest in the three TMPRSS2-ERG fusion genotypes. Averaged, concatenation and summation are consistently grouped together with increasing alpha from 0.05 to 0.20 in cohort 2. LMM approached convergence with the other methods in cohort 1 only in two genotypes (TMPRSS2-ERG fusion and TMPRSS2-ERG fusion/PTEN loss).
The clinical practice of determining an entire cancer being called PTEN-negative due to at least 5% of tumor cells showing PTEN reduction by IHC would be expected to have an effect on heterogeneity when multiple samples from the same tumor are captured without microdissection. Thus, when we assessed the similarities between the five different methods using unsupervised clustering of the differential expression output, we noted that LMM clustered away from other methods for all genotypes, in both cohorts (Figure 5A and B). Based on DEGs, we anticipated that PTEN loss would show the greatest difference by unsupervised clustering. However, the effect of PTEN loss was similar to 18q and all three TMPRSS2-ERG fusion genotypes in which accounting for multiple samples from the same individual as random effects has a stronger influence than any other method in which variance from repeated measures is ignored.
Figure 5.

The relatedness of the five methods were assessed by hierarchical clustering of log2 fold-change from the five methods in cohort 1 (A) and four methods in cohort 2 (B). The type of genomic comparator is indicated above each tree.
Because TMPRSS2-ERG fusion and losses in PTEN were commonly co-occurring in cohort 1, we specifically assessed which DEGs were spilling over from TMPRSS2-ERG into PTEN analysis in all five methods. The result revealed a total of 269, 283, 257, 282 and 5 genes that are common between TMPRSS2-ERG fusion and PTEN DEG analysis from counts averaging, RNA weighted averaging, concatenation of FASTQ files, counts summation and LMM methods, respectively. To better understand the impact of within-patient variability and remaining variance after accounting for each genomic alteration on gene expression, we performed variance decomposition using results from LMM for both cohorts (Figure 6). Indeed, between-patient variance was a significant source of transcriptomic heterogeneity, explained by a median of 31–38% percent of variance in TMPRSS2-ERG fusion, PTEN loss and 18q loss gene expression in cohort 1 and 55–59% for cohort 2, respectively. In contrast, genomic alterations explained a median of 0% variance in both cohorts when accounted for as a fixed effect.
Figure 6.

Decomposition of gene expression variation into patient, genotypic feature and residuals from cohort 1 (left) and cohort 2 (right). Patient, genomic features and residuals explain a median of 35%, 0% and 62% for TMPRSS2-ERG fusion; 37%, 0% and 62% for PTEN loss; 31%, 0% and 64% for TMPRSS2-ERG fusion/PTEN intact; 33%, 0% and 62% for TMPRSS2-ERG fusion/PTEN loss; and 38%, 0% and 62% for 18q loss, respectively. For cohort 2, patient, genomic features, purity and residuals can explain a median of 35%, 7%, 0% and 48% for TMPRSS2-ERG, 37%, 7%, 0% and 48% for PTEN loss; 30%, 9%, 0% and 49% for TMPRSS2-ERG fusion/PTEN intact; 36%, 7%, 0% and 44% for TMPRSS2-ERG fusion/PTEN loss; and 38%, 7%, 0% and 47% for 18q loss.
Comparison of DEGs to a standardized gene set
As blocking for genomic alterations reduced variance in the LMM, we still have observed that multiple methods for deriving DEGs for each genotype generally produced overlapping but distinct results. Thus, a key question is whether one approach arrives at a more accurate representation of gene expression changes driven by TMPRSS2-ERG, PTEN or 18q mutations. To benchmark these approaches, we next derived ‘ground-truth’ gene sets by performing an integrated analysis of the prostate adenocarcinoma cancer genome atlas (TCGA-PRAD) cohort to derive a DEG list of the same five genotypes described above: (1) TMPRSS2-ERG fusion-positive versus TMPRSS2-ERG fusion-negative, (2) PTEN loss versus PTEN intact, (3) TMPRSS2-ERG fusion-positive and PTEN intact versus TMPRSS2-ERG fusion-negative and PTEN intact, (4) TMPRSS2-ERG fusion-positive and PTEN loss versus TMPRSS2-ERG fusion-negative and PTEN intact and (5) chr18q loss versus chr18q intact. For each of these gene sets, up to 150 significantly up-regulated genes in the perturbed phenotype (e.g. TMPRSS2-ERG fusion-positive) were used to generate the gene set. Overall, TMPRSS2-ERG fusion calls were made in 478 cases (172 cases were positive for the fusion), PTEN status was determined in 427 cases (103 cases harbored PTEN loss-of-function mutations or copy number losses) and 18q status was called in 416 cases (with 61 cases harboring 18q losses). After determining the DEGs for each genotype, we selected the top 150 up-regulated genes for each gene set, as the average number of genes across the mSigDB Hallmark gene sets is also 150. The genes comprising these signatures are provided in Supplementary Table S11 available online at http://bib.oxfordjournals.org/.
Finally, we performed gene set enrichment analysis (GSEA) to assess the sensitivity of each aggregation approach for cohorts 1 and 2 based on known alteration status to each corresponding TCGA PRAD-derived gene set. Employing normalized enrichment score (NES) as a measure of sensitivity, the five methods produced a range of NES that was highly dependent on genomic alteration. As depicted in Figure 7, GSEA of TMPRSS2-ERG fusion/PTEN intact produced the highest and statistically significant NES for both cohorts, ranging from 3.1 to 3.4 (cohort 1) and 3.3 to 3.5 (cohort 2). Consistent with its ability to block for heterogeneous variables, LMM resulted in the highest statistically significant NES for three out of four alterations in cohort 1 and for two out of four alterations in cohort 2. Despite PTEN loss showing the fewest number of DEGs for both cohorts (Table 1), the NES for PTEN loss was greatest for LMM in both cohorts. As a negative control, 18q did not approach the biologically significant threshold of NES ± 2 in either cohort.
Figure 7.

Bubble plot illustrating the NES, colored from low (blue) to high (red) and −log10 adjusted P-value (size of the dot) derived from GSEA of TMPRSS2-ERG fusion, PTEN loss, TMPRSS2-ERG fusion/PTEN intact, TMPRSS2-ERG fusion/PTEN loss, and chr18q loss using differential expression analysis outputted from counts averaging, RNA weighting, concatenation, summation and LMM.
Discussion
With the increasing recognition of tumor heterogeneity as a significant prognostic factor for patient outcome [3, 8, 26], experiments that account for tumor heterogeneity show promise to improve the statistical power of differential gene expression analysis. As a practical matter, the more frequently a tumor is measured, within-tumor variance for particular genes can indicate relative importance toward salient phenotypes and genotypes. Any intra- and inter-tumoral transcriptomic variance could be assessed using statistical models, such as a LMM, which incorporate random and fixed effects for repeated measures and heterogeneity, respectively. However, the benefits to experimental designs such as these have yet to be fully explored, especially in the context of other aggregation methods that collapse multi-region samples into a single sample. Therefore, we investigated herein the impact of four methods for aggregation of RNA-seq counts and compared them to linear mixed modeling for detecting DEGs related to the important PCa genotypes of TMPRSS2-ERG fusion, PTEN loss and chr18q loss.
These three target genotypes range in frequency and relevance in PCa, with TMPRSS2-ERG fusion generally being a very early event in PCa tumorigenesis, followed by PTEN loss and 18q loss [18, 27]. This is reflected in part by the clonal or subclonal nature of these events, the frequent co-occurrence of TMPRSS2-ERG and PTEN loss in the same tumor and the potential ability to isolate pure populations of tumor cells by using laser capture microdissection based on stains detecting these events. Due to microdissection in cohort 1, the specificity of transcriptome underpinning the phenotypic differences between TMPRSS2-ERG fusion-positive and -negative tumors between cases was so great that all aggregation methods performed comparably (Figure 5A). In contrast, the loss of chromosome 18q, which is likely a passenger event, did not produce robust DEGs and GSEA NES from any methods. For PTEN loss in both cohorts, the DEGs derived by a LMM were substantially different than all other methods (Figure 5B). The simplest explanation for this observation is that a per-patient classification of a genotype such as ‘PTEN loss’ demonstrates more heterogeneity across the tumor despite apparent ‘spill over’ from TMPRSS2-ERG fusion, as PTEN-intact and PTEN-reduced tumor cells harbored strikingly different inter-lesion and intra-lesion gene expression profiles. Additional evidence from variance decomposition supports the notion that majority of gene expression variation is not due to genomic changes but is attributable to differences between patients or other unaccounted variables.
Indeed, when we used GSEA NES as a metric for detection sensitivity (Figure 7), DEGs associated with PTEN loss benefitted most from an LMM analysis that was capable of modeling the intra-patient or intra-lesion variability as a blocking effect. In addition, the co-occurrence of relatively homogeneous TMPRSS2-ERG fusion and PTEN loss within each focus from cohort 1 created additional challenges during the PTEN loss analysis for the four aggregation methods but were significantly reduced by LMM. Thus, inclusion of the foci variability using models such as LMM significantly increased the sensitivity and specificity of an analysis to arrive at per-patient phenotypes. While we included the addition of PTEN status as a parameter for TMPRSS2-ERG status to mitigate the strong effects of TMPRSS2-ERG fusion on transcription, doing so in standard practice would require its status known a priori, and patient-level transcriptomic studies often lack genomic and histologic annotations, and thus the inclusion of such parameters would not be always practical.
Compared to cohort 1, which was comprised of LCM tumor foci, cohort 2 was intrinsically more heterogeneous within-foci. The approach used for sampling prostate tumors used in cohort 2 more closely mirrors that used in clinical settings, whereby a large region expected to represent the most aggressive region is generally selected for molecular profiling [28]. Thus, when more than one area was selected in this manner, the variance in DEG profiling the entire patient was distinctive with an LMM, whereas averaging/summing counts or FASTQ concatenation arrived at similar profiles. However, when we considered NES as a basis for this comparison, generalized to all three target genotypes, no single method outperformed the other. Nonetheless, the advantage to LMM is the use of blocking factors for fixed effects and the ability to assess within-lesion or within-patient heterogeneity against one or more salient phenotypes.
Conclusions
The aggregation of multiple transcriptomes from a single tumor to estimate per-patient phenotypes will vary in its reliability as a direct function of heterogeneity. Tumors often harbor heterogeneous alterations within the same geographic region, resulting in vastly different gene expression profiles between nearly adjacent tumor cells. Experimental designs that include tumor resampling have improved the power to detect driver genotypes and the variance of gene expression or biomarker profiles related to phenotype. Although we found that a LMM was a robust aggregation method that performed consistently well in analyses with homogeneous and heterogeneous phenotypes within individual tumors, patient-level summarization methods, including RNA-seq count averaging and summation, showed benefit for detecting gene expression changes that were associated with more homogeneous genotypes. Consequently, the method of per-patient gene expression summarization is highly context-dependent on any salient target genotypes and the tolerance of variability for formulating robust conclusions.
Key Points
Within- and between-lesion tumor heterogeneities can exist within the same patient. Strategies to sampling the same tumor multiple times can improve detection of intratumoral phenotypic variability.
The performance of different methods to aggregate multiple transcriptomes into per-patient summaries was highly dependent upon the phenotype of interest.
Dominant or clonal transcriptomic signals in PCa, such as the TMPRSS2-ERG fusion, were robust to multiple aggregation methods, although subclonal alterations, such as PTEN loss, were more conducive to LMMs.
Comparison of different aggregation methods to benchmarked gene expression profiles can identify unappreciated sources of within-patient transcriptomic heterogeneity based on defined genomic alterations.
Supplementary Material
Acknowledgements
Portions of this work utilized the computational resources of the NIH HPC Biowulf cluster.
Anson T. Ku is a postdoctoral fellow at the Laboratory of Genitourinary Cancer Pathogenesis (LGCP) at the National Cancer Institute (NCI). His research interests focus on the contribution of tumor heterogeneity to cancer phenotype and patient outcome.
Scott Wilkinson is a postdoctoral fellow at the LGCP at the NCI. His research interests include histologic assessments of tumor heterogeneity and the development of multiplex diagnostic assays for prostate cancer.
Adam G. Sowalsky is an investigator at the LGCP at the NCI. His work focuses on prostate cancer evolution in response to hormone-directed therapy.
Contributor Information
Anson T Ku, Laboratory of Genitourinary Cancer Pathogenesis (LGCP) at the National Cancer Institute (NCI), NIH, 37 Convent Drive, Building 37, Room 1062B, Bethesda, MD 20892, USA.
Scott Wilkinson, Laboratory of Genitourinary Cancer Pathogenesis (LGCP) at the National Cancer Institute (NCI), NIH, 37 Convent Drive, Building 37, Room 1062B, Bethesda, MD 20892, USA.
Adam G Sowalsky, Laboratory of Genitourinary Cancer Pathogenesis (LGCP) at the National Cancer Institute (NCI), NIH, 37 Convent Drive, Building 37, Room 1062B, Bethesda, MD 20892, USA.
Data availability
The data underlying this article are available in: dbGaP at https://www.ncbi.nlm.nih.gov/gap/ and can be accessed with accession ID phs001938.v2.p1; SRA at https://www.ncbi.nlm.nih.gov/sra and can be accessed with accession ID PRJNA579899; and NCI GDC at http://portal.gdc.cancer.com and can be accessed with the project ID TCGA-PRAD.
Code availability
All codes used in the preparation of this work are available at https://github.com/CBIIT/lgcp.
Data availability
The data underlying this article are available in the Database of Genotypes and Phenotypes (dbGaP) and Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/gap/ and https://www.ncbi.nlm.nih.gov/sra, respectively, and can be accessed with PRJNA579899 (SRA) and phs001938.v2.p1 (dbGaP).
Funding
Prostate Cancer Foundation (Young Investigator Awards to S.W. and A.G.S.); Department of Defense Prostate Cancer Research Program (W81XWH-19-1-0712 to S.W. and W81XWH-16-1-0433 to A.G.S.); Intramural Research Program of the NIH, National Cancer Institute.
References
- 1. Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 Tumors from 33 types of cancer. Cell 2018;173(2):291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium . Pan-cancer analysis of whole genomes. Nature 2020;578(7793):82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. PCAWG Evolution & Heterogeneity Working Group, PCAWG Evolution & Heterogeneity Working Group, PCAWG Consortium, et al. The evolutionary history of 2,658 cancers. Nature 2020;578(7793):122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018;15(2):81–94. [DOI] [PubMed] [Google Scholar]
- 5. Salami SS, Hovelson DH, Kaplan JB, et al. Transcriptomic heterogeneity in multifocal prostate cancer. JCI Insight 2018;3(21): e123468 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sowalsky AG, Kissick HT, Gerrin SJ, et al. Gleason score 7 prostate cancers emerge through branched evolution of clonal Gleason pattern 3 and 4. Clin Cancer Res 2017;23(14):3823–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilkinson S, Harmon SA, Terrigino NT, et al. A case report of multiple primary prostate tumors with differential drug sensitivity. Nat Commun 2020;11(1):837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilkinson S, Ye H, Karzai F, et al. Nascent prostate cancer heterogeneity drives evolution and resistance to intense hormonal therapy. Eur Urol 2021. 10.1016/j.eururo.2021.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Purysko AS, Magi-Galluzzi C, Mian OY, et al. Correlation between MRI phenotypes and a genomic classifier of prostate cancer: preliminary findings. Eur Radiol 2019;29(9): 4861–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Black JRM, McGranahan N. Genetic and non-genetic clonal diversity in cancer evolution. Nat Rev Cancer 2021; 21(6):379–92. [DOI] [PubMed] [Google Scholar]
- 11. Liu J, Dang H, Wang XW. The significance of intertumor and intratumor heterogeneity in liver cancer. Exp Mol Med 2018;50(1):e416–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grzywa TM, Paskal W, Włodarski PK. Intratumor and intertumor heterogeneity in melanoma. Transl Oncol 2017;10(6):956–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43(7):e47–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bates D, Machler M, Bolker B, et al. Fitting linear mixed-effects models using lme4. Journal of Statistical Software 2015; 67(1):48. [Google Scholar]
- 17. Hoffman GE, Roussos P. Dream: powerful differential expression analysis for repeated measures designs. Bioinformatics 2020;37(2):192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sowalsky AG, Ye H, Bubley GJ, et al. Clonal progression of prostate cancers from Gleason grade 3 to grade 4. Cancer Res 2013;73(3):1050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Charmpi K, Guo T, Zhong Q, et al. Convergent network effects along the axis of gene expression during prostate cancer progression. Genome Biol 2020;21(1):302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li B, Ruotti V, Stewart RM, et al. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010;26(4):493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-Seq aligner. Bioinformatics 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McPherson A, Hormozdiari F, Zayed A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol 2011;7(5):e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4(1):2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Espiritu SMG, Liu LY, Rubanova Y, et al. The evolutionary landscape of localized prostate cancers drives clinical aggression. Cell 2018;173(4):1003–1013.e15. [DOI] [PubMed] [Google Scholar]
- 27. Cancer Genome Atlas Research Network . The molecular taxonomy of primary prostate cancer. Cell 2015;163(4):1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wheeler DA, Takebe N, Hinoue T, et al. Molecular features of cancers exhibiting exceptional responses to treatment. Cancer Cell 2021;39(1):38–53.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in: dbGaP at https://www.ncbi.nlm.nih.gov/gap/ and can be accessed with accession ID phs001938.v2.p1; SRA at https://www.ncbi.nlm.nih.gov/sra and can be accessed with accession ID PRJNA579899; and NCI GDC at http://portal.gdc.cancer.com and can be accessed with the project ID TCGA-PRAD.
The data underlying this article are available in the Database of Genotypes and Phenotypes (dbGaP) and Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/gap/ and https://www.ncbi.nlm.nih.gov/sra, respectively, and can be accessed with PRJNA579899 (SRA) and phs001938.v2.p1 (dbGaP).