

Abstract
Cryopyrin associated periodic syndrome (CAPS) is a rare inherited autoinflammatory disorder characterized by systemic, cutaneous, musculoskeletal, and central nervous system inflammation. Gain-of-function mutations in NLRP3 in CAPS patients lead to activation of the cryopyrin inflammasome resulting in the inappropriate release of inflammatory cytokines including IL-1β and CAPS related inflammatory symptoms. Several mechanisms have been identified that are important for the normal regulation of the cryopyrin inflammasome in order to prevent uncontrolled inflammation. Investigators have taken advantage of some of these pathways to develop and apply novel targeted therapies, which have resulted in improved quality of life for patients with this orphan disease.
Keywords: Familial cold autoinflammatory syndrome, Muckle Wells syndrome, Neonatal onset multisystem inflammatory disease, Inflammasome, Cryopyrin associated periodic syndromes
Introduction
In 1940, Kile and Rusk described a multi-generational family with several affected members exhibiting recurrent episodes of urticarial-like rash, limb pain, and fever following generalized cold exposure [1]. Over the next 60 years, many other patients and families from Europe and North America were reported with a similar inflammatory phenotype referred to variably as cold hypersensitivity, familial cold urticaria (FCU), and finally familial cold autoinflammatory syndrome (FCAS), in an attempt to differentiate this chronic systemic inflammatory disease from the more common acquired cold urticarial [2]. In 1962, Muckle and Wells investigated a family with a similar clinical picture of recurrent episodes of rash, limb pain, and fever, but not associated with cold exposure. Many of these patients developed progressive sensorineural hearing loss and end stage renal disease secondary to AA amyloidosis [3]. Over the next few decades additional reports of Muckle-Wells syndrome patients were published including some noting phenotypic overlap with FCAS. In 1980, Prieur described young patients with chronic severe urticarial-like rash, pain and fever with significant central nervous system involvement including developmental delay and seizures due to chronic sterile meningitis and increased intracranial pressure. These patients were also reported with distal femur arthropathy resulting in significant disability [4]. Over the next 20 years, patients with similar clinical features were reported and the disease was referred to as either chronic infantile neurologic cutaneous articular (CINCA) syndrome or neonatal onset multisystem inflammatory disease (NOMID) with rare mention of phenotypic overlap with MWS [5].
The identification of heterozygous mutations in the same novel gene (NLRP3) in patients from all 3 phenotypes established this as a spectrum of one monogenic disease [6]. Since NLRP3 codes for the protein cryopyrin, this disease continuum is now known as cryopyrin associated periodic syndrome (CAPS) or cryopyrinopathies. CAPS patients have been reported from all over the world with estimated prevalence ranging between 1–3 per million [7]. The distribution of CAPS subtypes varies around the world as a majority of CAPS patients in North America are classified as FCAS due to large families with a founder mutation [8], while MWS is the most common phenotype reported in Europe. CINCA/NOMID patients are less common since most cases are de novo. In this review, we will discuss the clinical features, genetics, pathogenesis, and therapy of CAPS and examine the role and function of cryopyrin in human disease in order to help clinical immunologists and immune disease researchers gain a better understanding of this fascinating disease and important immune regulatory protein.
Clinical description of CAPS
While FCAS, MWS, and CINCA/NOMID were described as unique disease entities, it is clear that these disorders are part of a clinical continuum with several shared features that differ in severity which is a determinant of level of therapy. There are also some unique features that are worth distinguishing since they may affect prognosis and clinical management. All of the subtypes have cutaneous, musculoskeletal, ocular, and central nervous system involvement to varying degrees. As more patients have been described, there are clearly patients with clinical features that overlap more than one subtype [6], which is consistent with the concept of a single disease spectrum (Table 1).
Table 1 –
Clinical features of CAPS
| FCAS | MWS | CINCA/NOMID | |
|---|---|---|---|
| Cutaneous | Urticaria-like rash | Urticaria-like rash | Urticaria-like rash |
| Systemic | Fever / Fatigue / Chills | Fever / Fatigue | Fever / Fatigue |
| Musculoskeletal | Arthralgia / Myalgia | Arthralgia / Myalgia / Arthritis | Arthralgia / Myalgia / Distal femur overgrowth |
| Ocular | Conjunctivitis / Keratitis | Conjunctivitis/Keratitis/Uveitis | Conjunctivitis / Keratitis / Uveitis / Papillitis |
| Auditory | Sensorineural hearing loss | Sensorineural hearing loss | |
| Central nervous system | Headache | Headache | Sterile meningitis, Elevated intracranial pressure |
| Morbidity | Amyloidosis (rare) | Amyloidosis | Amyloidosis, developmental delay |
| Episode Pattern | 12–24 hours | 1–3 days | Chronic with 1–3 day flares |
| Triggers | Generalized cold / pneumovax | Stress / exercise / infection / pneumovax | Stress / exercise / infection / pneumovax |
Cryopyrin associated periodic syndrome (CAPS), Familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), Chronic infantile neurologic cutaneous articular (CINCA) syndrome or Neonatal onset multisystemic inflammatory disease (NOMID)
Most CAPS patients present with symptoms early in life consistent with the inherited nature of the disease, although the observation that some patients develop symptoms at or even before birth and others have delayed onset for many years may suggest environmental influences including exposure to microbial organisms or antigens [9]. An urticaria-like rash is often the first indication of disease and the most prominent shared clinical sign in CAPS patients. Fever is a common clinical sign, although it is often not a primary clinical complaint, and the recorded body temperature may not meet actual criteria for fever. Most CAPS patients report myalgia, arthralgia, headache, and fatigue, although these symptoms are often difficult to quantify objectively. Conjunctivitis and keratitis, while less prevalent than rash, can be observed in patients from all 3 subtypes. As expected in a disease continuum, chronicity or severity of shared symptoms range from the most severe in CINCA/NOMID to the least in FCAS, however, quality in life is decreased in all patients [10].
Generalized cold exposure as a trigger for symptoms is the most prominent clinical feature of FCAS and often is their chief complaint to medical providers. Patients report that exposure to temperatures below 72°F for more than 30 minutes such as air conditioned rooms is sufficient to induce noticeable symptoms within a few hours of exposure. FCAS patients also often report chills in association with fever [2]. Cold can be one of many symptom triggers for MWS patients, but it is not reported consistently. FCAS and MWS patients have daily baseline symptoms of fatigue and flu-like malaise. A diurnal pattern of symptoms worsening in the afternoon and evening is common in many FCAS and MWS patients [2, 11]. Symptomatic flares in FCAS episodes usually last less than a day and MWS flares may last 1–3 days. Amyloidosis was reported in up to 30% of one MWS cohort prior to definitive therapy [3], but is much less common in FCAS patients. Hearing loss is common in MWS and CINCA/NOMID patients, but rarely seen in the FCAS subtype. Ocular findings including uveitis and papilledema can be observed in CINCA/NOMID patients and rarely in MWS patients [12, 13]. Central nervous system symptoms observed in many CINCA/NOMID patients including developmental delay and seizures are secondary to sterile meningitis and increased intracranial pressure that has only rarely been documented in MWS patients. Arthropathy involving the distal femur and dysmorphic features such as frontal bossing are also fairly unique to a subset of CINCA/NOMID patients [14].
Laboratory and pathologic findings
Chronic leukocytosis is common in most CAPS patients with acute increases of blood neutrophilia during symptom flares. Increased serum IL-6 levels have also been observed during flares [15]. In contrast, modest to significant elevations of acute phase reactants such as C reactive protein and erythrocyte sedimentation rates are common at baseline and may not change significantly during a flare. Microcytic anemia and thrombocytosis may also occur due to chronic inflammation. Cerebrospinal fluid analysis often reveals chronic increased intracranial pressure and leukocytosis [16]. Skin biopsy shows dermal edema primarily with neutrophil infiltration in the dermis, especially in the perivascular regions and near sweat glands [15]. Bone radiologic examinations show calcified physeal lesions and osteoporosis, and pathologic analysis demonstrates disorganized cartilage without inflammation [14].
CAPS as an autoinflammatory syndrome
The unique and multi-systemic clinical and laboratory features of this disease continuum and low incidence of this orphan disease have resulted in many patients with misdiagnosis and significant delays in diagnosis [10]. Additionally, these patients often present to a wide variety of medical providers such as primary care physicians and different specialists including dermatologists, rheumatologists, ophthalmologists, otolaryngologists, neurologists, infectious disease specialists, allergists and clinical immunologists resulting in the lack of a medical home. This clinical paradox is similar to the experience of patients with a group of rare inherited syndromes known as the hereditary recurrent fever disorders including familial Mediterranean fever, hyper IgD syndrome, and tumor necrosis factor receptor associated periodic syndrome. The identification of the genetic basis for these diseases led to the description of a new immune disease classification known as the autoinflammatory disorders to differentiate conditions that do not fit into the classical categories of immune dysregulation including immunodeficiency, allergy, or autoimmunity [17].
NLRP3 as a disease gene
The availability of large families with autosomal dominant inheritance of FCAS and MWS, combined with advances in human molecular genetics led to the identification of heterozygous mutations in NLRP3 (also known as CIAS1 or PYPAF1), a novel gene coding for a protein with initial unknown function referred to as cryopyrin [18–20]. Clinical similarities between MWS and NOMID prompted scientists to search for NLRP3 mutations in NOMID patients [21, 22]. While NLRP3 mutations were identified in these patients, many NOMID patients without significant CNS and cochlear inflammation and a few FCAS and MWS patients did not have identifiable mutations by standard Sanger sequencing. It was later determined that most of these “mutation negative” patients are somatic mosaics, often possessing a small percentage of mutant cells within the myeloid lineage resulting in difficult to detect mutations [23]. Somatic mosaicism is clinically relevant as it makes genetic diagnosis and counseling more challenging and could have implications for the use of more definitive cell-based therapies in the future [24]. Approximately 100 pathogenic NLRP3 mutations have been reported (Infevers accessed 3/4/19) in CAPS patients [25] with strong genotype-phenotype correlation along the disease continuum (Fig. 1). In addition, a few low penetrance variants in NLRP3 have also been reported in patients with typical or atypical CAPS phenotypes, but also in unaffected people with no significant symptomatology. [26]. These variants have been shown to have less in vitro functional consequences than classic CAPS mutations and have also been found to be risk alleles for more common diseases in genetic association studies [27].
Figure 1. NLRP3 mutations reported in CAPS.
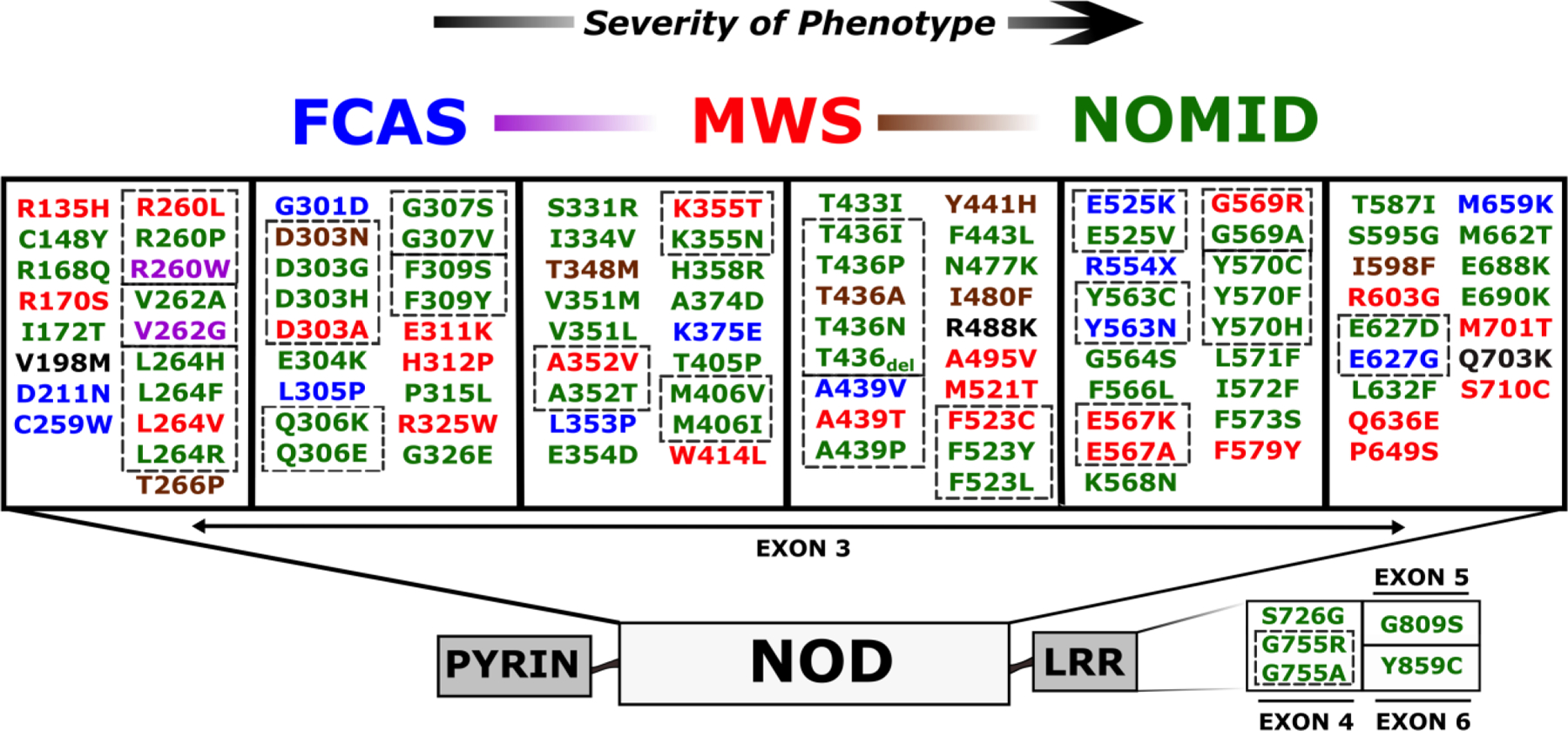
Most CAPS disease-associated mutations are located in exon 3, which codes for the NOD domain, and there are a few mutations in C terminal exons that code for the LRR domain. Infevers; an online database for autoinflammatory mutations - https://infevers.umai-montpellier.fr/) [25, 82–84] was accessed 3/4/19 and all potential CAPS associated mutations associated with a sub-phenotype were included. The presence of multiple mutations coding the same amino acid suggest mutational hotspots (indicated by interrupted line boxes). There is fairly consistent genotype-phenotype correlation indicated by colors: FCAS (blue), FCAS/MWS (purple), MWS (red), MWS/NOMID (brown), NOMID (green), and low penetrance mutations (black). All mutations are numbered according to the second methionine (although many mutation sequences utilize the first methionine which adds two amino acids to the reported variant). Familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), Neonatal onset multisystemic inflammatory disease (NOMID), nucleotide oligomerization domain (NOD), Leucine rich repeat (LRR)
Cryopyrin function
The success of the Human Genome Project allowed investigators to mine data for gene families based on structural domain similarities resulting in the discovery of a large group of innate immune sensor proteins known as nucleotide oligomerization domain (NOD)-like receptors (NLRs) [28]. NLRs contain PYRIN domains (like NLRP3) and/or caspase activation recruitment domains (CARDs), which promote self-assembly. In addition, NLRs possess central NOD domains where most disease mutations are located, suggesting an important functional role, and C-terminal Leucine-rich repeat domains. Some NLRs form the central structure of large intracellular multi-protein complexes known as inflammasomes that function to protect the cell from external and internal threats, but also regulate homeostasis [29]. Inflammasomes are comprised of adaptor molecules, interacting regulatory proteins, chaperone proteins, and enzymatic effector molecules (Fig. 2).
Figure 2. Normal and mutant cryopyrin function.
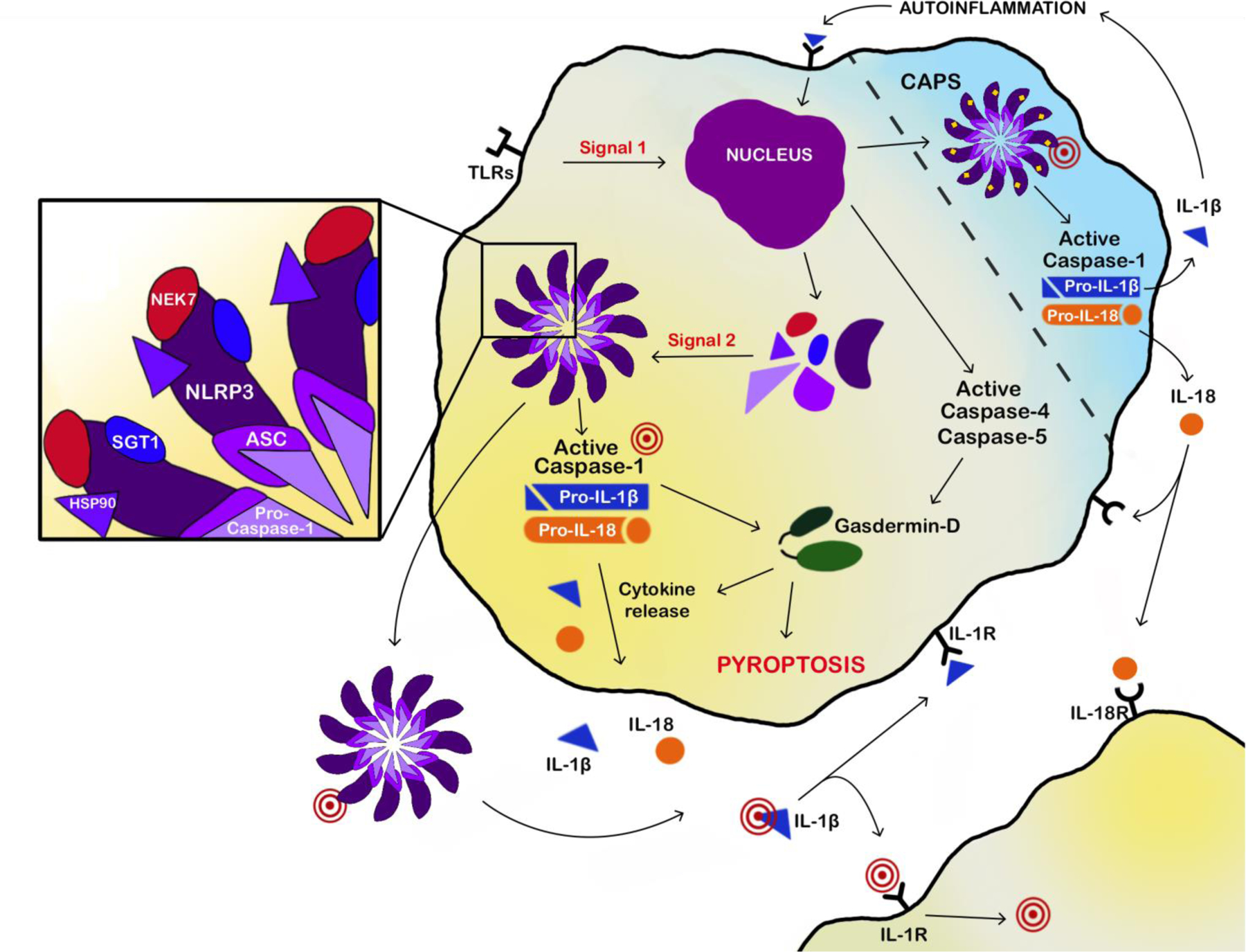
Normal activation of the cryopyrin inflammasome in monocytes and macrophages involves 2 signals: Signal 1 involves toll-like receptor (TLR) activation resulting in NFκB mediated expression of inflammasome protein components and pro-cytokines. Signal 2 involves various specific triggers such as nucleic acids, toxins, and crystals resulting in oligomerization of inflammasome protein components including cryopyrin, ASC, pro-caspase-1, NEK7, SGT1, and HSP90 into a multimeric ring like structure. Formation of the inflammation leads to cleavage of caspases, gasdermin-D, and subsequent cleavage pro-IL-1β and pro-IL-18 with release of mature and active cytokines (IL-1β and IL-18) as well as pyroptosis. IL-1β and IL-18 bind to their respective receptors on the same cell resulting in autoinflammation or other cells resulting in a cascade of inflammatory signaling. Inflammasomes may be released from the cells as ASC specks where they may continue to be functional or be taken up by other macrophages. Activation of mutant (yellow dots) cryopyrin inflammasome does not depend on signal 2 resulting inappropriate activation and inflammation. Several therapies (red targets) in the pathway are either available or in development.
Apoptosis speck-like protein containing CARD (ASC) is a multi-domain activating adapter molecule for caspase-1, which is mobilized during inflammasome assembly [30]. ASC is also referred to as PYCARD, representing the PYRIN domain at the N-terminus and CARD domain at the C-terminus [31–33]. The CARD domain facilitates caspase-1 binding and subsequent filament formation. ASC molecules congregate to form large masses near the nucleus resembling a ‘speck’ readily visualized via microscopy; thus, speck formation is a valuable indicator of inflammasome activation [34]. ASC specks can also be expelled from the cell and internalized by nearby macrophages, causing further propagation of inflammation. ASC specks can also continue to be functional in the extracellular space [35, 36].
Caspases are cysteine proteases that act as enzymatic effector molecules in the inflammasome complex. Notably, caspase-1 is central to the canonical, or classical, inflammasome pathway [37], while caspase-4 and caspase-5 drive the non-canonical inflammasome pathway [38]. The canonical inflammasome pathway is defined by the caspase-1 mediated production of interleukin-1β (IL-1β) and IL-18. Cleavage of caspase by either pathway results in cleavage of gasdermin-D resulting in pyroptosis, a unique inflammasome-specific cell death distinguishable from apoptosis and necrosis, marked by the combined release of caspase-1 and lactate dehydrogenase [39]. Gasdermin-D is also associated with cleavage of pro-forms of inflammatory cytokines and release of mature cytokines [37, 40] such as IL-1β and IL-18 [41]. Release of cytokines also function to further the inflammatory cascade by binding to IL-1 receptor or IL-18 receptor on the same, or neighboring cells and increasing expression of inflammasome component proteins or pro-cytokines.
Normal inflammasome activation is dependent on a two-signal process. In the absence of either signal, the cryopyrin inflammasome remains inactive. Signal 1 is a priming step required to initiate recruitment of necessary components for assembly. TLR activation by PAMPs signals transcriptional factor NFκ-B to upregulate expression of crucial components of the inflammasome and pro-cytokines. Signal 2 then coordinates protein assembly and activation of the inflammasome complex [29]. There are a wide variety of Signal 2 triggers including a myriad of DAMPs such as nucleic acids (ATP), aggregate proteins (amyloid), or crystals (monosodium urate or cholesterol) [42, 43]. Extracellular ATP can activate P2X7 receptor and promote ion efflux [44, 45]. Crystals or protein aggregates can cause lysosomal damage releasing proteases (cathepsins) or cell death generating further inflammasome stimuli. Several hypotheses have been proposed to elucidate the inherent ability of cryopyrin to respond to multiple stimuli including ion efflux, and reactive oxygen species (ROS). Recent studies indicate a chief role of mitochondrial DNA or mitochondrial ROS in inflammasome activation [46].
Inappropriate inflammasome activation can lead to excessive inflammation and tissue damage illustrating the importance of selective triggers and tight regulation of inflammasomes. Regulation occurs at many levels, including transcriptional and post-translational processes influencing expression and modification of sensor proteins, or other key inflammasome-associated proteins. For example, NLRP3 is alternatively spliced which can affect protein function. Additionally, TTP and miR223 regulate transcription by binding upstream of NLRP3 [47, 48]. Cytokines can also act like a positive feedback loop by binding to receptors and influencing transcriptional regulation of inflammasome components and pro-cytokines. Serine phosphorylation has also been implicated in regulating cryopyrin function [49, 50]. Some post-translational modifications can negatively regulate the inflammasome, such as S-nitrosylation by nitric oxide [51], or deubiquitination by E3 ligase, Ariadne homolog 2, or TRIM31 [52–54]. There are also endogenous cytokine receptor inhibitors that regulate downstream inflammation including IL-1 receptor antagonist (IL-1RA) and IL-18 binding protein which prevent cytokines from binding to their receptor [55].
CAPS disease pathogenesis
CAPS associated NLRP3 mutations are gain of function leading to a hyperactive cryopyrin inflammasome, increased myeloid cell derived pro-inflammatory cytokine release, and systemic and tissue inflammation leading to disease symptoms. This is supported by in vitro studies using cell lines expressing recombinant inflammasome proteins [33]. Mutant cryopyrin does not require a Signal 2 normally required for cryopyrin inflammasome assembly as observed in ex vivo studies using peripheral blood leukocytes isolated from CAPS patients and stimulated with LPS without ATP [56]. These cells demonstrate increased ASC speck formation, caspase-1 cleavage, IL-1β release, and pyroptosis. CAPS patient cells also demonstrate higher levels of reactive oxygen species due to elevated redox stress and ineffective anti-inflammatory mechanisms [57], and recently CAPS mutations have been shown to have increased cryopyrin phosphorylation leading to inflammasome overactivation [49]. While the mechanisms have not been elucidated, monocytes from FCAS, but not MWS or CINCA/NOMID patients produce IL-1β when cultured at 32°C without LPS [58]. The excellent response of CAPS patients to IL-1 targeted therapy supports a significant role for IL-1β in CAPS disease pathogenesis.
CAPS knockin mutant mouse models were generated with Nlrp3 mutations observed in FCAS, MWS, and CINCA/NOMID patients to further investigate mechanisms involved in CAPS pathogenesis. Bone marrow derived cells from these mice show similar indications of hyperactivation of the cryopyrin inflammasome with cytokine release and speck formation with addition of LPS alone, or exposure to cold temperature in cells from FCAS, but not MWS, or CINCA/NOMID mutant mice [59, 60]. The phenotype of the CAPS knockin mutant mice has many similarities to human CAPS in that the mice demonstrate systemic inflammation affecting the skin, eyes, joints, and central nervous system. Interestingly, the disease severity continuum is reversed in mice with FCAS mice being the most severe and CINCA/NOMID mice being the least affected [59, 61]. IL-1 targeted therapy in the mutant mice has not been as effective as observed in CAPS human patients [59]. Genetic studies utilizing various knockout mice in the inflammasome pathway show that the disease phenotype is dependent on ASC and caspase-1, partially dependent on IL-1β, IL-18, TNF, and pyroptosis, and independent of IL-6 and IL-17 [24, 59, 62–64]. The murine disease is primarily myeloid cell driven with some data supporting a pathogenic role for mast cells [65], and no significant role for lymphocytes [59].
NLRP3 in other diseases
NLRP3 has been implicated in a wide variety of common diseases based on genetic association studies, gene expression analysis, or recombinant mouse models [29]. Cryopyrin activation is also shown to mediate the inflammatory response in tumor necrosis factor associated periodic syndrome (TRAPS) patients, another monogenic auto inflammatory disorder displaying fever, rash, ocular, and musculoskeletal symptoms. Initially, TNF receptor shedding was reported as the pathogenic mechanism [17], but later data including mutant knockin mouse studies supported a role for cryopyrin inflammasome activation through a number of potential mechanisms, including elevated mitochondrial reactive oxygen species generation [66, 67]. Effective IL-1 targeted therapy in TRAPS also points to a role for a dysfunctional cryopyrin inflammasome.
Targeting the NLRP3 pathway in CAPS
The discovery of mutations in NLRP3 in CAPS patients and the elucidation of cryopyrin function prompted investigators to attempt IL-1 targeted therapy. Anakinra, a recombinant form of IL-1RA, was initially studied in sepsis without success, but later approved in rheumatoid arthritis in 2001. It was therefore available for proof-of-concept trials in CAPS patients. Hawkins et al. treated 2 MWS patients with daily anakinra injections and showed that it prevented all CAPS related symptoms and reduced serum amyloid A to normal levels [68]. We treated 3 FCAS patients with two injections prior to an environmental challenge and demonstrated prevention of all FCAS associated symptoms, blunted blood leukocytosis, and reduction in serum IL-6 levels [15]. Additional investigator-initiated trials using daily anakinra in patients with FCAS and MWS confirmed these translational therapeutic successes [69, 70], but it was the remarkable efficacy in patients with CINCA/NOMID [16], the most severe CAPS phenotype, that has had the most impact on patients, and later resulted in FDA and EMA approval (Table 2).
Table 2 -.
Currently Approved Therapies for CAPS
| Anakinra | Rilonacept | Canakinumab | |
|---|---|---|---|
| Pharmacology | IL-1 receptor antagonist | IL-1 receptor fusion protein | IL-1β monoclonal antibody |
| Half- life | ~4–6 hours | ~8 days | ~26 days |
| Approved Dosage (Ped) | 100 mg (1–8 mg/kg) sq | 160–320 mg (2.2–4.4 mg/kg) sq | 150 – 300 mg (2–4 mg/kg) sq |
| Dosage Frequency | 1 day | 1 week | 4–8 weeks |
| Side Effects | Infection, site reaction | Infection, site reaction | Infection, vertigo |
| FDA approval (Age) | CINCA/NOMID (8 mo) | FCAS / MWS (12 yrs) | FCAS / MWS (2 yrs) |
| EMA approval (Age) | CAPS (8 mo) | CAPS (2 yrs) |
Cryopyrin associated periodic syndrome (CAPS), Familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), Chronic infantile neurologic cutaneous articular (CINCA) syndrome or Neonatal onset multisystemic inflammatory disease (NOMID), Pediatric dosing (Ped), age approved for treatment (Age), milligram (mg), kilogram (kg), subcutaneous (sq).
The success of anakinra in CAPS suggested that inhibiting IL-1 mediated inflammation alone was sufficient to prevent CAPS associated symptoms and this was supported by similar clinical success with additional IL-1 targeted drugs in development. Rilonacept, a dimeric IL-1 receptor fusion protein with a longer half-life, provided similar efficacy in FCAS and MWS patients with favorable weekly dosing. A successful clinical trial supported Rilonacept as the first FDA approved therapy in CAPS in 2008 [71]. Canakinumab, a monoclonal antibody against IL-1β demonstrated similar efficacy with every 2 month dosing and was approved for MWS and FCAS in 2009 [72]. Early on, physicians realized that higher dosing of each of these treatments was required in more severely affected patients, while lower dosing was sometimes effective in milder patients further supporting the clinical spectrum of CAPS. While IL-1 targeted therapy may ameliorate clinical abnormalities including progressive hearing loss, brain MRI findings, and early renal disease from amyloidosis, the clinical response in patients with stable deafness, cartilage or bone hypertrophy, or chronic renal failure is often poor. [9, 73] Longer studies with these therapies showed continued efficacy over 1– 2 years [74, 75], but clinical experience over the last decade has illustrated that some CAPS patients are less responsive over time, and require higher or more frequent dosing or switching of therapies[76, 77] All of the IL-1 targeted therapies are associated with increased frequency of non-opportunistic infections prompting early use of antibiotics [71, 72, 74, 75]. Vaccination to prevent common bacterial and viral infections is warranted, but needs to be balanced with reports of significant local reactions or symptomatic episodes following pneumococcal vaccines. Vertigo has been reported in some CAPS patients on canakinumab [72, 75]. Efficacy, safety, and cost concerns and the desire for effective oral medicines have prompted a search for alternative or adjunctive treatments including higher potency IL-1 blockers as well as small molecule inhibitors targeting downstream or upstream in the NLRP3 pathway.
IL-1 receptor blockers showed efficacy so targeting IL-1 receptor signaling with specific small molecule inhibitors is a logical approach [78], but has not progressed to clinical studies in CAPS patients. Since enzymes like caspases are often amenable to pharmacologic targeting, and caspase-1 inhibitors were in clinical development, specific caspase-1 inhibitors were studied in CAPS ex vivo and in vivo models [79]. While the drugs demonstrated some efficacy in pre-clinical studies, it was challenging to achieve adequate serum drug levels and clinical efficacy. Recently, there has been tremendous interest in targeting the cryopyrin inflammasome directly due to the role of NLRP3 in so many common inflammatory and non-inflammatory diseases. Successful preclinical studies using MCC950 in a CAPS mouse model [80] suggest that similar drugs may provide a more reliable long-term therapy for CAPS patients in the near future. Current recommendations for monitoring organ inflammation and targeting symptom resolution may be referenced in other works focused on management of CAPS [81].
Conclusions
The journey from the initial description of FCAS in 1940 to the discovery of NLRP3 and function of the cryopyrin inflammasome, and finally to the application of effective targeted therapies in patients around the world is one of the best examples of translational medicine success. The field began with important detailed clinical descriptions and classification of patients with FCAS, MWS, and CINCA/NOMID by astute clinicians followed by methodical application of modern human genetics techniques. Crucial molecular studies that defined the structure and function of the cryopyrin inflammasome followed by pre-clinical and clinical studies using novel targeted therapies have made a significant impact on our understanding of the regulatory pathways of the innate immune system, but more importantly on the lives of patients with CAPS. Although current anti-IL-1 therapies have proven successful in CAPS, continued mechanistic and therapeutic investigations will further elucidate the normal and pathogenic functions of cryopyrin and are likely to provide more direct and effective treatments for these patients, but also for patients with more common diseases.
Acknowledgements:
NIH grants - RO1 HL126703, KD113592, HL140898
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest:
Dr. Hoffman has received speaking fees from Novartis, consulting fees from Novartis, SOBI, Regeneron, and IFM, and research funds from Glaxo Wellcome, Vertex, Burroughs Wellcome, and Jecure.
References
- 1.Kile RM, Rusk HA. A case of cold urticaria with an unusual family history. JAMA 1940;114:1067–8. [Google Scholar]
- 2.Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol 2001. October;108(4):615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muckle TJ, Wells M. Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. Q J Med 1962. April;31:235–48. [PubMed] [Google Scholar]
- 4.Prieur AM, Griscelli C. [Chronic meningo-cutaneo-articular syndrome in children]. Revue du rhumatisme et des maladies osteo-articulaires 1980. November;47(11):645–9. [PubMed] [Google Scholar]
- 5.Hassink SG, Goldsmith DP. Neonatal onset multisystem inflammatory disease. Arthritis Rheum 1983. May;26(5):668–73. [DOI] [PubMed] [Google Scholar]
- 6.Aksentijevich I, C P, Remmers EF, Mueller JL, Le J, Kolodner RD, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum 2007. April;56(4):1273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuisset L, Jeru I, Dumont B, Fabre A, Cochet E, Le Bozec J, et al. Mutations in the autoinflammatory cryopyrin-associated periodic syndrome gene: epidemiological study and lessons from eight years of genetic analysis in France. Annals of the rheumatic diseases 2011. March;70(3):495–9. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman HM, Gregory SG, Mueller JL, Tresierras M, Broide DH, Wanderer AA, et al. Fine structure mapping of CIAS1: identification of an ancestral haplotype and a common FCAS mutation, L353P. Hum Genet 2003. February;112(2):209–16. [DOI] [PubMed] [Google Scholar]
- 9.Neven B, Marvillet I, Terrada C, Ferster A, Boddaert N, Couloignier V, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum 2010. January;62(1):258–67. [DOI] [PubMed] [Google Scholar]
- 10.Stych B, Dobrovolny D. Familial cold auto-inflammatory syndrome (FCAS): characterization of symptomatology and impact on patients’ lives. Current medical research and opinion 2008. June;24(6):1577–82. [DOI] [PubMed] [Google Scholar]
- 11.Gerbig AW, Dahinden CA, Mullis P, Hunziker T. Circadian elevation of IL-6 levels in Muckle-Wells syndrome: a disorder of the neuro-immune axis? Q J Med 1998. July;91(7):489–92. [DOI] [PubMed] [Google Scholar]
- 12.Cekic S, Yalcinbayir O, Kilic SS. Ocular Involvement in Muckle-Wells Syndrome. Ocular immunology and inflammation 2018. December 17:1–9. [DOI] [PubMed] [Google Scholar]
- 13.Dollfus H, Hafner R, Hofmann HM, Russo RA, Denda L, Gonzales LD, et al. Chronic infantile neurological cutaneous and articular/neonatal onset multisystem inflammatory disease syndrome: ocular manifestations in a recently recognized chronic inflammatory disease of childhood. Archives of ophthalmology 2000. October;118(10):1386–92. [DOI] [PubMed] [Google Scholar]
- 14.Hill SC, Namde M, Dwyer A, Poznanski A, Canna S, Goldbach-Mansky R. Arthropathy of neonatal onset multisystem inflammatory disease (NOMID/CINCA). Pediatric radiology 2007. February;37(2):145–52. [DOI] [PubMed] [Google Scholar]
- 15.Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet 2004. November 13;364(9447):1779–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. The New England journal of medicine 2006. August 10;355(6):581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999. April 2;97(1):133–44. [DOI] [PubMed] [Google Scholar]
- 18.Aganna E, Martinon F, Hawkins PN, Ross JB, Swan DC, Booth DR, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002. September;46(9):2445–52. [DOI] [PubMed] [Google Scholar]
- 19.Dode C, Le Du N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet 2002. June;70(6):1498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001. November;29(3):301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002. December;46(12):3340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet 2002. July;71(1):198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito M, Nishikomori R, Kambe N, Fujisawa A, Tanizaki H, Takeichi K, et al. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood 2008. February 15;111(4):2132–41. [DOI] [PubMed] [Google Scholar]
- 24.McGeough MD, Wree A, Inzaugarat ME, Haimovich A, Johnson CD, Pena CA, et al. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. The Journal of clinical investigation 2017. December 1;127(12):4488–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat 2004. September;24(3):194–8. [DOI] [PubMed] [Google Scholar]
- 26.Kuemmerle-Deschner JB, Verma D, Endres T, Broderick L, de Jesus AA, Hofer F, et al. Clinical and Molecular Phenotypes of Low-Penetrance Variants of NLRP3: Diagnostic and Therapeutic Challenges. Arthritis & rheumatology 2017. November;69(11):2233–40. [DOI] [PubMed] [Google Scholar]
- 27.Verma D, Lerm M, Blomgran Julinder R, Eriksson P, Soderkvist P, Sarndahl E. Gene polymorphisms in the NALP3 inflammasome are associated with interleukin-1 production and severe inflammation: relation to common inflammatory diseases? Arthritis Rheum 2008. March;58(3):888–94. [DOI] [PubMed] [Google Scholar]
- 28.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR gene family: a standard nomenclature. Immunity 2008. March;28(3):285–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annual review of pathology 2015;10:395–424. [DOI] [PubMed] [Google Scholar]
- 30.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 2002. June 14;277(24):21119–22. [DOI] [PubMed] [Google Scholar]
- 31.Dowds TA, Masumoto J, Zhu L, Inohara N, Nunez G. Cryopyrin-induced interleukin 1beta secretion in monocytic cells: enhanced activity of disease-associated mutants and requirement for ASC. J Biol Chem 2004. May 21;279(21):21924–8. [DOI] [PubMed] [Google Scholar]
- 32.Gumucio DL, Diaz A, Schaner P, Richards N, Babcock C, Schaller M, et al. Fire and ICE: the role of pyrin domain-containing proteins in inflammation and apoptosis. Clin Exp Rheumatol 2002. Jul-Aug;20(4 Suppl 26):S45–53. [PubMed] [Google Scholar]
- 33.Manji GA, Wang L, Geddes BJ, Brown M, Merriam S, Al-Garawi A, et al. PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-kappa B. J Biol Chem 2002. March 29;277(13):11570–5. [DOI] [PubMed] [Google Scholar]
- 34.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem 1999. November 26;274(48):33835–8. [DOI] [PubMed] [Google Scholar]
- 35.Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 2014. August;15(8):738–48. [DOI] [PubMed] [Google Scholar]
- 36.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 2014. August;15(8):727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 2013. June;13(6):397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015. October 29;526(7575):666–71. [DOI] [PubMed] [Google Scholar]
- 39.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 2005. April;73(4):1907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 2015. December;25(12):1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015. October 29;526(7575):660–5. [DOI] [PubMed] [Google Scholar]
- 42.Chamaillard M, Girardin SE, Viala J, Philpott DJ. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol 2003. September;5(9):581–92. [DOI] [PubMed] [Google Scholar]
- 43.Inohara N, Ogura Y, Nunez G. Nods: a family of cytosolic proteins that regulate the host response to pathogens. Curr Opin Microbiol 2002. February;5(1):76–80. [DOI] [PubMed] [Google Scholar]
- 44.Barbera-Cremades M, Baroja-Mazo A, Gomez AI, Machado F, Di Virgilio F, Pelegrin P. P2X7 receptor-stimulation causes fever via PGE2 and IL-1beta release. FASEB J 2012. July;26(7):2951–62. [DOI] [PubMed] [Google Scholar]
- 45.He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci 2016. December;41(12):1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018. August;560(7717):198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haneklaus M, O’Neil JD, Clark AR, Masters SL, O’Neill LAJ. The RNA-binding protein Tristetraprolin (TTP) is a critical negative regulator of the NLRP3 inflammasome. J Biol Chem 2017. April 28;292(17):6869–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neudecker V, Haneklaus M, Jensen O, Khailova L, Masterson JC, Tye H, et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. The Journal of experimental medicine 2017. June 5;214(6):1737–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Molecular cell 2017. October 5;68(1):185–97 e6. [DOI] [PubMed] [Google Scholar]
- 50.Stutz A, Kolbe CC, Stahl R, Horvath GL, Franklin BS, van Ray O, et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. The Journal of experimental medicine 2017. June 5;214(6):1725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, et al. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol 2012. December 1;189(11):5113–7. [DOI] [PubMed] [Google Scholar]
- 52.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 2012. October 19;287(43):36617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawashima A, Karasawa T, Tago K, Kimura H, Kamata R, Usui-Kawanishi F, et al. ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J Immunol 2017. November 15;199(10):3614–22. [DOI] [PubMed] [Google Scholar]
- 54.Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nature communications 2016. December 8;7:13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dinarello CA. Overview of the interleukin-1 family of ligands and receptors. Seminars in immunology 2013. December 15;25(6):389–93. [DOI] [PubMed] [Google Scholar]
- 56.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004. March;20(3):319–25. [DOI] [PubMed] [Google Scholar]
- 57.Tassi S, Carta S, Delfino L, Caorsi R, Martini A, Gattorno M, et al. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1beta secretion. Proceedings of the National Academy of Sciences of the United States of America 2010. May 25;107(21):9789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosengren S, Mueller JL, Anderson JP, Niehaus BL, Misaghi A, Anderson S, et al. Monocytes from familial cold autoinflammatory syndrome patients are activated by mild hypothermia. J Allergy Clin Immunol 2007. April;119(4):991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity 2009. June 19;30(6):875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity 2009. June 19;30(6):860–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bonar SL, Brydges SD, Mueller JL, McGeough MD, Pena C, Chen D, et al. Constitutively activated NLRP3 inflammasome causes inflammation and abnormal skeletal development in mice. PloS one 2012;7(4):e35979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. The Journal of clinical investigation 2013. November;123(11):4695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McGeough MD, Pena CA, Mueller JL, Pociask DA, Broderick L, Hoffman HM, et al. Cutting edge: IL-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J Immunol 2012. September 15;189(6):2707–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD, et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 2017. September 13;67(2):736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakamura Y, Franchi L, Kambe N, Meng G, Strober W, Nunez G. Critical role for mast cells in interleukin-1beta-driven skin inflammation associated with an activating mutation in the nlrp3 protein. Immunity 2012. July 27;37(1):85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). The Journal of experimental medicine 2011. March 14;208(3):519–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simon A, Park H, Maddipati R, Lobito AA, Bulua AC, Jackson AJ, et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proceedings of the National Academy of Sciences 2010. May 25, 2010;107(21):9801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle-Wells syndrome. The New England journal of medicine 2003. June 19;348(25):2583–4. [DOI] [PubMed] [Google Scholar]
- 69.Leslie KS, Lachmann HJ, Bruning E, McGrath JA, Bybee A, Gallimore JR, et al. Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Archives of dermatology 2006. December;142(12):1591–7. [DOI] [PubMed] [Google Scholar]
- 70.Ross JB, Finlayson LA, Klotz PJ, Langley RG, Gaudet R, Thompson K, et al. Use of anakinra (Kineret) in the treatment of familial cold autoinflammatory syndrome with a 16-month follow-up. J Cutan Med Surg 2008. Jan-Feb;12(1):8–16. [DOI] [PubMed] [Google Scholar]
- 71.Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008. August;58(8):2443–52. [DOI] [PubMed] [Google Scholar]
- 72.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. The New England journal of medicine 2009. June 4;360(23):2416–25. [DOI] [PubMed] [Google Scholar]
- 73.Goldbach-Mansky R Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Current rheumatology reports 2011. April;13(2):123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoffman HM, Throne ML, Amar NJ, Cartwright RC, Kivitz AJ, Soo Y, et al. Long-term efficacy and safety profile of rilonacept in the treatment of cryopryin-associated periodic syndromes: results of a 72-week open-label extension study. Clinical therapeutics 2012. October;34(10):2091–103. [DOI] [PubMed] [Google Scholar]
- 75.Kuemmerle-Deschner J, Hofer F, Endres T, Kortus-Goetze B, Blank N, Weissbarth-Riedelet E, et al. Real-life effectiveness of canakinumab in cryopyrin-associated periodic syndrome. Rheumatology 2016;55(4):689–96. [DOI] [PubMed] [Google Scholar]
- 76.Caorsi R, Lepore L, Zulian F, Alessio M, Stabile A, Insalaco A, et al. The schedule of administration of canakinumab in cryopyrin associated periodic syndrome is driven by the phenotype severity rather than the age. Arthritis research & therapy 2013. February 26;15(1):R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Urien S, Bardin C, Bader-Meunier B, Mouy R, Compeyrot-Lacassagne S, Foissac F, et al. Anakinra pharmacokinetics in children and adolescents with systemic-onset juvenile idiopathic arthritis and autoinflammatory syndromes. BMC pharmacology & toxicology 2013. August 5;14:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang C, Hockerman S, Jacobsen EJ, Alippe Y, Selness SR, Hope HR, et al. Selective inhibition of the p38alpha MAPK-MK2 axis inhibits inflammatory cues including inflammasome priming signals. The Journal of experimental medicine 2018. May 7;215(5):1315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JC, et al. IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol 2005. August 15;175(4):2630–4. [DOI] [PubMed] [Google Scholar]
- 80.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature medicine 2015. March;21(3):248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Annals of the rheumatic diseases 2015. September;74(9):1636–44. [DOI] [PubMed] [Google Scholar]
- 82.Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, et al. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat 2008. June;29(6):803–8. [DOI] [PubMed] [Google Scholar]
- 83.Sarrauste de Menthiere C, Terriere S, Pugnere D, Ruiz M, Demaille J, Touitou I. INFEVERS: the Registry for FMF and hereditary inflammatory disorders mutations. Nucleic acids research 2003. January 1;31(1):282–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van Gijn ME, Ceccherini I, Shinar Y, Carbo EC, Slofstra M, Arostegui JI, et al. New workflow for classification of genetic variants’ pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). Journal of medical genetics 2018. August;55(8):530–7. [DOI] [PubMed] [Google Scholar]