

Abstract
Rationale:
Noonan syndrome (NS) is one of the most frequent genetic disorders. Bleeding problems are among the most common, yet poorly defined complications associated with NS. A lack of consensus on the management of bleeding complications in NS patients indicates an urgent need for new therapeutic approaches.
Objective:
Bleeding disorders have recently been described in NS patients harboring mutations of LZTR1, an adaptor for CULLIN3 ubiquitin ligase complex. Here, we assessed the pathobiology of LZTR1-mediated bleeding disorders.
Methods and Results:
Whole-body and vascular specific knockout of Lztr1 results in perinatal lethality due to cardiovascular dysfunction. Lztr1 deletion in blood vessels of adult mice leads to abnormal vascular leakage. We found that defective adherent and tight junctions in Lztr1-depleted endothelial cells are caused by dysregulation of vesicular trafficking. LZTR1 affects the dynamics of fusion and fission of recycling endosomes by controlling ubiquitination of the ESCRT-III component CHMP1B, whereas NS-associated LZTR1 mutations diminish CHMP1B ubiquitination. LZTR1-mediated dysregulation of CHMP1B ubiquitination triggers endosomal accumulation and subsequent activation of VEGFR2 and decreases blood levels of soluble VEGFR2 in Lztr1 haploinsufficient mice. Inhibition of VEGFR2 activity by cediranib rescues vascular abnormalities observed in Lztr1 knockout mice
Conclusions:
Lztr1 deletion phenotypically overlaps with bleeding diathesis observed in NS patients. ELISA screening of soluble VEGFR2 in the blood of LZTR1-mutated NS patients may predict both the severity of NS phenotypes and potential responders to anti-VEGF therapy. VEGFR inhibitors could be beneficial for the treatment of bleeding disorders in NS patients
Keywords: Angiogenesis, Animal Models of Human Disease, Genetically Altered and Transgenic Models, Vascular Disease, Noonan Syndrome, Ubiquitination, Cardiovascular disease, Vesicular trafficking, LZTR1
Introduction
The incidence of Noonan Syndrome (NS) is reported to be between 1 in 1000 and 1 in 2500 births1. NS is characterized by short stature, facial dysmorphia, and congenital heart defects2. NS is also the first cause of nuchal translucency after Down Syndrome. Infants suffering from NS present varying degrees of symptoms, ranging from mild to life-threatening, including polyhydramnios, pleural effusions and edema3,4.
Several studies have linked NS to lung lymphangiectasis, which is characterized by vessel dilatation, bronchiole collapse and hemorrhages5. It is usually associated with lung hypertension and can trigger pulmonary heart disease by affecting right ventricular function6. Between 50-89% of NS patients present bleeding disorders such as bruising and hemangioma7, whereas severe hemorrhages are observed in about 3% of cases8. Most of the studies describing the etiology of bleeding disorders in NS patients are incomplete. Coagulation defects, especially deficiencies of factors VIII, XI, XII and various combinations of these, have been documented in about 30% of NS patients. However, no consistent pattern of coagulation defects in NS has been established and many reports describe a poor correlation between bleeding history and clotting factor deficiencies7, indicating that the bleeding defects in NS patients are caused by other alterations.
An understanding of bleeding disorders in NS is clinically important because a large number of patients require surgery or take medication presenting bleeding as adverse effects, such as aspirin9. Currently, there is no consensus on the management of bleeding complications in patients with NS. Furthermore, conventional treatment including platelets and fresh frozen plasma do not appear to be efficient in NS10, demonstrating an urgent need for identification of new therapies for NS patients.
NS is an autosomal dominant disease11. In about half of the cases, the disease is caused by missense mutations in the PTPN11 gene, resulting in a gain-of-function of the tyrosine phosphatase SHP-212, whereas mutations in SOS1, SOS2, RAF1, KRAS, NRAS, BRAF, SHOC2, CBL, RIT1 are found in NS patients at low rates. LZTR1 has also been recently added to the list of genes causing NS and is present in up to 8% of NS patients13,14. NS patients harboring LZTR1 mutations presented the typical NS facial features, webbed neck, cardiovascular defects, and bleeding15.
LZTR1 serves as a substrate adaptor for CULLIN3 (CUL3) ubiquitin ligase complexes. In addition to NS, mutations in LZTR1 have also been associated with glioblastoma16, hepatocarcinoma17, familial Schwannomatosis18, and pediatric cancers19. In our recent study, we demonstrated that knockout of Lztr1 in mice leads to prenatal lethality, whereas Lztr1 haploinsufficiency partially recapitulates NS phenotypes including facial dimorphism and heart malformations20. As bleeding disorders are observed in NS patients with different genotypes, we took advantage of conventional and vascular specific Lztr1 knockout mouse models to characterize the vascular phenotypes triggered by Lztr1 loss. The results of our study provide a fundamental explanation underlying the molecular mechanisms of endothelial dysfunction and etiology of the bleeding disorders in Lztr1-mutated NS as well as identify anti-VEGF therapies as a potential treatment approach for NS patients.
Methods
Detailed experimental procedures, mouse models, cell culture and bioinformatics analysis are described in the Supplemental Materials in the online Supplement file.
Results
Lztr1 loss leads to abnormal cardiovascular development
To elucidate the etiology of bleeding disorders in LZTR1-mutated NS patients, we analyzed the whole-body Lztr1 and the PdgfbiCre ERT2, Lztr1fl/fl knockout mice. The PdgfbiCre ERT2 model allows to target genes in the endothelium21,22 and is widely used to study vascular development during embryogenesis or disease progression23,24. Vascular specific Lztr1 knockout was induced by treating PdgfbiCre ERT2, Lztr1fl/fl pregnant females with tamoxifen. Survival analysis demonstrated that both the whole-body and vascular specific knockouts of Lztr1 led to embryonic lethality between E14.5 and E18.5 (Online Figure Ia, b). At E14.5, Lztr1−/− embryos showed growth defects, hemorrhages, or significant hydrops, whereas Lztr1+/− embryos did not show any obvious phenotypes (Figure 1a, b). Similarly, vascular specific Lztr1 loss led to growth retardation, superficial hemorrhages, and edema (Figure 1c, d). In line with these findings, yolk sac vasculature of Lztr1−/− embryos at that stage was dysfunctional as indicated by the presence of enlarged vascular structures (Figure 1e). Abnormal vascular leakage was also present in the cephalic plexus and skin vasculature (Figure 1b, d, f).
Figure 1. Loss of Lztr1 leads to vascular defects during embryogenesis.
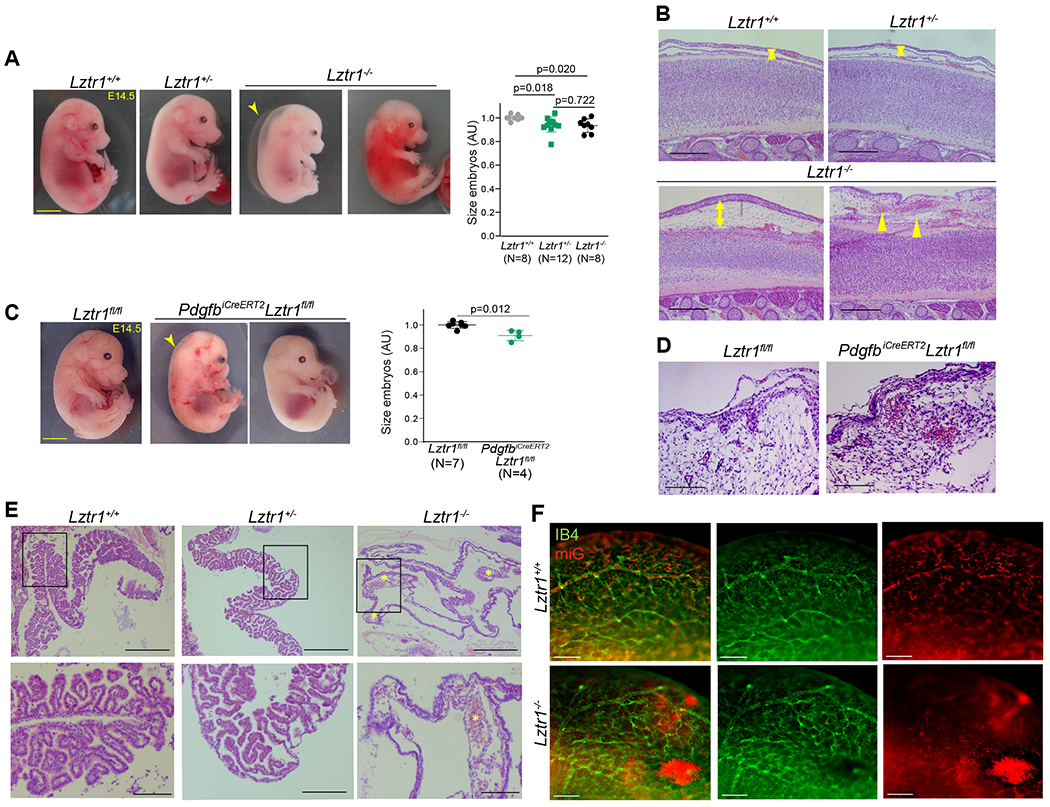
(a) Representative images of Lztr1+/+, Lztr1+/−, and Lztr1−/− embryos at E14.5. The arrowhead indicates hydrops. Scale bar 1 mm. Size of E14.5 embryos is shown as mean ± SEM; p-values were assessed by Kruskal-Wallis test with Dunn’s Multiple Comparison Test. (b) H&E-stained sections of the back skin of E14.5 Lztr1+/+, Lztr1+/−, and Lztr1−/− embryos. Arrowheads indicate hemorrhages; arrows denote dermis thickness. Scale bar 200 μm. (c) Representative images of Lztr1fl/fl and PdgfbiCreERT2, Lztr1fl/fl embryos at E14.5. The arrowhead indicates nuchal translucency. Scale bar 1 mm. Size of E14.5 embryos is shown as mean ± SEM; p-values were assessed by Wilcoxon-Mann-Whitney test. (d) H&E-stained sections of the head of Lztr1fl/fl and PdgfbiCreERT2, Lztr1fl/fl embryos at E14.5. Scale bar 100 μm (e) H&E-stained sections of Lztr1+/+, Lztr1+/− and Lztr1−/− yolk sacs. * indicates enlarged vascular structure. Scale bar 200 and 500 μm, respectively. (f) 3D vasculature of brain (cephalic plexus) imaged after whole mount-staining of E14.5 embryos with Isolectin B4 (IB4), mouse iG (miG), and clearing. Scale bar 200 μm.
Lztr1 loss also caused pulmonary dysplasia. Histological analysis showed that Lztr1−/− lungs presented narrower airspaces and discontinuous lung vessels (Online Figure Ic, d). SMA and CD31 immunostaining revealed disruption of the lung structure and vasculature in Lztr1−/− embryos at E14.5 (Online Figure Ie). Vascular dysfunction might be partially responsible for the dysmorphic lung phenotype as it was also observed in PdgfbiCre ERT2, Lztr1fl/fl embryos (Online Figure If, g).
Furthermore, we observed defective cardiac chamber maturation in E14.5 Lztr1−/− embryos as indicated by a moderate decrease in ventricular wall thickness (Online Figure IIa), suggesting that cardiac failure could also be a possible cause of the Lztr1−/− embryonic lethality. The defective cardiac maturation could also explain the cardiomyopathy phenotype previously described in adult Lztr1+/− mice20. Ventricular trabeculation and myocardial defects have been associated with endothelial dysfunction in other NS mice models25. Concordantly, the heart of PdgfbiCreERT2, Lztr1fl/fl embryos displayed a striking non-compaction phenotype, as indicated by the hyper-trabeculation and decreased ventricular wall thickness (Online Figure IIb). These data indicate that Lztr1 is essential for normal vascular function and subsequent coordination of lung alveolar and heart development.
Lztr1 loss in adult mice leads to cardiovascular dysfunction
To further examine the vascular phenotypes caused by Lztr1 loss, we focused on the analysis of the adult Lztr1+/− mice. We observed higher vessel density in the cardiac ventricles and relatively higher levels of phosphorylated Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) in cardiac ventricle vessels of adult Lztr1+/− mice (Online Figure IIc, d). Adult PdgfbiCreERT2, Lztr1fl/fl mice injected with tamoxifen at weaning presented severe fibrosis in the right ventricle (Online Figure IIe), demonstrating the endothelial contribution to the severe cardiac dysfunction previously described in Lztr1+/− mice20.
Histological analyses also demonstrated that Lztr1+/− animals presented multiple lung defects including vessel dilatation, pulmonary hemorrhages, and lung fibrosis (Figure 2a–d). Collectively, these results indicate that Lztr1 inactivation partially recapitulates pulmonary and cardiovascular phenotypes commonly observed in NS patients presenting LZTR1 mutations5,26, indicating that the Lztr1 knockout mice represent a clinically relevant model.
Figure 2. Loss of Lztr1 leads to pulmonary vascular disease.
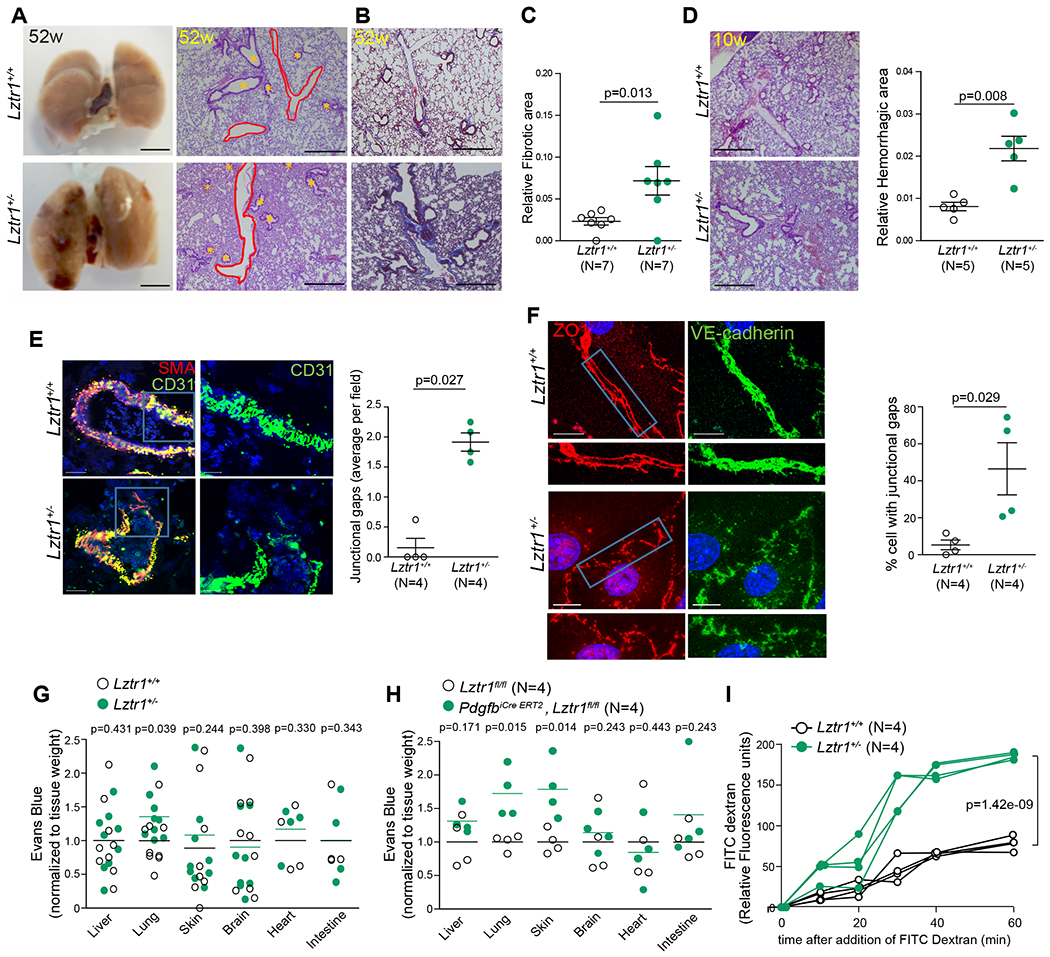
(a) Gross anatomy and H&E-staining of lungs of 52-week old Lztr1+/+ and Lztr1+/− mice. Scale bar 500 μm. * indicate bronchioles; pulmonary vessels are delineated in red. (b) Masson Trichrome–stained sections of Lztr1+/+ and Lztr1+/− lungs. Fibrosis is colored in blue. Scale bar 500 μm. (c) Fibrotic area in lungs of 52-week old Lztr1+/+ and Lztr1+/− mice. Data are shown as mean ± SEM; p-values were assessed by Wilcoxon-Mann-Whitney test. (d) H&E-staining of lungs of 10-week old Lztr1+/+ and Lztr1+/− mice. Scale bar 500 μm. Hemorrhagic area is shown as mean ± SEM; p-values were assessed by Wilcoxon-Mann-Whitney test. (e) Lungs of 52-week old Lztr1+/+ and Lztr1+/− mice immunostained with anti-SMA and anti-CD31 antibodies. Data are shown as mean±SEM; p-values were assessed by Wilcoxon-Mann-Whitney test. Scale bar 20 μm. (f) ECs isolated from Lztr1+/+ and Lztr1+/− mice immunostained with antibodies against VE-cadherin and ZO1. Blue rectangles delineate the magnified areas (below). Data are shown as mean ± SEM; p-values were assessed by Wilcoxon-Mann-Whitney test. Scale bar 20 μm. (g) Evans blue extravasation from lungs of Lztr1+/+ and Lztr1+/− mice. Lines shows mean; p-values were assessed by Wilcoxon-Mann-Whitney test. (h) Evans blue extravasation from Lztr1fl/fl and PdgfbiCre ERT2, Lztr1fl/fl tissues. Lines shows mean; p-values were assessed by Wilcoxon-Mann-Whitney test. (i) Paracellular permeability of Lztr1+/+ and Lztr1+/− ECs as measured by quantifying FITC Dextran fluorescence in a trans-well assay. P-values were assessed by two-way ANOVA with Bonferroni post-tests.
CD31 immunostaining showed that lung vessels in Lztr1+/− mice were thinner and discontinuous (Figure 2e). Immunostaining of isolated endothelial cells (ECs) demonstrated that Lztr1 haploinsufficiency led to abnormal adherent and tight junctions (Figure 2f; Online Figure IIf). The Evans blue extravasation assay revealed increased vascular permeability in the lungs of Lztr1+/− mice (Figure 2g). We observed an even more striking increase in vascular permeability in the lungs and skin of PdgfbiCre ERT2, Lztr1fl/fl mice injected with tamoxifen at weaning (Figure 2h). In line with these findings, Lztr1+/− sorted ECs were more permeable to 40 kDa Dextran when compared to Lztr1+/+ ECs (Figure 2i).
The observed phenotypes suggest a potential role of LZTR1 in the control of vascular function. Concordantly, aortic ring assay27 demonstrated that Lztr1+/− ECs were more neo-angiogenic as indicated by an increase in the sprouting area (Online Figure IIIa–c). To further assess the angiogenic potential of Lztr1+/− ECs in vivo, we implanted Lewis lung carcinoma (LLC) cells28 in the flanks of Lztr1+/+ and Lztr1+/− mice. LLC tumors appeared earlier in Lztr1+/− mice and displayed a higher weight at sacrifice (Online Figure IIId, e). Histological analyses showed that LLC tumors in Lztr1+/− mice were more vascularized and presented increased vascular leakage (Online Figure IIIf–i). Aortic ring assay using aortic rings isolated from PdgfbiCre ERT2, Lztr1fl/fl mice injected with tamoxifen at weaning also demonstrated that vascular specific loss of Lztr1 was sufficient to increase neo-angiogenesis (Online Figure IIIj). These data indicate that Lztr1 deletion disrupts the balance between vascular integrity and angiogenesis, leading to a more pro-angiogenic profile associated with less stable junctions.
LZTR1 controls endothelial function by regulating vesicular trafficking
We next explored a potential mechanism by which Lztr1 loss leads to a pro-angiogenic phenotype. In our recent study, we found that LZTR1 controls the activity of the RAS pathway20. Indeed, we observed increased phosphorylation of ERK1/2 and AKT1 when we deleted Lztr1 in Lztr1fl/fl ECs derived from different mice by overexpressing CreERT2 recombinase (Online Figure IVa, b). However, the MEK1/2 inhibitor pimasertib, the AKT inhibitor ipatasertib, or the combination of both inhibitors only partially rescued the embryonic lethality phenotype, as upon either of the treatments we recovered Lztr1−/− embryos at E18.5, but no pups at birth (Online Figure IVc). Treatment with pimasertib or ipatasertib did not rescue lung dysplasia in E18.5 Lztr1−/− embryos. E18.5 Lztr1−/− embryos treated with the combination of pimasertib and ipatasertib showed more moderate lung dysplasia, but still displayed massive lung hemorrhages, indicating that concurrent inhibition of MEK1/2 and AKT1 in Lztr1−/− embryos could not restore normal lung vascular function (Online Figure IVd, e). Moreover, pimasertib, ipatasertib, or the combination of both inhibitors did not rescue the formation of abnormal adherent and tight junctions in Lztr1fl/fl-CreERT2 ECs (Online Figure IVf). This suggests that Lztr1 loss may contribute to human disease by additional mechanisms rather than the sole activation of the RAS pathway.
Using mass-spectrometry based approaches, we explored how LZTR1 loss alters the proteome and ubiquitome landscapes. The Ingenuity Pathway Analysis (IPA) of either differentially expressed, or differentially ubiquitinated proteins in HeLa cells harboring wt-LZTR1 or LZTR1-indels demonstrated that endocytosis was one of the top canonical pathways altered by LZTR1 loss (Figure 3a, b). Several putative LZTR1 interactors identified by the Virotrap approach20 were also associated with vesicular trafficking (Figure 3c). Collectively, these data suggest a potential role of LZTR1 in the regulation of vesicular trafficking.
Figure 3. Lztr1 loss affects vesicular trafficking.
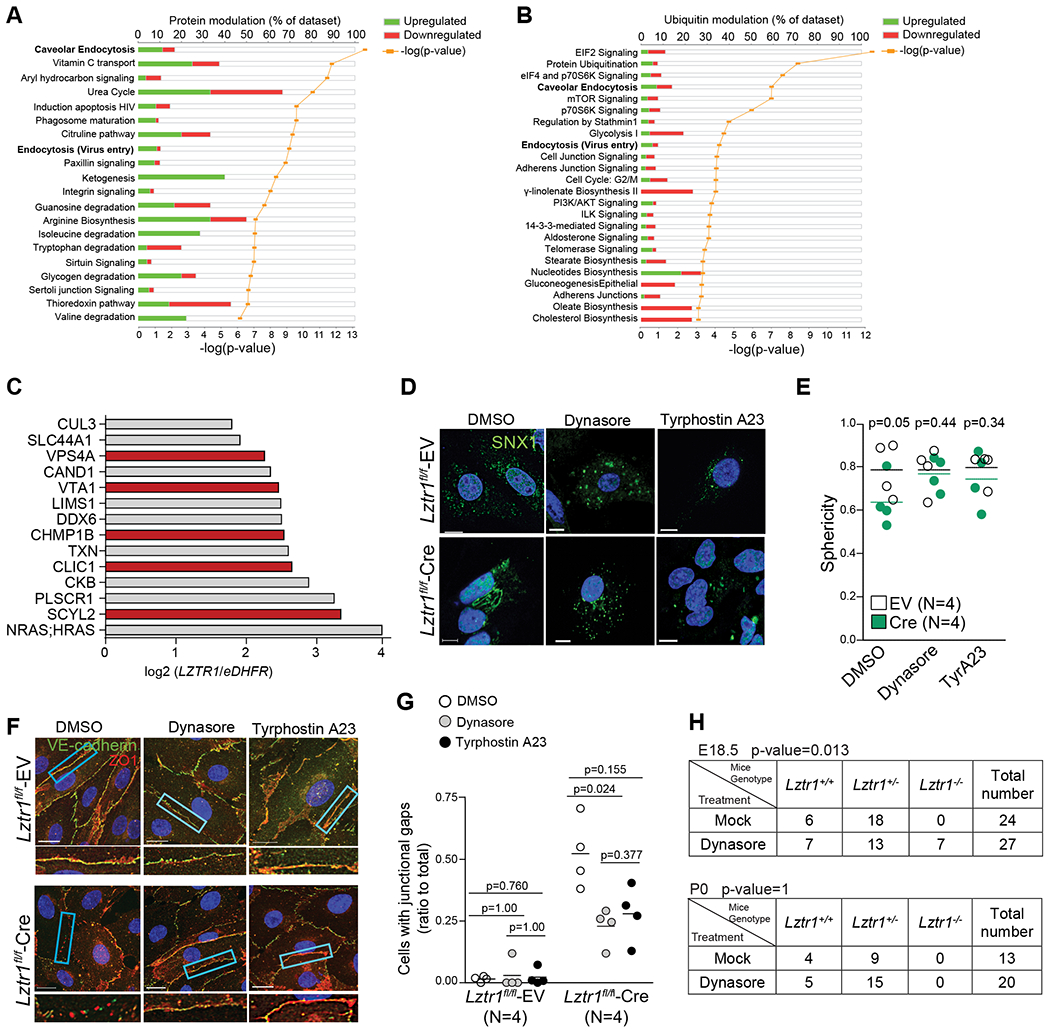
(a) IPA analysis of differentially expressed proteins in HeLa cells harboring wt-LZTR1 or LZTR1-indels. Differentially expressed proteins were identified by MS-based shot gun analysis using the FDR approach. N=3 (b) IPA analysis of differentially ubiquitinated peptides in HeLa cells harboring wt-LZTR1 or LZTR1-indels. GG-modified peptides were captured by anti-K-ε-GG antibody and quantified by MS. Differentially ubiquitinated proteins were identified by the FDR approach. N=3 (c) Putative LZTR1 interactors identified by the Virotrap approach as described in Steklov et al., 2018. Endocytosis-related proteins are shown in red. N=3 (d, e) Lztr1 fl/fl ECs expressing an empty vector (EV) or Cre recombinase were immunostained with anti-SNX1 antibody. Scale bar 10 μm. Each dot shows mean value for 25 ECs isolated from one mouse. Lines show mean±SEM; p-values were assessed by Wilcoxon-Mann-Whitney test. Sphericity ranges from 0 to 1 indicating if the structures are spheric (close to 1) or more tubular (close to 0.5). (f, g) Lztr1 fl/fl ECs transduced with an empty vector (EV) or Cre-recombinase coding virus (Cre) were treated with DMSO, Dynasore (40μM), or Tyrphostin A23 (10μM) for 12 hours and immunostained with anti-ZO1 and anti-VE-cadherin antibody. Scale bar 20 μm. Quantification of the number of junctional gaps per cells. N=4; 25 cells per group were analyzed; p-values were assessed by Kruskall Wallis test with Dunn’s Multiple Comparison Test (h) Progeny from Lztr1+/− matings at E18.5 and after birth in a mock group or a dynasore-treated group (1mg/kg; starting from E2.5). P-values were calculated using Fisher Exact test.
To test this idea, we performed immunostaining of LZTR1-deleted cells for sorting nexin-1 (SNX1), a protein essential for early to late endosome trafficking. Lztr1 knockout in Lztr1fl/fl ECs infected with a CreERT2 recombinase led to abnormal endosomal tubulation, a phenotype associated with vesicular trafficking impairment (Figure 3d, e). Concordantly, SNX1-positive structures appeared spherical in wt-LZTR1 HeLa cells, whereas LZTR1 loss led to the accumulation of endosomal tubules (Online Figure Va). These results implicate LZTR1 in the control of endosomal maturation and recycling29,30.
To ascertain the role of LZTR1 in vesicular trafficking, we performed a set of rescue experiments using the inhibitors of vesicular trafficking, dynasore31,32 and tyrphostin A23 (TyrA23)33,34. Both treatments abolished the difference in the appearance of endosomal tubules between wt-LZTR1 and LZTR1-deleted cells (Figure 3d, e; Online Figure Va). Furthermore, both inhibitors normalized adherent and tight junctions in Lztr1fl/fl-CreERT2 ECs (Figure 3f, g). Depletion of Dynamin I with a siRNA (Online Figure IVg) also restored normal junctions in Lztr1fl/fl-CreERT2 ECs (Online Figure IVg, h, i). Prenatal treatment with dynasore partially rescued the embryonic lethality phenotype caused by Lztr1 knockout, as we observed a nearly normal ratio of Lztr1−/− embryo at E18.5, but not at birth (Figure 3h). In contrast to the treatment with either pimasertib or ipatasertib, dynasore recovered lung dysplasia in E18.5 Lztr1−/− embryos (Online Figure IVd, e). Together, these results indicate that LZTR1-mediated vascular dysfunction could be explained by defects in vesicular trafficking.
LZTR1 controls ubiquitination of the ESCRT-III component CHMP1B
We next assessed the molecular mechanism by which LZTR1 is implicated in vesicular trafficking. The component of ESCRT-III (endosomal sorting complex required for transport III) trafficking machinery, CHarged Multivesicular Proteins 1B (CHMP1B), was present among both the putative LZTR1 interactors identified by the Virotrap screen20. CHMP1B also showed one of the highest scores in the fold change ranking when we compared ubiquitination landscapes of wt-LZTR1 and LZTR1-indel cells. Apart from the lysine 6 site, all CHMP1B ubiquitination sites (lysines 42, 59, 87, 90, 104, and 110) showed at least a two-fold decrease in LZTR1-indel cells (Figure 4a). This suggests that LZTR1 might regulate vesicle trafficking by controlling CHMP1B ubiquitination.
Figure 4. LZTR1 regulates CHMP1B ubiquitination.
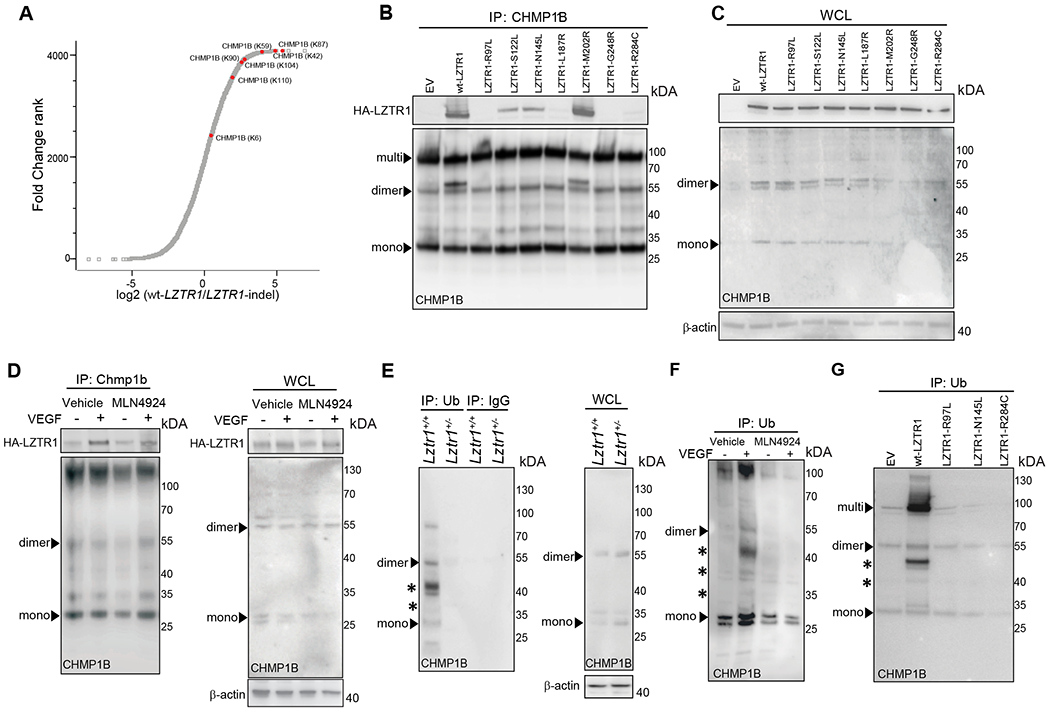
(a) Ubiquitinome analysis of wt-LZTR1 or LZTR1-indel HeLa cells. N=3. For each protein, log2 (fold change between both conditions) is plotted vs Fold Change Rank. CHMP1B ubiquitination sites are shown in red. (b, c) An empty vector (EV), HA-tagged wt-LZTR1, or LZTR1 mutants were overexpressed in HeLa cells. Endogenous CHMP1B was immunoprecipitated using anti-CHMP1B antibody conjugated to agarose. The amount of pulled-down LZTR1 was detected by immunoblotting with anti-HA antibody. (d) CHMP1B was pulled down from ECs expressing HA-wt-LZTR1 using an anti-CHMP1B antibody. The amount of pulled-down HA-wt-LZTR1 was detected by immunoblotting. (e) Ubiquitinated proteins were immunoprecipitated from Lztr1+/+ and Lztr1+/− ECs isolated using anti-ubiquitin FK2 antibody. The amount of pulled-down CHMP1B was detected by immunoblotting. (f) Ubiquitinated proteins were immunoprecipitated from ECs treated with VEGF (100ng/ml, 30 minutes) or MLN4924 (1μM, 12hours) using anti-Ubiquitin FK2 antibody. The amount of pulled-down CHMP1B was detected by immunoblotting. (g) Ubiquitinated proteins were immunoprecipitated from HeLa cells with Lztr1-indel overexpressing empty vector (EV), wt-LZTR1, or NS-associated LZTR1 mutants using anti-ubiquitin FK2 antibody conjugated to agarose. The amount of pulled-down CHMP1B was detected by immunoblotting. * indicates ubiquitinated CHMP1B.
We confirmed the interaction of endogenous CHMP1B and HA-tagged LZTR1 using co-immunoprecipitation (Figure 4b, c). Of note, CHMP1B forms homo- and heteromers and is detected in cell lysates as a monomer (28kDa), a dimer (55kDa), and a multimer even under denaturating conditions, which is consistent with previously published results35. Moreover, a recent report demonstrated that stimulation with growth factors or cytokines promotes CHMP1B ubiquitination35. Concordantly to this observation, we found increased binding of LZTR1 to CHMP1B in ECs upon stimulation with VEGF (Figure 4d). On the other hand, treatment with the CULLIN neddylation inhibitor MLN4924, which blocks the activity of the LZTR1/ CUL3 complex, did not affect the interaction between LZTR1 and CHMP1B, indicating that their binding does not depend on the ubiquitination status of CHMP1B (Figure 4d).
We then assessed LZTR1 contribution to the control of CHMP1B ubiquitination. CHMP1B was present in mono- or multi-ubiquitinated forms. This is consistent with the mass-spectrometry results showing ubiquitination of CHMP1B at seven different lysines. Substitution of 7 lysines to arginines abolished CHMP1B ubiquitination (Online Figure Vb). We found that LZTR1-indel in HeLa cells also led to decreased ubiquitination of V5-tagged wild-type and endogenous CHMP1B (Online Figure Vc). Moreover, we observed lower levels of CHMP1B ubiquitination upon Lztr1 knockout in mice when comparing Lztr1+/+ and Lztr1+/− yolk sacs; Lztr1+/+ and Lztr1+/− sorted ECs; and Lztr1fl/fl ECs expressing an empty vector (EV) or CreERT2 recombinase (Figure 4e; Online Figure Vd, e). Increased binding of LZTR1 to CHMP1B upon VEGF stimulation in ECs was associated with increased levels of ubiquitinated CHMP1B, whereas MLN4924 treatment abolished VEGF-induced CHMP1B ubiquitination (Figure 4f), further confirming that LZTR1 controls CHMP1B ubiquitination in a Cullin-dependent manner.
Noonan-associated LZTR1 mutations diminish the ability of LZTR1 to ubiquitinate CHMP1B
We examined whether Noonan Syndrome associated phenotypes could be also be associated with the impaired ability of mutated LZTR1 to control CHMP1B ubiquitination. Both truncated and missense mutations of LZTR1 have been reported in Noonan Syndrome. Missense mutations of LZTR1 are spread throughout the whole gene13,36,37. In our recent study, we found that LZTR1 mutations within the BTB (broad-complex, tramtrack and bric-a-brac)-BACK domains abolish the formation of the LZTR1/CUL3 complex, suggesting that the BTB-BACK LZTR1 mutations abolish ubiquitination of all LZTR1 substrates20. On the other hand, LZTR1 mutations within the Kelch domain, which is responsible for the substrate binding, might diminish the ability of LZTR1 to ubiquitinate CHMP1B by affecting the interaction between LZTR1 and CHMP1B. Therefore, we tested the ability of the Kelch domain LZTR1 mutants to bind and ubiquitinate CHMP1B. We found that all Kelch domain LZTR1 mutations, except for M202R, showed decreased ability to interact with CHMP1B (Figure 4b, c). Importantly, three LZTR1 mutations, R97L, N145L and R284C, observed in NS patients showing bleeding phenotype38,39, caused the inability of LZTR1 to ubiquitinate CHMP1B (Figure 4g). This suggests that dysregulation of CHMP1B ubiquitination could be associated with the bleeding disorders observed in LZTR1-mutated Noonan Syndrome patients.
Dysregulation of LZTR1-mediated ubiquitination of CHMP1B impedes vesicle trafficking
Our next question was how dysregulation of LZTR1-mediated ubiquitination of CHMP1B affects vesicular trafficking. The most well-described function of CHMP1B is to promote the formation of recycling tubules from endosomes40 by co-assembling with the other ESCRT-III component increased sodium tolerance 1 (IST1)41,42. Indeed, Chmp1b depletion led to the dysregulation of recycling trafficking as we detected an accumulation of RAB11-positive recycling endosomes (Figure 5a, b). Lztr1 knockout in ECs presented a similar accumulation of RAB11-positive endosomes. We also found an accumulation of CHMP1B/RAB11-positive recycling endosomes in Lztr1+/− ECs and Lztr1fl/fl ECs infected with a CreERT2 recombinase (Figure 5c, d, e). In contrast, we did not observe an increase of CHMP1B in early EAA1-positive or late LAMP1-positive endosomes (Figure 5d, e). On the other hand, suppression of Chmp1b in Lztr1−/− ECs did not affect the localization of RAB11 (Figure 5a, b), suggesting that the effect of CHMP1B on trafficking is LZTR1 dependent.
Figure 5. LZTR1-mediated CHMP1B ubiquitination modulates vesicle trafficking.
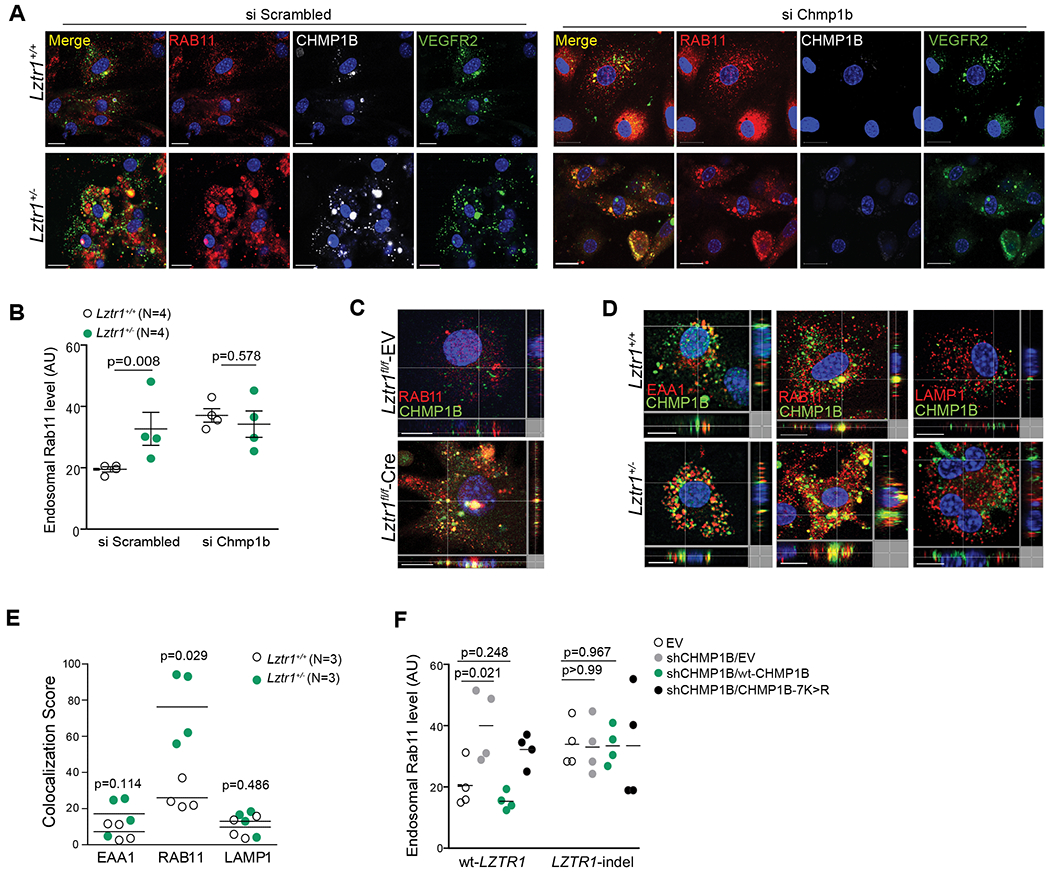
(a) Immunostaining of Lztr1+/+ and Lztr1+/− ECs transfected with a Scrambled siRNA or siRNA targetting Chmp1b with antibodies against RAB11, CHMP1B and VEGFR2. Scale bar 20 μm. (b) Quantification of endosomal RAB11 levels in Lztr1+/+ and Lztr1+/− ECs transfected with a Scrambled siRNA or siRNA targeting Chmp1b. Data shown as mean ± SEM; p-values were assessed by Wilcoxon Mann-Whitney test. >25 cells per mice were analyzed. (c) Lztr1fl/fl ECs expressing an empty vector (EV) or Cre recombinase were immunostained with the indicated antibodies. Scale bar 10 μm. (d) ECs isolated from Lztr1+/+ and Lztr1+/− mice immunostained with the indicated antibodies. Scale bar 10μm. (e) Quantification of co-localization of CHMP1B and endosomal markers in Lztr1+/+ and Lztr1+/− ECs is shown as mean ± SEM; p-values were assessed by Wilcoxon-Mann-Whitney test. 25 cells per mice were analyzed. (f) An empty vector (EV), V5-tagged wt-CHMP1B, or CHMP1B-7K>R mutant was overexpressed in wt-LZTR1 or LZTR1-indels HeLa cells expressing sh CHMP1B. Quantification of endosomal RAB11 is shown as mean ± SEM; p-values were assessed by Wilcoxon Mann-Whitney test. N=4.
We further confirmed the effect of LZTR1-mediated CHMP1B ubiquitination on vesicle trafficking by rescue experiments. Whereas suppression of CHMP1B led to the accumulation of RAB11-positive vesicles, overexpression of wild-type CHMP1B restored the trafficking of recycling endosomes. On the other hand, ubiquitination-deficient CHMP1B-7K>R mutant did not rescue the accumulation of RAB11-positive endosomes triggered by CHMP1B depletion. Finally, modulation of CHMP1B expression in LZTR1-indels cells showed no effect on recycling endosome trafficking (Figure 5f, Online Figure Vf, g), indicating that LZTR1-mediated ubiquitination of CHMP1B plays a crucial role in vesicular trafficking.
We also assessed the mechanism by which LZTR1-mediated ubiquitination of CHMP1B affects its functioning. Consistently to a recent study35, we did not observe any degradative poly-ubiquitination of CHMP1B (Figure 4e, f; Online Figure Vb–e). On the other hand, it was reported that USP8-mediated deubiquitination of CHMP1B promotes its co-assembly with IST135. In line with this observation, LZTR1 loss increased the interaction between CHMP1B and IST1 (Online Figure Vh). Collectively, these results indicate that LZTR1 loss affects recycling trafficking by abolishing CHMP1B ubiquitination and dysregulating the assembly of the CHMP1B/ IST1 complex, a key component of the ESCRTIII complex necessary for endosomal trafficking.
Lztr1 loss activates the VEGF pathway by dysregulating VEGFR2 trafficking
As a component of the ESCRT-III complex, CHMP1B is implicated in the trafficking of stimulated growth factor receptors, suggesting that LZTR1-mediated dysregulation of CHMP1B could affect receptor signaling. Consistent with this idea, the timing of VEGF-induced CHMP1B ubiquitination coincides with the onset of VEGFR2 internalization and activation (Figure 4d, f), suggesting that CHMP1B ubiquitination might regulate VEGFR2 trafficking. Moreover, we observed higher levels of phosphorylated VEGFR2 in Lztr1−/− heart vessels, suggesting that LZTR1 might contribute to the regulation of VEGFR2 activity (Online Figure IId). The IPA Upstream Regulator Analysis of genes differentially expressed in Lztr1+/+ and Lztr1+/− MEFs also predicted VEGF activation of in Lztr1 knockout cells (Online Figure VIa). Besides, the VEGF/ VEGFR2 pathway plays a central role in endothelial cell function; and among the angiogenic drugs, the VEGFR2 specific inhibitors have produced notable results in different diseases43–45. Therefore, we decided to specifically focus on VEGF signaling.
We assessed the effect of Lztr1 on the intracellular localization of VEGFR2 by performing immunocytochemistry analysis of VEGFR2 in ECs with or without permeabilization. Whereas VEGFR2 was strongly expressed on the cell surface of Lztr1+/+ ECs, we detected less VEGFR2 signal on the surface of Lztr1+/− ECs (Online Figure VIb, c). On the other hand, we observed an accumulation of VEGFR2 on RAB11/ CHMP1B-positive vesicles in Lztr1-deleted ECs (Figure 6a–c). A similar accumulation of VEGFR2 on endosomes was observed in Chmp1b-depleted cells, indicating abnormal trafficking of the receptor (Figure 5a; Figure 6c). It has been recently reported that the constitutive VEGFR2 internalization protects the receptor against ectodomain cleavage46, suggesting that Lztr1 loss might result in decreased VEGFR2 shedding. Indeed, ELISA analysis revealed lower levels of soluble VEGFR2 in the blood serum of Lztr1+/− mice (Figure 6d). Treatment with dynasore or tyrphostin A23 abolished endosomal accumulation of VEGFR2 in Lztr1+/− ECs (Figure 6a, b). Dynamin I suppression also reduced internalization of the receptor in Lztr1fl/fl ECs expressing CreERT2 (Online Figure VId, e). Altogether, these results indicate that dysregulation of LZTR1-mediated ubiquitination of CHMP1B could play a critical role in the regulation of VEGFR2 trafficking.
Figure 6. LZTR1-mediated VEGFR2 internalization affects blood vessel integrity.
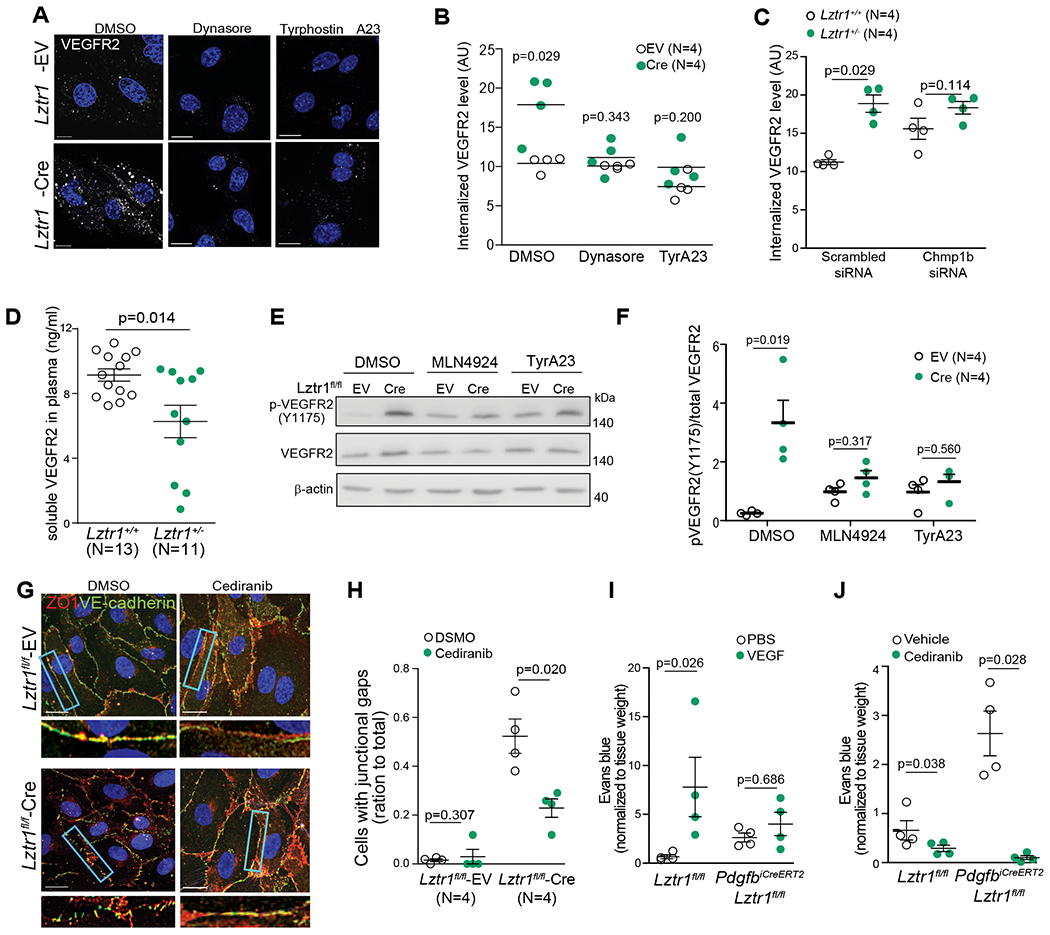
(a, b) Lztr1fl/fl ECs were infected with an empty vector (EV) or Cre-recombinase (Cre) and treated with DMSO, Dynasore (40μM), or Tyrphostin A23 (10μM) for 12 hours. After permeabilization, the cells were immunostained with anti-VEGFR2 antibody. Scale bar 20 μm. Data shown as mean ± SEM; p-values were assessed by Wilcoxon Mann-Whitney test. (c) Levels of internalized VEGFR2 were assessed by immunostaining of Lztr1+/+ and Lztr1+/− ECs transfected with Scrambled siRNA or siRNA targeting Chmp1b. Data are presented as mean ± SEM; p-values were assessed by Wilcoxon Mann-Whitney test. (d) ELISA analysis of soluble VEGFR2 in the plasma of Lztr1+/+ and Lztr1 +/− mice. Data are present as mean ± SEM; p-values were assessed by Wilcoxon Mann-Whitney test. (e, f) Immunoblotting analysis of phosphorylated and total VEGFR2 in Lztr1fl/fl ECs infected with an empty vector (EV) or Cre-recombinase (Cre) and treated with DMSO, MLN4924 (1μM), or Tyrphostin A23(10μM) for 12 hours. Data shown as mean ± SEM; p-values were assessed by Mann-Whitney test. (g, h) Lztr1 fl/fl ECs transduced with empty vector (EV) or Cre-recombinase (Cre), treated for 12 hours with DMSO or cediranib (10μM), and immunostained with antibodies against ZO1 and VE-cadherin. Scale bar 20 μm. Quantification of the number of cells with junctional gaps is shown as mean ± SEM; p-values were assessed by Wilcoxon Mann-Whitney test to compare efficiency of each specific treatment within each group. (i) Evans blue extravasation from PdgfbiCreERT2, Lztr1 fl/fl and Lztr1 fl/fl mice skin after treatment with PBS or VEGF. Miles assay performed on the ears. N=4 mice per group. Data are normalized to tissue weight and shown as mean ± SEM; p-values were assessed by Wilcoxon Mann-Whitney test to compare efficiency of treatment within group. (j) Evans blue extravasation from Pdgf iCreERT2, Lztr1fl/fl and Lztr fl/fl mice skin after vehicle or cediranib treatment. Miles assay. N=4 mice per group. Data are normalized to tissue weight and shown as mean ± SEM; p-values were assessed by Wilcoxon-Mann-Whitney test to compare efficiency of treatment within group.
Because internalization and phosphorylation of the VEGFR2 complex are hallmarks of VEGF signaling activation47,48, we hypothesized that LZTR1-mediated dysregulation of VEGFR2 trafficking might lead to its activation. Concordantly, we detected a higher level of phosphorylated VEGFR2 in Lztr1+/− ECs and Lztr1+/− yolk sacs (Online Figure VIf, g, i). Lztr1 knockout in Lztr1fl/fl ECs by overexpressing CreERT2 recombinase also led to increased levels of VEGFR2 phosphorylation (Online Figure VIh, i). Suppression of Dynamin I expression partially rescued increased levels of VEGFR2 phosphorylation in Lztr1-depleted cells (Online Figure VIk). VEGFR2 activation was CULLIN-dependent as MLN4924 abolished the accumulation of phosphorylated VEGFR2 (Figure 6d, e). TyrA23 treatment partially rescued VEGFR2 activation in Lztr1 knockout ECs, indicating that increased VEGFR2 activity is due to dysregulated endosomal trafficking (Figure 6e, f).
To assess whether enhanced VEGFR signaling is responsible for the vascular phenotypes in Lztr1 knockout mice, we blocked VEGFR2 activity in Lztr1fl-fl-CreERT2 ECs with cediranib49,50. Cediranib treatment normalized adherent and tight junctions (Figure 6g, h), confirming that Lztr1 deletion affects junctional integrity through VEGFR activation. Furthermore, VEGF treatment significantly increased Evans blue extravasation in the skin of Lztr1fl/fl mice, but only moderately in PdgfbiCreERT2, Lztr1fl/fl mice, confirming that the pathway is already activated in Lztr1 deleted cells (Figure 6i). Importantly, blocking of VEGFR2 activity with cediranib normalized tight and adherent junctions and rescued vascular leakage in the skin of PdgfbiCreERT2, Lztr1fl/fl mice (Figure 6j). Altogether, these data indicate that loss of Lztr1 leads to the up-regulation of VEGFR2 signaling by dysregulating vesicular trafficking, thus implying that blocking the catalytic activity of VEGFR could be advantageous for NS patients.
Discussion
Nearly two-thirds of NS patients exhibit clinical signs of a bleeding diathesis that may manifest with easy bruising, postoperative bleeding complications, and rarely spontaneous hemorrhages26. Here we explored the role of LZTR1, a gene recently linked to NS development, in the regulation of vascular function. We found that Lztr1 whole-body knockout mice died in utero, suffering from severe hemorrhages and in some cases, hydrops. This is in line with previous observations from other NS models 25,51. Similarly, Ptpn11D61G and Sos1E846K embryos show hemorrhages at E13.551,52. Lztr1 haploinsufficiency in adult mice also partially recapitulates bleeding phenotypes observed in NS patients, especially in the context of novel studies making the link between Lztr1 mutations and coagulation defects38,39.
Patients with PTPN11 mutations show an increased prevalence of bleeding disorders compared with those having other mutations53. On the other hand, bleeding disorders in NS appear to be least correlated with KRAS and RAF1 mutations1,54. This suggests that bleeding phenotypes might be associated with RAS-independent functions of the NS genes. Concordantly to this idea, PTPN11, which encodes the protein tyrosine phosphatase SHP-2, protects endothelial barrier through VE-cadherin stabilization55. Here, we also found that MEK1/2 inhibition does not rescue vascular dysfunction induced by Lztr1 loss. Unbiased proteome and ubiquitome analyses revealed that endocytosis is one of the top canonical pathways altered by Lztr1 loss. Importantly, treatment with either dynasore, tyrphostin A23 or dynamin I siRNA rescues endothelial dysfunction mediated by Lztr1 loss, confirming that LZTR1-mediated vascular phenotypes are caused by dysregulation of vesicular trafficking.
Unbiased interaction screen and ubiquitome analysis indicate that the key component of the ESCRT III complex CHMP1B could be a direct substrate of the LZTR1/ CUL3 complex. A previous study demonstrated that decreased ubiquitination of CHMP1B appears to block membrane fission by affecting its interaction with IST156. Concordantly, LZTR1 loss appears to reduce CHMP1B ubiquitination on lysine residues important for its interaction with IST141,42. CHMP1B and IST1 form a 3D structure that gives the plasticity to membrane invagination required for the fusion and fission of vesicles. In fact, LZTR1 loss strengthens the binding of CHMP1B to IST1, resulting in inhibition of membrane fission. Therefore, we could speculate that LZTR1-triggered defects in vesicular trafficking might be due to the dysregulation of the interaction between CHMP1B and IST1 that may affect the disassembly of the ESCRT-III complex. Importantly, all LZTR1 mutations found in NS patients with bleeding disorders led to a decreased ability to interact with CHMP1B. Nonetheless, we could not exclude the possibility that LZTR1 might modulate other regulators of vesicular trafficking.
Dysregulation of vesicle trafficking might lead to the activation of several receptor pathways57. Here, we demonstrated that Lztr1 loss activates VEGFR2 signaling, which is the primary tyrosine kinase receptor transmitting VEGF signals to control the balance between endothelial plasticity and junctional stability. Increased internalization and phosphorylation of VEGFR2 in Clathrin-dependent vesicles led to the activation of angiogenesis and increased endothelial permeability32,58. Therefore, the Lztr1-mediated bleeding phenotypes could be at least partially explained by the activation of VEGF signaling. Consistent with this idea, overexpression of VEGF-A results in severe endothelial dysfunction, such as abnormal angiogenesis and vascular leakage during development59,60, leading to the accumulation of endocytic vesicles containing active VEGFR2.
Even though inhibition of the MAPK and PI3K/AKT pathways did not rescue endothelial dysfunction observed in Lztr1 knockout mice, VEGFR2 led to the activation of multiple signaling pathways61. As an example, VEGFR2 recruits the adaptor protein Nck adaptor protein 1 (NCK1) and the SRC family tyrosine kinase FYN. NCK1/FYN complex formation regulates phosphorylation of PAK2, which in turn activates CDC42 and p38 MAPK, leading to actin remodeling. Recruitment of PLCγ1 and SH2-domain-containing adaptor protein B (SHB) by VEGFR2 facilitates the interaction with FAK (focal adhesion kinase) and contributes to endothelial cell migration and attachment. SHB activation of PI3K induces endothelial nitric oxide (NO) synthase (eNOS) that promotes cell survival and NO-induced vascular permeability respectively.
Current treatments for NS patients suffering from bleeding including platelets and fresh frozen plasma do not appear to be efficient in NS10. Several anti-VEGF approaches have been recently validated to limit blood loss, including angiogenesis inhibitors such as bevacizumab, an antibody targeting Vascular Endothelial Growth Factor (VEGF), cediranib62–63 or thalidomide64. Our results strongly indicate that inhibiting the catalytic activity of VEGF receptors could be beneficial to reduce bleeding in NS patients. Moreover, we found that Lztr1 loss leads to increased levels of soluble VEGFR2 in blood serum, suggesting that ELISA-based screening of NS patients for the blood levels of soluble VEGFR2 might allow predicting potential responders to anti-VEGF therapy. Collectively, our results provide a potential explanation for the role of LZTR1 in vascular function and suggest novel therapeutic approaches for NS patients.
Supplementary Material
Novelty and Significance.
What Is Known?
The Noonan Syndrome gene LZTR1 determines substrate specificity of the CULLIN3 ubiquitin ligase complexes
Cardiac injury and bleeding disorders have been recently described in Noonan Syndrome patients harboring LZTR1 mutations.
Currently, there are no therapeutic options for the management of bleeding complications in Noonan Syndrome patients.
What New Information Does This Article Contribute?
Loss of Lztr1 in mice leads to endothelial dysfunction, thus phenotypically overlapping with bleeding pathologies observed in Noonan Syndrome patients
LZTR1 affects the dynamics of vesicle trafficking by controlling ubiquitination of the ESCRT-III component CHMP1B
Lztr1-mediated dysregulation of CHMP1B ubiquitination triggers endosomal accumulation and subsequent activation of VEGFR2, causing disruption of endothelial junctions and bleeding.
Our findings provide a fundamental explanation underlying the molecular mechanism of endothelial dysfunction in Noonan Syndrome patients harboring LZTR1 mutations. By combining comprehensive analysis of Lztr1 knockout models and unbiased proteomics and transcriptomics approaches, we demonstrated that Lztr1 loss of function leads to vascular dysfunction by dysregulating vesicular trafficking and subsequent activation of VEGFR2. Our results indicate that ELISA screening of Noonan Syndrome patients for the levels of soluble VEGFR2 in blood plasma may predict both the severity of Noonan Syndrome phenotypes and potential responders to anti-VEGF therapy, whereas anti-VEGF therapies such as cediranib could be a potential treatment approach for managing bleeding disorders in LZTR1-mutated Noonan Syndrome patients.
Sources of Funding
This work was supported by H2020 European Research Council (ub-RASDisease) and Fonds Wetenschappelijk Onderzoek Research project G068715N
Non-standard Abbreviations and Acronyms:
- NS
Noonan Syndrome
- LLC
Lewis lung carcinoma
- ECs
Endothelial cells
- IB4
Isolectin B4
- miG
mouse iG
- EV
empty vector
- Cre
Cre-recombinase
- WT
Wild-type
- AU
arbitrary units
Footnotes
Conflicts of interest
The authors claim no conflict of interest.
References
- 1.Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333–342. doi: 10.1016/S0140-6736(12)61023-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sznajer Y, Keren B, Baumann C, et al. The spectrum of cardiac anomalies in Noonan syndrome as a result of mutations in the PTPN11 gene. Pediatrics. 2007;119(6). doi: 10.1542/peds.2006-0211 [DOI] [PubMed] [Google Scholar]
- 3.Bakker M, Pajkrt E, Mathijssen IB, Bilardo CM. Targeted ultrasound examination and DNA testing for Noonan syndrome, in fetuses with increased nuchal translucency and normal karyotype. Prenat Diagn. 2011;31(9):833–840. doi: 10.1002/pd.2782 [DOI] [PubMed] [Google Scholar]
- 4.Nisbet DL, Griffin DR, Chitty LS. Prenatal features of Noonan syndrome. Prenat Diagn. 1999;19(7):642–647. doi: [DOI] [PubMed] [Google Scholar]
- 5.Hernandez RJ, Rosenthal A. Pulmonary Lymphangiectasis in Noonan Syndrome. AJR. 1980;134. [DOI] [PubMed] [Google Scholar]
- 6.Witt DR, Hoyme HE, Zonana J, et al. Lymphedema in Noonan syndrome: Clues to pathogenesis and prenatal diagnosis and review of the literature. Am J Med Genet. 1987;27(4):841–856. doi: 10.1002/ajmg.1320270412 [DOI] [PubMed] [Google Scholar]
- 7.Nugent DJ, Romano AA, Sabharwal S, Cooper DL. Evaluation of bleeding disorders in patients with noonan syndrome: A systematic review. J Blood Med. 2018;9:185–192. doi: 10.2147/JBM.S164474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massarano A, Wood A, Tait R, Stevens R, Super M. Noonan syndrome: coagulation and clinical aspects. Acta Paediatr. 2010;85(10):1181–1185. doi: 10.1111/j.1651-2227.1996.tb18225.x [DOI] [PubMed] [Google Scholar]
- 9.Sharland M, Patton MA, Talbot S, Chitolie A, Bevan DH. Coagulation-factor deficiencies and abnormal bleeding in Noonan’s syndrome. Lancet. 1992;339(8784):19–21. doi: 10.1016/0140-6736(92)90141-O [DOI] [PubMed] [Google Scholar]
- 10.Tofil NM, Winkler MK, Watts RG, Noonan J. The use of recombinant factor VIIa in a patient with Noonan syndrome and life-threatening bleeding. Pediatr Crit Care Med. 2005;6(3):352–354. doi: 10.1097/01.PCC.0000160656.71424.D1 [DOI] [PubMed] [Google Scholar]
- 11.Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab. 2011;25(1):161–179. doi: 10.1016/j.beem.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468. doi: 10.1038/ng772 [DOI] [PubMed] [Google Scholar]
- 13.Johnston JJ, van der Smagt JJ, Rosenfeld JA, et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet Med. 2018;20(10):1175–1185. doi: 10.1038/gim.2017.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakaguma M, Jorge AAL, Arnhold IJP. Noonan syndrome associated with growth hormone deficiency with biallelic LZTR1 variants. Genet Med. 2018;0(0):41436. doi: 10.1038/s41436-018-0041-5 [DOI] [PubMed] [Google Scholar]
- 15.Aoki Y, Niihori T, Inoue SI, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2016;61(1):33–39. doi: 10.1038/jhg.2015.114 [DOI] [PubMed] [Google Scholar]
- 16.Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45(10):1141–1149. doi: 10.1038/ng.2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ally A, Balasundaram M, Carlsen R, et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169(7):1327–1341.e23. doi: 10.1016/j.cell.2017.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paganini I, Chang VY, Capone GL, et al. Expanding the mutational spectrum of LZTR1 in schwannomatosis. Eur J Hum Genet. 2015;23(7):963–968. doi: 10.1038/ejhg.2014.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruggieri M, Praticò AD, Evans DG. Diagnosis, Management, and New Therapeutic Options in Childhood Neurofibromatosis Type 2 and Related Forms. Semin Pediatr Neurol. 2015;22(4):240–258. doi: 10.1016/j.spen.2015.10.008 [DOI] [PubMed] [Google Scholar]
- 20.Steklov M, Pandolfi S, Baietti MF, et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science (80- ). 2018;362(6419):1177–1182. doi: 10.1126/science.aap7607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible cre-recombinase activation in vascular endothelium. Genesis. 2008;46(2):74–80. doi: 10.1002/dvg.20367 [DOI] [PubMed] [Google Scholar]
- 22.Hellström M, Gerhardt H, Kalén M, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;152(3):543–553. doi: 10.1083/jcb.153.3.543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sewduth RN, Jaspard-Vinassa B, Peghaire C, et al. The ubiquitin ligase PDZRN3 is required for vascular morphogenesis through Wnt/planar cell polarity signalling. Nat Commun. 2014;5:4832. doi: 10.1038/ncomms5832 [DOI] [PubMed] [Google Scholar]
- 24.Sewduth RN, Kovacic H, Jaspard-Vinassa B, et al. PDZRN3 destabilizes endothelial cell-cell junctions through a PKCζ-containing polarity complex to increase vascular permeability. Sci Signal. 2017;10(464). doi: 10.1126/scisignal.aag3209 [DOI] [PubMed] [Google Scholar]
- 25.Lauriol J, Cabrera JR, Roy A, et al. Developmental SHP2 dysfunction underlies cardiac hypertrophy in Noonan syndrome with multiple lentigines. J Clin Invest. 2016;126(8):2989–3005. doi: 10.1172/JCI80396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Artoni A, Selicorni A, Passamonti SM, et al. Hemostatic Abnormalities in Noonan Syndrome. Pediatrics. 2014;133(5):e1299–e1304. doi: 10.1542/peds.2013-3251 [DOI] [PubMed] [Google Scholar]
- 27.Baker M, Robinson SD, Lechertier T, et al. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2012;7(1):89–104. doi: 10.1038/nprot.2011.435 [DOI] [PubMed] [Google Scholar]
- 28.Futakuchi M Animal Model of Lung Metastasis of Hepatocellular Carcinoma: A Tool for the Development of Anti-Metastatic Therapeutics. J Cancer Ther. 2013;04(02):420–425. doi: 10.4236/jct.2013.42A051 [DOI] [Google Scholar]
- 29.Newton TM, Reid E. An automated image analysis system to quantify endosomal tubulation. PLoS One. 2016;11(12):1–11. doi: 10.1371/journal.pone.0168294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bryant DM, Kerr MC, Hammond LA, et al. EGF induces macropinocytosis and SNX1-modulated recycling of E-cadherin. J Cell Sci. 2007;120(10):1818–1828. doi: 10.1242/jcs.000653 [DOI] [PubMed] [Google Scholar]
- 31.Preta G, Cronin JG, Sheldon IM. Dynasore - Not just a dynamin inhibitor. Cell Commun Signal. 2015;13(1):1–7. doi: 10.1186/s12964-015-0102-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Basagiannis D, Zografou S, Galanopoulou K, Christoforidis S. Dynasore impairs VEGFR2 signalling in an endocytosis-independent manner. Sci Rep. 2017;7:1–11. doi: 10.1038/srep45035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stahlschmidt W, Robertson MJ, Robinson PJ, McCluskey A, Haucke V. Clathrin terminal domain-ligand interactions regulate sorting of mannose 6-phosphate receptors mediated by AP-1 and GGA adaptors. J Biol Chem. 2014;289(8):4906–4918. doi: 10.1074/jbc.M113.535211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson MJ, Deane FM, Stahlschmidt W, et al. Synthesis of the Pitstop family of clathrin inhibitors. Nat Protoc. 2014;9(7):1592–1606. doi: 10.1038/nprot.2014.106 [DOI] [PubMed] [Google Scholar]
- 35.Crespo-Yàñez X, Aguilar-Gurrieri C, Jacomin AC, et al. CHMP1B is a target of USP8/UBPY regulated by ubiquitin during endocytosis. PLoS Genet. 2018;14(6):1–24. doi: 10.1371/journal.pgen.1007456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghedira N, Kraoua L, Lagarde A, et al. Further Evidence for the Implication of LZTR1 , a Gene Not Associated with the Ras-Mapk Pathway , in the Pathogenesis of Noonan Syndrome. 2017;9(6):4–7. doi: 10.4172/0974-8369.1000414 [DOI] [Google Scholar]
- 37.Umeki I, Niihori T, Abe T, Nobuhiko K, Seiji O. Delineation of LZTR1 mutation-positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1 – PPP1CB complexes. Hum Genet. 2018;0(0):0. doi: 10.1007/s00439-018-1951-7 [DOI] [PubMed] [Google Scholar]
- 38.Pagnamenta AT, Kaisaki PJ, Bennett F, et al. Delineation of dominant and recessive forms of LZTR1-associated Noonan syndrome. Clin Genet. 2019;95(6):693–703. doi: 10.1111/cge.13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacquinet A, Bonnard A, Capri Y, et al. Oligo-astrocytoma in LZTR1-related Noonan syndrome. Eur J Med Genet. 2020;63(1). doi: 10.1016/j.ejmg.2019.01.007 [DOI] [PubMed] [Google Scholar]
- 40.Allison R, Lumb JH, Fassier C, et al. An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome. J Cell Biol. 2013;202(3):527–543. doi: 10.1083/jcb.201211045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCullough J, Clippinger AK, Talledge N, et al. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science (80- ). 2015;350(6267):1548–1551. doi: 10.1126/science.aad8305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bajorek M, Schubert HL, McCullough J, et al. Structural basis for ESCRT-III protein autoinhibition. Nat Struct Mol Biol. 2009;16(7):754–762. doi: 10.1038/nsmb.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park MS, Ravi V, Araujo DM. Inhibiting the VEGF-VEGFR pathway in angiosarcoma, epithelioid hemangioendothelioma, and hemangiopericytoma/solitary fibrous tumor. Curr Opin Oncol. 2010;22(4):351–355. doi: 10.1097/CCO.0b013e32833aaad4 [DOI] [PubMed] [Google Scholar]
- 44.Kodack DP, Chung E, Yamashita H, et al. Combined targeting of HER2 and VEGFR2 for effective treatmentof HER2-amplified breast cancer brain metastases. Proc Natl Acad Sci U S A. 2012;109(45). doi: 10.1073/pnas.1216078109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pandey AK, Singhi EK, Arroyo JP, et al. Mechanisms of VEGF (vascular endothelial growth factor) inhibitor-associated hypertension and vascular disease. Hypertension. 2018;71(2):E1–E8. doi: 10.1161/HYPERTENSIONAHA.117.10271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basagiannis D, Christoforidis S. Constitutive endocytosis of VEGFR2 protects the receptor against shedding. J Biol Chem. 2016;291(32):16892–16903. doi: 10.1074/jbc.M116.730309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sawamiphak S, Seidel S, Essmann CL, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465(7297):487–491. doi: 10.1038/nature08995 [DOI] [PubMed] [Google Scholar]
- 48.Genet G, Boyé K, Mathivet T, et al. Endophilin-A2 dependent VEGFR2 endocytosis promotes sprouting angiogenesis. Nat Commun. 2019;10(1):1–15. doi: 10.1038/s41467-019-10359-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Medinger M, Esser N, Zirrgiebel U, Ryan A, Jürgensmeier JM, Drevs J. Antitumor and antiangiogenic activity of cediranib in a preclinical model of renal cell carcinoma. Anticancer Res. 2009;29(12):5065–5076. [PubMed] [Google Scholar]
- 50.Brave SR, Ratcliffe K, Wilson Z, et al. Assessing the activity of cediranib, a VEGFR-2/3 tyrosine kinase inhibitor, against VEGFR-1 and members of the structurally related PDGFR family. Mol Cancer Ther. 2011;10(5):861–873. doi: 10.1158/1535-7163.MCT-10-0976 [DOI] [PubMed] [Google Scholar]
- 51.Araki T, Mohi MG, Ismat FA, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10(8):849–857. doi: 10.1038/nm1084 [DOI] [PubMed] [Google Scholar]
- 52.Uhlén P, Burch PM, Zito CI, Estrada M, Ehrlich BE, Bennett AM. Gain-of-function/Noonan syndrome SHP-2/Ptpn11 mutants enhance calcium oscillations and impair NFAT signaling. Proc Natl Acad Sci U S A. 2006;103(7):2160–2165. doi: 10.1073/pnas.0510876103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468. doi: 10.1038/ng772 [DOI] [PubMed] [Google Scholar]
- 54.Kroisel PM, Häusler M, Klaritsch P, et al. Targeted enrichment sequencing in two midterm pregnancies with severe abnormalities on ultrasound. Lancet. 2017;389(10081):1857–1858. doi: 10.1016/S0140-6736(17)31049-8 [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, Huang J, Qi T, et al. SHP2 protects endothelial cell barrier through suppressing VE-cadherin internalization regulated by MET-ARF1. FASEB J. 2019;33(1):1124–1137. doi: 10.1096/fj.201800284R [DOI] [PubMed] [Google Scholar]
- 56.Derivery E, Sousa C, Gautier JJ, Lombard B, Loew D, Gautreau A. The Arp2/3 Activator WASH Controls the Fission of Endosomes through a Large Multiprotein Complex. Dev Cell. 2009;17(5):712–723. doi: 10.1016/j.devcel.2009.09.010 [DOI] [PubMed] [Google Scholar]
- 57.Sadowski L, Pilecka I, Miaczynska M. Signaling from endosomes: Location makes a difference. Exp Cell Res. 2009;315(9):1601–1609. doi: 10.1016/j.yexcr.2008.09.021 [DOI] [PubMed] [Google Scholar]
- 58.Tian Y, Gawlak G, O’Donnell JJ, Birukova AA, Birukov KG. Activation of Vascular Endothelial Growth Factor (VEGF) receptor 2 mediates endothelial permeability caused by cyclic stretch. J Biol Chem. 2016;291(19):10032–10045. doi: 10.1074/jbc.M115.690487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127(18):3941–3946. [DOI] [PubMed] [Google Scholar]
- 60.Le Cras TD, Spitzmiller RE, Albertine KH, Greenberg JM, Whitsett JA, Akeson AL. VEGF causes pulmonary hemorrhage, hemosiderosis, and air space enlargement in neonatal mice. Am J Physiol - Lung Cell Mol Physiol. 2004;287(1 31-1). doi: 10.1152/ajplung.00050.2004 [DOI] [PubMed] [Google Scholar]
- 61.Smith GA, Fearnley GW, Tomlinson DC, Harrison MA, Ponnambalam S. The cellular response to vascular endothelial growth factors requires co-ordinated signal transduction, trafficking and proteolysis. Biosci Rep. 2015;35(5). doi: 10.1042/BSR20150171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peterson T, Kirkpatrick N, Huang Y, et al. Tmic-07. Dual Inhibition of Ang-2 and Vegf Receptors Normalizes Tumor Vasculature and Prolongs Survival in Glioblastoma By Altering Macrophages. Neuro Oncol. 2016;18(suppl_6):vi201–vi201. doi: 10.1093/neuonc/now212.847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kang S, Park KC, Yang KJ, Choi HS, Kim SH, Roh YJ. Effect of cediranib, an inhibitor of vascular endothelial growth factor receptor tyrosine kinase, in a mouse model of choroidal neovascularization. Clin Exp Ophthalmol. 2013;41(1):63–72. doi: 10.1111/j.1442-9071.2012.02813.x [DOI] [PubMed] [Google Scholar]
- 64.Bauditz J, Lochs H. Angiogenesis and vascular malformations: Antiangiogenic drugs for treatment of gastrointestinal bleeding. World J Gastroenterol. 2007;13(45):5979–5984. doi: 10.3748/wjg.13.5979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Araki T, Mohi MG, Ismat FA, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10(8):849–857. doi: 10.1038/nm1084 [DOI] [PubMed] [Google Scholar]
- 66.Steklov M, Pandolfi S, Baietti MF, et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science (80- ). 2018;362(6419):1177–1182. doi: 10.1126/science.aap7607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. Bioinforma Impact Accurate Quantif Proteomic Genet Anal Res. 2014:41–74. doi: 10.1201/b16589 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.