

Abstract
Rationale:
Abdominal aortic aneurysm (AAA) is a permanent and localized dilatation of abdominal aorta with potentially fatal consequence of aortic rupture. No effective pharmacological approach has been identified to limit AAA progression and rupture. AAA is characterized by extensive aortic wall matrix degradation that contributes to arterial wall remodeling and eventual rupture, in which smooth muscle cell (SMC) phenotypic transition and matrix metalloproteinases (MMP), especially MMP2 and MMP9, play critical roles.
Objective:
Our previous study showed that adenosine deaminases acting on RNA 1 (ADAR1) regulates SMC phenotype, which prompted us to study if ADAR1 is involved in AAA development.
Methods and Results:
We used angiotensin II (Ang II) infusion ApoE−/− mouse model combined with ADAR1 global and SMC-specific knockout to study the role of ADAR1 in AAA formation/dissection. Aortic transplantation was conducted to determine the importance of vascular cell ADAR1 in AAA development/dissection. Primary cultured SMC were used to study how ADAR1 regulates the inflammatory SMC phenotype and MMP production/activity. Patient specimens were obtained to investigate the relevance of ADAR1 expression to human AAA disease. ADAR1 was induced in abdominal aortic SMC in both mouse and human AAA tissues. Heterozygous knockout of ADAR1 diminished the Ang II-induced AAA/dissection in ApoE−/− mice. Mouse aortic transplantation showed that ADAR1 in vascular cells was essential for AAA formation. SMC-specific ADAR1 knockout reduced experimental AAA formation/dissection. Mechanistically, ADAR1 interacted with HuR to increase the stability of MMP2 and MMP9 mRNA, leading to increased MMP levels and activities.
Conclusion:
ADAR1 is novel regulator of AAA development/dissection, and thus may represent a potentially new therapeutic target to hinder AAA growth and rupture.
Keywords: Aneurysm, Basic Science Research, Smooth Muscle Proliferation and Differentiation, Vascular Biology, Vascular Disease
Graphical Abstract
INTRODUCTION
Abdominal aortic aneurysm (AAA) is defined as a permanent, localized dilatation of the abdominal aorta with potentially fatal consequence of aortic rupture 1. AAA is a common vascular disease with more than 150,000 new cases and 10,000 deaths annually in the United States, posing a particularly high risk to the elderly population due to high morbidity and mortality associated with rupture and dissection. Moreover, both open surgical and endovascular repair are associated with significant short- and long-term morbidity and mortality 2. Pharmacological approaches proposed to limit AAA progression and rupture have thus far proven ineffective 3, highlighting an essential need to understand the regulatory mechanisms contributing to AAA pathogenesis.
Inflammation of the arterial wall is one of the most important events in the initiation and progression of AAA 4. Smooth muscle cells (SMC) play a key role in vascular inflammation in AAA by producing biologically active cytokines, chemokines, and pro-inflammatory factors that attract and activate leukocytes and induce SMC phenotype alterations, ultimately leading to destruction of extracellular matrix (ECM) components and aneurysmal dilation. Histological analyses of both animal and human AAA have demonstrated phenotypically altered SMC in these specimens, which are proposed to contribute to the pathogenesis/progression of AAA 5–7. Proteolytic enzymes, mainly MMP2/9, produced by phenotypically-modulated SMC, disrupt the structural integrity of the vessel wall and degrade ECM, leading to AAA formation/dissection 8, 9. Increased MMP2/9 levels have been detected in aortic SMC from patients with AAA 10, 11.
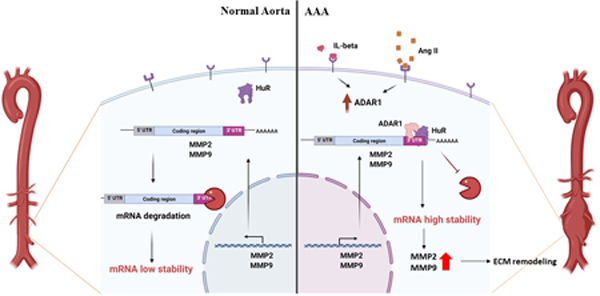
RNA editing (A→I editing) is a posttranscriptional mechanism affecting mRNA splicing and is catalyzed by adenosine deaminase acting on RNA (ADAR) 12, 13. ADAR1 has also been shown to regulate RNA stability, microRNA processing, and RNA storage 14. We have shown that ADAR1 plays an essential role in SMC phenotypic modulation and vascular remodeling 15. In view of the importance of SMC phenotypic modulation in AAA, we studied the role and mechanisms of ADAR1 in AAA development. Increased expression of ADAR1 was detected in SMC in AAA lesions, in association with an inflammatory SMC phenotype. ADAR1 deficiency attenuated AAA development/dissection. Moreover, SMC tissue-specific knockout of ADAR1 diminished AAA formation/dissection. Mechanistic studies identified a new role for ADAR1 to increase matrix metalloproteinase (MMP)-2 and MMP9 mRNA stability by interacting with HuR, leading to increased MMP levels and activities.
METHODS
Data Availability.
The detailed experimental materials and methods are available in the Data Supplement. The authors declare that the majority of supporting data are presented within this article and in the Data Supplement. The source data for the figures and the data that are not shown are available from the corresponding author upon reasonable request.
RESULTS
ADAR1 expression is increased in mouse and human AAA lesions.
To test if ADAR1 is involved in the development of AAA, we used a well-established experimental aneurysm model that was generated by chronic infusion of angiotensin II (Ang II) into ApoE knockout (ApoE−/−) mice 16. While immunostaining showed that ADAR1 was barely detectable in control aortas, consistent with our previous observation 15, it was significantly induced in AAA lesions (Fig 1A). Importantly, ADAR1 largely co-localized with ACAT2-positive cells in the media layer, indicating a potential role of SMC ADAR1 in AAA formation (Fig 1A–1B). Both ADAR1 isoforms (p150 and p110) were significantly upregulated in the AAA lesions as detected by Western blotting (Fig 1, C-D). More significantly, ADAR1 was also upregulated in human AAA tissues, and was largely co-localized with SMC (Fig 1, E-F). Similar to the expression pattern in mouse AAA, both ADAR1 isoforms were significantly elevated in human AAA lesions (Fig 1, G-H). These data suggest that ADAR1 may be involved in the AAA development in both mouse and human patients.
Figure 1: ADAR1 was induced in both mouse and human AAA lesions.
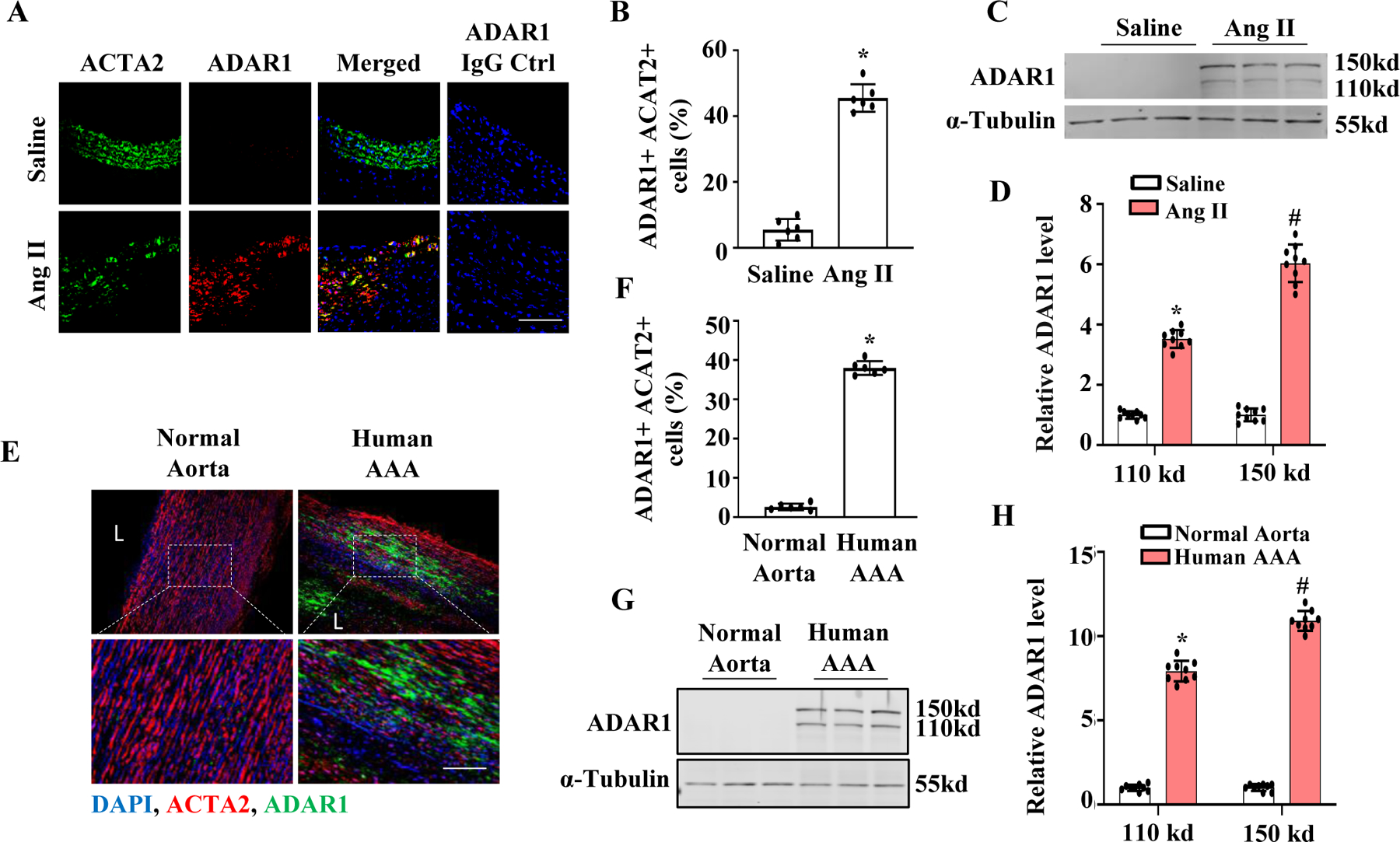
A-D, ApoE−/− Mice were infused with saline or angiotensin II (Ang II, 1000 ng/kg/min) for 28 days. Abdominal aorta sections were co-immunostained with ACTA2 and ADAR1 antibodies (A). The percentage of ADAR1+ACTA2+ cells relative to the total ACTA2+ cells were quantified from 10 sections each animal (B). ADAR1 was expressed predominantly in SMCs in mouse AAA lesions. *P = 4.4e-5 vs. saline-infused mouse aorta, n=6, Fisher’s exact test (two-tailed). (C-D) ADAR1 protein expression in abdominal aorta were examined by Western blotting (C) and quantified by normalizing to α-Tubulin for each ADAR1 isoform, respectively (D). *P = 1.9423e-2, #P = 1.7e-5 vs. saline-treated group for the corresponding isoform. Unpaired t test (two-tailed), n=9. E, Normal healthy human abdominal aorta or AAA sections were co-immunostained with ACTA2 and ADAR1 antibodies. Blue: DAPI, Red: ACTA2, Green: ADAR1. F, Quantitative analyses of the percentage of ADAR1+ ACTA2+ cells relative to the total ACTA2+ cells in each section. *P = 3e-6 vs. normal aorta, n=9, Fisher’s exact test (two-tailed). G-H, ADAR1 protein expression in human abdominal aorta or AAA lesions were detected by Western blotting (G) and quantified by normalizing to α-Tubulin for each ADAR1 isoform, respectively (H). *P = 1.9423e-2 and #P = 1.7e-5 vs. the normal aortas for each isoform, respectively, n=9, Unpaired t test (two-tailed). Shown are fold inductions relative to the mean value of saline-infused (B & D) or human normal aortas (F & H), which was set as 1.
ADAR1 deficiency attenuates AAA formation.
To determine if ADAR1 is important for AAA development, we generated ADAR1 deficient mice in ApoE−/− background. Due to embryonic lethality of the homozygous ADAR1 knockout, ADAR1 heterozygous knockout mice (ADAR1+/−;ApoE−/−) were compared with ApoE−/− mice to determine the role of ADAR1 in AAA. While ApoE−/− mice infused with Ang II for 28 days developed significant aortic dilation/dissection, ADAR1+/− mice were mostly protected against aneurysm formation (Fig 2, A-E); blood pressure was similar in Ang II-infused ApoE−/− and ADAR1+/−;ApoE−/− mice. The incidence of AAA was reduced from 75% in ApoE−/− mice to 20% in ADAR1+/−;ApoE−/− mice. There was a 10% mortality rate in ApoE−/− mice infused with Ang II, but no mortality was observed in ADAR+/−;ApoE−/− mice. To accurately measure the maximal aorta diameter in vivo, B mode ultrasound of abdominal aorta was performed (both longitudinally and transversely) in mice infused with saline or Ang II for 28 days. As shown in Fig 2B–2D, Ang II infusion increased aortic diameter in ApoE−/− mice infused with Ang II, which was significantly blunted in the ADAR1+/− mice. The alterations of diameters were also confirmed by measuring the aortas and AAA sizes ex vivo (online Fig I, A). These results demonstrated that ADAR1 plays an essential role in AAA development.
Figure 2: ADAR1 deficiency attenuated AAA formation in mice.
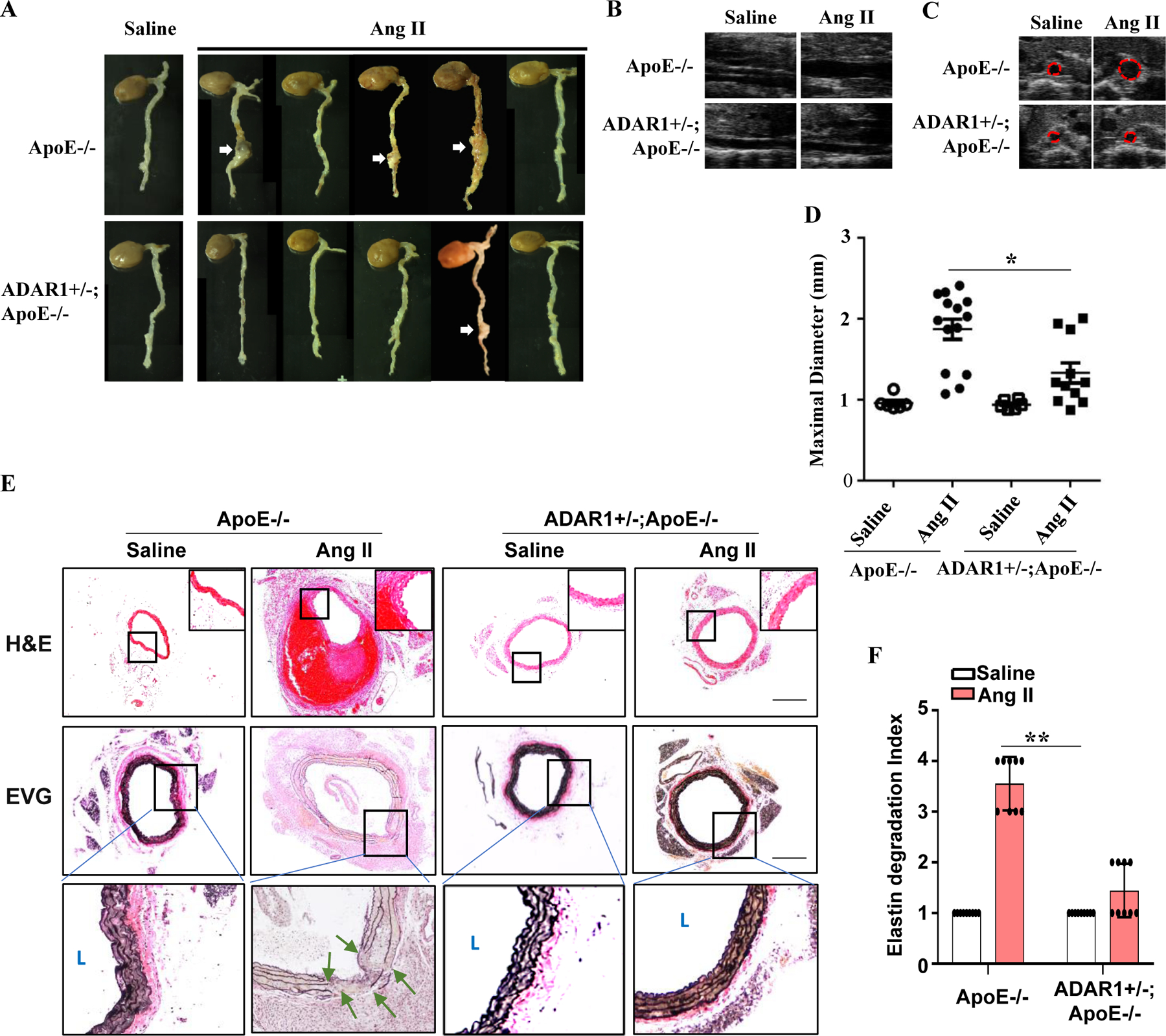
A, ADAR1 heterozygous deletion (AD1+/−) attenuated Ang II-induced AAA formation in ApoE−/− mice. Abdominal aortas were collected 28 days after Ang II infusion. White arrows indicate AAA. B-C, ADAR1+/− diminished Ang II infusion-caused aorta dilation in ApoE−/− mice as imaged by B mode ultrasound longitudinally (C) and transversely (D). D, Quantitative analyses of maximal external aortic diameters. *P = 2.71E-02, n=12. E, Representative images of hematoxylin and eosin (H&E) or Verhoeff’s elastic (EVG) staining of AAA tissues. Images in lower panels of the EVG staining show higher magnification images of the areas in the black boxes in the upper panels. Green arrows indicate breaks of elastic lamella. Scar bar = 100μm. F, Quantification of the elastin degradation indexes in abdominal aorta media. Shown are fold inductions relative to the mean value of saline-infused ApoE−/− mouse group, which was set as 1. **P = 2.05E-02, n=8. One-way Anova with Tukey’s multiple comparison tests were performed for D, and Kruskal-Wallis test with Dunn’s multiple comparisons test were performed for F.
Medial degeneration and elastin degradation are hallmarks for AAA development, contributing to both aortic dilation and dissection. Hematoxylin and eosin (H&E) staining showed that Ang II infusion caused marked aortic media degeneration/dissection and consequently intraluminal thrombus in ApoE−/− mice, but no statistical difference was observed in ADAR+/−;ApoE−/− mice (Fig 2E). Consistently, elastin fragmentation, determined by Verhoeff’s elastic (EVG) staining to quantify the elastin degradation index 17, was also markedly increased in ApoE−/− mice, but no statistical difference was observed the ADAR1+/− mice (Fig 2, E-F).
ADAR1 deficiency reduces the expression and activities of MMP2 and MMP9 in AAA lesions.
MMP2 and MMP9 play critical roles in AAA formation/dissection and are largely responsible for elastin degradation. Consistent with previous studies 16, 18, 19, we observed a marked increase in both pro- and cleaved MMP2 and MMP9 protein levels in the abdominal aortas of Ang II-infused ApoE−/− mice. However, Ang II-infused ADAR+/−;ApoE−/− mice exhibited no statistical difference in aortic MMP2 or MMP9 levels (Fig 3, A-C). Since MMPs, especially MMP2, are expressed by phenotypically modulated SMC 20, we evaluated SMC phenotype in AAA tissues. As shown in Fig 3, A & D-E, in ApoE−/− mice, Ang II infusion led to increased expression of vascular cell adhesion molecule-1 (VCAM-1) but decreased expression of smooth muscle markers ACTA2 and calponin in the abdominal aorta, consistent with SMC phenotypic modulation. However, the expression of these markers was unaffected by Ang II infusion into ADAR1+/−;ApoE−/− mice (Fig 3, A, D-E), suggesting that ADAR1 deficiency prevented inflammatory SMC phenotypic modulation during AAA development. In order to examine MMP activity in AAA tissues, in situ zymography was performed. Ang II infusion resulted in significantly increased MMP activity in abdominal aortas of ApoE−/− mice as indicated by the green signal (Fig 3F), but no statistical difference was observed in ADAR1+/−;ApoE−/− mice (Fig 3, F-G). To evaluate the effect of ADAR1 deficiency on MMP-2 and MMP-9 activities individually, gelatin gel zymography was performed on aortic lysates. The activity of both MMP2 and MMP9 was significantly increased by Ang II in ApoE−/− mice, but no statistical difference was observed in ADAR1+/−;ApoE−/− mice (Fig 3, H-J). These results suggest that ADAR1 deficiency modulated both MMP2 and MMP9 expression/activities in the context of AAA formation.
Figure 3: ADAR1 promoted MMP2 and MMP9 production and their activity in AAA lesions.
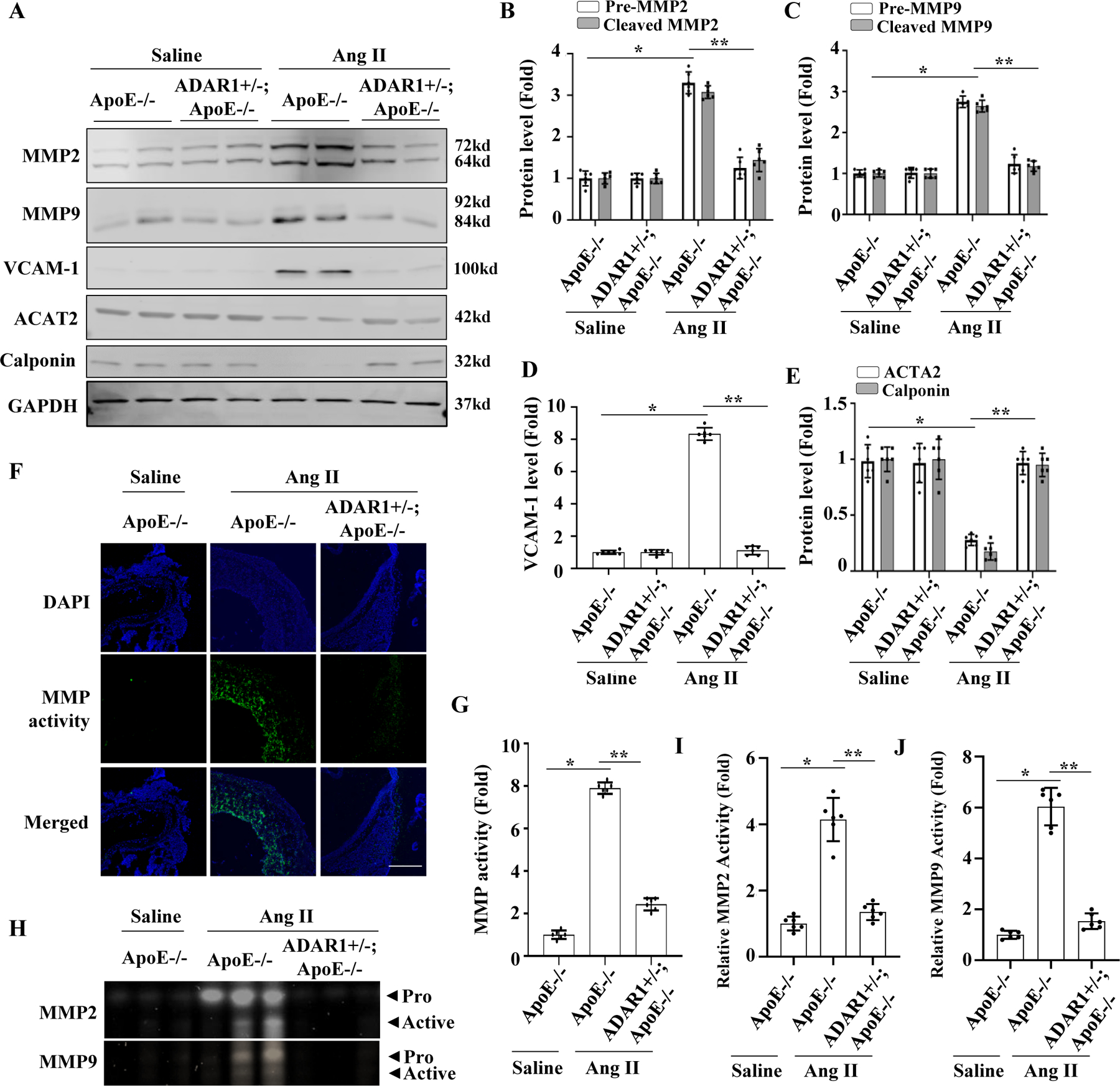
A-E, ApoE−/− and ADAR1+/− mice in ApoE−/− background were infused with saline or Ang II (1000ng/kg/min) for 14 days. The protein levels of pre- and active MMP2 (B), MMP9 (C), VCAM-1 (D), ACTA2 & Calponin (E) were detected by Western blot and quantified by normalizing to GAPDH. *P =2.34E-02 for pre-MMP2, 2.70E-02 for cleaved-MMP2 (B), 1.87E-02 for pre-MMP9, 2.87E-02 for cleaved-MMP9 (C), 1.80E-02 (D), 2.40E-05 for ACTA2, 4.40E-05 for Calponin (E) vs. WT with saline infusion, respectively (n=6); **P = 3.31E-02 for pre-MMP2, 2.40E-02 for cleaved-MMP2 (B), 8.70E-02 for pre-MMP9, 7.60E-02 for cleaved-MMP9 (C), 2.10E-02 (D), 3.20E-05 for ACTA2, 4.20E-05 for Calponin (E) vs. ApoE−/− mice with Ang II infusion, respectively (n=6). F-G, In situ zymography of aorta showing increased MMP activity in Ang II-infused ApoE−/− mouse aorta, which was mitigated in ADAR1+/− aorta. Green fluorescence indicates MMP activities, which were quantified by normalizing to the fluorescent signal intensity in saline-treated mice, Scale bar = 30 um. *P = 2.80E-04 vs. WT with saline infusion; **P = 2.40E-02 vs. ApoE−/− mice with Ang II infusion, n=6. H-J, Aorta homogenates of ApoE−/− and ADAR1+/−; ApoE−/− mice infused with saline or Ang II for 14 days were prepared, and MMP2 and MMP9 activities were examined by gelatin zymography (H), measured by densitometry, and quantified by normalizing to the saline-infused group (I-J). *P = 3.60E-03 (I) and 2.90E-03 (J) vs. ApoE−/− mice with saline infusion, respectively (n=6); **P = 3.30E-02 (I) and 1.30E-02 (J) vs. ApoE−/− mice with Ang II infusion, respectively (n=6). All protein levels or activities were shown as fold inductions relative to the mean value of saline-infused ApoE−/− mouse group, which was set as 1 in each comparison. Kruskal-Wallis test with Dunn’s multiple comparisons tests were performed to determine statistical difference for all panels.
Vascular cell ADAR1 is essential for AAA formation.
To test if ADAR1 expressed in vascular cells is essential for AAA formation, we generated a heterotopic transplantation model by grafting abdominal aorta of ApoE−/− or ADAR1+/−;ApoE−/− mice to recipient ApoE−/− mice (Fig 4A). The recipient aorta was ligated to detour the blood flow to the donor aorta as previously described 21. One week later, Ang II was infused for 28 days, and ultrasound was performed to measure the diameter of the donor aorta (Fig 4B). The diameter of transplanted ADAR+/− aortas was significantly less than ApoE−/− mouse aortas (Fig 4C). H&E and EVG staining of the aorta sections showed significant dilation, media degeneration, and elastin fragmentation in transplanted ApoE−/− mouse aorta. However, these defects were attenuated in the transplanted ADAR1+/− aorta (Fig 4, D-E). As a result, the elastin degradation index was significantly reduced in the transplanted ADAR1+/− aorta (Fig 4F). Moreover, the prevalence of AAA formation in the transplanted ADAR1+/− aorta was also significantly lower than the WT aorta (Fig 4G). These results indicate that vascular cell ADAR1 is vital for AAA formation in Ang II-infused ApoE−/− mice.
Figure 4: Vascular ADAR1 was essential for AAA formation.
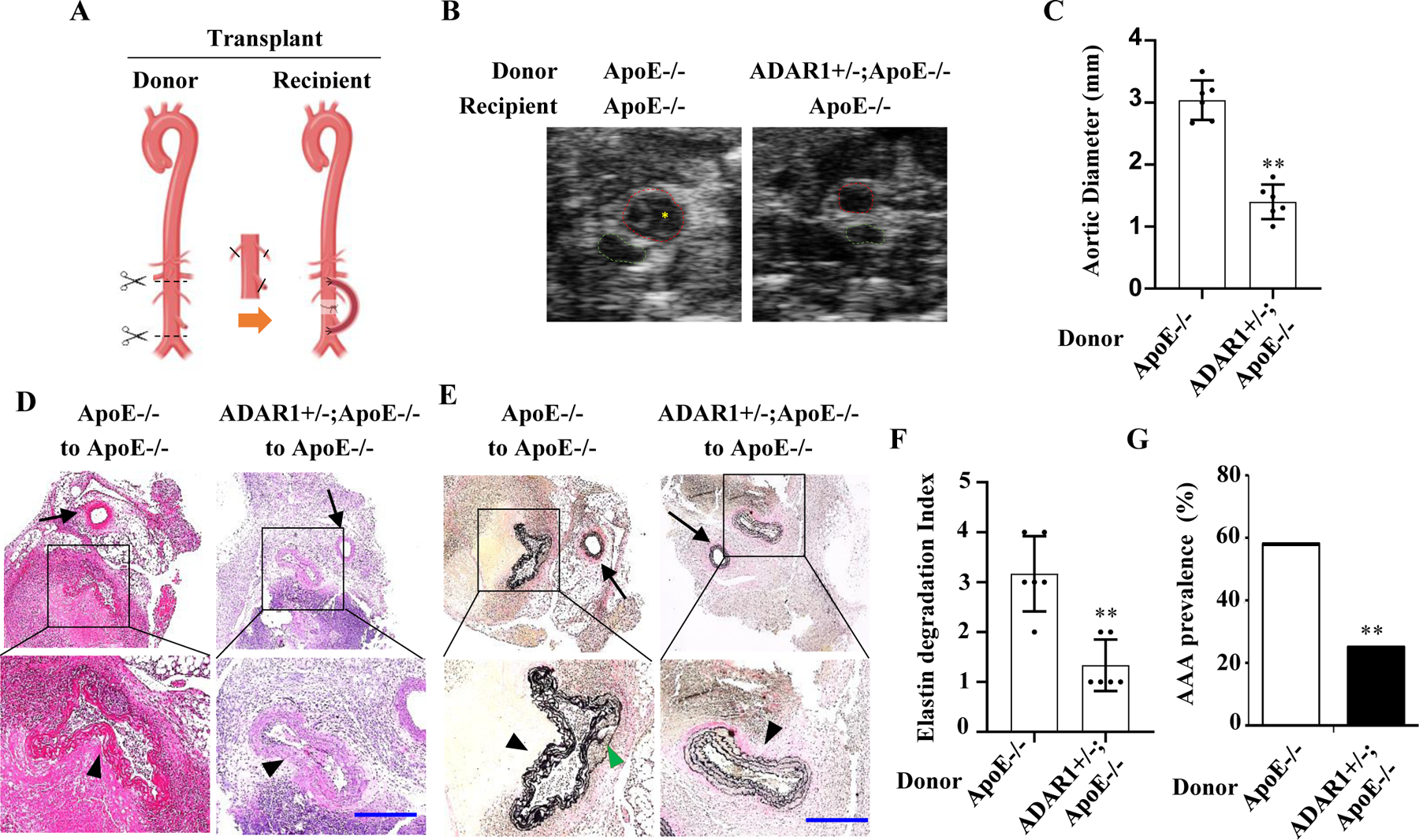
A, A graphic illustration of the mouse aortic transplant. Donor aortic segment was anastomosed to recipient abdominal aorta in an end-to-side manner. Recipient mice were then infused with Ang II (1000 ng/kg/min) for 28 days (generated by Biorender). B, Representative transverse ultrasound images of abdominal aorta 28 days after the transplantation. Red dash circles: donor aortas; Green dash circles: recipient mice’s own aortas. C, Maximal external diameters of donor aortas were measured by ultrasound imaging. One-way analysis of variance followed by Tukey’s test was performed to determine statistical difference. **P = 2.17E-03 vs. ApoE−/− mouse donor aortas, n=6, Mann-Whitney test (two-sided). D-E, Representative images of hematoxylin and eosin (H&E) (D) or Verhoeff’s elastic (EVG) staining (E) of the transplanted aortas with aneurysm. The areas in the black box were shown with a higher magnification in the lower part of the panel. Black arrowhead: donor aortas; Black arrow: recipient’s own aortas; Green arrowhead: elastic lamellae breaks. Scar bar= 100 μm. F: Quantification of elastin break indexes in the donor aortas of Ang II-infused ApoE−/− and ADAR1+/−;ApoE−/− recipient mice. Kruskal-Wallis test followed by Dunn’s post-hoc test was performed. **P = 2.165e-3 vs. ApoE−/− mouse donor aortas, n=6, Mann-Whitney test (two-sided). G, Prevalence of AAA. AAA was defined as a percentage increase in aortic diameter ≥100%. Unpaired Student t test was performed. **P = 1.72e-4 vs. ApoE−/− mouse donor aortas, n=6, Fisher’s exact test (two-sided).
ADAR1 deficiency in smooth muscle ameliorates AAA formation.
Since ADAR1 has been shown to modulate SMC phenotype and is prominently expressed in medial SMC in AAA tissues (Fig 1), we generated ADAR1 SMC-specific knockout mice by crossing ADAR1fl/fl mice with Mhy11-CreERT;ApoE−/− mice 22. Because homozygous ADAR1 deletion in SMC is lethal within 7 days after tamoxifen-induced Cre recombination (data not shown), ADAR1 SMC heterozygous knockout (ADAR1sm+/−) mice were employed. While Mhy11-CreERT;ApoE−/− mice injected with tamoxifen and infused with Ang II for 28 days readily developed AAA with a 10% mortality rate, ADAR1sm+/− mice were resistant to aneurysm formation without any lethality (Fig 5A). The AAA incidence was reduced from 75% in ApoE−/− mice to 25% in ADAR1sm−/− mice. These findings were corroborated by ultrasound data, which confirmed a reduction in maximal aortic diameter in Ang II-infused ADAR1sm+/− mice as compared to control ApoE−/− mice (Fig 5, B-D and online Fig I, B). Histological analyses showed that aortas from ADAR1sm+/− mice had significantly less elastin fragmentation as compared to ApoE−/− mice (Fig 5, E-F). These results indicated that SMC ADAR1 plays an essential role in AAA formation in Ang II-infused ApoE−/− mice.
Figure 5: ADAR1 deficiency in smooth muscle (ADAR1sm+/−) attenuated AAA formation.
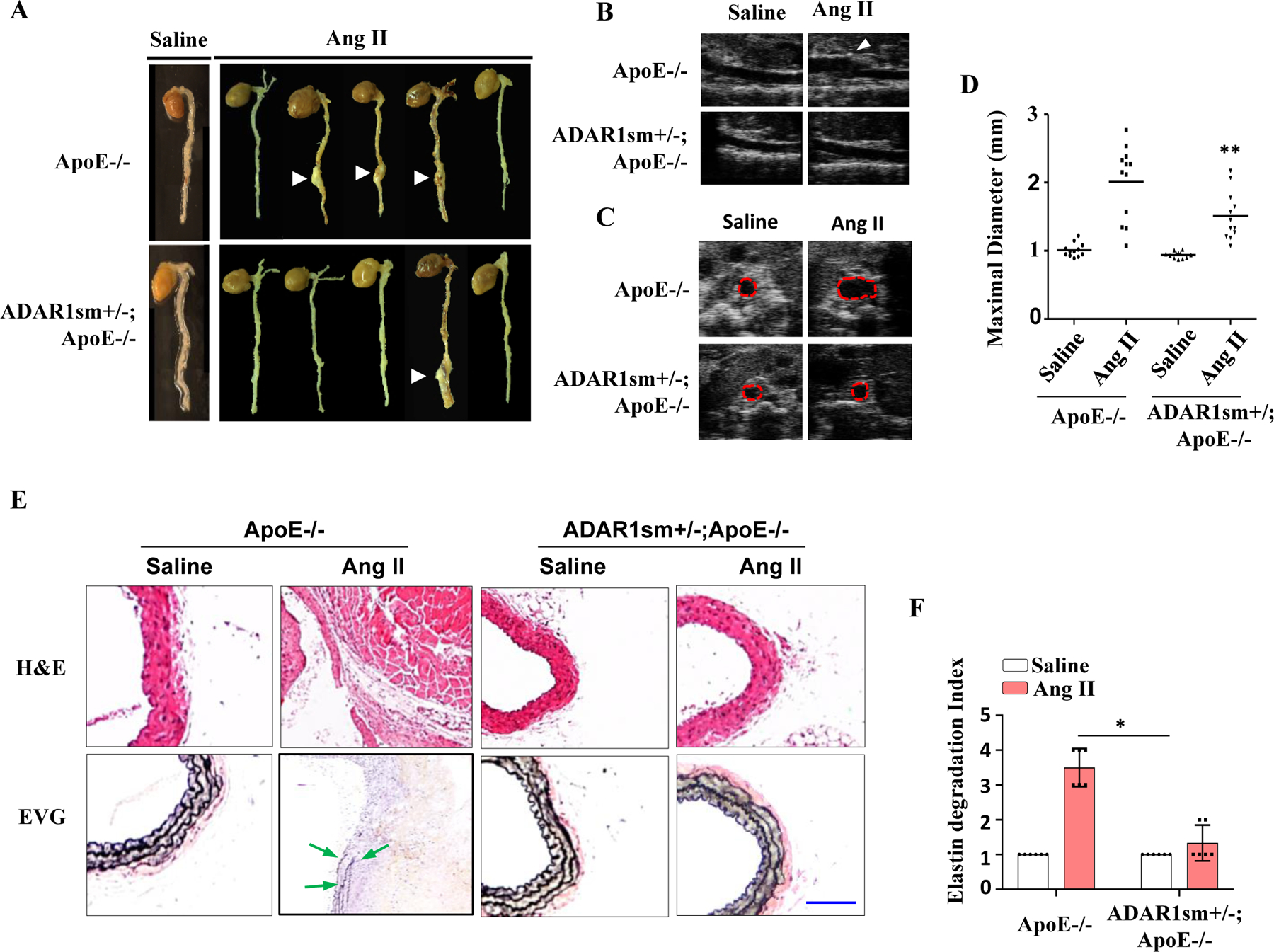
ApoE−/− and ADAR1sm+/−;ApoE−/− mice were infused with Ang II (1000 ng/kg/min) for 28 days. A, Representative aneurysm formation as marked by arrowheads. B-C, Representative longitudinal (B) and transverse (C) ultrasound images of abdominal aorta. Red dash circle indicates aorta lumen. D, Quantitative analyses of maximal aorta diameters. **P = 2.50E-02 vs. ApoE−/− mice with Ang II infusion, n=12. One-way Anova with Tukey’s multiple comparison test were performed to determine statistical difference. E, Hematoxylin and eosin (H&E) or Verhoeff’s elastic (EVG) staining of AAA tissues. Scale Bar: 30 μm. F, Quantification of elastin degradation indexes that were shown as fold induction relative to the mean value of saline-infused ApoE−/− mouse group, which was set as 1. *P = 4.56E-03, n=6. Kruskal-Wallis test with Dunn’s multiple comparisons tests were performed to determine statistical difference.
ADAR1 promotes the synthetic/inflammatory SMC phenotype and MMP expression independent of its RNA editing function.
SMC phenotypic modulation plays a role in the pathogenesis of AAA. The pro-inflammatory cytokine IL-1β is upregulated in AAA tissues and has been shown to induce synthetic/inflammatory SMC phenotype 23, 24. Interestingly, in cultured SMC, IL-1β induced the expression of ADAR1, vascular cell adhesion molecule1 (VCAM1), MMP2 and MMP9 while downregulating the SMC markers MYH11, calponin, and ACAT2 (Fig 6A). Knockdown of ADAR1 using shRNA prevented IL-1β -induced downregulation of SMC marker proteins and blunted the upregulation of VCAM1, MMP2 and MMP9 (Fig 6, A-H). To determine if ADAR1 regulates activities of secreted MMPs from IL-1β-treated cells, supernatants of cultured SMC were collected, and MMP activity was detected by zymography. Consistent with the protein expression data, IL-1β increased the activity of secreted MMPs, which was prevented by knockdown of ADAR1 (Fig 6, I). These results suggest that ADAR1 promotes the synthetic/inflammatory SMC phenotype and MMP production/activity.
Figure 6: ADAR1 regulated SMC phenotype and MMP level/activity independent of its editing function.
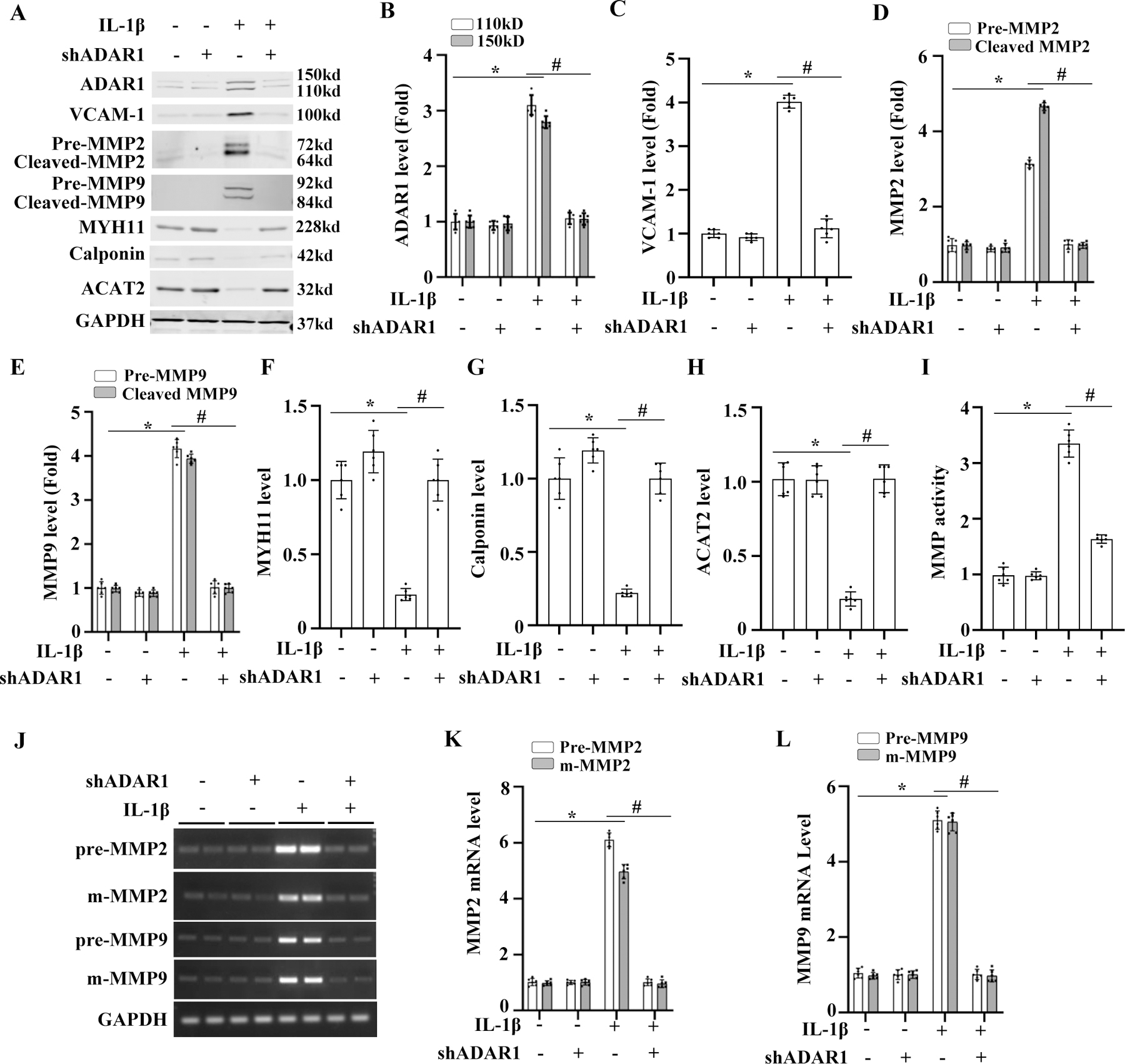
A-I, Mouse aortic SMCs were transduced with control (−) or ADAR1 shRNA (shADAR1) adenoviral vector and then treated with 10 ng/ml of IL-1β for 24 h. Protein expressions of ADAR1, inflammatory cell marker VCAM-1, MMP2, MMP9, and SMC markers were detected by Western blot and quantified by normalizing to GAPDH, respectively. MMP activity was measured by in situ zymography (I). *P =1.45E-02 for ADAR1 110 kD, 2.05E-02 for ADAR1 150 kD (B), 2.08E-03 (C), 2.85E-03 for pre-MMP2, 1.45E-03 for m-MMP2 (D), 1.85E-03 for pre-MMP9, 2.35E-03 for m-MMP9 (E), 2.11E-02 (F), 1.41E-02 (G), 8.41E-03 (H), 1.85E-03 (I) vs. vehicle-treated cells (−), respectively; #P = 1.98E-02 for ADAR1 110 kD, 2.48E-02 for ADAR1 150 kD (B), 3.11E-02 (C), 2.53E-02 for pre-MMP2, 1.83E-02 for m-MMP2 (D), 2.53E-02 for pre-MMP9, 3.23E-02 for m-MMP9 (E), 4.05E-02 (F), 2.05E-02 (G), 1.35E-02 (H), 2.53E-02 (I) vs. control adenovirus-transduced cells (−) with IL-1β treatment, respectively. n=6 for B-H; n=3 for I. J-L, Mouse aortic SMCs were transduced with control (−) or ADAR1 shRNA adenoviral vector (shADAR1) and then treated with vehicle (−) or 10 ng/ml of IL-1β for 24 h. The precursor (pre-) and mature (m)-mRNA levels of MMP2 and MMP9 were detected by RT-PCR (J). The pre- and m-mRNA levels of MMP2 (K) and MMP9 (L) were quantified by normalizing to GAPDH, respectively. *P = 4.92E-03 for pre-MMP2, 1.99E-02 for m-MMP2 (K) and 4.92E-03 for pre-MMP9, 9.22E-03 for m-MMP9 (L) vs. vehicle-treated cells (−); #P = 5.98E-03 for pre-MMP2, 3.40E-02 for m-MMP2 (K) and 2.98E-02 for pre-MMP9, 3.18E-02 for m-MMP9 (L) vs. control vector-transduced cells (−) with IL-1β treatment; n=6. All protein or mRNA levels were shown as fold induction relative to the mean value of vehicle (−) and control vector-transduced cells (−), which was set as 1 for each comparison. Kruskal-Wallis test with Dunn’s multiple comparisons tests were performed to determine statistical difference for all panels.
Since ADAR1 is an RNA editing enzyme, we tested whether it participates in MMP2 and MMP9 pre-mRNA splicing. As shown in Fig 6, J-L, both MMP2 and MMP9 pre-mRNAs were spliced normally in IL-1β-treated SMC. Knockdown of ADAR1 using its specific shRNA reduced both the pre- and mature mRNAs (Fig 6, J-L), suggesting that ADAR1 does not participate in MMP2 or MMP9 pre-mRNA splicing. Moreover, ADAR1 editing inhibitor 8-azaadenosine, which blocks ADAR1 editing function 25, also failed to alter IL-1β or ADAR1 regulation of MMP2, MMP9, or VCAM-1 mRNA expression (Online Fig II & III, A-D). However, 8-azaadenosine was able to block IL-1β- or ADAR1-induced abnormal splicing of ACTA2 pre-mRNA (online Fig II & III, A & E), consistent with our previous finding that ADAR1 regulates SMC marker genes through its RNA editing function. These results indicate that ADAR1 regulates MMP2/9 and inflammatory marker expression through an RNA editing-independent mechanism.
ADAR1 stabilizes MMP2 and MMP9 mRNA via interacting with human antigen R (HuR).
Although ADAR1 did not affect MMP2 and MMP9 pre-mRNA splicing, knockdown of ADAR1 increased the mRNA degradation rates of MMP2 and MMP9 while their gene transcription was blocked by actinomycin D (Fig 7, A-C), suggesting that ADAR1 promotes MMP2 and MMP9 mRNA stability. Since previous studies have shown that HuR, but not HuB, HuC, or HuD, stabilizes MMP2 and MMP9 mRNA by binding their 3′ UTRs 26, 27, we hypothesized that ADAR1 may interact with HuR to regulate MMP2 and MMP9 mRNA stability. To test this hypothesis, we first detected if ADAR1 interacts with HuR in SMC. Co-immunoprecipitation assay showed that ADAR1 physically interacted with HuR, and IL-1β treatment significantly enhanced the ADAR1-HuR interaction (Fig 7, D-E). In order to test if ADAR1 cooperates with HuR to stabilize MMP2/9 transcripts, we knocked down ADAR1 in HuR-overexpressing SMC. As shown in Fig 7, F-H, overexpression of HuR promoted MMP2 and MMP9 mRNA stability as evidenced by the reduced mRNA degradation rate. However, knockdown of ADAR1 diminished the ability of HuR in promoting MMP2 and MMP9 mRNA stability (Fig 7, F-H). These results suggest that ADAR1 interacts with HuR to promote MMP2 and MMP9 mRNA stability, thus contributing to the phenotypic modulation of SMC and AAA formation.
Figure 7: ADAR1 promoted HuR-dependent MMP2 and MMP9 mRNA stability.
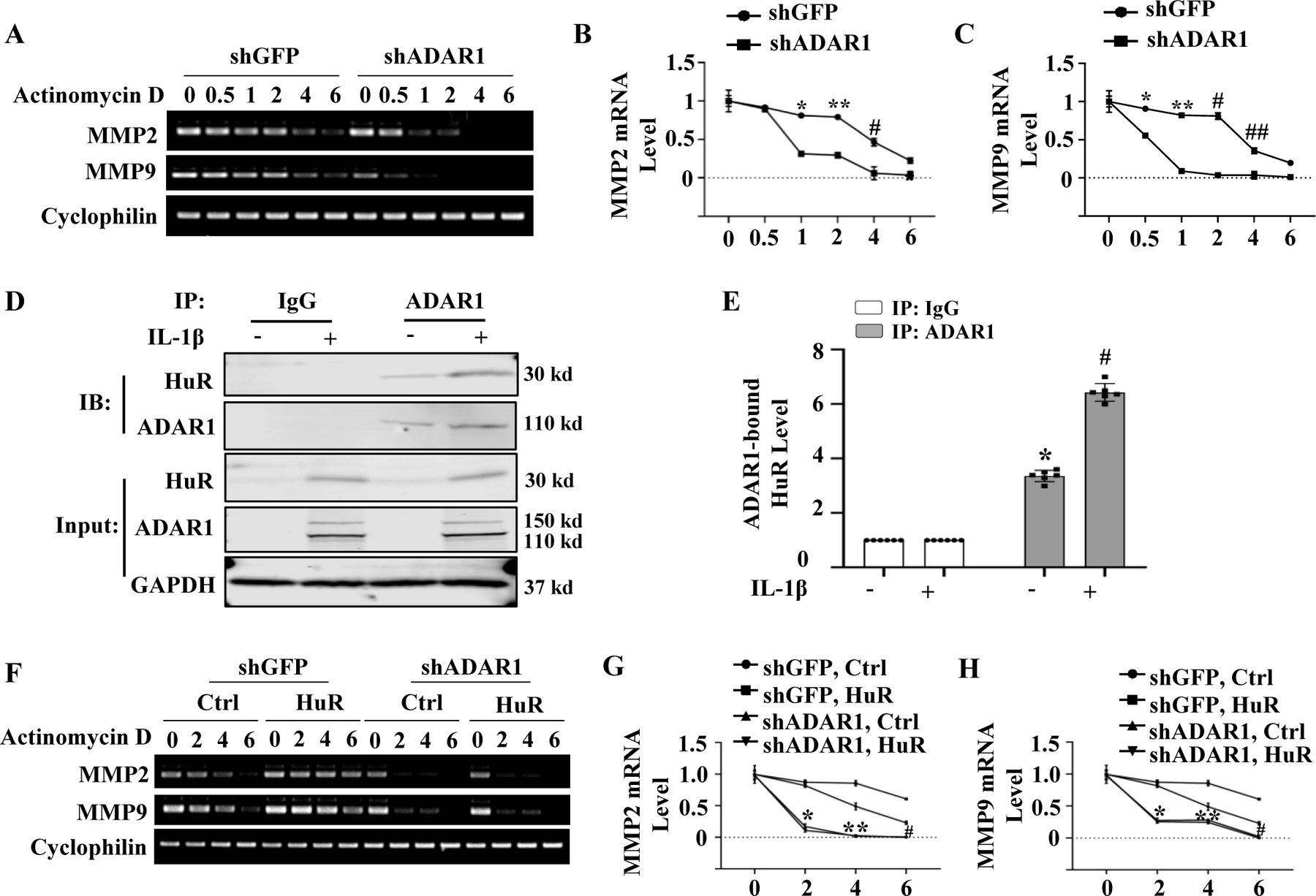
A-C, Mouse aortic SMCs were transduced with control (shGFP) or shADAR1. RNA synthesis was blocked with actinomycin D (5 μg/ml). MMP2 and MMP9 mRNA levels were detected by RT-PCR at 0, 0.5, 1, 2, 4 and 6 h after the treatment (A) and normalized to the mean level prior to the actinomycin treatment (0 h) (B-C), which was set as 1. *P = 2.17E-03 (B) and 1.40E-02 (C), **P = 1.50E-03 (B) and 2.30E-03 (C), #P = 2.21E-01 (B) and 1.40E-04 (C), ##P = 1.10E-02 vs. shGFP-transduced cells in each corresponding time point, respectively, n=6. D-E, Detection of ADAR1 interaction with HuR. Mouse aortic SMCs were treated with vehicle (−) or 10 ng/ml of IL-1β for 24 h. Co-immunoprecipitation assays were performed using IgG (control) or ADAR1 antibody. The presence of ADAR1 and HuR was detected by immunoblotting using individual antibody as indicated (D). ADAR1-bound HuR levels shown in D were quantified by normalizing to the input HuR level (E). *P = 5.64E-03 vs. IgG group; #P = 2.24E-02 vs. vehicle-treated group (−) immunoprecipitated (IP) with ADAR1 antibody, n=6. F-H, The effect of ADAR1 on HuR activity in stabilizing MMP2 and MMP9 mRNAs. Mouse aortic SMCs were transduced with shGFP or shADAR1 followed by transfection of empty vector (Ctrl) or HuR expression plasmid for 24 h. RNA synthesis was then blocked with actinomycin D (5 μg/ml). MMP2 and MMP9 mRNA levels were assessed by RT-PCR at 0, 2, 4 and 6 h following the treatment (F) and normalized to the mean level prior to the actinomycin treatment (0 h), which was set as 1 (G-H). *P =2.17E-02 (G) and 1.40E-02 (H), **P = 1.10E-02 (G) and2.20E-02 (H), and #P = 2.50E-02 (G) and 3.80E-02 (H) vs. shGFP-transduced cells with HuR at each corresponding time point, respectively, n=6. Mann-Whitney test (two-sided) was performed for B and C. Kruskal-Wallis test with Dunn’s multiple comparisons test were performed for E, G, and H.
ADAR1 mediates HuR interaction with MMP2 and MMP9 mRNAs in AAA lesions.
To test if ADAR1 is important for HuR binding to MMP2 and MMP9 transcripts in vivo, we performed RNA immunoprecipitation (RIP) assay using lysates from mouse aortas and AAA tissues (Fig 8A). Significantly increased MMP2 and MMP9 mRNA binding with HuR was observed in abdominal aortas of Ang II-infused ApoE−/− mice. However, the binding was significantly blocked in Ang II-infused ADAR1+/−;ApoE−/− mice (Fig 8, A-C), suggesting that ADAR1 is essential for HuR binding to MMP2/9 transcripts in AAA lesion. To determine if ADAR1 physically interacts with HuR in abdominal aorta during AAA formation, we performed Proximity Ligation Assay (PLA) assay. While ADAR1 barely interacts with HuR in saline-infused ApoE−/− mouse abdominal aorta, the interaction was significantly increased in Ang II-infused ApoE−/− mouse aortas, and attenuated in ADAR1 heterozygous mice (Fig 8, D). More importantly, PLA assay showed that ADAR1-HuR interaction was also significantly increased in human AAA lesion, especially in the media layer as compared to the healthy aortas (Fig 8, E), suggesting that ADAR1-HuR interaction could be also important for AAA development in human patients.
Figure 8: ADAR1 mediated HuR interaction with MMP2 and MMP9 mRNAs in AAA lesions.
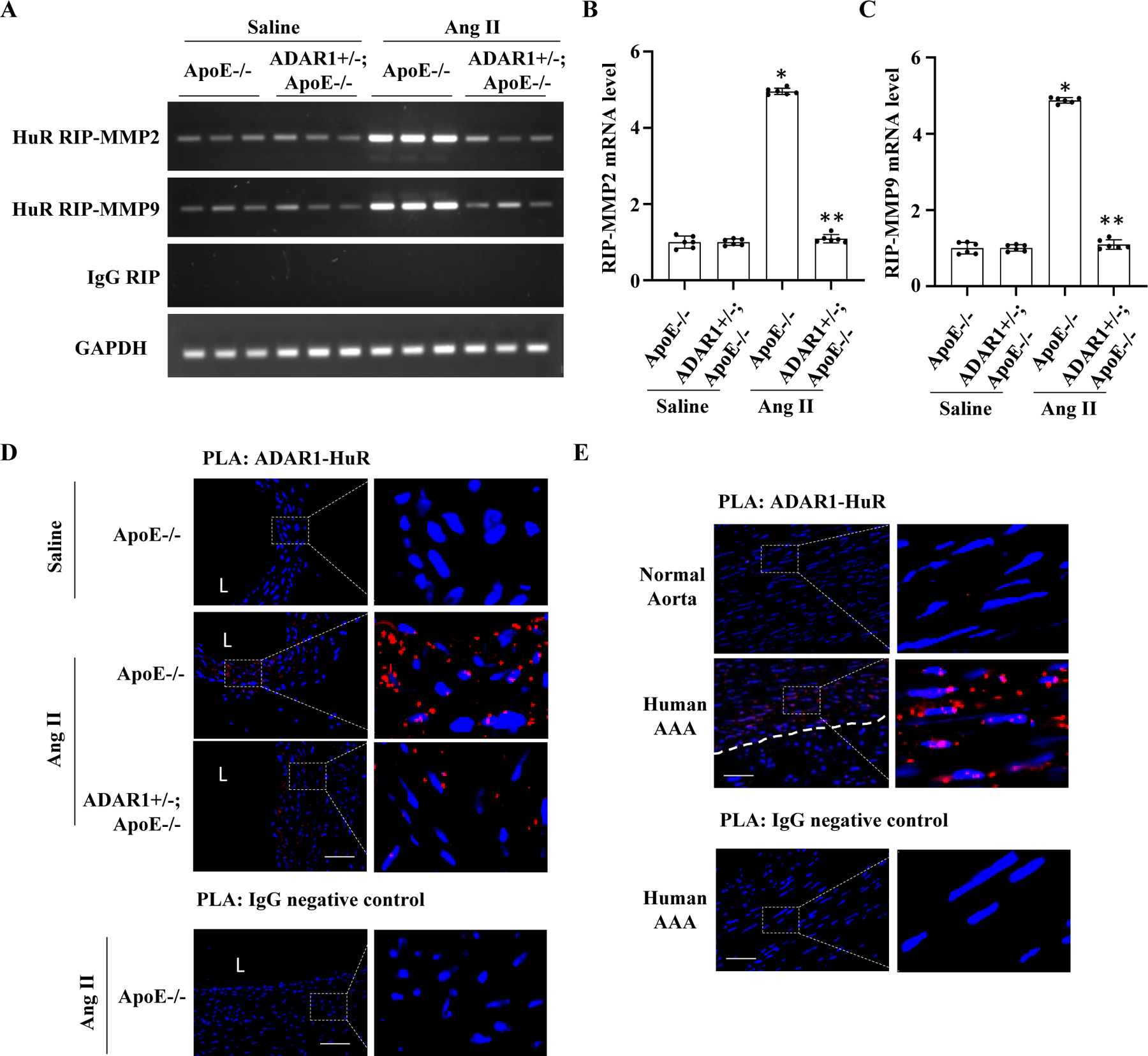
A-D, ADAR1 was essential for HuR interaction on MMP2 and MMP9 transcripts. ApoE−/− and ADAR1+/−;ApoE−/− mice were infused with saline or Ang II (1000 ng/kg/min) for 14 days. AAA tissue or control abdominal aorta were fixed with 1% formaldehyde, and RNA immunoprecipitation assay (RIP) was performed with control IgG or anti-HuR antibody. RNAs in the pulldown complexes were then extracted and reversely transcribed. MMP2 and MMP9 mRNA levels were detected by RT-PCR (A) and quantified by normalizing to the input GAPDH (B-C). All mRNA levels were shown as fold induction relative to the mean value of saline-infused ApoE−/− mice, which was set as 1. *P = 4.92E-03 (B) and 1.92E-02 (C) vs. ApoE−/− mice with saline infusion; **P = 5.98E-03 (B) and 3.98E-02 (C) vs. ApoE−/− mice with Ang II infusion, respectively n=6. Kruskal-Wallis test with Dunn’s multiple comparisons test were performed to determine statistical difference. D, ADAR1 interacted with HuR in mouse AAA lesion. Abdominal aorta or AAA sections were stained with both ADAR1 and HuR antibodies or IgG as a negative control. In situ Duolink proximity ligation assay (PLA) was performed. Nuclei were stained with DAPI. L: lumen, Scale bar: 10 µm. E, ADAR1 interacted with HuR in human AAA lesions as detected by In Situ Duolink PLA. Nuclei were stained with DAPI. Whit dash line denotes the outer elastin. Scale bar: 10 µm.
DISCUSSION
By using Ang II-infusion of ApoE−/− mouse model, we found that ADAR1 is a novel factor essential for AAA formation/dissection. ADAR1 was induced in both mouse and human AAA lesions. Heterozygous knockout of ADAR1 significantly attenuated the aneurysm formation/dissection, as shown by the significantly decreased AAA prevalence and reduction of aortic dilation and elastin fragmentation as well as thrombus formation. Moreover, ADAR1 deficiency decreased the expression and activities of elastin-degradation enzymes MMP2 and MMP9 in the abdominal aorta of Ang II-infused mice and restored SMC marker protein expression, indicating that ADAR1 promotes AAA formation by mediating SMC phenotypic modulation and facilitating the increased expression/activities of MMP2 and MMP9.
AAA formation/dissection is a complex process involving multiple molecular and cellular events including immune and vascular cell responses 28–30. Both immune and vascular cells play critical roles in AAA development and aorta dissection. ADAR1 in vascular cells appears to be required for the AAA formation because transplanting ADAR1+/− aorta to ApoE−/− mice significantly reduced the AAA formation and elastin degradation. These results are consistent with the predominant expression of ADAR1 in aortic media in both mouse and human aneurysm lesions. Indeed, heterozygous knockout of ADAR1 in SMC reduced AAA prevalence, attenuated aortic dilation, and decreased elastin degradation, demonstrating that SMC ADAR1 is essential for AAA formation/dissection. In addition to SMCs, ADAR1 has been shown to regulate immune cells, especially T cell migration and function. It is likely that T cell ADAR1 may also be important for AAA formation. However, since the majority of ADAR1-positive cells were SMC, ADAR1 is likely to promote aortic aneurysm/dissection in large part by regulating SMC phenotype. Future studies are necessary to determine if T cell or macrophage ADAR1 is important for AAA formation/dissection.
ADAR1 is an RNA editing enzyme, and our previous studies have shown that ADAR1 disrupts SMC marker gene pre-mRNA splicing through its editing function, leading to reduction of their mature mRNAs 15. However, neither knockdown of ADAR1 nor the ADAR1 editing inhibitor affected the pre-mRNA splicing of MMP2 or MMP9, suggesting that ADAR1 editing function was not involved in the increased MMP2 and MMP9 expression/activities in aneurysm lesions. Rather, our data suggest that ADAR1 promotes MMP2 and MMP9 mRNA stability. Knockdown of ADAR1 accelerated MMP2 and MMP9 mRNA degradation and abolished HuR-mediated mRNA stability, indicating that ADAR1 is essential for HuR stabilization of MM2 and MMP9 mRNAs. Indeed, ADAR1 is required for HuR binding to MMP2 and MMP9 transcripts in mouse aortas during AAA development in vivo. These finding are also supported by the physical interaction between ADAR1 and HuR, which was promoted by IL-1β. More importantly, the physical interaction between ADAR1 and HuR was significantly increased in both mouse and human AAA lesions, suggesting that ADAR1-facilitated HuR binding to MMP2 and MMP9 mRNA could be an essential mechanism for AAA formation in human patients.
Since MMP9 is mainly expressed in macrophages during AAA development 11, the effects of ADAR1 on MMP9 production/activity could be attributed to both macrophages and SMC because ADAR1 is present in both the phenotypically modulated SMC and immune cells. In vitro evidence also showed that SMC express MMP9 with IL-1β treatment. However, MMP9 function in AAA development is controversial because MMP9 deficiency inhibits CaCl2-induced AAA but enhances Ang II-induced AAA 9, 19. It appears that macrophage MMP9 plays a major role in CaCl2-induced AAA formation. Due to these discrepancies, we are unable to conclude how important the ADAR1 regulation of MMP9 is in AAA formation/dissection. Nevertheless, our data suggest that ADAR1, specifically SMC ADAR1, primarily regulates MMP2 production/activity to mediate AAA formation/dissection.
Taken together, our studies have identified a new role for ADAR1 in AAA formation/dissection. ADAR1 promotes inflammatory SMC phenotype and increases the expression/activities of MMP2 and MMP9. Mechanistically, ADAR1 interacts with HuR to promote MMP2 and MMP9 mRNA stability in inflammatory SMCs, leading to elastin degradation, aortic dilation/dissection, and aneurysm formation. Therefore, ADAR1 could be a novel potential target for hindering AAA development and/or rupture.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Abdominal aortic aneurysm (AAA) can have high morbidity and mortality associated with aorta rupture and dissection, especially in the elderly population. Pharmacological approaches to limit AAA progression and rupture have thus far proven ineffective.
Vascular smooth muscle cell (SMC) phenotypic modulation, particularly the inflammatory SMCs play critical roles in AAA development.
Adenosine deaminase acting on RNA 1 (ADAR1) regulates RNA stability, mRNA splicing, microRNA processing, and RNA storage, etc. ADAR1 is upregulated in vascular pathologies such as atherosclerosis and neointimal SMC. ADAR1 promotes neointima formation following vascular injury.
What New Information Does This Article Contribute?
ADAR1 is significantly upregulated in both mouse and human AAA lesions, especially in aortic SMCs, which contributes to the formation of the inflammatory SMC phenotype.
ADAR1 deficiency either globally or specifically in SMCs diminished angiotensin II-induced AAA formation/dissection in ApoE−/− mice.
ADAR1 promotes matrix metalloproteinase (MMP)-2 and MMP-9 production/activities by interacting with human antigen R to increase MMP-2 and MMP-9 mRNA stability, leading to elastin degradation and AAA formation/dissection.
AAA is a progressive vascular disease with more than 150,000 new cases and 10,000 deaths annually in the United States. Both open surgical and endovascular repair are associated with significant short- and long-term morbidity and mortality. Currently there is no effective pharmacological treatment for AAA. Our previous study discovered that ADAR1 promotes SMC phenotypic modulation and artery remodeling following vascular injury. However, the goal of these studies was focused on SMC marker gene expression without analyzing the function of ADAR1 in the induction of inflammatory SMCs and aneurysm formation. In this study, by using global and SMC-specific deficient mice, aorta transplantation and Angiotensin II-induced AAA model, we show that ADAR1 is a novel essential regulator for the inflammatory SMC phenotype and AAA development. We further show that ADAR1 promotes SMC inflammation and AAA formation/dissection by stabilizing MMP2 and MMP9 mRNA and consequently increasing MMP production and activities, causing extracellular matrix degradation and weakening aorta wall. These findings provide key insights into the mechanism of AAA development and identify a potentially new therapeutic target to hinder AAA growth and rupture.
SOURCES OF FUNDING
This work was supported by grants from National Institutes of Health (HL117247, HL119053, HL135854, and HL147313).
Nonstandard Abbreviations and Acronyms:
- ADAR1
Adenosine deaminases acting on RNA1
- AAA
Abdominal aortic aneurysm
- ANG II
Angiotensin II
- CHX
Cycloheximide
- MMP2
Matrix metalloproteinase 2
- MMP9
Matrix metalloproteinase 9
- HuR
Human antigen R
- ACTA2
Smooth muscle α-actin
- PLA
Proximity Ligation Assay
- RIP
RNA immunoprecipitation
Footnotes
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
DISCLOURES
None
REFERENCES
- 1.Force USPST, Owens DK, Davidson KW, Krist AH, Barry MJ, Cabana M, Caughey AB, Doubeni CA, Epling JW Jr., Kubik M, Landefeld CS, Mangione CM, Pbert L, Silverstein M, Simon MA, Tseng CW and Wong JB. Screening for Abdominal Aortic Aneurysm: US Preventive Services Task Force Recommendation Statement. JAMA. 2019;322:2211–2218. [DOI] [PubMed] [Google Scholar]
- 2.Schermerhorn ML, Buck DB, O’Malley AJ, Curran T, McCallum JC, Darling J and Landon BE. Long-Term Outcomes of Abdominal Aortic Aneurysm in the Medicare Population. N Engl J Med. 2015;373:328–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaikof EL, Dalman RL, Eskandari MK, Jackson BM, Lee WA, Mansour MA, Mastracci TM, Mell M, Murad MH, Nguyen LL, Oderich GS, Patel MS, Schermerhorn ML and Starnes BW. The Society for Vascular Surgery practice guidelines on the care of patients with an abdominal aortic aneurysm. J Vasc Surg. 2018;67:2–77 e2. [DOI] [PubMed] [Google Scholar]
- 4.Nordon IM, Hinchliffe RJ, Loftus IM and Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:92–102. [DOI] [PubMed] [Google Scholar]
- 5.Koch AE, Haines GK, Rizzo RJ, Radosevich JA, Pope RM, Robinson PG and Pearce WH. Human abdominal aortic aneurysms. Immunophenotypic analysis suggesting an immune-mediated response. Am J Pathol. 1990;137:1199–213. [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Candales A, Holmes DR, Liao S, Scott MJ, Wickline SA and Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. 1997;150:993–1007. [PMC free article] [PubMed] [Google Scholar]
- 7.Patel MI, Melrose J, Ghosh P and Appleberg M. Increased synthesis of matrix metalloproteinases by aortic smooth muscle cells is implicated in the etiopathogenesis of abdominal aortic aneurysms. J Vasc Surg. 1996;24:82–92. [DOI] [PubMed] [Google Scholar]
- 8.Abdul-Hussien H, Hanemaaijer R, Kleemann R, Verhaaren BF, van Bockel JH and Lindeman JH. The pathophysiology of abdominal aortic aneurysm growth: corresponding and discordant inflammatory and proteolytic processes in abdominal aortic and popliteal artery aneurysms. J Vasc Surg. 2010;51:1479–87. [DOI] [PubMed] [Google Scholar]
- 9.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N and Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowther M, Goodall S, Jones JL, Bell PR and Thompson MM. Increased matrix metalloproteinase 2 expression in vascular smooth muscle cells cultured from abdominal aortic aneurysms. J Vasc Surg. 2000;32:575–83. [DOI] [PubMed] [Google Scholar]
- 11.Thompson RW, Holmes DR, Mertens RA, Liao S, Botney MD, Mecham RP, Welgus HG and Parks WC. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J Clin Invest. 1995;96:318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benne R. RNA editing: how a message is changed. Current Opinion in Genetics & Development. 1996;6:221–231. [DOI] [PubMed] [Google Scholar]
- 13.Benne R, Van den Burg J, Brakenhoff JP, Sloof P, Van Boom JH and Tromp MC. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell. 1986;46:819–26. [DOI] [PubMed] [Google Scholar]
- 14.George CX, Gan Z, Liu Y and Samuel CE. Adenosine deaminases acting on RNA, RNA editing, and interferon action. J Interferon Cytokine Res. 2011;31:99–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fei J, Cui XB, Wang JN, Dong K and Chen SY. ADAR1-Mediated RNA Editing, A Novel Mechanism Controlling Phenotypic Modulation of Vascular Smooth Muscle Cells. Circ Res. 2016;119:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daugherty A, Manning MW and Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, Sun C, Zhang Y, Liu J, Ennis TL, Knispel R, Xiong W, Thompson RW, Baxter BT and Shi GP. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest. 2007;117:3359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Findeisen HM, Gizard F, Zhao Y, Cohn D, Heywood EB, Jones KL, Lovett DH, Howatt DA, Daugherty A and Bruemmer D. Telomerase deficiency in bone marrow-derived cells attenuates angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2011;31:253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howatt DA, Dajee M, Xie X, Moorleghen J, Rateri DL, Balakrishnan A, Da Cunha V, Johns DG, Gutstein DE, Daugherty A and Lu H. Relaxin and Matrix Metalloproteinase-9 in Angiotensin II-Induced Abdominal Aortic Aneurysms. Circ J. 2017;81:888–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, Lau CL and Owens GK. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. 2009;138:1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, Wang Q, Ren J, Assa CR, Morgan S, Giles J, Han Q and Liu B. Murine abdominal aortic aneurysm model by orthotopic allograft transplantation of elastase-treated abdominal aorta. J Vasc Surg. 2015;62:1607–14.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AAC, Greene ES, Straub AC, Isakson B, Randolph GJ and Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isoda K, Akita K, Kitamura K, Sato-Okabayashi Y, Kadoguchi T, Isobe S, Ohtomo F, Sano M, Shimada K, Iwakura Y and Daida H. Inhibition of interleukin-1 suppresses angiotensin II-induced aortic inflammation and aneurysm formation. Int J Cardiol. 2018;270:221–227. [DOI] [PubMed] [Google Scholar]
- 24.Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y, Owens GK, Upchurch GR Jr. and Ailawadi G. Genetic and pharmacologic disruption of interleukin-1β signaling inhibits experimental aortic aneurysm formation. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramírez-Moya J, Baker AR, Slack FJ and Santisteban P. ADAR1-mediated RNA editing is a novel oncogenic process in thyroid cancer and regulates miR-200 activity. Oncogene. 2020;39:3738–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grinan-Ferre C, Izquierdo V, Otero E, Puigoriol-Illamola D, Corpas R, Sanfeliu C, Ortuno-Sahagun D and Pallas M. Environmental Enrichment Improves Cognitive Deficits, AD Hallmarks and Epigenetic Alterations Presented in 5xFAD Mouse Model. Front Cell Neurosci. 2018;12:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong J, Zhang Y, Liu S, Li H, Liu S, Wang J, Qin X, Jiang X, Yang J, Zhang C and Zhang W. Melatonin attenuates angiotensin II-induced abdominal aortic aneurysm through the down-regulation of matrix metalloproteinases. Oncotarget. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jana S, Hu M, Shen M and Kassiri Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Experimental & Molecular Medicine. 2019;51:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuivaniemi H, Ryer EJ, Elmore JR and Tromp G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther. 2015;13:975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quintana RA and Taylor WR. Cellular Mechanisms of Aortic Aneurysm Formation. Circ Res. 2019;124:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS and Smith JD. Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies-Statement From ATVB Council. Arterioscler Thromb Vasc Biol. 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu H, Howatt DA, Balakrishnan A, Moorleghen JJ, Rateri DL, Cassis LA and Daugherty A. Subcutaneous Angiotensin II Infusion using Osmotic Pumps Induces Aortic Aneurysms in Mice. J Vis Exp. 2015:53191. [DOI] [PMC free article] [PubMed]
- 33.Wang Q, Liu Z, Ren J, Morgan S, Assa C and Liu B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ Res. 2015;116:600–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawashima Y, Kodera Y, Singh A, Matsumoto M and Matsumoto H. Efficient extraction of proteins from formalin-fixed paraffin-embedded tissues requires higher concentration of tris(hydroxymethyl)aminomethane. Clinical Proteomics. 2014;11:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolff C, Schott C, Porschewski P, Reischauer B and Becker K-F. Successful Protein Extraction from Over-Fixed and Long-Term Stored Formalin-Fixed Tissues. PLOS ONE. 2011;6:e16353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo X, Shi N, Cui XB, Wang JN, Fukui Y and Chen SY. Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling. Circ Res. 2015;116:e71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang JN, Shi N, Xie WB, Guo X and Chen SY. Response gene to complement 32 promotes vascular lesion formation through stimulation of smooth muscle cell proliferation and migration. Arterioscler Thromb Vasc Biol. 2011;31:e19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toth M and Fridman R. Assessment of Gelatinases (MMP-2 and MMP-9 by Gelatin Zymography. Methods Mol Med. 2001;57:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The detailed experimental materials and methods are available in the Data Supplement. The authors declare that the majority of supporting data are presented within this article and in the Data Supplement. The source data for the figures and the data that are not shown are available from the corresponding author upon reasonable request.