

Abstract
Background:
The management of clinical heart failure (HF) with a preserved ejection fraction (EF) (HFpEF) is often complicated by concurrent renal dysfunction, known as the cardiorenal syndrome (CRS). This, combined with the notable lack of evidence-based therapies for HFpEF, highlights the importance of examining mechanisms and targetable pathways in HFpEF with the CRS.
Methods:
HFpEF was induced in mice by uninephrectomy, infusion of d-aldosterone (HFpEF; N=10) or saline (Sham; N=8) and given 1% NaCl drinking water for 4 weeks. Renal fibrosis and endothelial-mesenchymal transition (endo-MT) were evident once HFpEF developed. Human aortic endothelial cells (HAECs) were treated for 4 days with 10% serum obtained from patients with chronically stable HFpEF with the CRS (N=12) and compared to serum-treated HAECs from control subjects (no cardiac/renal disease; N=12) to recapitulate the in vivo findings.
Results:
Kidneys from HFpEF mice demonstrated hypertrophy, interstitial fibrosis (1.9-fold increase; p<0.05) with increased expression of endo-MT transcripts, including platelet-derived growth factor receptor beta (pdgfrβ), snail, fibronectin, fibroblast-specific protein 1 (fsp1) and vimentin by 1.7- (P= 0.004), 1.7- (P=0.05), 1.8- (P=0.005), 2.6- (P=0.001) and 2.0-fold (P=0.001) versus Sham. Immunostaining demonstrated co-localization of CD31 and ACTA2 in kidney sections suggesting evidence of endo-MT. Similar to the findings in HFpEF mice, comparable endo-MT markers were also significantly elevated in serum-treated HAECs from HFpEF patients compared to serum-treated HAECs from control subjects.
Conclusions:
These translational findings demonstrate a plausible role for endo-MT in HFpEF with CRS, and may have therapeutic implications in drug development for patients with HFpEF and concomitant renal dysfunction.
Journal subject terms: Cardiorenal Syndrome, Heart Failure, Fibrosis, Translational Studies
INTRODUCTION
Heart failure (HF) with preserved ejection fraction (EF) (HFpEF) accounts for nearly half of all clinical HF presentations; and because of the aging population and the ongoing epidemics of metabolic disorders and hypertension, the prevalence of HFpEF is projected to increase exponentially. However, unlike HF with reduced EF (HFrEF) there are no evidence-based therapies that reduce morbidity and mortality. HFpEF is a multiorgan, heterogeneous disorder that often occurs concomitantly with comorbidities, thus phenotyping HFpEF into subgroups underscores its importance to identify new mechanisms for therapeutic targets in this complicated disease.
Chronic kidney disease (CKD) is ubiquitous in HF and its presence in HFpEF signifies a poorer prognosis than its presence in HFrEF. About 25–50 % of HFpEF patients have CKD, and the prevalence of both HFpEF and CKD increases with aging1. In a large cohort study of HF patients, aimed at identifying non-cardiac comorbidities, the most common was CKD, with the greatest prevalence in the HFpEF subgroup. Regardless of left ventricular (LV) EF (LVEF), renal dysfunction was associated with increased mortality in all HF subgroups2.
HFpEF and CKD coexist partly due to shared risk factors, such as hypertension, diabetes and obesity. Additionally, myocardial dysfunction directly impacts kidney function, and vice versa3. Thus, these deleterious effects are reinforced in a feedback cycle, accelerating the progression of both cardiac and renal damage4, a process known as the cardiorenal syndrome (CRS). Presently there is limited mechanistic insight into the CRS in HFpEF5. It was previously suggested that the relationship between HF and CKD was predominantly cardiocentric, driven by hemodynamic dysfunction and poor forward flow or renal venous congestion leading to renal hypoperfusion, activation of the renin-angiotensin-aldosterone system, and arginine and vasopressin hypersecretion6. However, recent evidence indicates that additional pathways are involved in the CRS in HFpEF including bidirectional crosstalk between the kidneys and the heart. It has been proposed that systemic inflammation leads to endothelial dysfunction which may explain, in part, the coexistence of renal dysfunction and HFpEF7.
Fibrosis, often a consequence of inflammation-related endothelial dysfunction, is a common feature in HF and CKD and interstitial fibrosis may play a key pathophysiological role in the CRS8. Although initially considered an adaptive response to injury, fibrosis also occurs in chronic disease states where myofibroblast activation and collagen deposition are also present. The precise signaling pathways and processes involved in the CRS and fibrosis are unknown. It is evident that during fibrosis, in addition to the presence of resident interstitial fibroblasts, activated myofibroblasts may arise from epithelial cells via epithelial-to-mesenchymal transition (EMT) or are recruited from the bone marrow9. Additionally, endothelial cells are also capable of acquiring a mesenchymal phenotype through a process known as endothelial-to-mesenchymal transition (endo-MT), thus providing another source of activated myofibroblasts in fibrotic diseases10. Since endo-MT may play a pathogenic role in cardiac and kidney fibrosis11, we therefore sought to investigate the presence of endo-MT in the CRS in HFpEF and the clinical relevance of these findings.
METHODS
The data that support the findings of this study, including methods and study materials are available from the corresponding author upon reasonable request.
In vivo murine model:
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1985) and was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine.
Experimental model of HFpEF:
Eight-week old male C57BL/6J mice (Jackson Laboratories) were anesthetized with 80–100 mg/Kg ketamine and 5–10 mg/Kg xylazine intraperitoneally, as described previously12–15. All mice (20–25 g) underwent uninephrectomy and received either a continuous infusion of saline (Sham, N=8) or d-aldosterone (0.30 μg/h, Sigma-Aldrich, St. Louis, MO, USA; HFpEF, N=10) for 4 weeks via osmotic minipumps (Alzet, Durect Corp., Cupertino, CA, USA). All mice were given 1.0% sodium chloride drinking water ad libitum. After 4 weeks mice were euthanized and LV weight, renal weight and wet-to-dry lung ratio were measured.
Physiological measurements:
Blood pressure measurement was performed at the end of the 4 experimental weeks using a noninvasive tail-cuff blood pressure analyzer (BP-2000 Blood Pressure Analysis System; Visitech Systems, Inc., Apex, NC, USA).
Histopathological analyses:
Transverse sections of kidney tissue were obtained by cutting through the renal pelvis. The samples were fixed in formalin, embedded in paraffin, mounted, sectioned (5 μm), and subsequently stained with hematoxylin and eosin (H&E, Sigma-Aldrich) and Pricosirius red staining (Sigma-Aldrich) to determine renal morphology and fibrosis, respectively. 15–20 image fields per kidney were obtained using a BZ-9000 BioRevo microscope (Keyence Corp. of America, Itasca, IL, USA) at 20X magnification and were analyzed blinded to group identity using Adobe Photoshop (Adobe, San Jose, CA, USA) measuring software. To assess glomeruli size, 25 glomeruli were selected randomly across 10 fields, traced to calculate the cross-sectional area and averaged per sample. The percent fibrosis per field was averaged for each sample using Adobe Photoshop.
Endo-MT immunostaining:
Kidney sections were stained with rat anti-cluster of differentiation 31 (CD31, Dianova, Hamburg, Germany) and rabbit anti-smooth muscle actin alpha 2 (ACTA2, Cell Signaling, Danvers, MA, USA) to determine co-localization of endothelial and myofibroblasts markers. ImPRESS-AP (alkaline phosphatase) polymer anti-rat IgG and ImPRESS-HRP anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA), were used as secondary antibodies. Highdef Blue IHC chromogen AP (Enzo, Farmingdale, NY, USA) and ImmPACT AMEC Red peroxidase HRP substrate (Vector Laboratories) were used for developing. Microscopy images were acquired on an Olympus BH-2 microscope, and captured with an attached Olympus 12.5 million pixel, DP70 Digital Camera. Images were analyzed blinded to group identity by two independent investigators.
Gene expression analysis by quantitative PCR:
Total RNA was extracted from kidney and cardiac tissue samples using a Qiagen RNeasy Tissue Mini Kit (QIAGEN, Valencia, CA, USA) and the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) was used for cDNA synthesis. qPCR was performed with PerfeCta SYBR® Green FastMix (Quanta Biosciences, Gaithersburg, MD, USA) in a ViiA7 PCR system (Life Technologies, Carlsbad, CA). Primers sequences are available upon request. Results were analyzed with the ΔΔ Ct method using GAPDH expression as reference for normalization.
Western blot analysis:
Renal protein extracts were obtained using ice-cold lysis buffer (Cell Signaling) supplemented with protease and phosphatase inhibitors (Cell Signaling). Equal amounts of protein were subjected to electrophoresis in SDS-polyacrylamide gel under reducing conditions and blotted to nitrocellulose membrane using the Bio-Rad Transblot Turbo Transfer System (Hercules, CA, USA). The membranes were blocked in 5% BSA, 0.1% Tween-20 in Tris-buffered saline (TBST) for 1 hour at room temperature and then incubated overnight at 4°C with primary antibodies (1:1000). Membranes were then washed with TBST and incubated with horseradish peroxidase-conjugated secondary antibodies (R&D Systems, Minneapolis, MN, USA) for 1 hour at room temperature; immune complexes were detected with the enhanced chemiluminescence ECL detection system (Bio-Rad) in the ImageQuant LAS 4000 biomolecular imaging system (GE Healthcare, Pittsburg, PA, USA). The intensity of bands for each protein was normalized to the loading control alpha-tubulin (Cell Signaling, #A2148). The specific primary antibodies used for immunoblotting were: anti-fibroblast-specific protein 1 (aka S100A4, Cell Signaling, #13018); anti-platelet-derived growth factor receptor beta (PDGFRβ, Sigma-Aldrich, #06–498-I); anti-snail (Cell Signaling, #3879); and anti-vimentin (Sigma-Aldrich, #V2258).
In vitro experiments:
Endothelial cell culture and treatment:
Human aortic endothelial cells (HAECs; Lonza Inc, Walkersville, MD, USA) were maintained with endothelial growth medium‐2 containing 5 mmol/L glucose in a standard incubator (37°C, 5% CO2). Cells from passages 4 to 7 and 90% confluence were used for the experiments. After being starved for 3 hours in endothelial cell growth basal medium-2 (Lonza), cells were cultured using the same medium but supplemented with 10% human serum from HFpEF patients or control subjects for 4 days. Medium was changed every other day and by day 4 of experiment cells were washed with PBS and collected in 1 mL of Qiazol reagent (QIAGEN) for RNA isolation following the same protocol described under Gene expression analysis by qPCR section.
Serum samples:
Blood samples were obtained from patients with chronically stable HFpEF in the ambulatory HF Clinic at Boston Medical Center (N=12). Study approval: Written informed consent was obtained from all subjects prior to the collection of blood samples. The study was approved by the Boston University Medical Center Institutional Review Board and conducted according to Declaration of Helsinki principles. Chronic patients with HFpEF were included if they had been admitted with HF within the prior year (but not within the last 3 months), had a LVEF >50% as measured by echocardiogram within 6 months prior to enrollment, and showed evidence of renal dysfunction or the CRS i.e., elevated levels of creatinine and glomerular filtration rate (GFR) <60 mL/min/1.73 m2. The MDRD formula was used to calculate GFR at our institution. (Patient characteristics are included in Table 1). Patients were included if they had ≥ 2 consecutive measurements of GFR <60 mL/min/1.73 m2. Gender and age-matched, healthy control subjects with no known HF, cardiac disease and renal disease were also included (N=12; Table 2). After informed consent, blood samples were obtained using routine venipuncture procedure. Samples were centrifuged at 2000 g for 15 min within 1 h from collection and aliquoted and stored at −80°C.
Table 1:
Characteristics of chronic stable ambulatory HFpEF patients with cardiorenal syndrome (CRS) used to investigate endo-MT activation in vitro
| Chronic HFpEF (N=12) | Values | Median IQR | Normal values |
|---|---|---|---|
| Age (years) | 68 ± 11 | 65.5 [58.5, 75.5] | |
| Sex: Male/Female (%) | 3/9 (25/75) | ||
| Race: White/Black/Other (%) | 3/8/1 (25/67/8) | ||
| Systolic blood pressure (mmHg) | 141± 17 | 144.5 [127.5, 153.3] | |
| Comorbidities (%) | |||
| Hypertension | 12 (100) | ||
| Obesity: BMI>30 kg/m2 | 11 (92) | ||
| Type 2 diabetes | 8 (67) | ||
| Atrial fibrillation/Atrial flutter | 5 (42) | ||
| Chronic kidney disease (stage) | |||
| 3 (eGFR 30–59 mL/min/1.73m2) | 9 (75) | ||
| 4 (eGFR 30–59 mL/min/1.73m2) | 3 (25) | ||
| NYHA Functional Class I/II/III/IV (%) | 2/9/1/0 (17/75/8/0) | ||
| Echocardiographic parameters | |||
| IVS (mm) | 11.0 ± 1.9 | 11.0 [9.3, 12.8] | 6–10 |
| LVEF (%) | 60.9 ± 7.3 | 61.5 [55.5, 65.0] | ≥50 |
| LVEDD (mm) | 47.9 ± 7.2 | 49.0 [45.3, 53.0] | <57 |
| LVESD (mm) | 31.8 ± 5.8 | 33.5 [27.5, 35.8] | 21–40 |
| Posterior wall thickness (mm) | 10.9 ± 1.5 | 10.0 [10.0, 12.0] | 6–10 |
| Relative wall thickness | 0.47 ± 0.10 | 0.44 [0.40, 0.48] | 0.22–0.42 |
| Calculated LV mass (g) | 195.8 ± 60.6 | 173.0 [144.3, 233.2] | 67–162 |
| Normal diastolic function (%) | 2 (17) | ||
| Grade I diastolic dysfunction (%) | 7 (58) | ||
| Grade 2 diastolic dysfunction (%) | 2 (17) | ||
| Grade 3 diastolic dysfunction (%) | 1 (8) | ||
| Medications (%) | |||
| Diuretic (loop) | 11 (92) | ||
| ACE-I/ARB | 5 (42) | ||
| Beta-blocker | 10 (83) | ||
| Calcium channel blocker | 3 (25) | ||
| Statin | 11 (92) | ||
| Insulin/OHG | 7 (58) | ||
| Aspirin | 6 (50) | ||
| Biomarkers | |||
| Troponin (ng/mL) | 0.020 [0.006, 0.033] | <0.033 | |
| BNP (pg/mL) | 257.5 [75.0, 500.3] | 0 – 176 | |
| BUN (mg/dL) | 36.3 ± 21.0 | 28.0 [22.5, 51.5] | 7 – 25 |
| Creatinine (mg/dL) | 1.89 ± 1.0 | 1.56 [1.28, 2.61] | 0.7 – 1.3 |
| MDRD eGFR (mL/min/1.73m2) | 38.8 ± 13.9 | 38.70 [28.6, 51.7] | >60 |
| Glucose (mg/dL) | 147.9 ± 73.6 | 121.0 [92.3, 199.8] | 70–100 |
| Sodium (mmol/L) | 140.0 ± 3.1 | 139 [138, 143] | 135–145 |
| Potassium (mmol/L) | 4.2 ± 0.5 | 4.2 [3.9, 4.5] | 3.1 – 5.3 |
Data are expressed as mean ± SD for continuous variables or numbers or percent (%) for categorical variables. BNP: brain natriuretic peptide; BUN: blood urea nitrogen; BMI: body mass index; MDRD eGFR: glomerular filtration rate by Modification of Diet in Renal Disease equation; IQR: interquartile range; IVS: Intraventricular septal thickness; LV: Left ventricular; LVEF: Left Ventricular Ejection Fraction; LVEDD: LV End Diastolic Diameter; LVESD: LV End Systolic Diameter; OHG: Oral hypoglycemic agents; ACE-I: angiotensin-converting enzyme-inhibitor; ARB: angiotensin receptor blocker; NYHA: New York Heart Association; TnI: troponin I. Grade I diastolic dysfunction: impaired relaxation; Grade 2 diastolic dysfunction: pseudo-normal filling pattern; Grade 3 diastolic dysfunction: reversible restrictive filling pattern.
Table 2:
Characteristics of Control subjects used to investigate endo-MT activation in vitro
| Control (N=12) | Values |
|---|---|
| Age (years) | 57 ± 14 |
| Sex: Male/Female (%) | 5/7 (42/58) |
| Race: White/Black/Other (%) | 9/1/2 (75/8/17) |
| BMI kg/m2 | 26.9 ± 1.5 |
| Comorbidities (0%) | |
| Hypertension | 3 (25%) |
| Obesity: BMI>30 kg/m2 | 0 |
| Type 2 diabetes | 0 |
| Atrial fibrillation/Atrial flutter | 0 |
| Medications (%) | |
| Diuretic | 2 (17) |
| ACE-I/ARB | 2 (17) |
| Beta-blocker | 0 |
| Calcium channel blocker | 0 |
| Aspirin | 2 (17) |
| Statin | 5 (42) |
Data are expressed as mean ± SD for continuous variables or numbers or percent (%) for categorical variables. BMI: body mass index; ACE-I: angiotensin-converting enzyme-inhibitor; ARB: angiotensin receptor blocker.
Statistics:
Data are shown as mean ± SEM for experimental studies. For analysis of human HFpEF characteristics, continuous variables were expressed as mean ± standard deviation, and categorical variables were expressed as number of patients and percentages or median (interquartile range). Statistical significance of differences was assessed using the Student t test (2-sided). In those cases when data were not sampled as a normal distribution, nonparametric Mann–Whitney U test was used. P≤0.05 values were considered significant. All statistical tests were performed using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA).
RESULTS
Renal hypertrophy and fibrosis in mice with HFpEF
We and others previously demonstrated that 1% NaCl drinking water, unilateral nephrectomy, and chronic infusion of aldosterone induces HFpEF in mice (this is also termed the SAUNA model13 -an acronym for salty drinking water, unilateral nephrectomy, and chronic aldosterone). These HFpEF mice recapitulate human HFpEF by demonstrating exercise intolerance16, 17, lung congestion12, 13, LV hypertrophy14, 15, and hemodynamic evidence of diastolic dysfunction characterized by higher LV end-diastolic pressure, reduced diastolic relaxation (–dP/dt), and prolonged time constant of pressure decay (τ)18, 19 while LVEF remains preserved (i.e. > 50%).
Similar to prior studies12–15, HFpEF mice showed LV hypertrophy, as measured by the LV weight-to-total body weight ratio, (4.5 ± 0.2 mg/g in HFpEF vs. 3.3 ± 0.1 mg/g in Sham; P=0.0003) and increased lung congestion, defined as the increase in wet-to-dry lung ratio, in HFpEF vs. Sham mice (4.3 ± 0.1 vs. 4.1 ± 0.1; P=0.04). Systolic blood pressure was increased in HFpEF (152.3 ± 5.2 mmHg) compared to Sham mice (123.8 ± 3.2 mmHg; P=0.0009).
Kidney size and morphology in HFpEF mice demonstrated renal hypertrophy, as shown by an increase in relative kidney weight compared to Sham mice (12.0 ± 0.2 vs. 8.3 ± 0.2 mg/g of body weight, P=0.0000004; Figure 1A–B). Glomerular area was also increased 28 % in HFpEF mice (P=0.004 vs. Sham; Figure 1B), as previously shown20. There were no differences in tubular size between HFpEF and Sham mice. Collagen deposition by Pricosirius red staining showed significantly increased periglomerular and interstitial fibrosis by 1.9-fold in the kidney of HFpEF mice (P=0.03 vs. Sham; Figure 2A–B). These results were accompanied by a parallel and significant increase in the expression of renal fibrotic transcripts including transforming growth factor beta1 (tgf-β1), collagen 1a and collagen 3a by 1.7- (P= 0.0008), 4.8- (P=0.005) and 3.0-fold (P=0.0004) respectively vs. Sham (Figure 2C).
Figure 1: Renal size and morphology in HFpEF mice.
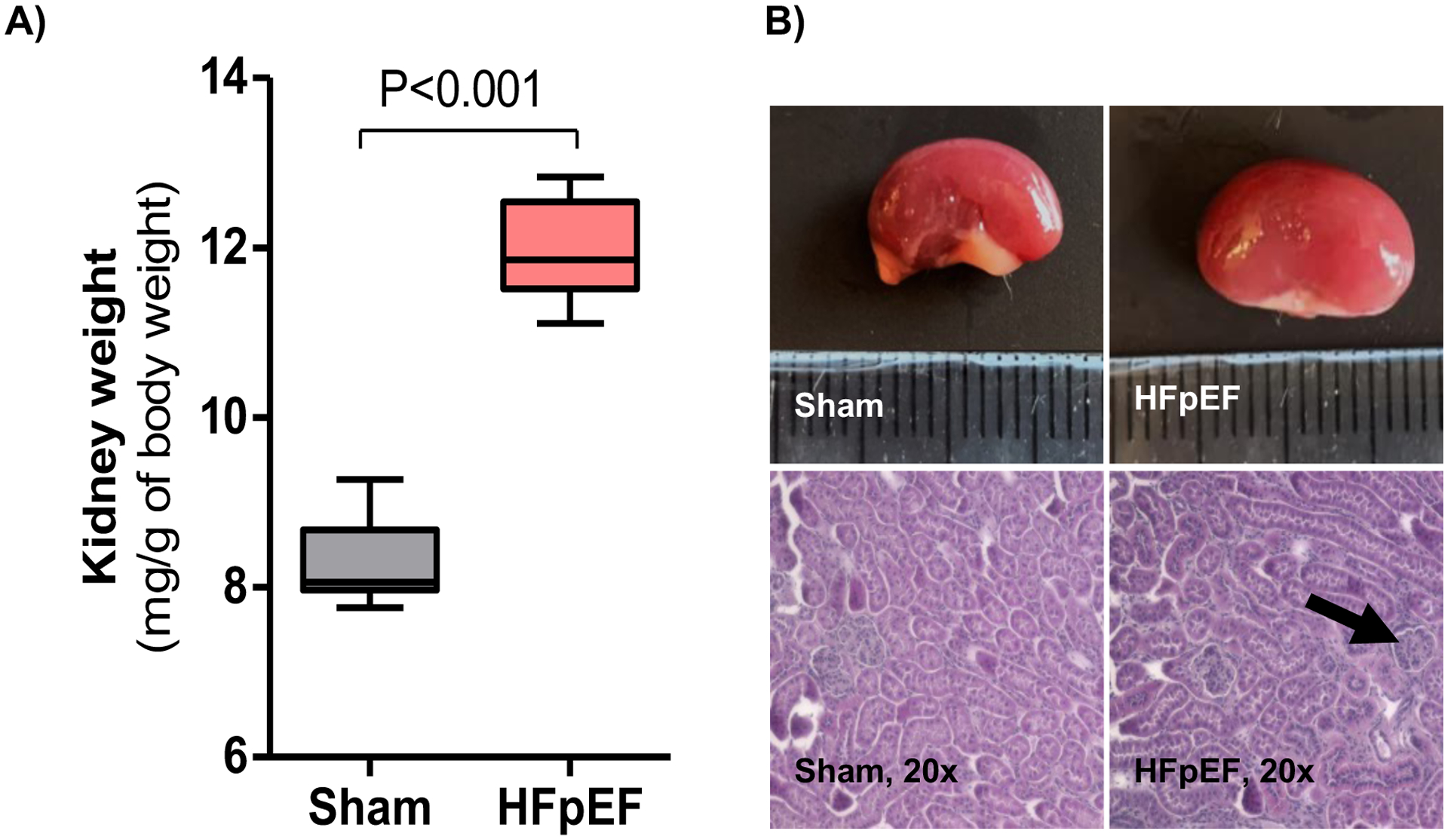
(A) Kidney weight relative to body weight. n = 7–10/group. (B) Representative macroscopic and microscopic (hematoxylin-eosin staining, ×20) images of the kidneys from Sham and HFpEF mice (black arrow indicates hypertrophied glomerulus). Statistical analysis by 2-tailed Student t-test.
Figure 2: Renal fibrosis in HFpEF mice.
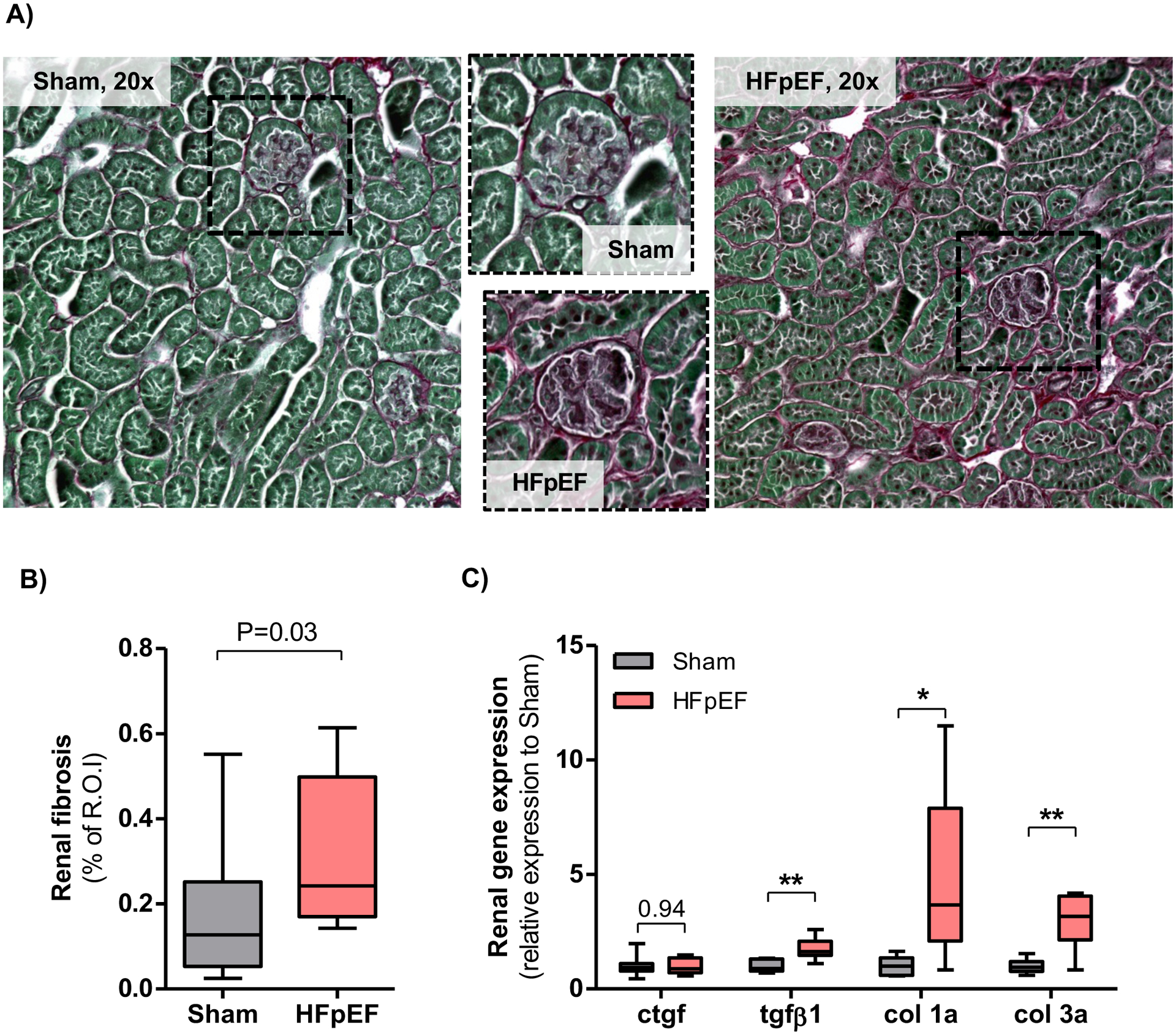
(A) Representative staining of collagen deposition (Picrosirius red, ×20) in kidneys from Sham and HFpEF mice. (B) Quantification of renal fibrosis with Pricosirius red staining in Sham and HFpEF mice. n = 10/group; R.O.I is region of interest. (C) Analysis of gene expression of fibrotic markers (connective tissue growth factor [ctgf], transforming growth factor beta1 [tgf-β1], collagen 1a [col 1a] and collagen 1a [col 3a]) in the kidneys of Sham and HFpEF mice. n = 8–10/group. Statistical analysis by 2-tailed Student t test for normally distributed data or Mann-Whitney U for those variables that were non-normally distributed. *P<0.01; **P<0.001.
Renal endo-MT activation in mice with HFpEF
To examine potential activators of the observed fibrotic phenotype, molecular analysis and immunostaining were used to determine the presence of endo-MT activation. There were marked phenotypic differences in endo-MT mRNA transcripts in the kidneys of HFpEF mice vs. Sham. These included a significant increase of platelet-derived growth factor receptor beta (pdgfrβ), snail (snail), fibronectin (fn), fibroblast-specific protein 1 (fsp1) and vimentin by 1.7- (P=0.004), 1.7- (P=0.05), 1.8- (P=0.005), 2.6- (P=0.001) and 2.0-fold (P=0.0001), respectively as compared to Sham (Figure 3A). Significant results were confirmed by western blot and showed an increase in FSP1 and vimentin protein expression in HFpEF by 1.8- (P=0.03) and 2.9-fold (P=0.008), respectively vs. Sham (Figure 3B).
Figure 3: Endothelial-mesenchymal transition signature in kidneys from HFpEF mice.
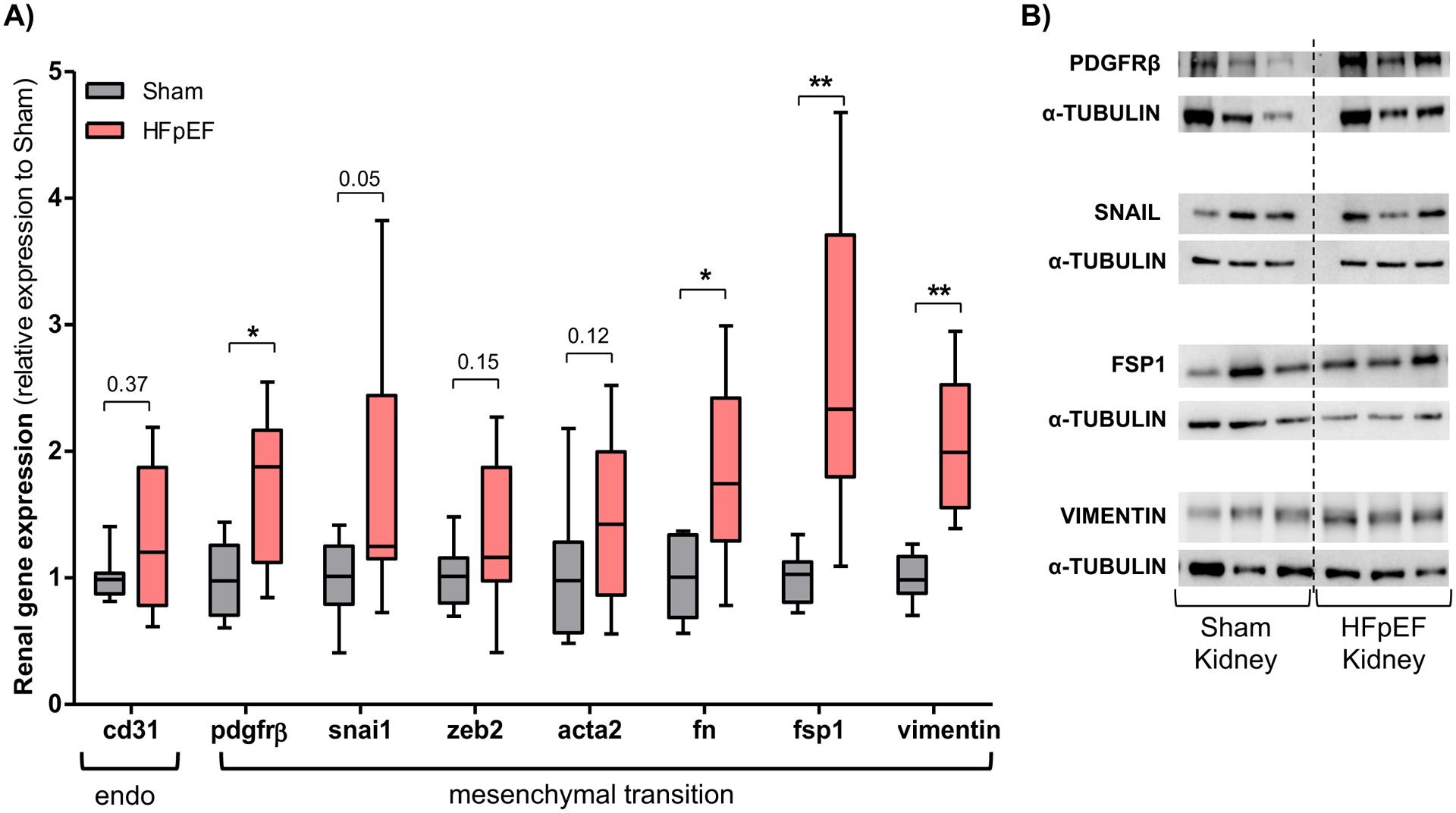
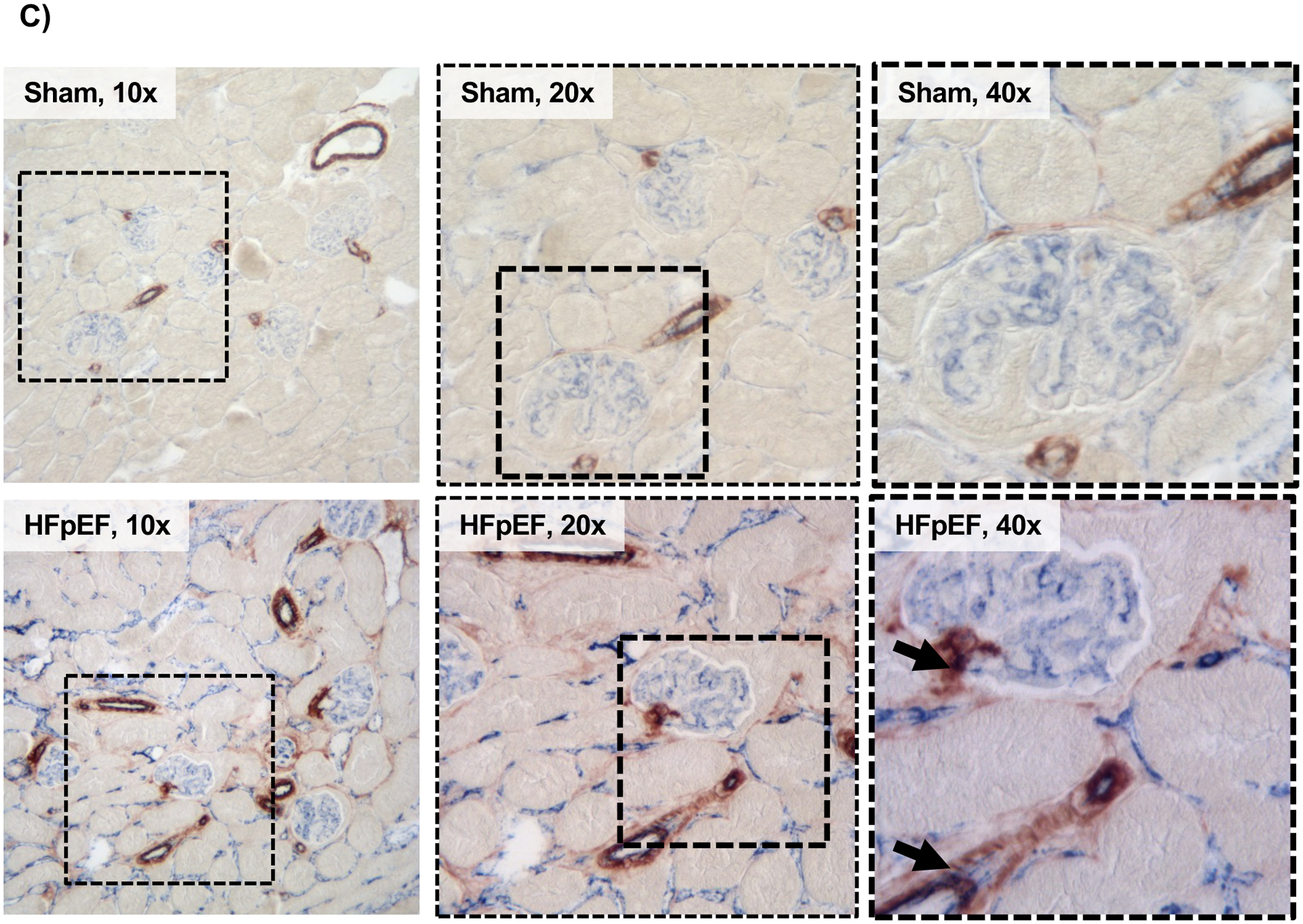
(A) Gene expression analysis of cluster of differentiation 31 (cd31), platelet-derived growth factor receptor beta (pdgfrβ), snail (snai1), zinc finger E-box-binding homeobox 2 (zeb2), smooth muscle actin alpha 2 (acta2), fibronectin (fn), fibroblast-specific protein 1 (fsp1) and vimentin in the kidneys of Sham and HFpEF mice. n = 8–9/group (B) Representative blots of PDGFRβ, snail, FSP1 and vimentin. (C) Renal immunostaining of CD31 and smooth muscle actin alpha 2 (ACTA2) in Sham and HFpEF mice, showing CD31 (blue) and ACTA2 (brown) staining but only in HFpEF were there double-stained cells (black arrows; bottom panel, 40X). Squared area shows magnification. Statistical analysis by 2-tailed Student t test for normally distributed data or Mann-Whitney U for those variables that were non-normally distributed. *P<0.01; **P<0.001.
Immunostaining showed a significant increase in the endothelial marker CD31 and the myofibroblast marker ACTA2 in the kidneys of HFpEF mice compared to Sham. Importantly co-localization of CD31 and ACTA2 staining was seen only in HFpEF mice (Figure 3C). These findings suggested that endo-MT may account for a considerable portion of the fibrotic renal phenotype in HFpEF.
Cardiac fibrosis and endo-MT activation in mice with HFpEF
Similarly to the kidney, cardiac fibrosis was increased in HFpEF as seen by a significant increase in collagen 1a and collagen 3a transcript expression by 1.6- and 1.4-fold, respectively in HFpEF mice vs. Sham (P= 0.04 for both). These results were accompanied by a similar increase in tgf-β1 mRNA expression by 1.2-fold (P=0.04) vs. Sham. However, when we examined the presence of endo-MT in the hearts of HFpEF mice by qPCR we found that only the myofibroblast markers fsp1 and vimentin were increased by1.4- (P=0.05) and 1.9-fold (P=0.04) respectively, whereas the remaining levels of endothelial and mesenchymal transition markers in the heart (CD31, pdgfrβ, snai1, zeb2, acta2, fn) were no different between HFpEF and Sham mice (Figure 4).
Figure 4: Endothelial-mesenchymal transition cardiac expression in HFpEF mice.
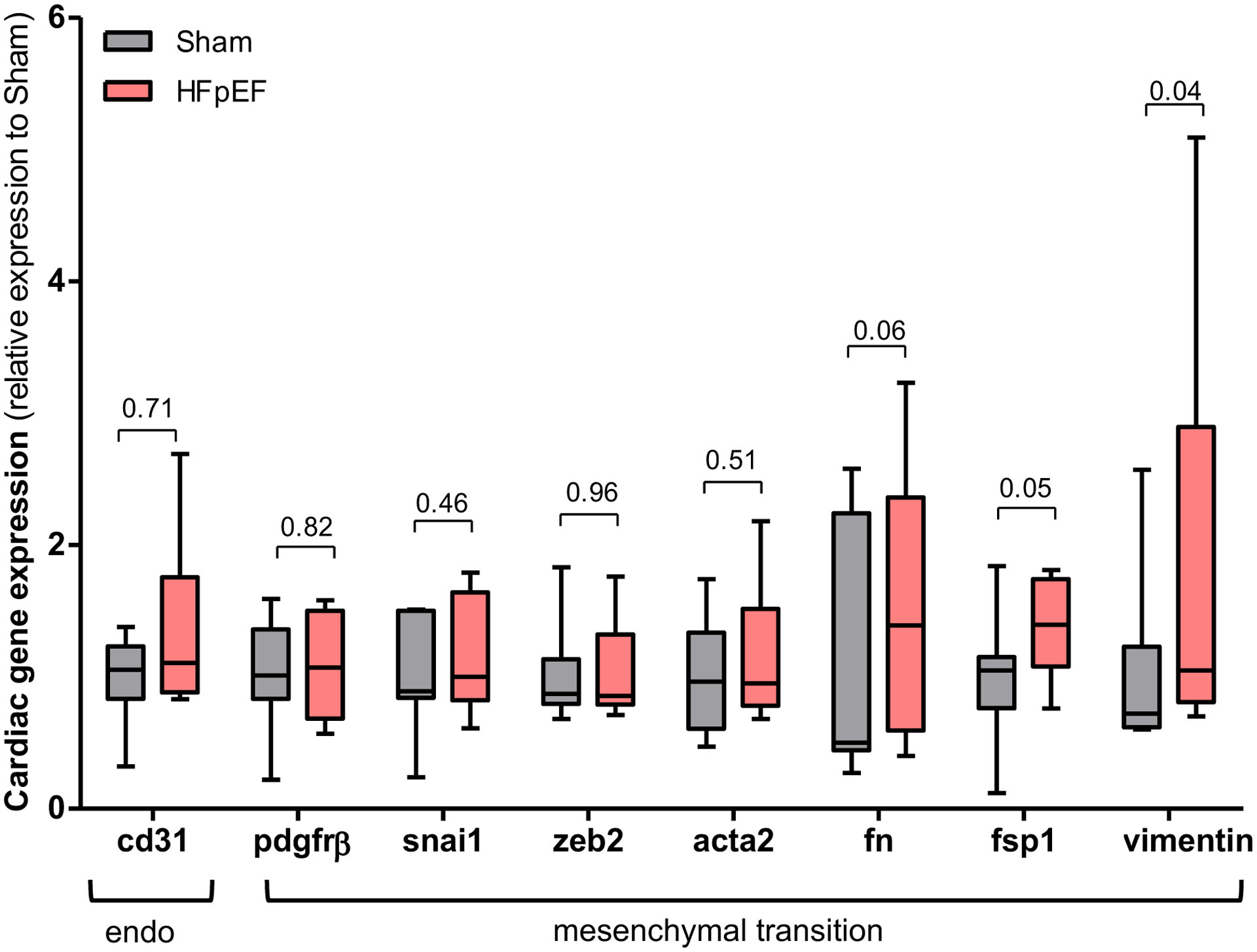
Gene expression analysis of cluster of differentiation 31 (cd31), platelet-derived growth factor receptor beta (pdgfrβ), snail (snai1), zinc finger E-box-binding homeobox 2 (zeb2), smooth muscle actin alpha 2 (acta2), fibronectin (fn), fibroblast-specific protein 1 (fsp1) and vimentin in the hearts of Sham and HFpEF mice. n = 7–11/group. Statistical analysis by 2-tailed Student t test for normally distributed data or Mann-Whitney U for those variables that were non-normally distributed.
Serum from patients with HFpEF and CRS induces endo-MT in vitro
Having shown that endo-MT was involved in kidney fibrosis in HFpEF mice, we sought to further investigate the translational and clinical implication of these findings. To better understand whether endo-MT was similarly activated in patients with HFpEF and CRS, human aortic endothelial cells (HAECs) were treated with either 10% serum from HFpEF patients with CRS or age-matched control subjects without cardiovascular or renal disease (Figure 5A).
Figure 5: Endothelial-mesenchymal transition is activated in HAECs treated with serum from HFpEF patients.
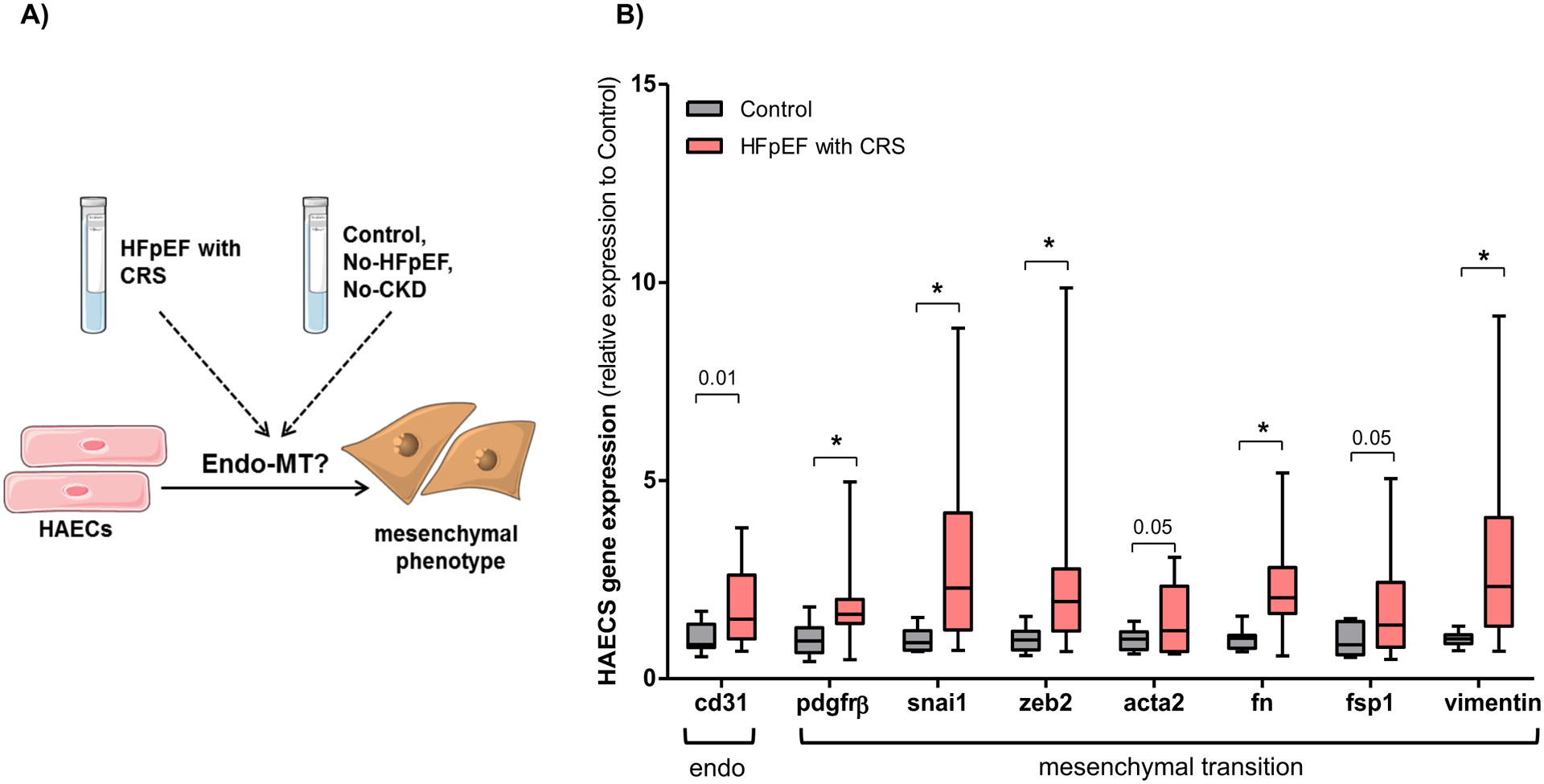
(A) Experimental design: HAECs were treated with 10% serum obtained from HFpEF patients with cardiorenal syndrome (CRS) and healthy control subjects without known cardiovascular or renal diseases for 4 days. (B) Gene expression analysis of cluster of differentiation 31 (cd31), platelet-derived growth factor receptor beta (pdgfrβ), snail (snai1), zinc finger E-box-binding homeobox 2 (zeb2), smooth muscle actin alpha 2 (acta2), fibronectin (fn), fibroblast-specific protein 1 (fsp1) and vimentin in HAECs treated with HFpEF patients (n = 12) and controls (n = 12) serum. Statistical analysis by 2-tailed Student t test for normally distributed data or Mann-Whitney U for those variables that were non-normally distributed. *P<0.01.
Chronic, stable HFpEF patients were recruited from an ambulatory, outpatient HF clinic (Table 1). The mean age was 68 ± 11 years, 75 % were women and 67 % and were black. Comorbidities included hypertension (100 %), obesity (92 %), type 2 diabetes mellitus (67 %) and atrial fibrillation/atrial flutter (42 %). The mean New York Heart Association (NYHA) functional class was 1.9 ± 0.5 at the time of enrollment. Impaired diastolic function was present in 83 % of these patients. Mean LVEF and LV mass were 60.9 ± 7.3 % and 195.8 ± 60.6 g, respectively. Evidence of cardiac remodeling included increased mean posterior wall thickness (10.9 ± 1.5 mm) and relative wall thickness (0.47 ± 0.1). Impaired renal function and the CRS were characterized by a decreased MDRD eGFR (glomerular filtration rate by modification of diet in renal disease equation) of 38.8 ± 13.9 mL/min/1.73 m2 and an increased serum creatinine. Controls subjects were healthy, with a mean age of 58 ± 14 years, and 58 % were women. Hypertension was present in 25 % of control subjects (Table 2).
Similar to the in vivo results obtained in the kidneys of HFpEF mice, HAECs treated with serum from patients with chronic HFpEF and CRS demonstrated a significant increase in the mesenchymal transition markers, pdgfrβ, snail and zeb2 by 2.0- (P=0.0075), 2.9- (P=0.0095) and 2.6-fold (P=0.0074), respectively, and the fibroblast specific markers acta2, fibronectin, fsp1 and vimentin by 1.5- (P=0.05), 2.3- (P=0.003), 1.9- (P=0.05), 2.9-fold (P=0.006), respectively as compared to HAECs-treated with control serum (Figure 5B). The endothelial marker CD31 was also markedly increased by 1.9-fold in HAECs treated with serum from HFpEF patients compared to control serum (P=0.01).
DISCUSSION
Complex, bidirectional, dysfunctional cross-talk between the heart and the kidneys occurs in HFpEF with CRS. Despite the presence of other comorbidities in HFpEF, hypertension is the most common comorbidity in HFpEF patients and it is implicated in both the pathogenesis and the prognosis of the disease21, 22. In the present study, renal hypertrophy with increased glomerular size was evident in a murine model of hypertension-associated HFpEF. This was accompanied by increased fibrosis and augmented expression of collagen 1a, 3a and tgf-β1 transcripts in both the kidneys and hearts of HFpEF mice. Although there was an increase in both mesenchymal transition markers (pdgfrβ and snai1) and myofibroblast markers (FN, fsp1 and vimentin) in the HFpEF kidneys, there was only an increase in myofibroblast markers (fsp1 and vimentin) in the HFpEF heart. Importantly, translational findings showed that HAECs treated with serum from patients with HFpEF and CRS were comparable with in vivo findings obtained in the HFpEF kidney. There was an increase in the mesenchymal transition markers (pdgfrβ, snail and zeb2), the fibroblast specific markers (acta2, fibronectin, fsp1 and vimentin) and the endothelial marker CD31.
Myofibroblasts are mediators of fibrosis in many organs including the kidney11. As such they are rarely found in healthy human physiology but are markedly up-regulated after injury23. Activated myofibroblasts are comprised of a unique population of mesenchymal cells that express ACTA2 and exhibit a marked profibrotic phenotype with increased production of collagens leading to enhanced ECM deposition10. We sought to investigate the potential pathophysiological mechanisms implicated in the increased fibrosis seen in HFpEF kidneys. Myofibroblasts originate from several sources such as (i) expansion and activation of quiescent resident fibroblasts, (ii) migration and tissue accumulation of bone marrow-derived fibrocytes, (iii) phenotypic transition of epithelial cells by EMT, or from (iv) other cell types such as pericytes, adipocytes and macrophages10. Additionally, the contribution of endothelial cells to fibrosis has become increasingly recognized as endo-MT is implicated in fibrosis in other organs24. In the present study, we investigated the role of endothelial cells in the pathogenesis of HFpEF with CRS and found that fibrosis seen in the HFpEF kidney was temporally associated with the transdifferentiation of endothelial cells to myofibroblasts. We found that in HFpEF kidneys there was an increase in the transcripts of endo-MT activation pdgfrβ, and snai1, and this increase was associated with an elevation of myofibroblast markers FN, fsp1 and vimentin suggesting that increased fibrosis may be related to ongoing endo-MT. Importantly, the translational implications of these findings cannot be underscored as serum obtained from HFpEF patients with CRS induced similar activation of endo-MT in HAECs.
In injured kidneys, endothelial cells undergoing endo-MT are characterized by the coexpression of endothelial (CD31) and mesenchymal markers (such as ACTA2 or FSP1)25. We showed that immunostaining of CD31 and ACTA2 were both increased in the kidneys of HFpEF mice with CRS, and there was substantial co-localization of CD31-positive and ACTA2–positive cell populations, which were not observed in Sham mice. These results indicate that molecular changes seen in the HFpEF kidneys were associated with phenotypical cellular changes, demonstrating that endo-MT plays at least some role in renal fibrosis in HFpEF. Therefore, these data align well with prior reports in several mouse models of CKD showing that endo-MT contributes to the accumulation of activated fibroblasts and myofibroblasts in kidney fibrosis11, and suggests that mitigating endo-MT might decrease renal fibrosis26, 27.
Likewise renovascular hypertension, which causes both renal and cardiac remodeling and damage, associates with markers of endo-MT in both the kidneys and the heart28. We previously characterized cardiac remodeling and cardiac fibrosis in mice with hypertension-associated HFpEF13. Similar to the kidney, tgf-β1, a major stimulus of endo-MT29, was also increased in the heart in HFpEF mice13, 20 as were collagen 1a and collagen 3a levels. Myofibroblasts function to facilitate tissue remodeling and fibrosis and its presence provides a compelling rationale to consider the role endo-MT in the pathophysiology of HFpEF with CRS. Noticeable, at the time-point studied there was a lack of cardiac endo-MT activation in hypertension-associated HFpEF. These data suggest that fibrosis might be motivated by different mechanisms in the kidney versus the heart in HFpEF18 with CRS. Similarly, a preclinical study in obese diabetic Zucker fatty/spontaneously hypertensive heart failure rats demonstrated a distinct endothelial cell response with microvascular fibrotic changes in the kidney and heart in HFpEF with CRS30. Our findings provide further evidence that endo-MT is a source of myofibroblasts, which is crucial in tissue fibrosis in the kidney but unclear in the heart in HFpEF (and additional studies are warranted). Interestingly, a small human study showed that successful kidney transplantation was associated with improvement in the cardiac MRI parameters of extracellular volume (ECV) and native T1 (which correlates with histological myocardial fibrosis), and maybe associated with the regression of myocardial interstitial fibrosis31.
In addition to increasing the myofibroblast pool, endothelial cells participate in the inception and progression of fibrosis by also promoting the recruitment of immune cells, participating in vascular rarefaction, and secreting pro-fibrotic mediators24. In the present study, we explored the potential role of endothelial cells in the pathogenesis of HFpEF with CRS in an in vitro system by treating HAECS with serum obtained from chronic, stable, ambulatory HFpEF patients with CRS. This allowed for consideration of the circulating milieu and the associated complex pathophysiology of the HFpEF phenotype in a cellular system. To our knowledge this is the first translational study simulating HFpEF with CRS in vitro.
HAECS treated with serum from HFpEF patients with CRS were characterized by the acquisition of myofibroblasts markers acta2, fn, fsp1 and vimentin, as well as increased levels of pdgfrβ, snail and zeb2 transcripts compared to HAECs treated with serum from control subjects (with no known cardiovascular or renal disease). These results were similar to our in vivo kidney data, providing further evidence of the importance of endo-MT activation in the kidney in mice and human with HFpEF and CRS.
The interplay between HFpEF and renal dysfunction in patients with CRS leads to highly complex and challenging clinical scenarios and thus, identifying therapeutic interventions to improve outcomes is needed. It was recently postulated that fibrosis is not only a common feature but also a primary promoter of the pathophysiology in the CRS8. Importantly, there is increasing recognition of the coexistence of renal impairment in HFpEF but clinical trials enrolling these patients are still lacking as patients with renal dysfunction are often excluded32. Unlike HFrEF with CRS, the involvement of the kidney may be underdiagnosed in HFpEF patients and optimal therapeutic strategies have yet to be clarified4.
Interestingly, drugs that inhibit endo-MT target a broad range of signaling molecules such as DPP-4, Smad, TGF-β, AMPK etc. but none of these drugs have been studied in HFpEF with CRS. These include nintedanib which inhibits endo-MT by inhibiting kinases and cell proliferation in pulmonary fibrosis33. Macitentan, which abrogated adverse cardiac remodeling and tgf-β13, also inhibited TGF-β- and ET-1-mediated endo-MT in HAECs from patients with systemic sclerosis34. Diabetic complications and end-organ damage, appear to involve endo-MT. Vildagliptin, for diabetes, regulates DPP-4-dependent endo-MT and demonstrated anti-fibrotic effects in murine sepsis35. Liraglutide, a GLP-1 receptor agonist approved for treating diabetes and its complications, similarly inhibited endo-MT in diabetic mice and during in vitro conditions36. Additionally, imatinib, a PDGF receptor antagonist, regulated endo-MT in pulmonary artery remodeling in pulmonary artery hypertension37.
Our findings indicate that fibrosis is a hallmark of HFpEF with CRS. Both the kidneys and hearts from HFpEF mice showed evidence of increased fibrosis which may be due to an increase of the myofibroblast pool. We demonstrated that endo-MT in the kidney plays a role in this increased myofibroblast pool but cannot rule out the contribution of additional cell sources, such as pericytes, fibrocytes, resident fibroblasts, etc., or the role of EMT. However, in contrast, we did not observe the presence of endo-MT in the heart in chronic HFpEF. Future studies are therefore warranted to investigate if this is a temporal limitation (i.e. at the time point studied there is no longer evidence of endo-MT in the heart) or if there are other factors at play in cardiac myofibroblast proliferation in HFpEF. For example, we previously demonstrated the contribution of bone-marrow derived monocytes and macrophages to the cardiac fibrosis phenotype in HFpEF18. Importantly, since our translational in vitro experiments showed transdifferentiation of endothelial cells to myofibroblast, endo-MT likely also plays a pivotal role in tissue fibrosis in other organs (because of the presence of endothelial cells) during HFpEF with CRS (Figure 6).
Figure 6: Central Figure. Fibrosis is a hallmark of HFpEF with CRS.
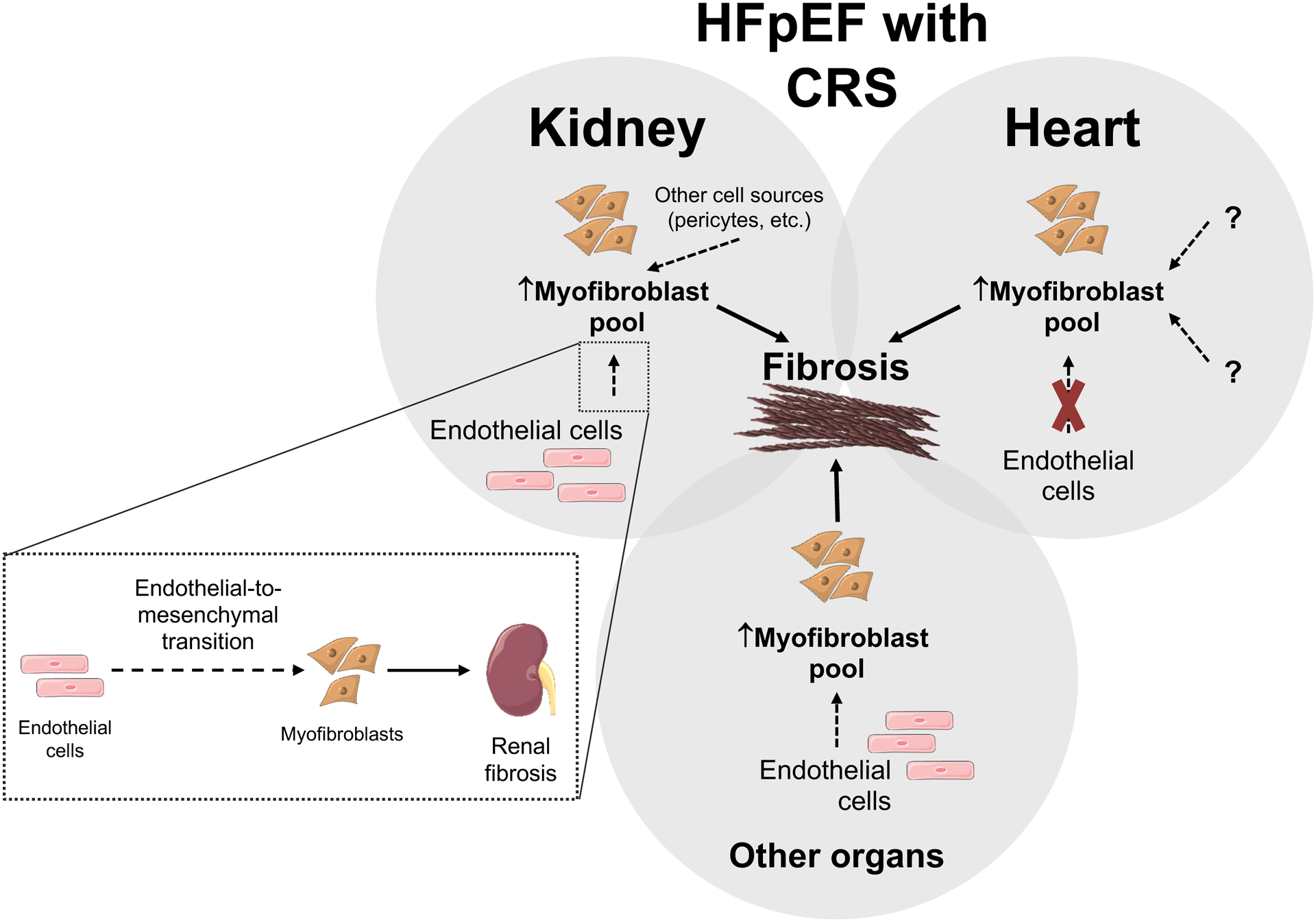
Both heart and kidneys from HFpEF mice showed evidence of increased fibrosis (compared to mice without HFpEF) which appears due to an increase of the myofibroblast pool. Endo-MT plays a role in this increased myofibroblast pool in the kidney in HFpEF, however the contribution from other sources, such as pericytes, fibrocytes, resident fibroblasts, etc. cannot be excluded. In contrast, at the time-point studied, there was no evidence of endo-MT activation in the heart in chronic HFpEF. Future studies are warranted to investigate if this lack of endo-MT in the heart is a temporal limitation or that other factors might be implicated in myofibroblast transition and proliferation in the heart in chronic HFpEF. Importantly, the in vitro translational experiments showed evidence of transdifferentiation of endothelial cells to myofibroblast, indicating that endo-MT likely plays a role in tissue fibrosis in HFpEF with CRS.
In conclusion, there is a relentless pursuit for HFpEF therapies, but limited understanding of the mechanisms that underlie HFpEF, particularly in HFpEF with CRS, which hampers drug discovery and ultimately treatment options for this sub-phenotype in HFpEF. In the present study we provide insight into the potential role of endo-MT in HFpEF with CRS, which may provide mechanistic insight for future target-directed therapies in HFpEF with CRS.
Limitations of the study:
The present study supports the hypothesis of a kidney-heart axis in HFpEF. However, the relationship between cardiac and renal failure and how one may affect the other remains unknown. As with all animal models of disease, there are some limitations of animal models of HFpEF38; however, the SAUNA model of HFpEF has been widely validated as a model of clinical HFpEF13, 14, 16, 19, 39 and is not just simply LV hypertrophy with CKD. It also shares multiple features with other purported HFpEF murine models such as mild transverse aortic constriction model12, 17 and the HFpEF aging model18. Finally, incubating HAECs with serum from HFpEF patients with CRS, remains observational. Therefore, future studies are warranted to provide mechanistic insights into the role of endo-MT in CRS in HFpEF. Additionally, therapeutic approaches should be tested in other models of HFpEF, including larger animal species (e.g. swine and sheep)40. In addition to hypertension other comorbidities such as weight gain, insulin resistance and hyperlipidemia should be considered as they combine hemodynamic load with metabolic stress41.
SHORT COMMENTARY.
What is new?
Cardiorenal Syndrome (CRS) describes the concomitant pathophysiological organ dysfunction of both the heart and the kidney that poses a significant challenge to the management of heart failure, particularly heart failure with preserved ejection fraction (HFpEF).
Our findings indicate that Endo-MT may play a pathogenic role in kidney fibrosis in HFpEF through bidirectional, crosstalk between the heart and the kidneys.
What are the clinical implications?.
Current therapeutic strategies for clinical management of HFpEF are limited to symptom control without evidenced based therapies to slow or reverse the disease progression.
Renal dysfunction, present in approximately 25–50% of patients with HFpEF, hampers medical management of HFpEF and chronic kidney disease (CKD) in HFpEF is an important independent predictor of poor clinical outcomes.
Elucidating the mechanisms of disease in CRS in HFpEF has the potential to uncover targetable pathways and opportunities for pharmacologic intervention to improve outcomes in HFpEF.
SOURCES OF FUNDING
This work was supported by the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Numbers RO1HL145985 to FS. AO was supported by T32HL125232 and FA by T32HL007224.
Non-standard Abbreviations and Acronyms
- ACTA2
actin alpha 2 (smooth muscle)
- AMPK
AMP-activated protein kinase
- BNP
brain natriuretic peptide
- BUN
blood urea nitrogen
- BMI
body mass index
- CD31
cluster of differentiation 31
- CRS
cardiorenal syndrome
- CKD
chronic kidney disease
- DPP-4
dipeptidyl peptidase-4
- EMT
epithelial-to-mesenchymal transition
- Endo-MT
endothelial-to-mesenchymal transition
- ET-1
endothelin 1
- FSP1
fibroblast-specific protein 1
- FN
fibronectin
- GLP-1
glucagon-like peptide 1
- HAECs
human aortic endothelial cells
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- IVS
Intraventricular septal thickness
- LV
left ventricular
- LVEF
left ventricular ejection fraction
- LVEDD
left ventricular end diastolic diameter
- LVESD
left ventricular end systolic diameter
- MDRD eGFR
glomerular filtration rate by modification of diet in renal disease equation
- OHG
oral hypoglycemic agents
- ACE-I
angiotensin-converting enzyme-inhibitor
- ARB
angiotensin receptor blocker
- NYHA
New York Heart Association
- PDGFRβ
platelet-derived growth factor receptor beta
- SAUNA
salty drinking water, unilateral nephrectomy, and chronic aldosterone
- SNAI1
snail family transcriptional repressor 1
- TGFβ1
transforming growth factor beta 1
- TnI
troponin I
- ZEB2
zinc finger E-box-binding homeobox 2
Footnotes
DISCLOSURES
The authors have declared that there are no conflicts of interest.
REFERENCES
- (1).Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- (2).Streng KW, Nauta JF, Hillege HL, Anker SD, Cleland JG, Dickstein K, Filippatos G, Lang CC, Metra M, Ng LL et al. Non-cardiac comorbidities in heart failure with reduced, mid-range and preserved ejection fraction. Int J Cardiol 2018;271:132–139. [DOI] [PubMed] [Google Scholar]
- (3).Brouwers FP, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, Hillege HL, van Veldhuisen DJ, van Gilst WH. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J 2013;34:1424–1431. [DOI] [PubMed] [Google Scholar]
- (4).Lazzeri C, Valente S, Tarquini R, Gensini GF. Cardiorenal syndrome caused by heart failure with preserved ejection fraction. Int J Nephrol 2011;2011:634903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Agrawal A, Naranjo M, Kanjanahattakij N, Rangaswami J, Gupta S. Cardiorenal syndrome in heart failure with preserved ejection fraction-an under-recognized clinical entity. Heart Fail Rev 2019;24:421–437. [DOI] [PubMed] [Google Scholar]
- (6).Damman K, Testani JM. The kidney in heart failure: an update. Eur Heart J 2015;36:1437–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- (8).Zannad F, Rossignol P. Cardiorenal Syndrome Revisited. Circulation 2018;138:929–944. [DOI] [PubMed] [Google Scholar]
- (9).Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J Clin Med 2016;5. 11;5(4):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 2008;19:2282–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Valero-Munoz M, Li S, Wilson RM, Hulsmans M, Aprahamian T, Fuster JJ, Nahrendorf M, Scherer PE, Sam F. Heart Failure With Preserved Ejection Fraction Induces Beiging in Adipose Tissue. Circ Heart Fail 2016;9:e002724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Valero-Munoz M, Li S, Wilson RM, Boldbaatar B, Iglarz M, Sam F. Dual Endothelin-A/Endothelin-B Receptor Blockade and Cardiac Remodeling in Heart Failure With Preserved Ejection Fraction. Circ Heart Fail 2016;9:e003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tanaka K, Wilson RM, Essick EE, Duffen JL, Scherer PE, Ouchi N, Sam F. Effects of adiponectin on calcium-handling proteins in heart failure with preserved ejection fraction. Circ Heart Fail 2014;7:976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tanaka K, Valero-Munoz M, Wilson RM, Essick EE, Fowler CT, Nakamura K, van den Hoff M, Ouchi N, Sam F. Follistatin like 1 Regulates Hypertrophy in Heart Failure with Preserved Ejection Fraction. JACC Basic Transl Sci 2016;1:207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wilson RM, De Silva DS, Sato K, Izumiya Y, Sam F. Effects of fixed-dose isosorbide dinitrate/hydralazine on diastolic function and exercise capacity in hypertension-induced diastolic heart failure. Hypertension 2009;54:583–590. [DOI] [PubMed] [Google Scholar]
- (17).Yoon S, Kim M, Lee H, Kang G, Bedi K, Margulies KB, Jain R, Nam KI, Kook H, Eom GH. S-Nitrosylation of Histone Deacetylase 2 by Neuronal Nitric Oxide Synthase as a Mechanism of Diastolic Dysfunction. Circulation 2021. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- (18).Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med 2018;215:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yang HJ, Kong B, Shuai W, Zhang JJ, Huang H. MD1 deletion exaggerates cardiomyocyte autophagy induced by heart failure with preserved ejection fraction through ROS/MAPK signalling pathway. J Cell Mol Med 2020; 24(16):9300–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sam F, Duhaney TA, Sato K, Wilson RM, Ohashi K, Sono-Romanelli S, Higuchi A, De Silva DS, Qin F, Walsh K et al. Adiponectin deficiency, diastolic dysfunction, and diastolic heart failure. Endocrinology 2010;151:322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, Chirinos JA, Collins S, Deo RC, Gladwin MT et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 2020;141:1001–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tromp J, Claggett BL, Liu J, Jackson AM, Jhund PS, Kober L, Widimsky J, Boytsov SA, Chopra VK, Anand IS et al. Global Differences in Heart Failure With Preserved Ejection Fraction: The PARAGON-HF Trial. Circ Heart Fail 2021;14:e007901. [DOI] [PubMed] [Google Scholar]
- (23).Gabbiani G The myofibroblast in wound healing and fibrocontractive diseases. J Pathol 2003;200:500–503. [DOI] [PubMed] [Google Scholar]
- (24).Sun X, Nkennor B, Mastikhina O, Soon K, Nunes SS. Endothelium-mediated contributions to fibrosis. Semin Cell Dev Biol 2020;101:78–86. [DOI] [PubMed] [Google Scholar]
- (25).Lovisa S, Fletcher-Sananikone E, Sugimoto H, Hensel J, Lahiri S, Hertig A, Taduri G, Lawson E, Dewar R, Revuelta I et al. Endothelial-to-mesenchymal transition compromises vascular integrity to induce Myc-mediated metabolic reprogramming in kidney fibrosis. Sci Signal 2020;13:eaaz2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chen X, Ge W, Dong T, Hu J, Chen L, Fan X, Gong Y, Zhou H. Spironolactone inhibits endothelial-mesenchymal transition via the adenosine A2A receptor to reduce cardiorenal fibrosis in rats. Life Sci 2019;224:177–186. [DOI] [PubMed] [Google Scholar]
- (27).Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, Yamamoto H, Bertram JF. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 2010;59:2612–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ferraz LR, Moreira BC, de Queiroz GSR, Formigari RF, Esquisatto MAM, Felonato M, Alves AA, Thomazini BF, de Oliveira CA. Tissue-specific transcriptional regulation of epithelial/endothelial and mesenchymal markers during renovascular hypertension. Mol Med Rep 2019;20:4467–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ma J, Sanchez-Duffhues G, Goumans MJ, Ten DP. TGF-beta-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front Cell Dev Biol 2020;8:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).van Dijk CG, Oosterhuis NR, Xu YJ, Brandt M, Paulus WJ, van HL, Duncker DJ, Verhaar MC, Fontoura D, Lourenco AP et al. Distinct Endothelial Cell Responses in the Heart and Kidney Microvasculature Characterize the Progression of Heart Failure With Preserved Ejection Fraction in the Obese ZSF1 Rat With Cardiorenal Metabolic Syndrome. Circ Heart Fail 2016;9:e002760. [DOI] [PubMed] [Google Scholar]
- (31).Contti MM, Barbosa MF, Del C,V, Nga HS, Valiatti MF, Takase HM, Bravin AM, de Andrade LGM. Kidney transplantation is associated with reduced myocardial fibrosis. A cardiovascular magnetic resonance study with native T1 mapping. J Cardiovasc Magn Reson 2019;21:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Coca SG, Krumholz HM, Garg AX, Parikh CR. Underrepresentation of renal disease in randomized controlled trials of cardiovascular disease. JAMA 2006;296:1377–1384. [DOI] [PubMed] [Google Scholar]
- (33).Tsutsumi T, Nagaoka T, Yoshida T, Wang L, Kuriyama S, Suzuki Y, Nagata Y, Harada N, Kodama Y, Takahashi F et al. Nintedanib ameliorates experimental pulmonary arterial hypertension via inhibition of endothelial mesenchymal transition and smooth muscle cell proliferation. PLoS One 2019;14:e0214697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cipriani P, Di BP, Ruscitti P, Capece D, Zazzeroni F, Liakouli V, Pantano I, Berardicurti O, Carubbi F, Pecetti G et al. The Endothelial-mesenchymal Transition in Systemic Sclerosis Is Induced by Endothelin-1 and Transforming Growth Factor-beta and May Be Blocked by Macitentan, a Dual Endothelin-1 Receptor Antagonist. J Rheumatol 2015;42:1808–1816. [DOI] [PubMed] [Google Scholar]
- (35).Suzuki T, Tada Y, Gladson S, Nishimura R, Shimomura I, Karasawa S, Tatsumi K, West J. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-mesenchymal transition. Respir Res 2017;18:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tsai TH, Lee CH, Cheng CI, Fang YN, Chung SY, Chen SM, Lin CJ, Wu CJ, Hang CL, Chen WY. Liraglutide Inhibits Endothelial-to-Mesenchymal Transition and Attenuates Neointima Formation after Endovascular Injury in Streptozotocin-Induced Diabetic Mice. Cells 2019;8:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Song S, Zhang M, Yi Z, Zhang H, Shen T, Yu X, Zhang C, Zheng X, Yu L, Ma C et al. The role of PDGF-B/TGF-beta1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell Signal 2016;28:1489–1501. [DOI] [PubMed] [Google Scholar]
- (38).Valero-Munoz M, Backman W, Sam F. Murine Models of Heart Failure with Preserved Ejection Fraction: a “Fishing Expedition”. JACC Basic Transl Sci 2017;2:770–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Garcia AG, Wilson RM, Heo J, Murthy NR, Baid S, Ouchi N, Sam F. Interferon-gamma ablation exacerbates myocardial hypertrophy in diastolic heart failure. Am J Physiol Heart Circ Physiol 2012;303:H587–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Charles CJ , Rademaker MT , Scott NJA , Richards AM. Large Animal Models of Heart Failure: Reduced vs. Preserved Ejection Fraction. Animals (Basel). 2020;10:1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kass DA. Understanding HFpEF With Obesity: Will Pigs Come to the Rescue? JACC Basic Transl Sci 2021;6:171–173. [DOI] [PMC free article] [PubMed] [Google Scholar]