

Summary
An accurate understanding of biomolecular mechanisms and diseases requires information on protein quaternary structure (QS). A critical challenge in inferring QS information from crystallography data is distinguishing biological interfaces from fortuitous crystal-packing contacts. Here, we employ QS conservation across homologs to infer the biological relevance of hetero-oligomers. We compare the structures and compositions of hetero-oligomers, which allow us to annotate 7,810 complexes as physiologically relevant, 1,060 as likely errors, and 1,432 with comparative information on subunit stoichiometry and composition. Excluding immunoglobulins, these annotations encompass over 51% of hetero-oligomers in the PDB. We curate a dataset of 577 hetero-oligomeric complexes to benchmark these annotations, which reveals an accuracy >94%. When homology information is not available, we compare QS across repositories (PDB, PISA, and EPPIC) to derive confidence estimates. This work provides high-quality annotations along with a large benchmark dataset of hetero-assemblies.
Keywords: protein quaternary structure, Protein Data Bank, hetero-oligomers, physiological assembly, crystal interfaces, structural similarity, protein interactions, protein structure, structure prediction, protein complexes benchmark
Graphical abstract
Highlights
-
•
QSalignHET compares the quaternary structure (QS) of hetero-oligomeric complexes
-
•
QS conservation of a complex reliably predicts its physiological relevance
-
•
QSalignHET annotates ~50% of hetero-oligomers in PDB at an error rate of ~6%
-
•
We introduce a manually curated benchmark dataset of hetero-oligomeric structures
The quaternary structure (QS) that a protein adopts in the cell is difficult to determine experimentally. Dey et al. report an automated approach based on the evolutionary conservation of the QS, which analyzes crystallographic data of hetero-oligomeric complexes and identifies their physiologically relevant QS with great accuracy.
Introduction
In the crowded environment of living cells, proteins and other biomolecules continuously interact with each other, forming multi-component complexes. The Protein Data Bank (PDB [Armstrong et al., 2020; Berman et al., 2000]) contains structural information about such complexes, of which a large fraction was solved by X-ray crystallography. However, quaternary structure (QS) information is not readily available from crystallography data because biological contacts between subunits need to be distinguished from crystal lattice contacts.
Interactions mediated by biological and crystal contacts are known to differ in interface size, amino acid composition, and evolutionary sequence conservation (Bahadur et al., 2003; Elcock and McCammon, 2001; Conte et al., 1999; Janin and Rodier, 1995; Chothia and Janin, 1975). Several methods have relied on these properties to discriminate between both types of interfaces, including CFPScore, EPPIC, PreBI, and COMP (Duarte et al., 2012; Liu et al., 2006; Tsuchiya et al., 2006, 2008). Other knowledge-based potentials, including information on B factor and inter-atomic distances, were used in PITA and CFPScore (Liu et al., 2006, 2014; Ponstingl et al., 2003). Alternatively, PISA (Krissinel and Henrick, 2007) and CLusPro (Yueh et al., 2017) have used an energy-based score for predictions. Several works also combined multiple features in machine-learning classifiers, as implemented in Dimovo, IPAC, IchemPic, NOXclass, RPAIAnalyst, PRODIGY-CRYSTAL, or PIACO (Bernauer et al., 2008; Fukasawa and Tomii, 2019; Hu et al., 2018; Jiménez-García et al., 2019; Mitra and Pal, 2011; Silva et al., 2015; Zhu et al., 2006). ProtCID has taken another approach by searching interfaces observed across multiple crystal forms of a protein or its homologs (Xu and Dunbrack, 2020; Xu et al., 2008).
While numerous methods and resources discriminate crystal interfaces from physiologically relevant interfaces, as recently reviewed (Capitani et al., 2016; Dey and Levy, 2018; Elez et al., 2020; Xu and Dunbrack, 2019), it is noteworthy that only a few methods make predictions on the whole protein assembly. PQS first addressed this challenge (Henrick and Thornton, 1998), and currently, the primary such resources are PISA (Krissinel and Henrick, 2007), EPPIC (Bliven et al., 2018), and QSalign (Dey et al., 2018). The latter relies on evolutionary conservation of QS geometry, which was a powerful means to distinguish between crystal lattice and physiological interfaces. Indeed, the method reached a high accuracy, superior to 95% (Dey et al., 2018). Although QSalign was limited to annotating homo-oligomers, the strategy applies to hetero-oligomers as well. However, comparing hetero-oligomers is more complicated than comparing homo-oligomers for several reasons now described.
Previous works involving the comparison of hetero-oligomeric complexes were aimed at measuring their similarity for the purpose of data mining (Berman et al., 2000; Madej et al., 2014) and classification, e.g., for generating non-redundant sets (Bertoni and Aloy, 2018; Koike and Ota, 2012; Levy et al., 2006; Mukherjee and Zhang, 2009; Sippl and Wiederstein, 2012). Here, we carry out such comparisons and integrate their results to evaluate the physiological relevance of a QS. We developed QSalignHET, which analyzes hetero-oligomers with conserved QS geometry. It is noteworthy that comparing the QS of hetero-oligomers raises several challenges that are absent when analyzing homo-oligomers. First, point group symmetries of homo-oligomers mean that different QS states (e.g., monomer versus dimer or dimer versus tetramer) necessarily yield low structural similarity scores. In contrast, different QSs may show high overall structural similarity among hetero-oligomers, e.g., if the difference in structure comes from a subunit that is small relative to other subunits. Second, homo-oligomers composition can be compared readily using a single sequence alignment. In contrast, hetero-oligomers contain multiple gene products so that composition heterogeneity must be considered. Third, the availability of manually curated datasets of physiologically relevant hetero-oligomers for benchmarking purposes is limited. Indeed, the atomic coordinates of two previously published datasets (Chakrabarti and Janin, 2002; Ponstingl et al., 2003) are not available. Other resources such as Docking Benchmark 5 (Vreven et al., 2015) and DOCKGROUND (Kundrotas et al., 2018) provide coordinates for 230 and 396 non-redundant complexes, respectively, but due to their intended use these complexes consist mainly of heterodimers with few larger complexes and no very large assemblies such as the proteasome (Lowe et al., 1995).
In this work, we tackled these challenges. We compared the structure and composition of hetero-oligomers across the PDB and integrated the comparisons with a framework we call QSalignHET. Using QSalignHET, we annotated 10,302 hetero-oligomeric QSs. Among these, we validated 7,810 complexes and identified 1,060 requiring a possible correction. We annotated an additional 1,432 complexes with a different set of relationships, such as the inclusion of a complex into another. To assess the performance of QSalignHET, we curated a benchmark dataset encompassing 577 non-redundant (2,337 total) structures (Table S1). Using this dataset, we benchmarked QSalignHET and subsequently derived confidence estimates across the PDB based on the consensus of PISA, EPPIC, and QSalignHET predictions.
Results and discussion
Comparing the structure of hetero-oligomeric assemblies to infer their physiological relevance
The evolutionary conservation of a QS is a powerful means to assess its physiological relevance. Indeed, we previously employed this concept to annotate homo-oligomeric structures in the PDB (Dey et al., 2018). Theoretically, the same principle of QS geometry conservation (Figure 1) can be used to annotate hetero-oligomeric proteins, but this requires more sophisticated comparisons. For example, calculating the similarity between two homo-oligomers involves comparing two sequences only. In contrast, to compare hetero-oligomers we must first establish subunit-subunit correspondences.
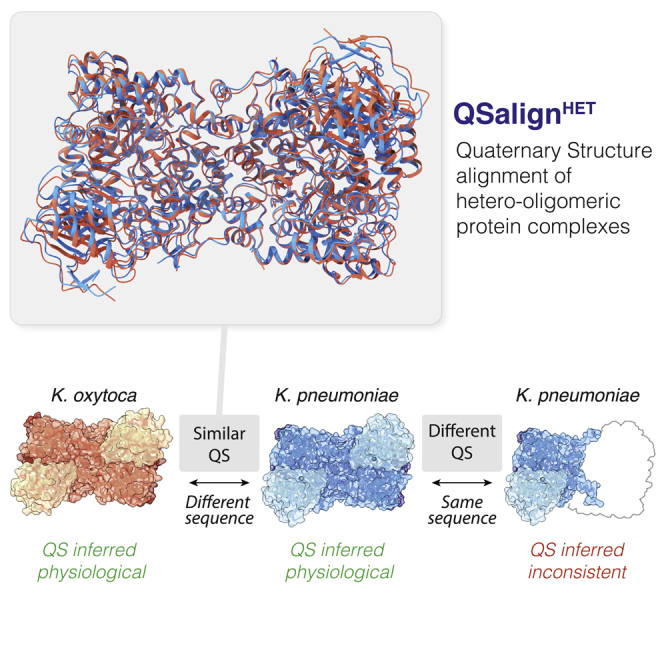
Figure 1.

Principle of QSalignHET to annotate the physiological relevance of hetero-oligomeric protein quaternary structures
Hydroxynitrile lyase is a heterohexameric enzyme in Klebsiella oxytoca (PDB: 1EEX). A glycerol dehydratase from Klebsiella pneumoniae shows a similar quaternary structure (PDB: 1IWP, TM-score 0.9, root-mean-square deviation 1.14 Å), although they share 63% sequence identity on average. Such conservation suggests that the quaternary structure (QS) of both of these hexamers is physiologically relevant. This information enables inferring that the QS of a different entry (PDB: 1MMF), which shares 100% sequence identity with 1IWP, may have missing subunits.
Therefore, we initially compared the subunit composition of complexes sharing at least one chain with the same domain architecture as defined by PFAM (El-Gebali et al., 2019) or ECOD (Schaeffer et al., 2017), yielding a table containing 40 million pairs of complexes (see STAR Methods). For each pair, we recorded chain-chain correspondences, minimum, maximum, and average sequence identities between matching chains, as well as information on missing subunits between the query and the target complexes.
We then carried out structural superpositions using a heuristic based on Kpax (Ritchie, 2016) as we did with homo-oligomers. From each superposition, we recorded the structural similarity between QSs (TM-score) as well as local TM-scores for individual chains. To annotate physiologically relevant assemblies, we used complex pairs where all subunits of the query existed in the target and showed less than 80% average sequence identity. We first searched for structurally similar homologs of the largest oligomeric form of each complex, so we processed query complexes by decreasing number of subunits. We identified pairs of homologous complexes where all subunits matched both at the sequence and structural level. Such pairs showed conservation of QS, and we therefore annotated them as physiologically relevant (Figure 1). We optimized the structural similarity cut-off to be used in this process, as described later in the section “benchmarking annotations of QSalignHET.”
Structures annotated as being physiologically relevant were subsequently used as starting points to predict inconsistent assemblies by transitivity. In other words, if two complexes show an identical composition and a different structure (e.g., PDB: 1IWP, 1MMF, Figure 1) and if we know that one of the structures is physiologically relevant (here 1IWP), we inferred that the other structure was inconsistent (e.g., 1MMF may have missing subunits). Our strategy assumes that the largest conserved QS is correct. Importantly, in certain cases, different QSs can co-exist in cells. For example, Allophycocyanins (PDB: 1ALL, 1KN1) are found as heterodimers, hetero-hexamers with A3B3 stoichiometry, and heterododecamers with A6B6 stoichiometry depending on solvent, pH, and protein concentration (Brejc et al., 1995; Liu et al., 1999). In such a case, the lower stoichiometry forms of this complex (AB and A3B3) would be deemed inconsistent by QSalignHET unless homologs with matching stoichiometries are also identified.
Relationship types between structurally similar complexes
In the previous section, we saw two types of relationships between protein complexes: equivalence in composition and structure (PDB: 1EEX, 1IWP) and equivalence in composition with different structures (PDB: 1IWP, 1MMX). We observed several additional types of relationships, which we summarize in Table S2 and Figure 2.
Figure 2.

Examples of assemblies for different types of annotation made by QSalignHET
Annotations are based on the structural similarity of the QS as well as the similarity in subunit number and composition. The number of structures with each annotation type is given in parentheses. A comprehensive description of annotation types is provided in STAR Methods and Table S2. In brief, Annotation #1 describes complexes where two homologous assemblies share the same interaction geometry, i.e., their QS is conserved. Annotation #2 is assigned when PDB and PISA QSs are different for the query and the PISA QS is supported by structural conservation, so the query PDB is annotated as incorrect. Annotation #3 arises when two complexes have the same composition but subunits are in lower stoichiometry, and Annotation #4 is assigned when one complex is included in the other and there exists a difference in composition. Annotation #5 is the opposite of Annotation #3: it is assigned to a complex when it shares the same composition as another validated complex and shows a higher subunit stoichiometry not found in homologs. Annotation #6 arises when a complex includes another one and shows structural differences, hinting at a possible crystal interface or a conformational change. Here, the difference in composition/stoichiometry may be associated with the structural differences detected. Annotation #7 is assigned when the composition and the number of subunits are the same between the two complexes although they are not structurally similar.
Overall, we annotated 10,302 biological assemblies from the PDB, of which 7,810 QSs were predicted “Physiologically relevant (#1)” (Table S2). We corrected the QS annotation of 1,060 assemblies owing to the presence of structurally similar homologs that are either of higher stoichiometry or show different conserved interfaces; these are likely errors and were tagged as “Sub-stoichiometry (#2),” “Crystal interface (#3),” and “Crystal interface or large conformational change (#6).” Also, there were 84, 989, 96, and 263 assemblies in categories assumed to be either probable errors or inconclusive. These were tagged, respectively, “Sub-composition (#4),” “Excessive stoichiometry (#5),” “Crystal interface or large conformational change (#7),” and “Ambiguous (#8, #9).” We provide one concrete example for each category of annotation and the number of entries corresponding to the category (Figure 2 and Table S2).
Manually curating a dataset of physiologically relevant hetero-oligomers
We manually curated a total of 2,337 hetero-oligomeric assemblies, corresponding to 577 non-redundant complexes at a cut-off of 90% sequence identity. Based on literature evidence, each assembly was annotated as being physiologically relevant (1,486 and 293 high and medium confidence, respectively), erroneous (259 and 159 high and medium confidence, respectively), or undefined (140 assemblies). The process of curation is described in STAR Methods. In brief, we searched the primary reference of the query structure for experimental evidence supporting the corresponding QS. If no evidence was found, we searched the primary references of similar structures (>97% sequence identity). In some cases, subunit annotation from Swiss-Prot and the latest Affinity and Docking Benchmark (UniProt Consortium, 2018; Vreven et al., 2015) were used. In the process, high-confidence cases were supported by experimental evidence. For example, the 2:1 trimeric complex of NGF-p75 (PDB: 1SG1) is supported by gel filtration, multi-angle light scattering, and isothermal titration calorimetry (He and Garcia, 2004). Annotations with medium confidence reflected cases such as the complex of Nuclear transport factor 2 and the Ras-family GTPase Ran (PDB: 5BXQ) for which the authors are confident about a particular QS (e.g., A2B2), although no direct experimental evidence is provided. We incorporated these annotations into the PiQSi web server. We call this curated dataset of hetero-oligomers PiQSiHET and use it to benchmark the predictions of QSalignHET.
Benchmarking annotations of QSalignHET
We scanned a range of cut-off values of the TM-score used to infer QS geometry conservation, from 0.4 to 0.9. We ran the annotation pipeline for each value and benchmarked the resulting annotations based on the manually curated dataset PiQSiHET (Figure 3A). The number of entries used from the benchmark dataset (203 positives and 79 negatives) was lower than the total (577 entries), as curated entries without homologs were not annotated by QSalignHET. We found the error rate in confirming “physiological assemblies” to be largely independent of the TM-score cut-off used to infer QS conservation (Figure 3B). This independence exists because we compare complexes with matching composition and only a small fraction of these pairs have a low TM-score (Figure S1). However, the number of annotated structures decreased for TM-scores above 0.6; thus, we used this value. Overall, we validated 7,810 assemblies from the PDB with an estimated error rate of 4.4% (Figure 3B). Subsequently, we used these validated assemblies to correct annotations where the protein complex is identical but the QS is different. Several scenarios were possible, as depicted in Figure 2. We only benchmarked the categories “Crystal interface (#2),” “Sub-stoichiometry (#3),” and “Crystal interface or large conformational change (#6),” as the others are either not errors (e.g., sub-composition), are unclear (e.g., excessive stoichiometry, ambiguous), or might originate in large conformational changes. Overall, we were able to correct the annotations of 1,060 assemblies with an error rate estimated at 10.2% (Figure 3).
Figure 3.

TM-score optimization and benchmark of predictions
(A) Number of non-redundant structures in the manually curated benchmark dataset. Positives are correct structures and negatives are likely errors. The number of redundant structures is given in parentheses.
(B) The structural similarity score (TM-score) cut-off determines the minimum value at which two QSs are considered conserved and thereby inferred “physiologically relevant.” We scanned different TM-score cut-offs, calculated the error rate (green line), and recorded the total number of QSs annotated (blue line) for each.
(C) Starting from validated QSs, QSalignHET then searches for conflicting QSs that have identical composition and different structures (i.e., TM-score below the cut-off). We annotated such cases as likely errors and show the accuracy of these predictions (purple line) as well as the number of structures annotated for different cut-off values (light blue).
Next, we compared the performance of QSalignHET with two state-of-the-art methods for QS annotation, PISA and EPPIC. Using PiQSiHET as a benchmark dataset, we found that PISA and EPPIC predict heterodimers with an error rate of 25% and 33%, respectively. At the same time, conservation of QS geometry appeared reliable on the same structures, with an error rate of 6%. The performance of predictions decreased further for larger hetero-oligomers, with error rates equal to 39% and 45% for PISA and EPPIC, respectively (Figure S2). This increased error rate is likely due to both methods making independent predictions for different interfaces within an assembly. Thus, larger complexes require more interfaces to be predicted correctly together for the whole assembly to be predicted correctly. In contrast, QSalignHET compares the QS conservation of entire complexes rather than individual interfaces. As a result, the accuracy of the predictions is not negatively affected by the size of the complex. Indeed, the error of QSalignHET was equal to 6% for those larger complexes as well (i.e., with three or more subunits). The results of the benchmark are shown in Figure 4 for all complexes and in Figure S2 for dimers and larger complexes separately. Examples of prediction differences across methods and cases in which QSalignHET fails are illustrated in Figure 5.
Figure 4.

Principle and benchmark of QSbio
(A) The integration of QSalignHET, PISA, and EPPIC is carried out by comparing the structure of predicted assemblies. Consensus between methods increases the confidence in a particular assembly (e.g., PDB: 3U7Q in the top row), whereas disagreement between methods yields lower confidence (e.g., PDB: 4UBP in the lower row).
(B) Benchmarking the individual methods and their combination into QSbio. Receiver-operating characteristic curves show the area under the curve for all assemblies together (dimers and higher-order oligomers).
(C) Statistics derived from the benchmark: FPR, false-positive rate; TPR, true-positive rate; AUC, area under the curve. We provide detailed information on the benchmark in Table S3. The number of true positives, false negatives, true negatives, and false positives for each method are provided in Table S4.
Figure 5.

Examples of prediction differences across methods
(A) EPPIC predicts human hemoglobin to be a dimer and PISA predicts it to be an octamer, whereas the conservation of the tetramer yields a correct annotation by QSalignHET.
(B) EPPIC and PISA predict half of the thermosome to be the physiologically relevant assembly, likely due to the inter-ring interface being small. However, the conservation of the two-ring structure is detected and yields a correct annotation with QSalignHET.
(C) PISA and EPPIC predict an ɑβ form for 2nqo instead of the ɑ2β2 form.
(D) The octameric structure of FGF2-FGFR1 is predicted by all methods as the correct assembly. Nevertheless, this complex is described as a tetramer in the primary reference.
(E) The dimeric structure of GluK2/GluK5 is predicted by all methods to be physiologically relevant, but the primary reference (Kumar et al., 2011) describes tetramers in solution. Based on these experimental data and considering that the tetramer's interface is observed in seven crystal forms according to PROTCID (Xu and Dunbrack, 2020), we included the tetrameric form in the PiQSiHET benchmark dataset.
(F) The dimeric structure of FGF1:FGFR2 is predicted by all the methods except QSalignHET to be physiologically relevant. However, its close homolog (99% identical) FGF2:FGFR1 has an octameric structure, and the octameric QS is found to be conserved. The octamer is observed in three crystal forms according to PROTCID.
QSBio combines predictors to infer confidence estimates on a PDB-wide scale
QSalignHET annotated ~51% of hetero-oligomers in the PDB. To increase coverage, we combined annotations from PISA and EPPIC for consensus-based predictions, as was done previously for homo-oligomers (Dey et al., 2018). We derived weighted scores and estimated error probabilities for each assembly as follows: for entries where the annotation is available from all three sources (QSalignHET, PISA, EPPIC), a weighted score is derived from all three. Otherwise, the score is derived from the combined predictions of PISA and EPPIC only. In this way, we could annotate all hetero-oligomeric assemblies in the PDB. The integration allows us to obtain a consensus prediction of high or low confidence depending on the agreement between methods (Figure 4A). We benchmarked the individual methods and their combination using PiQSiHET. Considering dimers and oligomers together, the areas under the curve (AUCs) are 0.71, 0.61, 0.73, 0.92, and 0.93 for PISA, EPPIC, PISA + EPPIC, QSalignHET, and QSbio, respectively. QSalignHET alone performs well, and integrating PISA and EPPIC in a “consensus prediction” approach improved the AUC moderately, by 0.01. The combination of PISA and EPPIC does not increase the AUC significantly relative to PISA alone but yields conservative predictions with a false-positive rate twice as low as when using PISA alone. This improvement comes at the cost of lower sensitivity, with the true-positive rate decreasing. However, such a compromise would be desirable if one's goal is to gather a high-confidence set of hetero-oligomers. In these analyses, we benchmarked PISA and EPPIC on the subset of structures also annotated by QSalignHET. The results do not change significantly when adding structures of the benchmark not annotated by QSalignHET (Figure S3).
QSbio provides error estimates to each assembly, and we grouped them into five classes of confidence (very high, high, medium, low, very low) depending on estimated error probabilities based on the benchmark and corresponding to 0%–2%, 2%–5%, 5%–15%, 15%–50%, and 50%–100%, respectively. The number of assemblies in each class is 3,626, 1,541, 5,759, 8,094, and 1,060, respectively (Table S5). The PDB provides multiple biological assemblies for about 30% of its entries (Xu and Dunbrack, 2019), so QSbio is useful in providing error estimates for each assembly, enabling end-users to choose the highest-confidence assemblies for analysis.
Conclusion
For structures solved by X-ray crystallography, the coordinates of the asymmetric unit (ASU) are deposited in the PDB for all entries. However, information about the physiological assembly is not always provided by the authors. The ASU is the physiological assembly for only about 40% of structures deposited (Xu and Dunbrack, 2019). Therefore, methods are needed to identify such assemblies. Here we showed that conservation of QS geometry provides reliable information for prediction of the physiological relevance of hetero-oligomeric complexes in X-ray crystallography data. We used this information in an automated strategy called QSalignHET and annotated 51% of hetero-oligomeric structures across the PDB. To assess the accuracy of our predictions, we manually curated a dataset of hetero-oligomers. The benchmark with this dataset showed that annotations inferred by QSalignHET were largely accurate. Finally, we integrated annotations from EPPIC and PISA to infer a confidence estimate for hetero-oligomeric complexes not covered by QSalignHET. We hope that these annotations will help the scientific community to focus on physiologically relevant complexes when carrying out global analyses of hetero-oligomers in the PDB.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Annotations of hetero-oligomers | This manuscript | www.piqsi.org |
| PDB coordinate files of the manually curated benchmark dataset of hetero-oligomers | This manuscript | https://doi.org/10.6084/m9.figshare.13801304 |
| Software and algorithms | ||
| 3DComplex | Levy et al., 2006 | https://shmoo.weizmann.ac.il/elevy/3dcomplexV6/Entry.cgi |
| PDB | Berman et al., 2000 | https://www.rcsb.org |
| PISA | Krissinel and Henrick, 2007 | https://www.ebi.ac.uk/pdbe/pisa/ |
| EPPIC | Duarte et al., 2012 | https://www.eppic-web.org/ewui/ |
| KPAX | Ritchie, 2016 | http://kpax.loria.fr/ |
Resource availability
Lead contact
Further information and requests for resources and code should be directed to and will be fulfilled by the lead contact, Emmanuel D Levy (emmanuel.levy@weizmann.ac.il).
Materials availability
This study did not generate new unique reagents.
Data and code availability
PDB coordinate files of the manually curated benchmark dataset have been deposited on Figshare and are publicly available. DOIs are listed in the key resources table. All annotations are available in supplementary information and can be browsed on the PiQSi website (www.piqsi.org). The pseudocode is available in this paper's supplementary information file (Methods S1). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
All data are generated from the datasets provided in the key resources table.
Method details
Dataset
The dataset of protein structures is based on 3DComplex (Levy et al., 2006) as of April 2017 and consists of 20,080 ′biological assemblies' of hetero-oligomers defined by the Protein DataBank, excluding immunoglobulins. The dataset is available on the 3DComplex (version 6) website: http://shmoo.weizmann.ac.il/elevy/3dcomplexV6/Home.cgi.
For each structure, we use the top prediction from PISA (Krissinel and Henrick, 2007) as of April 2017, and EPPIC predictions of assemblies (Version 3) were downloaded using the REST API (json format) on October 17th, 2019 (Bliven et al., 2018).
Hetero-oligomer Benchmark dataset
Using the web interface of PiQSi (Levy, 2007), we manually annotated 2,337 (577 NR) structures of hetero-oligomers, of which 1779 (406 NR) are annotated as physiologically relevant, 418 (135 NR) are annotated as erroneous, and 140 (36 NR) as undefined as we found no clear evidence of a specific oligomeric state. To curate oligomeric state information, we searched the primary reference of a structure for keywords such as “oligomeric,” “solution,” “chromatography,” “gel,” “dynamic light,” “monomer,” “tetramer,” “dimer,” etc. The curation process took place in several steps. First, we focused on structures present in the docking benchmark dataset (Vreven et al., 2015) and searched their associated reference, yielding 212 annotated complexes. Next, we examined structures that are well known, such as tryptophan synthase or hemoglobin. Third, we curated complexes exhibiting different quaternary structure states according to our “composition similarity table” to identify potential errors. At this stage, the dataset contained about 300 structures. To increase the size further, we went through a list of hetero-oligomers sharing no more than 30% average sequence identity across matched subunits and excluding antibodies. We curated complexes in this list until the benchmark dataset contained 485 and 577 structures at redundancy levels of 30% and 90% respectively, with a total of 2,337 structures when including all redundancies. The redundancy arose from an annotation transfer process. Once an entry was annotated, its annotation was transferred to close homologs sharing the same subunit composition and stoichiometry as well as a minimum of 95% sequence identity across all subunits. Furthermore, the transfer of annotations for all entries was manually verified to confirm that they correspond to the same protein complex. We call this manually curated benchmark dataset of hetero-oligomers PiQSiHET. For each curated entry, we provide the following information:
(i) The error status as “NO” (physiologically relevant structures with high confidence), “PROBNOT” (physiologically relevant structures with medium confidence), “YES” (erroneous structures with high confidence), “PROBYES” (erroneous structures with medium confidence) or “NA” (undefined), (ii) the symmetry and number of subunits for the original assembly and for the correct assembly, (iii) the PubMed identifier in which information was found for the annotation and (iv) a sentence that describes the annotation and supporting evidence from the literature.
Comparing the composition of hetero-oligomers
The annotation process of QSalignHET required comparing the composition of hetero-oligomers and homo-oligomers. We used sequence homology from 3DComplex and created a table with pairwise comparative information on hetero-oligomers sharing at least one subunit. For each pair, we computed chain-chain correspondences and recorded minimum, maximum and average sequence identities as well as “gaps,” if any. Correspondences between chains were established by comparing their sequences. For each chain in the query complex, we selected a matching chain in the target complex. The matching chain was the one with the highest sequence identity. Therefore, when complexes containing paralogs were compared such as hemoglobin, each chain was only matched once to its closest homolog (ɑ with ɑ, β with β). Gaps arose when one or more chains from the query complex were missing in the target complex. For these cases, we identify differences in subunit composition of the complex. Chains that did not share detectable sequence homology but showed identical PFAM (El-Gebali et al., 2019) or ECOD (Schaeffer et al., 2017) domain architecture were assigned an arbitrary sequence identity of 20%. Ultimately, this process yielded a table ('composition similarity table') containing 43,893,877 QS pairs stored as part of the 3DComplex MySQL database.
Structure comparison
To save computation time, we carried out structural alignments between potential matches only. That is, pairs of structures sharing sequence homology (>20% average sequence identity), with at least half of the subunits of the target matching the query. Ultimately, we measured the structural similarity of 13,549,218 QS pairs using Kpax (Ritchie, 2016) and the heuristic previously employed (Dey et al., 2018). We recorded the TM-score between both QSs, as well as all the individual chain-chain TM-scores that are stored in our MySQL database as the “structural similarity table.”
Annotation procedure
The overall workflow is illustrated in Figure S4, and detailed explanations with examples for the different annotations are given in Table S2. The information gathered from the sequence, and structural comparisons were used to develop an annotation inference methodology described in the pseudocode “QSinferHET” (Supplementary Note) to subsequently infer the most likely 'physiological' (annotation id #1) QS of hetero-oligomers. Briefly, a query QS is annotated as correct if a homologous QS (maximum sequence identity between two chains <80%) has the same composition and a similar structure (TM-score > 0.6 and all chain-chain TM-scores above 0.45). The sequence identity cut-off for homology was set to 80% to reduce the chance that the same crystal packing is formed on account of protein-surface similarity. The annotation process was carried out for each symmetry group separately, by decreasing number of subunits. This order is to ensure that lowest order oligomers (e.g., AB) are annotated last if no evidence for the formation of a higher-order structure is found (e.g., A2B2). Once all QSs from a symmetry group were processed, those annotated as correct were used to search for possible errors or sub-complexes among structures not yet annotated. The step of using correctly annotated structures to infer erroneous ones is described in the pseudocode “QSpropagateHET” (Supplementary Note). This propagation step involves identifying proteins with an identical sequence and dissimilar QS/composition to that of the other QS annotated as correct in the QSinfer step.
Annotations arising from the propagation step correspond to annotation groups 2–9, and they depend on:
-
•
The number of subunits of the QS inferred as correct (QS1) and the one to be annotated (QS2),
-
•
Differences in composition/stoichiometry between QS1 and QS2. Here, a complex “including” another complex means that all subunits of the smaller complex match a respective subunit (sequence identity >95%) in the larger one.
-
•
The TM-score between the two QSs being considered, as well as local (chain-chain) TM-scores between chains overlapping in the QS.
-
1
As we saw above, when QS1 and QS2 share the same structure, their QS is conserved and is annotated as likely physiological (annotation id #1). In contrast, when two QSs share the same subunits but differ in structure, stoichiometry, or show additional subunit types, we annotate them as follows:
-
2
QS2 and QS1 share the same composition and number of subunits, but their TM-score is < 0.6. However, the PISA prediction for QS2 matches QS1, indicating that the conserved QS does exist in QS2's crystal lattice. Thus, the PDB-form of QS2 is annotated with 'Crystal interface'; (annotation id #2)
-
3
If QS2 and QS1 share the same composition, but QS2 shows a lower stoichiometry, then QS2 is annotated with 'Sub-stoichiometry'; (annotation id #3)
-
4
If QS2 and QS1 have a different composition and QS2 is included in QS1, i.e., QS2 has missing subunits types, then QS2 is annotated as 'Sub-composition'; (annotation id #4)
-
5
If QS2 includes QS1 and shows a TM-score > 0.9, then QS2 is annotated as 'Excessive stoichiometry'; (annotation id #5)
-
6
If QS2 includes QS1 and shows a TM-score < 0.9, QS2 is annotated as 'Crystal interface or large conformational change'; (annotation id #6)
-
7
If QS2 and QS1 share the same composition and number of subunits, but their TM-score is < 0.65, then QS2 is annotated as 'Crystal interface or large conformational change'; (annotation id #7)
-
8
If two different assemblies of the same PDB code are supported by structural similarity, they are tagged as 'Ambiguous' (annotation id #8).
-
9
If the total number of structurally similar homologs supporting a particular QS is low compared to the total number of homologs (i.e., <5%), the QS is also tagged as 'Ambiguous' (annotation id #9).
Benchmarking predictions
We benchmarked the automated annotations from QSalignHET against the manually annotated dataset PiQSiHET. After the annotation procedure, we counted the number of structures in the following categories: TP (true positives), annotated as correct by both QSalignHET and PiQSiHET; FP (false positives), annotated as correct by QSalignHET and as incorrect by PiQSiHET; FN (false negatives), annotated as incorrect by QSalignHET and as correct by PiQSiHET; and TN (true negatives), annotated as incorrect by both QSalignHET and PiQSiHET. We calculated the error rate of 'correct' annotations by the false discovery rate FDR = FP/(TP + FP) and the error rate of 'incorrect' annotations by the false omission rate FOR = FN/(FN + TN). The dataset of structures on which these rates were calculated was filtered at a level of 90% sequence identity.
Integrating QS information into QSbio
QS predictions from PISA and EPPIC were integrated along with QSalignHET annotations to create a meta-predictor QSbio. For this, we compared the structure of assemblies predicted by both methods. Two assemblies were considered identical when the TM-score was above 0.9. We then estimated the confidence of different categories of agreement between methods using PiQSiHET, as done previously for homo-oligomers (Dey et al., 2018).
Quantification and statistical analysis
Statistical details are provided in the methods section where applicable.
Acknowledgments
We thank David Ritchie for his help with Kpax and Harry Greenblatt for helping with the computer infrastructure. This work was supported by the Israel Science Foundation (grant no. 1452/18), by the European Research Council under the European Union Horizon 2020 research and innovation program (grant agreement no. 819318), by a research grant from A.-M. Boucher, by research grants from the Estelle Funk Foundation, the Estate of Fannie Sherr, the Estate of Albert Delighter, the Merle S. Cahn Foundation, Mrs. Mildred S. Gosden, the Estate of Elizabeth Wachsman, and the Arnold Bortman Family Foundation. E.D.L. is incumbent of the Recanati Career Development Chair of Cancer Research. S.D. received support from the Koshland Foundation.
Author contributions
S.D. and E.D.L. designed the experiments. S.D. performed the experiments. S.D. and E.D.L. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 13, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.07.012.
Contributor Information
Sucharita Dey, Email: sdey@iitj.ac.in.
Emmanuel D. Levy, Email: emmanuel.levy@weizmann.ac.il.
Supplemental information
PDB coordinate files of the manually curated benchmark dataset are also available on Figshare at https://doi.org/10.6084/m9.figshare.13801304
References
- Armstrong D.R., Berrisford J.M., Conroy M.J., Gutmanas A., Anyango S., Choudhary P., Clark A.R., Dana J.M., Deshpande M., Dunlop R. PDBe: improved findability of macromolecular structure data in the PDB. Nucleic Acids Res. 2020;48:D335–D343. doi: 10.1093/nar/gkz990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadur R.P., Chakrabarti P., Rodier F., Janin J. Dissecting subunit interfaces in homodimeric proteins. Proteins. 2003;53:708–719. doi: 10.1002/prot.10461. [DOI] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernauer J., Bahadur R.P., Rodier F., Janin J., Poupon A. DiMoVo: a Voronoi tessellation-based method for discriminating crystallographic and biological protein-protein interactions. Bioinformatics. 2008;24:652–658. doi: 10.1093/bioinformatics/btn022. [DOI] [PubMed] [Google Scholar]
- Bertoni M., Aloy P. DynBench3D, a web-resource to dynamically generate benchmark sets of large heteromeric protein complexes. J. Mol. Biol. 2018;430:4431–4438. doi: 10.1016/j.jmb.2018.09.011. [DOI] [PubMed] [Google Scholar]
- Bliven S., Lafita A., Parker A., Capitani G., Duarte J.M. Automated evaluation of quaternary structures from protein crystals. PLoS Comput. Biol. 2018;14:e1006104. doi: 10.1371/journal.pcbi.1006104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K., Ficner R., Huber R., Steinbacher S. Isolation, crystallization, crystal structure analysis and refinement of allophycocyanin from the cyanobacterium Spirulina platensis at 2.3 A resolution. J. Mol. Biol. 1995;249:424–440. doi: 10.1006/jmbi.1995.0307. [DOI] [PubMed] [Google Scholar]
- Capitani G., Duarte J.M., Baskaran K., Bliven S., Somody J.C. Understanding the fabric of protein crystals: computational classification of biological interfaces and crystal contacts. Bioinformatics. 2016;32:481–489. doi: 10.1093/bioinformatics/btv622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti P., Janin J. Dissecting protein-protein recognition sites. Proteins. 2002;47:334–343. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- Chothia C., Janin J. Principles of protein-protein recognition. Nature. 1975;256:705–708. doi: 10.1038/256705a0. [DOI] [PubMed] [Google Scholar]
- Lo Conte L., Chothia C., Janin J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- Dey S., Levy E.D. In: Protein Complex Assembly: Methods and Protocols. Marsh J.A., editor. Springer; 2018. Inferring and using protein quaternary structure information from crystallographic data; pp. 357–375. [DOI] [PubMed] [Google Scholar]
- Dey S., Ritchie D.W., Levy E.D. PDB-wide identification of biological assemblies from conserved quaternary structure geometry. Nat. Methods. 2018;15:67–72. doi: 10.1038/nmeth.4510. [DOI] [PubMed] [Google Scholar]
- Duarte J.M., Srebniak A., Schärer M.A., Capitani G. Protein interface classification by evolutionary analysis. BMC Bioinformatics. 2012;13:334. doi: 10.1186/1471-2105-13-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock A.H., McCammon J.A. Identification of protein oligomerization states by analysis of interface conservation. Proc. Natl. Acad. Sci. U S A. 2001;98:2990–2994. doi: 10.1073/pnas.061411798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elez K., Bonvin A.M.J.J., Vangone A. Biological versus crystallographic protein interfaces: an overview of computational approaches for their classification. Crystals. 2020;10:114. [Google Scholar]
- El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa Y., Tomii K. Accurate classification of biological and non-biological interfaces in protein crystal structures using subtle covariation signals. Sci. Rep. 2019;9:12603. doi: 10.1038/s41598-019-48913-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.-L., Garcia K.C. Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science. 2004;304:870–875. doi: 10.1126/science.1095190. [DOI] [PubMed] [Google Scholar]
- Henrick K., Thornton J.M. PQS: a protein quaternary structure file server. Trends Biochem. Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- Hu J., Liu H.-F., Sun J., Wang J., Liu R. Integrating co-evolutionary signals and other properties of residue pairs to distinguish biological interfaces from crystal contacts. Protein Sci. 2018;27:1723–1735. doi: 10.1002/pro.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J., Rodier F. Protein-protein interaction at crystal contacts. Proteins. 1995;23:580–587. doi: 10.1002/prot.340230413. [DOI] [PubMed] [Google Scholar]
- Jiménez-García B., Elez K., Koukos P.I., Bonvin A.M., Vangone A. PRODIGY-crystal: a web-tool for classification of biological interfaces in protein complexes. Bioinformatics. 2019;35:4821–4823. doi: 10.1093/bioinformatics/btz437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike R., Ota M. SCPC: a method to structurally compare protein complexes. Bioinformatics. 2012;28:324–330. doi: 10.1093/bioinformatics/btr654. [DOI] [PubMed] [Google Scholar]
- Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kumar J., Schuck P., Mayer M.L. Structure and assembly mechanism for heteromeric kainate receptors. Neuron. 2011;71:319–331. doi: 10.1016/j.neuron.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundrotas P.J., Anishchenko I., Dauzhenka T., Kotthoff I., Mnevets D., Copeland M.M., Vakser I.A. Dockground: a comprehensive data resource for modeling of protein complexes. Protein Sci. 2018;27:172–181. doi: 10.1002/pro.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E.D. PiQSi: protein quaternary structure investigation. Structure. 2007;15:1364–1367. doi: 10.1016/j.str.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Levy E.D., Pereira-Leal J.B., Chothia C., Teichmann S.A. 3D complex: a structural classification of protein complexes. PLoS Comput. Biol. 2006;2:e155. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.Y., Jiang T., Zhang J.P., Liang D.C. Crystal structure of allophycocyanin from red algae Porphyra yezoensis at 2.2-A resolution. J. Biol. Chem. 1999;274:16945–16952. doi: 10.1074/jbc.274.24.16945. [DOI] [PubMed] [Google Scholar]
- Liu Q., Li Z., Li J. Use B-factor related features for accurate classification between protein binding interfaces and crystal packing contacts. BMC Bioinformatics. 2014;15(suppl. 16):S3. doi: 10.1186/1471-2105-15-S16-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Li Q., Lai L. A combinatorial score to distinguish biological and nonbiological protein-protein interfaces. Proteins. 2006;64:68–78. doi: 10.1002/prot.20954. [DOI] [PubMed] [Google Scholar]
- Lowe J., Stock D., Jap B., Zwickl P., Baumeister W., Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- Madej T., Lanczycki C.J., Zhang D., Thiessen P.A., Geer R.C., Marchler-Bauer A., Bryant S.H. MMDB and VAST+: tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014;42:D297–D303. doi: 10.1093/nar/gkt1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra P., Pal D. Combining Bayes classification and point group symmetry under boolean framework for enhanced protein quaternary structure inference. Structure. 2011;19:304–312. doi: 10.1016/j.str.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Mukherjee S., Zhang Y. MM-align: a quick algorithm for aligning multiple-chain protein complex structures using iterative dynamic programming. Nucleic Acids Res. 2009;37:e83. doi: 10.1093/nar/gkp318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponstingl H., Kabir T., Thornton J.M. Automatic inference of protein quaternary structure from crystals. J. Appl. Crystallogr. 2003;36:1116–1122. [Google Scholar]
- Ritchie D.W. Calculating and scoring high quality multiple flexible protein structure alignments. Bioinformatics. 2016;32:2650–2658. doi: 10.1093/bioinformatics/btw300. [DOI] [PubMed] [Google Scholar]
- Schaeffer R.D., Liao Y., Cheng H., Grishin N.V. ECOD: new developments in the evolutionary classification of domains. Nucleic Acids Res. 2017;45:D296–D302. doi: 10.1093/nar/gkw1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva F.D., Da Silva F., Desaphy J., Bret G., Rognan D. IChemPIC: a random forest classifier of biological and crystallographic protein-protein interfaces. J. Chem. Inf. Model. 2015;55:2005–2014. doi: 10.1021/acs.jcim.5b00190. [DOI] [PubMed] [Google Scholar]
- Sippl M.J., Wiederstein M. Detection of spatial correlations in protein structures and molecular complexes. Structure. 2012;20:718–728. doi: 10.1016/j.str.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y., Kinoshita K., Ito N., Nakamura H. PreBI: prediction of biological interfaces of proteins in crystals. Nucleic Acids Res. 2006;34:W320–W324. doi: 10.1093/nar/gkl267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y., Nakamura H., Kinoshita K. Discrimination between biological interfaces and crystal-packing contacts. Adv. Appl. Bioinform. Chem. 2008;1:99–113. doi: 10.2147/aabc.s4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018;46:2699. doi: 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreven T., Moal I.H., Vangone A., Pierce B.G., Kastritis P.L., Torchala M., Chaleil R., Jiménez-García B., Bates P.A., Fernandez-Recio J. Updates to the integrated protein-protein interaction benchmarks: docking benchmark version 5 and affinity benchmark version 2. J. Mol. Biol. 2015;427:3031–3041. doi: 10.1016/j.jmb.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Dunbrack R.L., Jr. Principles and characteristics of biological assemblies in experimentally determined protein structures. Curr. Opin. Struct. Biol. 2019;55:34–49. doi: 10.1016/j.sbi.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Dunbrack R.L., Jr. ProtCID: a data resource for structural information on protein interactions. Nat. Commun. 2020;11:711. doi: 10.1038/s41467-020-14301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Canutescu A.A., Wang G., Shapovalov M., Obradovic Z., Dunbrack R.L. Statistical analysis of interface similarity in crystals of homologous proteins. J. Mol. Biol. 2008;381:487–507. doi: 10.1016/j.jmb.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yueh C., Hall D.R., Xia B., Padhorny D., Kozakov D., Vajda S. ClusPro-DC: dimer classification by the cluspro server for protein-protein docking. J. Mol. Biol. 2017;429:372–381. doi: 10.1016/j.jmb.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Domingues F.S., Sommer I., Lengauer T. NOXclass: prediction of protein-protein interaction types. BMC Bioinformatics. 2006;7:27. doi: 10.1186/1471-2105-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB coordinate files of the manually curated benchmark dataset are also available on Figshare at https://doi.org/10.6084/m9.figshare.13801304
Data Availability Statement
PDB coordinate files of the manually curated benchmark dataset have been deposited on Figshare and are publicly available. DOIs are listed in the key resources table. All annotations are available in supplementary information and can be browsed on the PiQSi website (www.piqsi.org). The pseudocode is available in this paper's supplementary information file (Methods S1). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.